

Abstract
Experimental autoimmune encephalomyelitis (EAE) is a widely used animal model for multiple sclerosis (MS). EAE is typically initiated by CD4+ T helper cell type 1 (Th1) autoreactivity directed against a single priming immunodominant myelin peptide determinant. Recent studies have shown that clinical progression of EAE involves the accumulation of neo-autoreactivity, commonly referred to as epitope spreading, directed against peptide determinants not involved in the priming process. This study directly addresses the relative roles of primary autoreactivity and secondary epitope spreading in the progression of both EAE and MS. To this end we serially evaluated the development of several epitope-spreading cascades in SWXJ mice primed with distinctly different encephalitogenic determinants of myelin proteolipid protein. In a series of analogous experiments, we examined the development of epitope spreading in patients with isolated monosymptomatic demyelinating syndrome as their disease progressed to clinically definite MS. Our results indicate that in both EAE and MS, primary proliferative autoreactivity associated with onset of clinical disease invariably regresses with time and is often undetectable during periods of disease progression. In contrast, the emergence of sustained secondary autoreactivity to spreading determinants is consistently associated with disease progression in both EAE and MS. Our results indicate that chronic progression of EAE and MS involves a shifting of autoreactivity from primary initiating self-determinants to defined cascades of secondary determinants that sustain the self-recognition process during disease progression.
Keywords: multiple sclerosis, autoimmunity, epitope spreading, demyelination, myelin proteolipid protein
Multiple sclerosis (MS)1 and its related animal model, experimental autoimmune encephalomyelitis (EAE), are often characterized by a relapsing–remitting disease course with chronic-progressive disability (1). Typically, EAE is initiated by CD4+ Th1 autoreactivity directed against a single immunodominant myelin protein determinant (2, 3). However, the basis for disease progression is less clear. In the case of EAE, progression of disease may be attributed either to sustained autoreactivity directed against an immunodominant priming determinant or to acquired autoreactivity directed against determinants not involved in the initiation of disease. This acquisition of neo-autoreactivity, commonly referred to as epitope spreading, presumably results from endogenous priming with new self-antigens generated from damaged tissue over the course of disease (4–7). Similarly, in the case of MS, disease progression may be due to either a sustained primary autoreactivity or to a secondary acquisition of neo-autoreactivity as a result of epitope spreading.
Our study was designed to define the roles of primary autoreactivity and epitope spreading on the progression of autoimmune demyelinating disease. To this end, EAE was induced in SWXJ mice by immunization with a panel of distinct priming determinants of myelin proteolipid protein (PLP), and autoreactivity was assessed over time to known encephalitogenic determinants of PLP, myelin basic protein (MBP) and myelin oligodendrocyte glycoprotein (MOG). In complementary experiments, autoreactivity to an overlapping PLP peptide series was evaluated over a 3-yr period in patients with isolated monosymptomatic demyelinating syndrome (IMDS), a group of related neurologic disorders with variable rates of progression to clinically definite MS (CDMS; 8–10).
Our results indicate that during progression of EAE, proliferative responses to priming determinants invariably decline with time and frequently disappear in both the periphery and in the central nervous system (CNS). The regression of primary autoreactivity is succeeded by a sequential accumulation of neo-autoreactivity to defined spreading determinants. Similarly, autoreactivity associated with onset of neurologic symptoms in IMDS patients consistently wanes with time and becomes undetectable as disease progresses to CDMS. The spontaneous waning and frequent disappearance of self-recognition involved in the initiation of autoimmunity indicates that the natural history of autoimmune disease involves a shifting of responses from primary initiating determinants to defined cascades of secondary determinants that sustain the self-recognition process during disease progression.
Materials and Methods
Mice.
Female SWXJ (H-2q,s) mice were bred at the Biological Resources Facility of the Lerner Research Institute by mating SWR/J (H-2q) females with SJL/J (H-2s) males purchased from The Jackson Laboratory. Mice were immunized at 7–12 wk of age. All protocols for animal research met with prior approval of the Institutional Animal Care and Use Committee (IACUC) of the Cleveland Clinic Foundation in compliance with the Public Health Service policy on humane care and use of laboratory animals.
Bulk Peptide Synthesis.
The PLP peptides 104–117 KTTICGKGLSATVT, 139–151 HSLGKWLGHPDKF (serine for cysteine at residue 140), and 178–191 NTWTTCQSIAFPSK, as well as MBP 87–99 VHFFKNIVTPRTP and MOG 92–106 DEGGYTCFFRDHSYQ, were either purchased commercially (Bio-Synthesis) or synthesized at the Protein Core Facility of the Lerner Research Institute with standard solid phase methodology using amino acids with 9-fluorenylmethoxycarbonyl (FMOC) side chain protection. Peptides were purified >90% by reverse phase HPLC using a 22 × 250 mm C-18 column (Vydac Separations Group). The identity of each purified peptide was confirmed by mass spectrometry.
Epitope-Mapping PLP Peptide Series.
A PLP pin peptide series representing a walk-through of the entire 276-amino acid primary sequence of mouse PLP (11, 12) was purchased from Chiron Mimotopes. A total of 265 overlapping 12-mers were synthesized on high-density polyethylene rod tips assembled into holders designed in 96-well microtiter plate format (13). Each successive peptide differed from the previous 12-mer by sequential NH2-terminal deletion and COOH-terminal addition of PLP amino acids. Upon arrival, 1 mg of each PLP pin peptide was dissolved in 500 μl of a solution of 40% acetonitrile (Aldrich Chemical) in 10 mM Hepes buffer (GIBCO BRL). Working aqueous concentrations of pin peptides were prepared at 150 μg/ml in PBS, pH 7.2, and 20 μl of each working solution was distributed sequentially into individual wells of 96-well flat-bottomed microtiter Falcon plates (Becton Dickinson). The plates were stored at −20°C until ready for use.
Induction of EAE.
EAE was induced as previously described (14). SWXJ mice were immunized by subcutaneous injection in the abdominal flanks on day 0 with 100 nmol of PLP peptides p139–151 (154 μg), p178–191 (158 μg), or p104–117 (138 μg), and 400 μg Mycobacteria tuberculosis H37RA (Difco Labs.) in 200 μl of an emulsion of equal volumes of water and IFA (Difco Labs.). On days 0 and 3 each mouse also received intravenously 6 × 109 Bordetella pertussis bacilli (Michigan Department of Public Health, Lansing, MI). In the study presented here, all mice developed clinical EAE within 24 d of immunization.
Clinical Evaluation of EAE.
All mice were weighed and examined daily for neurologic signs as previously described (14) according to the following criteria: 0, no disease; l, decreased tail tone or slightly clumsy gait; 2, tail atony and/or moderately clumsy gait and/or poor righting ability; 3, limb weakness; 4, limb paralysis; 5, moribund state. The presence of relapse was determined when mice showed an increase in observed neurologic disability of at least one clinical score unit. The increased neurologic deficit was typically accompanied by an abrupt and substantial (>7%) weight loss.
Histologic and Immunocytochemical Evaluation of EAE.
Brains and spinal cords were fixed in 10% phosphate-buffered formalin, and paraffin-embedded tissue sections were cut (10 mm each) for immunostaining as previously described (15, 16). Sections were pretreated with 0.04% OsO4 and 1% H2O2 in 10% Triton (Electron Microscopy Sciences) and blocked with 5% normal goat serum (Vector Labs.) and 5% nonfat dehydrated milk for 60 min. Sections were treated sequentially with PLP monoclonal IgG2a antibody (Harlan) at a 1:200 dilution for 14 h at 4°C, biotinylated goat anti–mouse IgG2a (Southern Biotechnology Associates) at a 1:500 dilution for 30 min at 22°C, and avidin-peroxidase complex (Vector Labs.) for 1 h at 1:1,000 dilution. Sections were then treated with diaminobenzidine and 0.01% H2O2 for 8 min, 0.04% OsO4 for 30 s, and washed in PBS. Images were digitized using the AlphaImager 2000 System (Alpha Innotech) at 640 × 480 pixel resolution. Images were captured at 10× magnification with the black level scale set at 0, white level scale at 255, and gamma level scale at 1.0. All images were normalized by adjusting background gray matter stain to the same mean intensity value using Adobe Photoshop (Adobe Systems). The presence of demyelination in CNS meninges and parenchyma was determined visually as well as by digitized image analysis using NIH image software (version 1.57; National Institutes of Health, Bethesda, MD).
Evaluation of Epitope Spreading during EAE.
At wk 2, 4, 8, 12, and in some cases 16 after immunization of SWXJ mice with a PLP determinant, splenocytes were tested for proliferative responses to PLP determinants p104–117 (14, 17), p139–151 (18), and p178–191 (19), as well as MBP 87–99, an immunodominant encephalitogenic determinant for both SJL/J (20, 21) and SWR/J (22) mice, and MOG 92–106, an immunodominant encephalitogen for SJL/J mice (23). Mononuclear cells were purified by centrifugation on Lympholyte-M (Accurate Chemical Co.) for 20 min at 2,500 rpm. Cells collected from the interface were washed three times in HBSS and resuspended in DME (GIBCO BRL) supplemented with 10% FBS (Hyclone Labs.), 2 mM fresh l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 30 mM Hepes buffer (GIBCO BRL), and 5 × 10−5 M 2-ME. Triplicate test cultures containing peptide at 20 μg/ml were evaluated at 3 × 105 splenocytes/well in a total volume of 200 μl in 96-well flat-bottomed microtiter Falcon plates (Becton Dickinson). Triplicate positive control wells contained mouse CD3 mAb at 5 μg/ml (PharMingen), or 0.005 tuberculin units/ml of tuberculin purified protein derivative (PPD; Connaught), or 20 μg/ml Mycobacteria tuberculosis H37RA (Difco). Dose responses to whole PLP (0.1–100 μg/ml) were also assessed in each experiment. The PLP was prepared from a washed total lipid extract of bovine white matter (24) and was purified and converted to aqueous as previously described (14). Negative control wells contained no peptide. Cultures were incubated at 37°C in humidified air containing 5% CO2. At 4 d, cultures were pulsed with methyl-[3H]thymidine (l.0 μCi/well, specific activity 6.7 Ci/mmol; NEN), and the cells were harvested after 16 h by aspiration onto glass fiber filters. Levels of incorporated radioactivity were determined by scintillation spectrometry. Results are expressed as stimulation index (SI) defined as mean cpm of triplicate experimental cultures with Ag divided by mean cpm of cultures without Ag.
Evaluation of Autoreactivity by CNS-infiltrating Mononuclear Cells.
Mononuclear cells were recovered from the CNS at various clinical stages of EAE according to the method of Ford et al. (25). In brief, SWXJ EAE mice were killed by CO2 inhalation and perfused with 20 ml of HBSS to remove hematogenous leukocytes. Brain tissue was teased and digested with 1 mg/ml of collagenase D (Boehringer Mannheim) and 50 Kunitz U/ml of DNase (Sigma Chemical Co.) at 37°C for 60 min. After washing, cells were resuspended in Percoll (Amersham Pharmacia Biotech) adjusted with HBSS to a specific gravity of 1.030 and layered on Percoll/ HBSS at a specific gravity of 1.095. After centrifugation for 30 min at 1,250 g, cells were removed from the interface, washed, and counted for total yield. Percentages of T cells (FITC-labeled anti-CD3 or anti-CD4; PharMingen) and microglia (FITC-labeled anti-CD11b; PharMingen) were determined by flow cytometry analysis. Proliferation assays in response to peptides were performed as described above using 2 × 105 CNS harvested cells/microtiter well.
Selection and Clinical Evaluation of IMDS Patients.
We have previously reported that monocentric monophasic IMDS patients with no evidence by history or exam of prior subclinical CNS disease typically show fully sustained proliferative responses over a 1-yr period to defined PLP regions (26). The development and progression of myelin self-recognition was further evaluated over a total period of 3 yr in three of the original monocentric monophasic IMDS patients: VS, a 28-yr-old female with inflammatory internal capsule syndrome; DL, a 21-yr-old female with partial transverse myelitis; and JB, a 34-yr-old male with acute brainstem syndrome. During the course of the study reported here, both VS and DL showed progression to CDMS at 154 and 60 wk, respectively, after initial onset of their neurologic symptoms. Thus far, JB has not shown progression to CDMS. The three monocentric monophasic IMDS subjects were seen by the study neurologist (R.P. Kawczak) within 2 wk of the onset of their neurologic symptoms and were repeatedly examined over the course of the current study for the evaluation of new symptoms. Disease activity in IMDS patients was defined as the occurrence of interval clinical or MRI activity as described previously (26). CDMS was defined as a relapse involving an anatomically different area of the nervous system compared with onset symptoms (27). Magnetic resonance imaging (MRI) was performed as described previously (26) in a 1.5 Tesla superconducting whole body imaging system (Siemens Medical System). Image analysis was performed by two neuroradiologists blinded to the clinical disposition of the patients. All patients were able to understand informed consent and comply with the study protocol approved by the Institutional Review Board of the Cleveland Clinic Foundation.
Evaluation of Epitope Spreading during Progression of IMDS to CDMS.
PBMCs from each IMDS subject were serially evaluated for proliferative responses to the 265 overlapping PLP epitope-mapping peptides as previously described (26). PBMCs were separated by centrifugation on Ficoll-Paque (Amersham Pharmacia Biotech), washed three times in HBSS (GIBCO BRL), and resuspended in serum-free HL-1 media (Hycor) supplemented with 2 mM fresh l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 30 mM Hepes buffer (GIBCO BRL). Each well contained 105 PBMCs with 15 μg/ml pin peptide in a total volume of 200 μl. Triplicate positive control wells contained human CD3 mAb at 10 μg/ml (Ortho Biotech), tetanus toxoid at 1:1,000 dilution (Lederle Laboratories), 0.005 tuberculin U/ml of tuberculin purified protein derivative (PPD; Connaught), or 20 μg/ml Mycobacteria tuberculosis H37RA (Difco Labs.) although negative control wells contained either no peptide or one of 30 irrelevant pin peptides of myohemerythrin, a protein having minimal sequence homology with myelin proteins (28). Dose responses to whole bovine PLP (0.1–100 μg/ml) were also assessed in each experiment. Cultures were incubated at 37°C in humidified air containing 5% CO2. At 72 h, cultures were pulsed with methyl-[3H]thymidine (NEN) and harvested and processed as described above. Test wells containing 15 μg/ml of a single PLP peptide were considered positive with an SI > 2.0 and with a Δcpm > 1,000 and at least three standard deviations above the mean of nonstimulated control wells. Identification of PLP antigenic determinants required that positive proliferative responses be generated to at least three adjacent overlapping 12-mers.
Results
Chronic EAE Induced in SWXJ Mice with Three Different PLP Peptides.
SWXJ mice develop acute EAE ∼3 wk after conventional immunization with either of the three encephalitogenic PLP determinants p104–117, p178–191, and p139–151 (Fig. 1). Affected mice typically undergo an incomplete recovery from the initial attack with residual neuroparalytic signs. Recovery is soon followed by a series of multiple relapse/remission cycles with each neuroparalytic episode leaving the mice progressively more impaired. By 8–12 wk after immunization, the incremental accumulation of neurologic deficit makes it increasingly more difficult to observe relapses. This chronic-progressive stage of EAE is characterized by sustained limb paresis or paralysis and marked CNS demyelination particularly pronounced in the spinal cord (Fig. 2).
Figure 1.
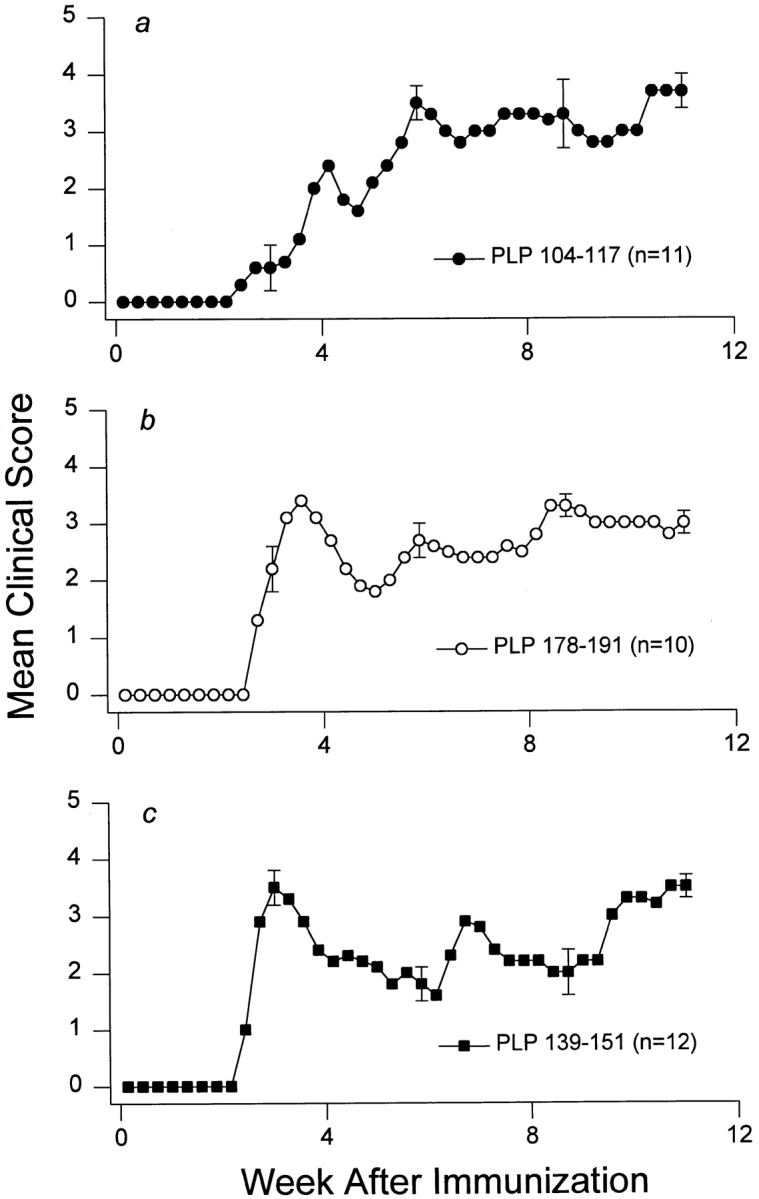
Chronic EAE induced in SWXJ mice with three different PLP peptides. Immunization of SWXJ mice with either of the three PLP determinants (p104–117, p178–191, and p139–151) induced an acute form of EAE that eventually progressed to chronicity. Error bars show ± SEM.
Figure 2.
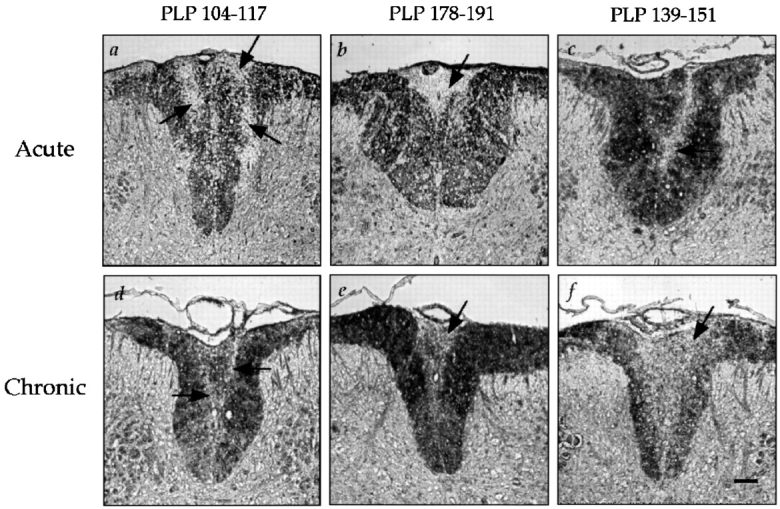
CNS demyelination induced by immunization of SWXJ mice with PLP determinants. Spinal cord sections from EAE mice were immunostained for detection of PLP. Arrows indicate demyelinated tissue appearing as unstained areas in digitized images of dorsal columns. Marked demyelination of the spinal cord was evident during the acute phase of EAE onset induced within three wk after immunization with (a) PLP 104–117, (b) PLP 178–191, and (c) PLP 139–151. Demyelination was also evident in mouse spinal cords during the chronic phase of EAE 12 wk after immunization with (d) PLP 104–117, (e) PLP 178–191, and (f ) PLP 139–151. Sections were taken from mice with either severe paresis or paralysis. Original magnification: 11.7×. Bar, 50 μm.
Regression of Primary Autoreactivity during Progression of EAE.
To evaluate the changes occurring in self-recognition after immunization with different encephalitogenic peptides, three groups of SWXJ mice were immunized with each of the PLP determinants (p104–117, p178–191, and p139–151), and splenocytes were tested at various times thereafter for proliferative responses to the three PLP determinants as well as to the defined encephalitogens MBP 87–99 (20–22) and MOG 92–106 (23). Three to five independent experiments were performed for each time point. We found that the kinetics of the response to the PLP priming determinants p104–117 (Fig. 3 a) and p178–191 (Fig. 3 b) were similar, with peak responses occurring at 2–4 wk, a decline in responses by 8 wk, and a virtual absence of detectable proliferative responses to the priming immunogens by 12 wk after immunization. Although the profile of reactivity to the p139–151 priming determinant also showed a rise, peak, and decline, the kinetics of the p139–151 response were notably distinct from those of the other PLP determinants, in that proliferation took longer to peak (8 wk versus 2–4 wk) and decline (12–16 wk versus 4–8 wk) and reached the level of control responses in only two out of five mice (Fig. 3 c). Nevertheless, by 12–16 wk after immunization, proliferative responses to three distinct PLP priming determinants invariably declined and were often undetectable despite the concurrent development of chronic-progressive disease (Fig. 1).
Figure 3.

Regression of primary autoreactivity during progression of EAE. By 12 wk after immunization, splenocyte proliferative responses to all priming determinants invariably declined from peak levels reached between 2 and 8 wk and were not evident in most animals immunized with either (a) PLP 104–117 or (b) PLP 178–191. (c) As shown in the graph, PLP 139–151–immunized mice showed a delayed pattern of declining responsiveness to the priming determinant. By 16 wk after immunization responses to PLP 139–151 were detectable but at levels substantially lower than those observed at 2–12 wk. Each experimental time point represents the mean response of 5–6 mice. Error bars show ± SEM.
Regression of Primary Autoreactivity Occurs in both Splenocytes and CNS-infiltrating Mononuclear Cells.
Since it was possible that the observed regression of splenocyte responsiveness to priming determinants may simply reflect a selective sequestration of autoreactive T cells from the periphery into the CNS, we simultaneously compared splenic and CNS autoreactivities during the course of disease. At distinct clinical stages of EAE, mononuclear cells were isolated from the brain and tested along with splenocytes for proliferative responses to priming determinants. In SWXJ mice immunized with both PLP 104–117 (Fig. 4 a) and PLP 178–191 (Fig. 4 b), concurrent declines in responses to priming determinants were evident in both splenocytes and CNS-infiltrating cells during the course of disease. By the second relapse, complete regression of primary autoreactivity was clearly evident in both splenocytes and CNS-infiltrating cells despite the presence of similar numbers of infiltrating cells compared with onset and first relapse (Fig. 4 c). These data indicate that regression of primary autoreactivity occurs simultaneously in both the periphery and CNS.
Figure 4.
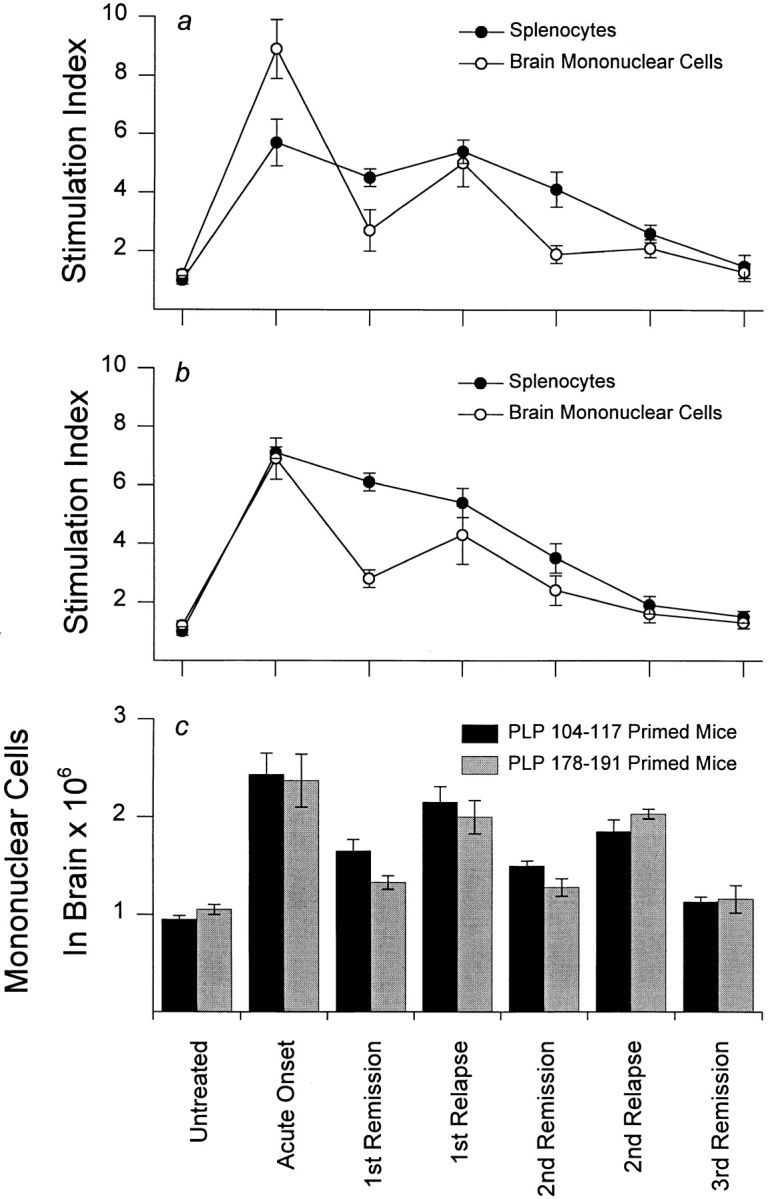
Regression of primary autoreactivity occurs in both splenocytes and CNS-infiltrating mononuclear cells. In SWXJ mice immunized with both (a) PLP 104–117 and (b) PLP 178–191, concurrent declines in responsiveness to priming determinants were evident in both splenocytes and CNS-infiltrating cells during the course of disease. (c) As depicted in the graph, the numbers of mononuclear cells recovered from the CNS during onset and relapse were consistently greater than numbers recovered during stages of remission. This observation was evident even during late relapse and remission stages when complete regression of primary autoreactivity was clearly evident in CNS-infiltrating cells. Typically, CD3+ T cells comprised 70%, 15%, and 40% of total recovered mononuclear cells in acute EAE, remissions, and relapses, respectively. The remaining cells were predominantly CD11b+ microglia. Error bars show ± SEM.
Emergence of Epitope Spreading Cascades during Progression of EAE.
During the regression of primary autoreactivity in EAE, responses invariably emerged to self-determinants not involved in the initial priming process. By 4 wk after immunization with PLP 104–117 (Fig. 5 a), proliferative responses to PLP 139–151 (SI = 2.5) and MBP 87–99 (SI = 2.3) became apparent, peaked by 8 wk (SI = 3.9 and 3.5, respectively), and remained markedly elevated at 12 wk when acquired responses to PLP 178–191 (SI = 3.6) first became evident and responses to the p104–117 priming determinant declined toward baseline (SI = 1.6). In contrast to the plasticity observed in self-recognition, responses to the priming adjuvant Mycobacteria tuberculosis H37RA showed little fluctuation throughout the testing period (SI = 6.13 ± 0.24 SEM). Thus, a cascading emergence of neo-autoreactivity accompanied the regression of primary autoreactivity during the development of chronic-progressive disease induced by immunization with PLP 104–117.
Figure 5.

Emergence of epitope spreading cascades during progression of EAE. Progression of EAE coincided with regression of primary autoreactivity and with the concurrent emergence of epitope spreading cascades. (a) SWXJ mice primed with PLP 104–117 showed neo-autoreactivity to MBP 87–99 and PLP 139–151 followed by responsiveness to PLP 178–191. (b) Mice primed with PLP 178–191 showed epitope spreading to PLP 139–151 and MBP 87–99. (c) Mice primed with PLP 139–151 showed sequential recognition of MBP 87–99 and PLP 178– 191. In all cases, variations in positive control responses to H37RA showed no correlation with the changes observed in primary and spreading autoreactivities. Error bars show ± SEM.
Epitope spreading also accompanied disease progression in SWXJ mice immunized with PLP 178–191 as indicated by the appearance of proliferative responses to PLP 139–151 (SI = 3.2) and MBP 87–99 (SI = 2.2) 4 wk after priming with the PLP 178–191 immunogen (Fig. 5 b). Moreover, the observed neo-autoreactivity remained elevated at 12 wk (SI = 3.3 and 3.2, respectively), when proliferative responses to the priming PLP 178–191 determinant were virtually undetectable (SI = 1.4). Responses to H37RA were similar at all times tested (SI = 4.4 ± 0.21 SEM).
As described in our previous report (6), a cascading epitope spreading pattern occurred after immunization of SWXJ mice with PLP 139–151. A readily detectable response to MBP 87–99 occurred at 8 wk (SI = 2.6) and remained elevated at 12 wk (SI = 2.9) when additional neo-autoreactivity to PLP 178–191 (SI = 3.6) became clearly evident (Fig. 5 c). Responses to H37RA showed little fluctuation throughout the testing period (SI = 4.25 ± 0.13 SEM).
Regression of Primary Autoreactivity during Progression of IMDS to CDMS.
To determine whether the concurrent processes of regressing primary autoreactivity and emerging epitope spreading occurs during progression of MS, a related series of experiments was performed using monocentric monophasic IMDS patients who showed no evidence of prior subclinical disease activity as determined by T2-weighted MRI. Such patients often progress to CDMS (8–10) and have been shown in our previous report to develop sustained autoreactivity to defined PLP peptides (26). In serial evaluation of PBMC-proliferative responses to an overlapping PLP peptide series, three IMDS patients (VS, DL, and JB) showed sustained autoreactivity to PLP determinants (p210–244, p116–150, and p117–152, respectively) coinciding with or appearing soon after the diagnosis of IMDS (Fig. 6, a, b, and c, respectively). Serial testing over a 3-yr period showed that in each case the established autoimmune responses associated with the early onset stage of the disease process invariably declined with time and eventually became undetectable.
Figure 6.

Regression of primary autoreactivity during progression of IMDS to CDMS. Three monocentric monophasic IMDS patients (a) VS, (b) DL, and (c) JB showed sustained autoreactivity to PLP peptides coinciding with or soon after initial onset of neurologic symptoms. In each case, the established primary autoreactivity uniformly declined with time and eventually disappeared. Error bars show ± SEM.
Emergence of Epitope Spreading during Progression of IMDS to CDMS.
The two IMDS patients, VS and DL, showed progression to CDMS, and their disease progression was accompanied by the emergence of PLP neo-autoreactivity (Fig. 7). At 154 wk after initial onset of neurologic symptoms, VS was diagnosed with CDMS after developing a cervical myelitis with a new enhancing MRI lesion of the cervical spinal cord that corresponded to new symptoms. The progression to CDMS was associated with the acquisition of newly acquired secondary responses directed against PLP 50–69 (SI = 4.7) at 154 wk and both PLP 167–185 (SI = 2.4) and PLP 258–271 (SI = 3.4) at 170 wk (Fig. 7 a). At 154 wk and after, there were no detectable proliferative responses to the primary PLP 210–244 region associated with the early disease stage.
Figure 7.

Emergence of epitope spreading during progression of IMDS to CDMS. Two IMDS patients (VS and DL) eventually showed progression to CDMS accompanied by the emergence of neo-autoreactivity. (a) VS showed secondary responses to PLP 50–69 at 154 and 170 wk and to both PLP 167–185 and PLP 258–272 at 170 wk. Epitope spreading at onset of CDMS at 154 wk was unaccompanied by detectable proliferative responses to the PLP 210–244 region implicated in earlier stages of the disease process. (b) In the case of DL, disease progression at 60 wk involved a clear sustained response to the newly acquired PLP 167–182 at 82 and 162 wk in the absence of responsiveness to the PLP 116–150 region associated with initial onset of symptoms. (c) Thus far, JB has not shown any progression to CDMS. However, at 148 wk JB showed the first signs of neo-autoreactivity with responses to PLP 261–274 unaccompanied by detectable responses to the primary PLP 117–152 associated with onset of IMDS. Error bars show ± SEM.
Patient DL showed progression from IMDS to CDMS 60 wk after initial onset of symptoms after developing symptoms consistent with a brainstem syndrome. Disease progression was associated with the development of a secondary spreading response to PLP 49–62 (SI = 2.4) at 54 wk equal in magnitude to the autoreactivity directed against PLP 116– 150 (SI = 2.4), a determinant that once generated a vigorous sustained autoreactivity during the onset stage of IMDS (Fig. 7 b). At 82 wk, the response to the spreading determinant PLP 167–182 (SI = 2.7) was greater than that elicited by PLP 116–150 (SI = 2.3) and was even greater by 162 wk (SI = 3.2) when the response to the initial autoreactive PLP 116–150 was undetectable (SI = 1.1).
Thus far, patient JB has not progressed to CDMS. However, at 148 wk after initial onset of symptoms, JB developed the first indications of neo-autoreactivity with responses directed against PLP 261–274 (SI = 2.4) unaccompanied by detectable responses to PLP 117–152 associated with the early onset stage of IMDS (SI = 1.1; Fig. 7 c). Throughout the experimental period, fluctuations occurring in positive control responses of VS, DL, and JB showed no correlation with changes observed in primary and spreading autoreactivities (data not shown).
Discussion
Our results indicate that progression of both EAE and MS is consistently accompanied by the spontaneous decrease and frequent disappearance of the established primary autoreactivity and the concurrent emergence of the epitope-spreading cascade. Our findings are consistent with the view that progression of autoimmune disease involves the sequential appearance and regression of responses to a cascading series of self-determinants so that at any given time the response to one of several distinct determinants may appear to be predominant. Indeed, based on the proliferative responses at 12–16 wk after immunization, it would be most difficult if not impossible to identify the determinant used as the priming immunogen for EAE induction in every case examined (Fig. 5). Thus, the concept of immunodominance as it pertains to the natural development of self-recognition during autoimmune disease may best be understood when considered in the temporal context of an “epitope du jour” perspective.
Our study directly challenges the widely held view that EAE and most notably MS are initiated and maintained by autoreactivity directed against a single predominant myelin protein or determinant. Several studies have claimed that autoreactivity in MS is directed in a predominant manner against specific myelin proteins and their defined immunodominant determinants. Predominant MHC class II–restricted responses have been described for peptides in the MBP 80–105 region (29–32) as well as for PLP peptides p40–60 (33), p89– 106 (34), p30–49, and p180–199 (35). Recent studies also claim a predominance of autoreactivity to MOG over other myelin proteins in MS patients (36). However, conclusions from these studies are based invariably on data taken at one time point from patients with long-standing disease. Thus, it is not surprising that a one-dimensional view of self-recognition would prevail from such a static perspective. Serial evaluation of self-recognition over a period of time sufficient to observe progression of clinical disease clearly reveals shifts from one predominant autoreactive pattern to another.
It is likely that the level of self-recognition remodeling observed in the study reported here during progression of EAE and MS effectively under-represents the dynamic level of the changes that actually occur. This view is proposed in light of the fact that PLP served as the sole autoantigen evaluated in this study and that proliferation was the only assay used for measuring autoreactivity. The actual level of self-recognition plasticity may best be estimated by serial assessment of the autoreactive changes to overlapping peptides of several myelin proteins implicated in MS autoreactivity, such as PLP, MBP, and MOG, as well as by the incorporation of more sensitive assays such as the ELISPOT capable of providing a 10–200-fold increased sensitivity over conventional methods for detecting immunoreactivity (37, 38) and the use of soluble peptide–MHC tetramers that enable frequency analysis of antigen-specific T cells by flow cytometry (39).
It seems reasonable to speculate that shifts from one epitope response pattern to another may also be accompanied by a broadening in usage of MHC class II molecules restricting the autoreactivity. Moreover, it may be possible to sustain responses to a given determinant by expanding the autoreactive repertoire with clones that utilize restriction elements not involved in the primary response. A recent report from our laboratory showed that during progression from IMDS to CDMS, a patient homozygous for DRB1*04 responded in a DPB1*0301-restricted manner to PLP 43–64 (40), a peptide region shown in other studies to be DRB1*04 restricted (33). Thus, autoimmune spreading may involve diversification of restriction elements in addition to broadening of epitopes recognized. From this perspective, both epitope spreading and “MHC spreading” may participate together to sustain autoreactivity and thereby facilitate chronic progression of autoimmune disease.
Although shifts from primary to secondary autoreactive profiles were observed in both MS and EAE, each step in the process was markedly prolonged in MS. Typically, primary autoreactivity was detectable for over 1 yr in MS compared with 12 wk for PLP 104–117 and PLP 178–191 in murine EAE. Highly developed secondary patterns of autoreactivity occurred within weeks or months of onset of clinical symptoms in murine EAE but often required years to develop during progression of IMDS to MS. However, it should be noted that the contrasting self-recognition kinetic profiles of EAE versus MS were accompanied by corresponding differences in the development and progression of clinical symptoms, i.e., progression of murine EAE occurs within a few months, whereas progression of IMDS to CDMS often develops over many months or years. Thus, it appears that the shifting autoreactivity observed in SWXJ EAE mice represents an accelerated version of the same underlying processes responsible for the development of self-recognition in MS.
The shifting patterns of self-recognition shown in this study functionally reveals the fundamental underlying instability of the autoreactive T cell repertoire in MS. This view is supported by other studies showing changes in T cell repertoire restriction in IMDS patients who progress to CDMS (41). The inherent plasticity of the autoreactive repertoire has implications with regard to therapeutic applications, perhaps most notably evident in clonotypic therapies targeting specific TCR genes. Such T cell vaccination approaches result in the emergence of new autoreactive repertoires using TCR genes distinct from those used by the original responding T cells (42). Thus, the inherent capacity of the immune system to provide a continual source of neo-autoreactivity may serve ultimately as a basis for undermining the effectiveness of TCR-based therapies that fail to target the secondary spreading repertoires.
Recently, it has been suggested that it may be possible to harness epitope spreading in such a manner as to facilitate the spread of immune suppression during autoimmune disease (43). Prior reports have shown that chronic progression of EAE can be inhibited by inducing intramolecular (5) or intermolecular (6) tolerance to determinants of the epitope spreading cascade. Recent studies in our laboratory indicate that adoptive Th2 immunotherapy targeting spreading determinants results in a marked long-term inhibition of EAE progression even when transfer occurs before the development of endogenous self-priming (44). Thus, stacking the T cell repertoire to favor an active Th2 response to spreading determinants may subvert the neo-autoreactive process and produce a long-lasting therapeutic outcome. In this regard, peptide-based therapies such as those incorporating the HFFK amino acid motif of the putative human immunodominant MBP determinant (45, 46) or those involving altered peptide ligand strategies (47, 48) may prove to be most effective if the repertoire capable of responding therapeutically to the selected peptide has undergone minimal spontaneous regression.
It is evident from this study that progression of both EAE and MS may occur in the absence of primary initiating autoreactivity. However, thus far the basis for regression of primary autoreactivity is unclear. Our data indicate that the disappearance of primary self-recognition is not due to sequestration of autoreactive T cells in the CNS thereby creating an apparent loss of systemic autoreactivity. Therefore, more intricate explanations are needed to account for the spontaneous disappearance of self-recognition. Chronic self-stimulation may result in T cell exhaustion and peripheral clonal deletion (42), perhaps through apoptosis (49, 50). Alternatively, autoreactive T cells may be present but unreactive as a result of T cell anergy (51–54) or suppression (55, 56). Studies designed to determine the underlying mechanism(s) responsible for the regression of primary autoreactivity and the emergence of epitope spreading are currently in progress.
Acknowledgments
This work was supported by National Multiple Sclerosis Society grant RG-2768 (V.K. Tuohy) and by National Institutes of Health grant NS36054 (V.K. Tuohy).
Footnotes
1 Abbreviations used in this paper: CDMS, clinically definite multiple sclerosis; CNS, central nervous system; EAE, experimental autoimmune encephalomyelitis; IMDS, isolated monosymptomatic demyelinating syndrome; MBP, myelin basic protein; MOG, myelin oligodendrocyte glycoprotein; MRI, magnetic resonance imaging; MS, multiple sclerosis; PLP, myelin proteolipid protein; SI, stimulation index.
References
- 1.Alvord, E.C., Jr., M.W. Kies, and A.J. Suckling, editors. 1984. Experimental Allergic Encephalomyelitis—A Useful Model for Multiple Sclerosis. Alan R. Liss, New York.
- 2.Martin R, McFarland HF, McFarlin DE. Immunological aspects of demyelinating diseases. Annu Rev Immunol. 1992;10:153–187. doi: 10.1146/annurev.iy.10.040192.001101. [DOI] [PubMed] [Google Scholar]
- 3.Steinman L. Multiple sclerosis: a coordinated immunological attack against myelin in the central nervous system. Cell. 1996;85:299–302. doi: 10.1016/s0092-8674(00)81107-1. [DOI] [PubMed] [Google Scholar]
- 4.Lehmann PV, Forsthuber T, Miller A, Sercarz EE. Spreading of T cell autoimmunity to cryptic determinants of an autoantigen. Nature. 1992;358:155–157. doi: 10.1038/358155a0. [DOI] [PubMed] [Google Scholar]
- 5.McRae BL, Vanderlugt CL, Dal MC, Canto, Miller SD. Functional evidence for epitope spreading in the relapsing pathology of experimental autoimmune encephalomyelitis. J Exp Med. 1995;182:75–85. doi: 10.1084/jem.182.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu M, Johnson JM, Tuohy VK. A predictable sequential determinant spreading cascade invariably accompanies progression of experimental autoimmune encephalomyelitis: a basis for peptide-specific therapy after onset of clinical disease. J Exp Med. 1996;183:1777–1788. doi: 10.1084/jem.183.4.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vanderlugt CL, Miller SD. Epitope spreading. Curr Opin Immunol. 1996;8:831–836. doi: 10.1016/S0952-7915(96)80012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beck RW, Cleary PA, Trobe JD, Kaufman DI, Kupersmith MJ, Paty DW, Brown CH. The effect of corticosteroid for acute optic neuritis on the subsequent development of multiple sclerosis. N Engl J Med. 1993;329:1764–1769. doi: 10.1056/NEJM199312093292403. [DOI] [PubMed] [Google Scholar]
- 9.Morrissey SP, Miller DH, Kendall BE, Kingsley DP, Kelly MA, Francis DA, MacManus DG, McDonald WI. The significance of brain magnetic resonance imaging abnormalities at presentation with clinically isolated syndromes suggestive of multiple sclerosis. Brain. 1993;116:135–146. doi: 10.1093/brain/116.1.135. [DOI] [PubMed] [Google Scholar]
- 10.Filippi M, Horsfield MA, Morrissey SP, MacManus DG, Rudge P, McDonald WI, Miller DH. Quantitative brain MRI lesion load predicts the course of clinically isolated syndromes suggestive of multiple sclerosis. Neurology. 1994;44:635–641. doi: 10.1212/wnl.44.4.635. [DOI] [PubMed] [Google Scholar]
- 11.Milner RJ, Lai C, Nave K-A, Lenoir D, Ogata J, Sutcliffe JG. Nucleotide sequences of two mRNAs for rat brain myelin proteolipid protein. Cell. 1985;42:931–939. doi: 10.1016/0092-8674(85)90289-2. [DOI] [PubMed] [Google Scholar]
- 12.Macklin WB, Campagnoni CW, Deininger PL, Gardinier MV. Structure and expression of the mouse myelin proteolipid protein gene. J Neurosci Res. 1987;18:383–394. doi: 10.1002/jnr.490180302. [DOI] [PubMed] [Google Scholar]
- 13.Geysen HM, Rodda SJ, Mason TJ, Tribbick G, Schoofs PG. Strategies for epitope analysis using peptide synthesis. J Immunol Methods. 1987;102:259–274. doi: 10.1016/0022-1759(87)90085-8. [DOI] [PubMed] [Google Scholar]
- 14.Tuohy VK, Lu Z, Sobel RA, Laursen RA, Lees MB. Identification of an encephalitogenic determinant of myelin proteolipid protein for SJL mice. J Immunol. 1989;142:1523–1526. [PubMed] [Google Scholar]
- 15.Mathisen PM, Yu M, Johnson JM, Drazba JA, Tuohy VK. Treatment of experimental autoimmune encephalomyelitis with genetically modified memory T cells. J Exp Med. 1997;186:159–164. doi: 10.1084/jem.186.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawczak JA, Mathisen PM, Drazba JA, Fuss B, Macklin WB, Tuohy VK. Digitized image analysis shows diffuse abnormalities of normal appearing white matter during acute experimental autoimmune encephalomyelitis. J Neurosci Res. 1998;54:364–372. doi: 10.1002/(SICI)1097-4547(19981101)54:3<364::AID-JNR7>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 17.Tuohy VK, Lu Z, Sobel RA, Laursen RA, Lees MB. A synthetic peptide from myelin proteolipid protein induces experimental allergic encephalomyelitis. J Immunol. 1988;141:1126–1130. [PubMed] [Google Scholar]
- 18.Tuohy VK, Thomas DM. Sequence 104–117 of myelin proteolipid protein is a cryptic encephalitogenic T cell determinant for SJL/J mice. J Neuroimmunol. 1995;56:161–170. doi: 10.1016/0165-5728(94)00143-c. [DOI] [PubMed] [Google Scholar]
- 19.Greer JM, Kuchroo VK, Sobel RA, Lees MB. Identification and characterization of a second encephalitogenic determinant of myelin proteolipid protein (residues 178–191) for SJL mice. J Immunol. 1992;149:783–788. [PubMed] [Google Scholar]
- 20.Kono DH, Urban JL, Horvath SJ, Ando DG, Saavedra RA, Hood L. Two minor determinants of myelin basic protein induce experimental allergic encephalomyelitis in SJL/J mice. J Exp Med. 1988;168:213–227. doi: 10.1084/jem.168.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakai K, Zamvil SS, Mitchell DJ, Lim M, Rothbard JB, Steinman L. Characterization of a major encephalitogenic T cell epitope in SJL/J mice with synthetic oligopeptides of myelin basic protein. J Neuroimmunol. 1988;19:21–32. doi: 10.1016/0165-5728(88)90032-x. [DOI] [PubMed] [Google Scholar]
- 22.Cross AH, Hashim GA, Raine CS. Adoptive transfer of experimental allergic encephalomyelitis and localization of the encephalitogenic epitope in the SWR mouse. J Neuroimmunol. 1991;31:59–66. doi: 10.1016/0165-5728(91)90087-n. [DOI] [PubMed] [Google Scholar]
- 23.Amor S, Groome N, Linington C, Morris MM, Dornmair K, Gardinier MV, Matthieu JM, Baker D. Identification of epitopes of myelin oligodendrocyte glycoprotein for the induction of experimental allergic encephalomyelitis in SJL and Biozzi AB/H mice. J Immunol. 1994;153:4349–4356. [PubMed] [Google Scholar]
- 24.Folch J, Lees M, Sloane GH, Stanley A simple method for the isolation and purification of total lipids from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 25.Ford AL, Goodsall AL, Hickey WF, Sedgwick JD. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+T cells compared. J Immunol. 1995;154:4309–4321. [PubMed] [Google Scholar]
- 26.Tuohy VK, Yu M, Weinstock-Guttman B, Kinkel RP. Diversity and plasticity of self-recognition during the development of multiple sclerosis. J Clin Invest. 1997;99:1682–1690. doi: 10.1172/JCI119331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poser CM, Paty DW, Scheinberg L, McDonald WI, Davis FA, Ebers GC, Johnson KP, Sibley WA, Silberberg DH, Tourtellotte WW. New diagnostic criteria for multiple sclerosis: guidelines for research protocols. Ann Neurol. 1983;13:227–231. doi: 10.1002/ana.410130302. [DOI] [PubMed] [Google Scholar]
- 28.Novotny J, Bruccoleri RE, Carlson WD, Handschumacher M, Haber E. Antigenicity of myohemerythrin. Science. 1987;238:1584–1586. doi: 10.1126/science.2446393. [DOI] [PubMed] [Google Scholar]
- 29.Baxevanis CN, Reclos GJ, Servis C, Anastasopoulos E, Arsenis P, Katsiyiannis A, Matikas N, Lambris JD, Papamichail M. Peptides of myelin basic protein stimulate T lymphocytes from patients with multiple sclerosis. J Neuroimmunol. 1989;22:23–30. doi: 10.1016/0165-5728(89)90005-2. [DOI] [PubMed] [Google Scholar]
- 30.Ota K, Matsui M, Milford EL, Mackin GA, Weiner HL, Hafler DA. T-cell recognition of an immunodominant myelin basic protein epitope in multiple sclerosis. Nature. 1990;346:183–187. doi: 10.1038/346183a0. [DOI] [PubMed] [Google Scholar]
- 31.Martin R, Utz U, Coligan JE, Richert JR, Flerlage M, Robinson E, Stone R, Biddison WE, McFarlin DE, McFarland HF. Diversity in fine specificity and T cell receptor usage of the human CD4+cytotoxic T cell response specific for the immunodominant myelin basic protein peptide 87–106. J Immunol. 1992;148:1359–1366. [PubMed] [Google Scholar]
- 32.Valli A, Sette A, Kappos L, Oseroff C, Sidney J, Miescher G, Hochberger M, Albert ED, Adorini L. Binding of myelin basic protein peptides to human histocompatibility leukocyte antigen class II molecules and their recognition by T cells from multiple sclerosis patients. J Clin Invest. 1993;91:616–628. doi: 10.1172/JCI116242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pelfrey CM, Trotter JL, Tranquill LR, McFarland HF. Identification of a novel T cell epitope of human proteolipid protein (residues 40–60) recognized by proliferative and cytolytic CD4+T cells from multiple sclerosis patients. J Neuroimmunol. 1993;46:33–42. doi: 10.1016/0165-5728(93)90231-m. [DOI] [PubMed] [Google Scholar]
- 34.Pelfrey CM, Trotter JL, Tranquill LR, McFarland HF. Identification of a second T cell epitope of human proteolipid protein (residues 89–106) recognized by proliferative and cytolytic CD4+T cells from multiple sclerosis patients. J Neuroimmunol. 1994;53:153–161. doi: 10.1016/0165-5728(94)90025-6. [DOI] [PubMed] [Google Scholar]
- 35.Markovic-Plese S, Fukaura H, Zhang J, Al-Sabbagh A, Southwood S, Sette A, Kuchroo VK, Hafler DA. T cell recognition of immunodominant and cryptic proteolipid protein epitopes in humans. J Immunol. 1995;155:982–992. [PubMed] [Google Scholar]
- 36.Kerlero de Rosbo N, Milo R, Lees MB, Burger D, Bernard CC, Ben-Nun A. Reactivity to myelin antigens in multiple sclerosis. Peripheral blood lymphocytes respond predominantly to myelin oligodendrocyte glycoprotein. J Clin Invest. 1993;92:2602–2608. doi: 10.1172/JCI116875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanguay S, Killion JJ. Direct comparison of ELISPOT and ELISA-based assays for detection of individual cytokine-secreting cells. Lymphokine Cytokine Res. 1994;13:259–263. [PubMed] [Google Scholar]
- 38.Forsthuber T, Yip HC, Lehmann PV. Induction of TH1 and TH2 immunity in neonatal mice. Science. 1996;271:1728–1730. doi: 10.1126/science.271.5256.1728. [DOI] [PubMed] [Google Scholar]
- 39.Altman JD, Moss PAH, Goulder PJR, Barouch DH, McHeyzer-Williams MG, Bell JI, McMichael AJ, Davis MM. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996;274:94–96. [PubMed] [Google Scholar]
- 40.Yu M, Kinkel RP, Weinstock-Guttman B, Cook DJ, Tuohy VK. HLA-DP: a class II restriction molecule involved in epitope spreading during the development of multiple sclerosis. Hum Immunol. 1998;59:15–24. doi: 10.1016/s0198-8859(97)00252-8. [DOI] [PubMed] [Google Scholar]
- 41.Soderstrom M, Link H, Fredrikson S, Sun J-B. Optic neuritis and multiple sclerosis: the T cell repertoires to myelin proteins and MBP peptides change with time. Acta Neurol Scand. 1994;90:10–18. doi: 10.1111/j.1600-0404.1994.tb02673.x. [DOI] [PubMed] [Google Scholar]
- 42.Zhang J, Vandevyver C, Stinissen P, Raus J. In vivo clonotypic regulation of human myelin basic protein- reactive T cells by T cell vaccination. J Immunol. 1995;155:5868–5877. [PubMed] [Google Scholar]
- 43.Steinman L, Conlon P. Viral damage and the breakdown of self-tolerance. Nat Med. 1997;3:1085–1087. doi: 10.1038/nm1097-1085. [DOI] [PubMed] [Google Scholar]
- 44.Tuohy VK. Optimizing the therapeutic potential of autoreactive T cells by genetic modification. J Neuroimmunol. 1998;90:10. doi: 10.1016/s0923-2494(99)80012-1. . (Abstr.) [DOI] [PubMed] [Google Scholar]
- 45.Wucherpfennig KW, Catz I, Hausmann S, Strominger JL, Steinman L, Warren KG. Recognition of the immunodominant myelin basic protein peptide by autoantibodies and HLA-DR2–restricted T cell clones from multiple sclerosis patients. Identity of key contact residues in the B cell and T cell epitopes. J Clin Invest. 1997;100:1114–1122. doi: 10.1172/JCI119622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Warren KG, Catz I, Wucherpfennig KW. Tolerance induction to myelin basic protein by intravenous synthetic peptides containing epitope P85 VVHFFKNIVTP96 in chronic progressive multiple sclerosis. J Neurol Sci. 1997;152:31–38. doi: 10.1016/s0022-510x(97)00130-5. [DOI] [PubMed] [Google Scholar]
- 47.Nicholson LB, Greer JM, Sobel RA, Lees MB, Kuchroo VK. An altered peptide ligand mediates immune deviation and prevents autoimmune encephalomyelitis. Immunity. 1995;3:397–405. doi: 10.1016/1074-7613(95)90169-8. [DOI] [PubMed] [Google Scholar]
- 48.Gaur A, Boehme SA, Chalmers D, Crowe PD, Pahuja A, Ling N, Brocke S, Steinman L, Conlon PJ. Amelioration of relapsing experimental autoimmune encephalomyelitis with altered myelin basic protein peptides involves different cellular mechanisms. J Neuroimmunol. 1997;74:149–158. doi: 10.1016/s0165-5728(96)00220-2. [DOI] [PubMed] [Google Scholar]
- 49.Pelfrey CM, Tranquill LR, Boehme SA, McFarland HF, Lenardo MJ. Two mechanisms of antigen-specific apoptosis of myelin basic protein (MBP)-specific T lymphocytes derived from multiple sclerosis patients and normal individuals. J Immunol. 1995;154:6191–6202. [PubMed] [Google Scholar]
- 50.Tanaka, H., K. Ota, M. Ikusaka, M. Ejima, and S. Maruyama. 1995. [Expression of Fas-antigen on T cells in multiple sclerosis]. Rinsho Shinkeigaku. 35:299–301. (Japanese.) [PubMed]
- 51.Schwartz RH. A cell culture model for T lymphocyte clonal anergy. Science. 1990;248:1349–1356. doi: 10.1126/science.2113314. [DOI] [PubMed] [Google Scholar]
- 52.DeSilva DR, Urdahl KB, Jenkins MK. Clonal anergy is induced in vitro by T cell receptor occupancy in the absence of proliferation. J Immunol. 1991;147:3261–3267. [PubMed] [Google Scholar]
- 53.Correale J, Gilmore W, Lopez J, Li SQ, McMillan M, Weiner LP. Defective post-thymic tolerance mechanisms during the chronic progressive stage of multiple sclerosis. Nat Med. 1996;2:1354–1360. doi: 10.1038/nm1296-1354. [DOI] [PubMed] [Google Scholar]
- 54.van Parijs LV, Perez VL, Biuckians A, Maki RG, London CA, Abbas AK. Role of interleukin 12 and costimulators in T cell anergy in vivo. J Exp Med. 1997;186:1119–1128. doi: 10.1084/jem.186.7.1119. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55.Vandenbark AA, Hashim G, Offner H. Immunization with a synthetic T-cell receptor V-region peptide protects against experimental autoimmune encephalomyelitis. Nature. 1989;341:541–544. doi: 10.1038/341541a0. [DOI] [PubMed] [Google Scholar]
- 56.Offner H, Hashim GA, Vandenbark AA. T cell receptor peptide therapy triggers autoregulation of experimental encephalomyelitis. Science. 1991;251:430–432. doi: 10.1126/science.1989076. [DOI] [PubMed] [Google Scholar]