

Abstract
The islet-infiltrating and disease-causing leukocytes that are a hallmark of insulin-dependent diabetes mellitus produce and respond to a set of cytokine molecules. Of these, interleukin 1β, tumor necrosis factor (TNF)-α, and interferon (IFN)-γ are perhaps the most important. However, as pleiotropic molecules, they can impact the path leading to β cell apoptosis and diabetes at multiple points. To understand how these cytokines influence both the formative and effector phases of insulitis, it is critical to determine their effects on the assorted cell types comprising the lesion: the effector T cells, antigen-presenting cells, vascular endothelium, and target islet tissue. Here, we report using nonobese diabetic chimeric mice harboring islets deficient in specific cytokine receptors or cytokine-induced effector molecules to assess how these compartmentalized loss-of-function mutations alter the events leading to diabetes. We found that islets deficient in Fas, IFN-γ receptor, or inducible nitric oxide synthase had normal diabetes development; however, the specific lack of TNF- α receptor 1 (p55) afforded islets a profound protection from disease by altering the ability of islet-reactive, CD4+ T cells to establish insulitis and subsequently destroy islet β cells. These results argue that islet cells play a TNF-α–dependent role in their own demise.
Keywords: autoimmunity, diabetes, tumor necrosis factor α receptor, T cells, insulitis
Insulin-dependent diabetes mellitus (IDDM)1 is an autoimmune disease caused by the T cell-mediated destruction of the insulin-producing β cells of the pancreatic islets of Langerhans (1, 2). The nonobese diabetic (NOD) mouse spontaneously develops IDDM remarkably similar to that seen in humans (2, 3). The disease process is characterized by an initial, silent and nondestructive accumulation of a heterogeneous mixture of CD4+ and CD8+ T lymphocytes, B lymphocytes, macrophages, and dendritic cells (4, 5) circumscribing the islet mass, termed peri-insulitis. An invasive and malignant phase follows, termed insulitis, during which the immune infiltrate invades the islet and induces the specific apoptotic destruction of β cells (6–8). The events leading to insulitis induction, its perpetuation, and the subsequent destruction of β cells are poorly understood.
Cytokines produced by the immune infiltrate itself are clearly involved in the propagation of insulitis, but their pluripotentiality has made it difficult to pinpoint their specific roles in diabetes. Nevertheless, TNF-α, IFN-γ, and IL-1β have all been implicated as critical players in the disease process. As these are all potent proinflammatory molecules with the capability of affecting events critical to insulitis such as the expression of adhesion molecules (9), the upregulation of MHC molecules (10), and the localized production of chemoattractants (11), it is not surprising to see a varied, and often conflicting, set of effects ascribed to them during the course of IDDM development.
TNF-α, which is secreted principally by activated macrophages and CD4+ Th1 cells (for review see references 12, 13), can, for example, either retard or exacerbate the development of IDDM in NOD mice depending on when and where it acts (14–20). Thus, although these experiments reveal the potent ability of TNF-α to alter the development of autoimmune diabetes, the specific role TNF-α normally plays in diabetes development has not yet been fully elucidated.
The effect of IFN-γ on IDDM is less clear. On the one hand, a number of studies have revealed a critical role for IFN-γ in the induction of insulitis and subsequent development of diabetes (21, 22). On the other hand, an absolute deficiency of the IFN-γ gene in NOD mice had little effect on the overall development of diabetes (23).
To further complicate matters, many proinflammatory cytokines not only promote inflammation but may also facilitate the localized destruction of target tissue. For example, IFN-γ, TNF-α, and IL-1β have all been implicated in the cytolysis of a number of cell types, including pancreatic β cells (24, 25). TNF-α and IFN-γ have been shown to directly induce apoptosis (24–26), whereas IL-1β may act indirectly through the induction of reactive nitric oxide (NO) intermediates to islet cell destruction (27–29). Here again, the pluripotent nature of cytokines has made it difficult to dissect and ascribe precisely what role they play in the actual destruction of β cells in vivo.
Similarly, understanding β cell death has been hampered by the inability to study individual apoptotic pathways in isolation. This is perhaps best exemplified by recent studies on Fas (CD95) as a potential inducer of β cell apoptosis (30, 31). Although Fas-deficient NOD mice (NODlpr/lpr) do not develop diabetes, the global loss of Fas expression in these mice affects not only the pancreatic β cell but the critical T and B lymphocyte populations as well. Therefore, it is difficult to assign the protection from diabetes seen in these mice to the inability to kill Fas-deficient β cells or to the gross abnormalities found in adaptive immunity.
How then does one root out the multiple effects cytokines have on a complex autoimmune disease such as IDDM? Here, we report the use of chimeric NOD mice carrying islets deficient in one of several defined cytokine receptors or cytokine-induced effector molecules as a means of specifically and physiologically isolating the target islet tissue from the effects of locally produced cytokines. In this way, we can examine the effect islet-specific deficiencies in Fas, IFN-γR, inducible NO synthase (iNOS), and TNFR have on the development of IDDM in NOD mice when confronted by a population of diabetogenic CD4+ T cells. Contrary to previous reports, we found that Fas, IFN-γR, and iNOS deficiencies did not alter the development of diabetes. However, the specific deficiency of the TNF-α receptor 1 (p55) rendered these islets profoundly impermeable to islet-reactive T cells. In fact, in the absence of an islet response to locally produced TNF-α, infiltrating T cells failed to proliferate, establish insulitis, and subsequently destroy islet β cells. Not only do these results argue that TNF-α is a key player in the development of diabetes, they argue that this molecule must act in part upon the target tissue. Therefore, the islet must play an active, and TNF-α–dependent, role in its own demise.
Materials and Methods
Mice.
BDC2.5 TCR transgenic mice have been described previously (32). B6.lpr mice were obtained from Dr. John Russell (Washington University, St. Louis, MO), who originally obtained congenic breeding pairs from The Jackson Laboratory. These mice were backcrossed >12 generations to C57Bl/6 (B6) and were maintained by brother/sister mating. IFN-γR–deficient mice (33) were obtained on a 129 background from Dr. M. Aguet (Institut de Recherche sur le Cancer, Epalinges, Switzerland). TNFR p55-deficient (34) and p75-deficient (35) mice were obtained as doubly deficient mice on a mixed 129 × B6 background from Dr. R.D. Schreiber (Washington University; with permission from Drs. W. Lesslauer, Roche, Basel, Switzerland, and M. Moore, Genentech, South San Francisco, CA). A control TNF-α wild-type line was derived from negative littermates and used as controls in all TNF-α receptor experiments. Mice deficient in iNOS (36) were obtained from Taconic Farms on a pure 129/SvEv background by permission of Dr. J. Mudgett (Merck Research, Rahway, NJ). All mice were bred and housed under specific pathogen free conditions. All donor islets were derived from the original knockout or mutant lines as indicated above, unless specifically noted in the text. Mice deficient in the p55 receptor were backcrossed onto NOD.scid for seven generations and intercrossed to generate p55-deficient NOD.scid mice.
Flow Cytometry.
Flow cytometry was performed on a Becton Dickinson FACScan®. We purchased PE-conjugated anti-CD4 (Caltag Labs.), anti-B220 (PharMingen) and goat anti–mouse IgM (Jackson ImmunoResearch Labs.). The mAb to the β chain of the transgenic TCR, anti-Vβ4 (37), was conjugated with FITC. List mode data was collected on 105 cells and reanalyzed on a PC using WinMDI (version 2.7) software written by J. Trotter (http://facs.scripps.edu).
Diabetes.
Diabetes was assessed by measurement of venous blood using a Bayer Glucometer Elite one-step blood glucose meter. Animals were considered diabetic after two consecutive measurements ≥250 mg/dl (13.75 mM). Onset of diabetes was dated from the first consecutive reading.
Streptozotocin Treatment.
Streptozotocin was prepared fresh for each set of injections in sodium citrate–buffered saline. NOD.scid mice were weighed and streptozotocin was injected intravenously at a dose of 180 mg/kg, after which most mice became diabetic (>400 mg/dl) within 48–72 h. Diabetic mice were transplanted with islets within 48–72 h of the onset of diabetes.
Islet Isolation and Culture.
Mouse islets were isolated by collagenase technique (38) and purified on Ficoll gradient. Individual clean islets were selected and cultured overnight at 24°C in 5% CO2 in DMEM supplemented with 5% FCS (Hyclone), 10 mM Hepes, 5 × 10−5 M 2-ME, penicillin (100 U/ml), streptomycin sulfate (100 μg/ml), and glutamine (2 mM).
Islet Transplantation.
250 islets from mutant or control mice (all H-2b on either a 129 or B6 background) were transplanted under the renal capsule of streptozotocin-induced diabetic NOD.scid mice as previously described (39). In brief, under anesthesia (87 mg/kg of sodium pentobarbital), islets were transplanted under the renal capsule by exposing the left kidney through a flank incision, pushing the kidney through the incision, and holding it in place with small clamp attached to fatty tissue; with the aid of a dissecting microscope the capsule was cut with a needle and islets were then delivered through the incision by a Hamilton syringe fitted with a polyethylene catheter. After the catheter was withdrawn and the capsule was sealed by a small, pen-size eyecautery, the kidney was returned to the abdomen and the incision was closed. Normoglycemia was reestablished within 24 h of successful transplantation. Mice were then rested for 10–14 d to allow for vascularization of the graft by host vascular endothelium before the introduction of diabetic T cells. In the mixed islet grafts, the number of islets was 300 (200 p55−/− and 100 wild-type). Transplants were performed in a similar manner on bilateral kidneys with a total of 300 islets being transplanted per mouse. In all experiments, mice not receiving diabetogenic T cells remained normoglycemic (80–110 mg/dl) for >180 d. Confirmation of the graft's support of normoglycemia was verified by nephrectomy of engrafted kidney. All transplants and manipulation of the mice were performed following written protocols that had the prior approval of the Washington University School of Medicine Animal Studies Committee. All necessary steps were taken to minimize the discomfort of the transplanted animals.
Nephrectomy.
Mice were anesthetized with 87 mg/kg of sodium pentobarbital and the engrafted kidney was exposed by a flank incision as above. The engrafted kidney was raised and freed from fatty tissue as before. The renal artery and vein along with the ureter were clamped off with a mosquito hemostat and were sutured distal to the kidney with 4-0 silk. The kidney was cut free with a scalpel. The clamp was released slowly and the suture was inspected for leaks and the incision closed.
Immunohistochemistry.
Kidney graft sections were stained with antibodies against Vβ4 (KT4), macrophages (ERMP-23; reference 40), dendritic cells (NLDC-145; Harlan sera labs), mad-CAM (MECA-367; reference 41), and peripheral node addressin (MECA-79; reference 41) as previously described (42).
T Cell Proliferation.
In vitro T cell proliferation to islet cell antigen was performed as previously described (32). In vivo T cell proliferation was performed using the cell surface dye, 5,6-carboxy-succinimidyl-fluorescein-ester (CSFE). Preparation and labeling of T cells with CSFE was performed as described in reference 43. CSFE-labeled BDC2.5 T cells (107 cells) were transferred intravenously in to NOD.scid mice carrying kidney grafts of either 250 p55-deficient or 250 p55-sufficient islets. T cells from the draining lymph nodes (peri-renal) were collected on the indicated day after transfer and analyzed by flow cytometer for evidence of cell division.
Results and Discussion
The ability to target loss-of-function mutations to specific organ systems remains a major challenge. With the exception of certain mutations targeted to the immune system using the Rag-mutant complementation system (44) or the inducible knockouts (45), it has been difficult to study the effects of broadly expressed or broadly acting mutations on specific organ systems or disease models. This is particularly true of the ubiquitously expressed cytokine receptors and their pluripotent ligands, the cytokine molecules themselves.
Several experimental models have long suggested that cytokines influence the development of autoimmune diabetes (for review see reference 46). Although these models have been informative, it has remained difficult to attribute particular stage dependency to any given cytokine, especially under physiological conditions. Moreover, it has been difficult to determine what cellular conduit channels the action of each cytokine; is it through the effector lymphocyte, the APC, the vascular epithelium, or the islet tissue itself? Therefore, we undertook to develop a novel approach that compartmentalizes the action of cytokines and their receptors to specific cellular targets in an effort to establish the dependency of particular phases of diabetes development to the local effect of these mediators.
We did this by creating chimeric NOD mice. Unlike prior models, these mice harbored wild-type NOD APCs that express the disease-associated MHC class II, I-Ag7, and wild-type NOD vascular endothelium. Moreover, they carried a defined population of diabetogenic CD4+ T cells (BDC2.5 TCR transgenic T cells) that respond to pancreatic β cell antigen, presented uniquely by NOD APCs, and are capable of mediating the autoimmune destruction of pancreatic β cells (32, 47–49). What distinguishes our current approach was the prior replacement of the endogenous β cells with those derived from one of several mouse strains deficient in key cytokine receptor or proapoptotic effector molecules. In so doing, we created chimeric NOD mice containing altered β cell target tissue. This allowed us to assess the potential impact of each genetic alteration on the islet cell's ability to serve as a target for autoimmune-mediated destruction by altering the host effector lymphocyte, APC, or vascular endothelium.
As shown schematically in Fig. 1, NOD.scid mice were treated with streptozotocin, a β cell toxin, to destroy endogenous β cells, producing a chemically-induced diabetes (≥400 mg/dl). Normoglycemia (<100 mg/dl) was rapidly returned with the transplantation of ∼250 islets under the left kidney capsule. (It should be noted that the donor islets can be from any strain of mouse as NOD.scid do not reject allogeneic tissue. We routinely used islets of H-2b donors.) The rescued mice were then rested for 7–10 d, allowing for host-derived vascularization of the graft. At this point, splenic T cells from diabetic BDC2.5/NOD.scid mice were transferred into these chimeric NOD.scid mice, and the mice were followed for onset of diabetes.
Figure 1.

Schematic representation of transplantation protocol. 6–8-wk-old healthy NOD.scid mice were injected with Streptozotocin to render them chemically diabetic (>400 mg/dl). Within 48–72 h of the onset of diabetes, the mice were engrafted with 250–300 islets under the left kidney capsule. Normoglycemia returned within 24 h of islet transplantation. The mice were then rested 7–10 d, during which time host-derived vascular support for the grafts developed. Splenic T cells from overtly diabetic BDC2.5/NOD.scid mice were transferred into the engrafted-NOD.scid mice (day 0). Mice were followed thereafter for the onset of diabetes. At day 28, normoglycemic mice were nephrectomized.
These experiments are predicated on the following observations. First, as mentioned above, the recipient mice are scid, hence they do not reject allogeneic islet grafts, as confirmed by control experiments where each series of donor islets are engrafted under the kidney capsule and the mice are left unmanipulated for >180 d. None of these mice develop diabetes during this period. Moreover, the transplanted islets are functional, and are responsible for the maintenance of normoglycemia, as removal of the engrafted kidney results in hyperglycemia (data not shown). Second, the BDC2.5 T cells do not recognize the β cells directly but rather require the transfer of islet antigen to NOD (H-2g7) APCs, which are supplied by the NOD.scid recipient mice. Third, although the recognition of antigen is MHC restricted, all strains of mice express the relevant antigen in their pancreatic β cells (48). And finally, once activated by antigen, BDC2.5/NOD.scid T cells can mediate the destruction of islet β cells in NOD.scid mice (6).
Destruction of Pancreatic β Cells in the Absence of Fas.
Pancreatic β cell death during the course of T cell–mediated diabetes is by apoptosis (6–8). One potential mediator of β cell apoptosis is Fas (CD95). The engagement of Fas, a proapoptotic member of the TNFR family, on the surface of target cells by Fas ligand–expressing T lymphocytes leads to the apoptotic destruction of the Fas-expressing target cell (for review see reference 50). Treatment with IFN-γ induces the expression of Fas on the surface of a variety of cell types including β cells (24). Moreover, islet-infiltrating T cells can induce Fas expression on β cells through localized production of IFN-γ (31). Additionally, Fas-deficient, NODlpr/lpr mice do not develop diabetes (30, 31); however, these mice had substantially altered T and B cell immunity (30). These results notwithstanding, it has not been formally demonstrated that Fas expression on β cells is required for their autoimmune-mediated destruction.
To test whether Fas expression on β cells is obligatory, we used our chimeric NOD mice model to create mice specifically lacking Fas expression on their islet cells. This was done by either eliminating the islet's ability to respond to IFN-γ by replacing the existing islet mass with islets lacking the IFN-γR or by using islets from B6.lpr/lpr mice that lack functional Fas expression as islet donors. After transplant, T cells from diabetic BDC2.5/NOD.scid mice were transferred into these mice and diabetes onset was followed. We found that BDC2.5 T cells destroyed B6.lpr/lpr islets as efficiently as control B6 islets, as shown in Fig. 2 a, indicating that Fas does not play an obligatory role as the critical inducer of β cell destruction, at least with respect to disease transferred by diabetogenic CD4+ T cells. Similarly, when NOD.scid mice were transplanted with islets deficient in IFN-γR, the chimeric mice developed diabetes at the same rate as control islet grafts (129/SvJ; Fig. 2 b). These results clearly demonstrate that islet cell Fas expression, either induced or constitutive, is not required for islet destruction by diabetogenic CD4+ T cells. Moreover, this would suggest that much of the protection seen in Fas-deficient NOD mice results from altered lymphoid development in the absence of Fas expression on T and B lymphocytes. Parenthetically, these results help to clarify the role IFN-γ may play in diabetes development. Wang et al., in describing the introduction of systemic IFN-γ receptor deficiency onto the NOD background, found that both NOD and BDC2.5/NOD mice lacking IFN-γR had severely retarded insulitis development (21). Our results would suggest that this is probably due to an effect IFN-γ has on the T cells, APCs, or host endothelium but not on the islet mass itself.
Figure 2.

TNF-αRI (p55)–deficient islets are protected from destruction by diabetogenic CD4+ T cells, whereas Fas-deficient, IFN-γR–deficient, iNOS-deficient, and TNF-αR p75 islets are destroyed. (a) 250 B6.lpr/lpr (•, n = 12) or control B6 islets (○, n = 10) transplanted under the kidney capsule are destroyed. □, diabetes in control NOD.scid mice transferred with the same population of T cells (n = 9). (b) 250 IFN-γR– deficient (♦, n = 7), iNOS-deficient (▪, n = 6) or control 129 islets (▵, n = 8) transplanted under the kidney capsule are destroyed. (c) Transplanted p55−/− islets (▾, n = 9) and p55−/−p75−/− doubly deficient islets (▴, n = 10) are protected from destruction by transferred T cells from diabetic BDC2.5/NOD.scid mice, whereas p75−/− islets (▿, n = 11) are destroyed. Diabetes is measured by sampling venous blood using a standard one-step glucometer. Mice are considered diabetic after two successive readings ≥250 mg/dl. In all cases, engrafted mice that did not receive diabetogenic T cells remained normoglycemic throughout the experimental period (>180 d); moreover, the engrafted islets were responsible for normoglycemia, as the hemi-nephrectomy of engrafted kidneys returned mice to hyperglycemia.
Islet Cell Production of Reactive Nitric Oxide Intermediates Is Not Required for β Cell Destruction.
Previous studies have indicated that IL-1 stimulates the production of nitric oxide (NO) either by priming for Fas-mediated apoptosis or by inducing the inducible form of the NO synthase gene (iNOS or NOS2; references 51, 52). iNOS-mediated NO production can lead to β cell death in vitro (27–29). NO can be produced by the islets themselves or by the infiltrating macrophage/dendritic population. We found that the in vivo neutralization of IL-1β with a cocktail of antibodies and soluble receptor did not prevent NOD mice from becoming diabetic (data not shown), leading us to question its role in β cell death. However, to examine the effect that the targeted iNOS deficiency in islets had on β cell destruction, we tested the ability of iNOS-deficient islets to resist T cell–mediated destruction. As shown in Fig. 2 b, iNOS-deficient islets were destroyed with similar kinetics and magnitude as wild-type islets, indicating that iNOS gene expression is not critical for islet cell apoptosis. Although this result does not rule out a role for NO production by infiltrating macrophage/dendritic cells, it clearly demonstrates that islets themselves do not need to produce intracellular NO to undergo T cell–mediated elimination.
TNF-αR Deficiency Affords Islets Protection from Diabetogenic CD4+ T Cell–mediated Destruction.
TNF-α, which is secreted principally by activated macrophages and CD4+ Th1 cells (12, 13), can both retard and exacerbate the development of IDDM in NOD mice largely dependent on the time of its administration (14–16). Thus, when TNF-α is given to NOD mice from birth to 3 wk of age, diabetes is accelerated, and conversely the administration of neutralizing antibody to TNF-α during this period markedly reduces both insulitis and diabetes (16). Yet when administered to adult NOD mice with established insulitis, TNF-α attenuates the disease process, and its antibody neutralization exacerbates diabetes (16, 53). Moreover, the transgenic expression of TNF-α in the islets of adult NOD mice leads to insulitis without disease (17–19) and produces a state of T cell tolerance to islet cell antigens (19, 20). Thus, although these experiments reveal the potent ability of TNF-α to alter the development of autoimmune diabetes, the physiological role played by TNF-α has yet to be fully elucidated.
To investigate the role that localized production of TNF-α can have on the development of diabetes, we transplanted streptozotocin-treated NOD.scid mice with islets rendered doubly deficient for both TNF-αRs (TNF-αR1, p55; TNF-αR2, p75). As before, the transfer of diabetogenic T cells led to the rapid destruction of wild-type islet grafts ( 7 out of 8); however, doubly deficient islets (p55−/−p75−/−) remained functional (Fig. 2 c). Mice engrafted with p55−/− p75−/− islets remained normoglycemic for up to 52 d after the transfer of T cells. To confirm that the introduced islets were responsible for the maintenance of blood glucose, normoglycemic p55−/−p75−/− islet recipients were hemi-nephrectomized at day 28 to remove the engrafted kidney. As shown in Fig. 2 c, these mice became hyperglycemic within 24 h of nephrectomy, proving that the transplanted p55−/−p75−/− islets were indeed responsible for the maintenance normoglycemia. Interestingly, the mice carrying p55−/−p75−/− islets contained BDC2.5 T cells as measured by flow cytometric analysis of spleen and lymph node. In addition, these T cells were phenotypically normal in that they could still transfer disease to unmanipulated NOD.scid mice (data not shown).
To assess which receptor conferred the protection, we produced chimeric mice carrying islets defective in either the p55 receptor or the p75 receptor. Fig. 2 c shows that p55−/− islets were protected from T cell–mediated destruction, whereas the p75−/− islets were efficiently destroyed. All the p75−/− transplanted mice (11 out of 11) became diabetic by day 12, whereas the p55−/− transplanted mice (9 out of 9) remained normoglycemic until end of the assay (≥28 d). We therefore concluded that the engagement of the p55 receptor by locally produced TNF-α was critical in the subsequent destruction of β cells.
p55−/− Islets and p75−/− Islets Are Equally Antigenic to BDC2.5 T Cells.
One explanation for the lack of islet destruction of the p55−/− grafts is that the p55−/− islets are non- or poorly antigenic. To test this, BDC2.5 T cells were cocultured with NOD APCs in the presence of dispersed p55−/− and p75−/− islet cells for 72 h under standard conditions, and T cell proliferation was measured. As shown in Fig. 3, BDC2.5 T cells proliferated equally well to both receptor-deficient islet cells, indicating that islet cell antigenicity does not depend on TNF-αR expression. We performed a similar assay with intact islets in vitro in the presence and absence of exogenous recombinant TNF-α and were unable to detect a difference in the induced proliferation of BDC2.5 T cells (data not shown). We therefore concluded that at least in vitro, there is no difference in the antigenicity of p55−/− and p75−/− islet cells. This is somewhat discordant with recent results by Green et al., who reported that the localized production of TNF-α in β cells of transgenic NOD mice enhanced autoantigen presentation to BDC2.5 T cells in vitro (54).
Figure 3.

TNFR-deficient islet cells are antigenic to BDC2.5 T cells. 2 × 104 BDC2.5 T cells were cocultured with 2.5 × 105 irradiated NOD splenic APCs and varying numbers of either dispersed islet cells from control 129 (▵), p55-deficient (▾) or p75-deficient (▿) donors. After 72 h, the assay was pulsed with [3H]TdR for 6 h to measure T cell proliferation to antigen. Data are depicted as mean ± SD.
The CD4+ T Cell Infiltration of p55−/− Islet Grafts Does Not Progress into Destructive Insulitis.
An alternative explanation for the p55-deficient islets' resistance to T cell–mediated destruction resides with a fundamental modification in the cellular constituency of the infiltrate. To evaluate this possibility, we performed an immunohistochemical analysis of both p55−/− and wild-type (or p75−/−) islet grafts after T cell transfer. Engrafted kidneys were sampled at day 5, 7, and 9 after T cell transfer as well as at the time of diabetes or in the case of normoglycemic mice at day 28. As seen in Fig. 4, there was no infiltration in either p55-deficient or wild-type islet grafts at day 5. At day 7, however, the wild-type graft showed distinct signs of peri-islet accumulation of leukocytes, with some grafts showing evidence of intra-islet infiltration and destruction. The same was not true for the p55-deficient islet grafts, which showed only modest peri-islet infiltration and no intra-islet infiltration. As seen in Fig. 4, the most dramatic difference was revealed at day 9 when the wild-type islet grafts were completely infiltrated. These islets showed discrete foci of apoptotic β cells as revealed by TUNEL analysis (data not shown). In contrast, the p55-deficient islet grafts were only mildly infiltrated at day 9 (Fig. 4) and showed no signs of apoptosis (data not shown). Moreover, by day 13, the wild-type islet grafts were destroyed and the mice were overtly diabetic. Surprisingly, the mild infiltration of the p55−/− grafts failed to progress, and in fact resolved, so that by day 28 they were nearly indistinguishable from those seen on or before day 5. We therefore concluded that the p55−/− islet grafts could not sustain the infiltrating CD4+ T cells and that the propagation of destructive insulitis requires a TNF-α– dependent response on part of the islets.
Figure 4.
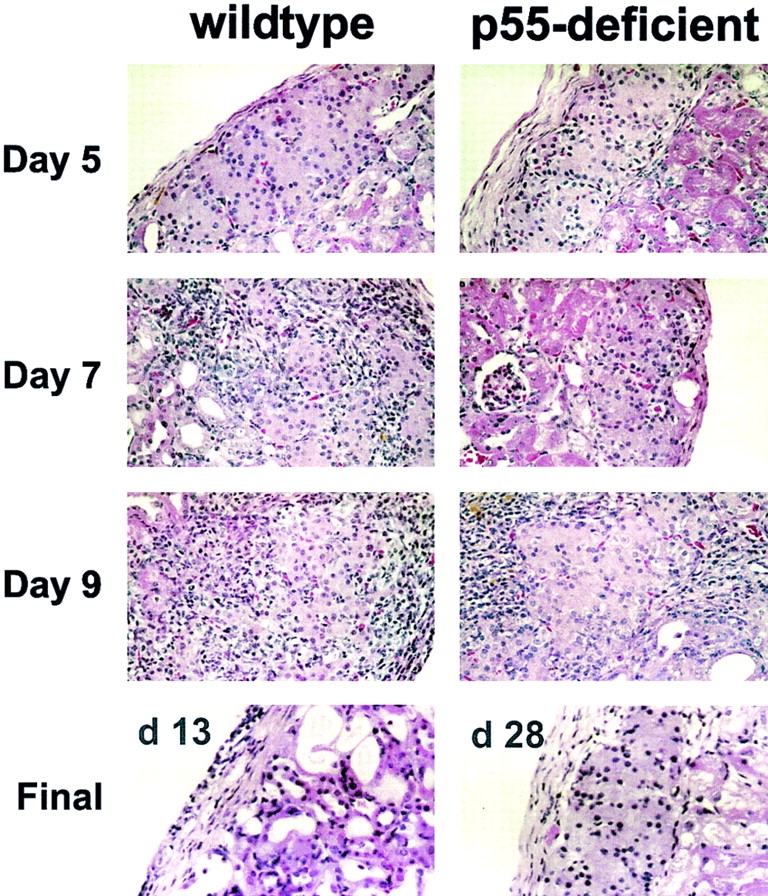
TNFR (p55) expression on the islet mass is necessary for sustained infiltration of islets. Histological analysis of the progression of insulitis in p55-deficient islet grafts and control islet grafts (p75-deficient or 129-negative littermates) at the indicated time after transfer of T cells from diabetic BDC2.5/NOD.scid mice. Engrafted kidneys were fixed in buffered 10% formalin, embedded, and sectioned (4 μm). Sections were stained with hematoxylin and eosin to show infiltration and islet architecture. A representative image from one of three mice at each time point is shown. Original magnification: ×400.
We then asked if the composition of the infiltrate was modified between the p55−/− and wild-type lesions. We compared the cellular components of the transient infiltration of p55-deficient islet grafts at day 9 with those of the wild-type grafts. In general, the composition of the infiltrate was similar to that seen in the pancreata of NOD.scid recipients of T cells from diabetic BDC2.5 mice (42). Moreover, we saw no difference between the p55−/− and the wild-type grafts in the activation state of the high endothelial venule (HEV) as revealed by staining for madCAM (MECA 367) and peripheral node addressin (MECA 79, data not shown). We were also able to identify the presence of roughly equal numbers of BDC2.5 T cells in both infiltrates as revealed by Vβ4 (KT4) and anti-CD4 staining. Both lesions contained similar subsets of macrophage (F4/80, MOMA 1, MOMA 2) and dendritic cells (NLDC-145) as well. Despite these similarities, there was one striking difference between the p55−/− and wild-type grafts: the complete absence of β cell apoptosis in the p55-deficient grafts (data not shown). We therefore concluded that apart from the lack of continued progression of the lesions and the lack of β cell apoptosis, there was little difference in the nature of the infiltrates and the vascular endothelium between wild-type and p55-deficient islet grafts.
NOD.scid Mice Lacking the p55 Receptor, but Carrying Wild-type Islets, Develop Diabetes upon BDC2.5 T Cell Transfer.
To verify that the resistance to diabetes resided with the p55-deficient islets rather than with the host endothelium or APC populations, reciprocal transplants were performed in which wild-type islets (from 129 mice) were transplanted under the kidney capsule of streptozotocin-treated p55-deficient NOD.scid mice (N7 generation). Under these conditions, both the host vasculature and APC population lacked p55 receptor expression, whereas the engrafted islet mass retained full p55 functionality. When purified diabetogenic CD4+ T cells were transferred into these mice, diabetes developed with similar kinetics in both these mice and control recipients (Fig. 5 a). This indicated that functional expression of the p55 receptor on the islet mass alone was sufficient to drive β cell destruction regardless of the p55 receptor expression status of the host APCs and the vascular endothelium.
Figure 5.

Reciprocal islet transplants and mixed transplants of p55- deficient and p55-sufficient islets are destroyed upon BDC2.5 T cell infiltration. (a) Reciprocal transplants of p55-sufficient islets transplanted under the kidney capsule of p55−/− NOD.scid mice (•, n = 9) are destroyed as efficiently as p55+/+ islets transplanted under the kidney capsule of p55+/− NOD.scid mice (○, n = 4). (b) Both control islet grafts (○; 300 islets; n = 4) and mixed islet grafts (•; 200 p55-deficient islets and 100 control islets; n = 8) are destroyed with similar kinetics upon transfer of T cells from diabetic BDC2.5/NOD.scid mice. Diabetes is measured by sampling venous blood using a standard one-step glucometer. Mice are considered diabetic after two successive readings ≥250 mg/dl.
An Islet Graft Containing both p55-deficient and p55-sufficient Islet Mass Is Destroyed upon Diabetogenic BDC2.5 T Cell Transfer.
Although the p55-deficient islets were no less antigenic than the p55-sufficient islets and were equally capable of attracting similar subsets of infiltrating leukocytes, they were clearly incapable of providing a microenvironment that supported the maturation of the immune response to a point were β cell death could occur. This could be for one of two reasons. First, the propagation of insulitis may require TNF-α–mediated β cell death. In this case, the TNF-α produced locally by the infiltrating T cells and macrophages would fail to kill the p55-deficient β cells and insulitis would subside due to a lack of β cell damage. This would be consistent with our failure to observe β cell apoptosis in p55-deficient islets during the early phase of infiltration, yet it also seemed unlikely as ectopic expression of super-physiologic levels of TNF-α by the islets of transgenic mice did not lead to β cell death or diabetes (17–19, 55). Alternatively, the evolution of insulitis from a benign accumulation of leukocytes to a destructive infiltrate may require a TNF-α–dependent change in the islet mass—either the release of an islet cell–produced chemoattractant or activation factor or an alteration in the secretion or production of antigen (something we are unable to mimic in vitro, but which has been observed by others, see reference 54). Either way, the net result would be the full activation of the infiltrating BDC2.5 T cell population such that it can now act to target β cells for destruction in a TNF-α–independent fashion.
To distinguish between these two possibilities, we designed and produced chimeric NOD.scid mice that carried mixed grafts containing both varying amounts of p55-deficient and p55-sufficient islets. If TNF-α acted strictly as a cytolytic agent, only the p55-sufficient islets would be destroyed upon transfer of diabetogenic T cells, while the p55-deficient islets would be spared and normoglycemia would be maintained, provided that adequate amounts of p55-deficient islets were included in the mixed graft. On the other hand, if TNF-α acted to alter the local environment in favor of T cell activation, the presence of even a modicum of TNF-α–responsive islets would result in T cell activation and the destruction of both the p55-deficient and -sufficient islets and diabetes.
We first ascertained the minimum number of islets required in our grafts to maintain a persistent state of normoglycemia in our Streptozotocin-treated NOD.scid mice. We found that as few as 100 islets could maintain blood glucose levels at ≤100 mg/dl (data not shown). Therefore, for our initial experiments we chose to mix ≥200 p55-deficient islets with ∼100 wild-type islets per graft. In this way, the “protected” p55-deficient islets were in sufficient excess to assure normoglycemia if all of the wild-type islets were destroyed. As before, mixed islet recipients received diabetogenic T cells 7–10 d after islet transplantation. As depicted in Fig. 5 b, both the mixed islet recipients and the control mice engrafted with 300 wild-type islets developed diabetes with comparable kinetics (between days 16 and 18 after transfer). In subsequent experiments, the numbers of wild-type islets were reduced until as few as 10 wild-type islets were mixed with ∼300 p55-deficient islets, yet the results (islet graft destruction and diabetes) were the same (data not shown). Therefore, we concluded from these experiments that the stimulation of islet cells through their p55 receptor altered the local environmental conditions favoring the development of a productive BDC2.5 T cell infiltrate.
The Physical Separation of p55-deficient and p55-sufficient Islet Grafts on Opposing Kidneys Still Allows for the In Vivo Activation of Diabetogenic CD4+ T Cells, the Destruction of β Cells and the Development of Diabetes.
Having determined that a small number of islet cells can, in response to locally produced TNF-α, support the transition from benign to destructive insulitis, it was now critical to determine if this was a purely localized effect. To address this, we performed kidney grafts on streptozotocin-treated NOD.scid mice in which wild-type islets (100 islets) were engrafted under the right kidney capsule and p55-deficient islets (250 islets) were engrafted under the left kidney capsule of the same animal. By physically separating the grafts we sought to minimize the effects between the wild-type and p55-deficient graft. If, upon transfer of diabetogenic T cells, the p55-deficient graft survived in these mice, despite the destruction of the wild-type islet grafts, then the original destruction of the p55-deficient islets in the mixed islet grafts described above (Fig. 5 b) resulted merely by virtue of their intimate proximity to the wild-type islets. On the other hand, if the distant p55-deficient grafts were likewise destroyed, it is more likely that the transferred T cells were altered by an encounter with wild-type islet cells.
We found that the twin-kidney engrafted NOD.scid recipient mice did, in fact, become diabetic 12–16 d after receiving BDC2.5 T cells, at a rate coincidental with the development of diabetes in mice harboring dual wild-type grafts (Fig. 6 a). As before, those mice engrafted with only p55-deficient islets did not develop disease. Additionally, histological analysis of the p55-deficient bilateral graft showed signs of β cell apoptosis within 1 d of the onset of destructive infiltration of the p55-sufficient graft (∼day 5–7 after transfer, Fig. 6 b). The ability of wild-type grafts to influence the outcome of the contralateral p55-deficient grafts indicated that TNF-α responsiveness on the part of the wild-type islets led to the activation of the transferred BDC2.5 T cells such that they were now capable of homing to and destroying the p55-deficient graft on the opposite kidney.
Figure 6.

Spatial separation of the p55-deficient and p55-sufficient islet grafts on opposing kidneys does not protect the p55-deficient grafts from destruction. (a) In dual-kidney grafts, control NOD.scid mice were engrafted with p55-sufficient islet grafts under both kidneys (○; 200 islets on left kidney and 100 islets on the right) and experimental mice were engrafted with 200 p55-deficient islets on the left kidney and 100 p55-sufficient islets on the right kidney (•). Both sets of mice received T cells from BDC2.5/NOD.scid mice. In both cases the engrafted islets were destroyed with similar kinetics. Mice engrafted with p55-deficient islets only (▴) did not develop diabetes. Diabetes is measured by sampling venous blood using a standard one-step glucometer. Mice are considered diabetic after two successive readings ≥250 mg/dl. (b) Photomicrographs of the contralateral kidney grafts 14 d after transfer of T cells. Engrafted kidneys were fixed in buffered 10% formalin, embedded, and sectioned (4 μm). Sections were stained with hematoxylin and eosin to show infiltration and islet architecture. Note that the grafts are destroyed and only show residual scar tissue remains of both the p55-deficient graft, left, and the p55-sufficient graft, right. Representative images are from one of four mice analyzed at this time point. Original magnification: ×400.
These results led us to assess the in vivo proliferative status of BDC2.5 T cells after transfer. We reasoned that the lack of progression in the p55-deficient islet engrafted mice may be due to the inability of the p55−/− islets to fully activate the transferred BDC2.5 T cells. In the mixed and twin-kidney graft experiments, the wild-type islets would provide an environment capable of furnishing this activation and therefore of leading to the subsequent destruction of the p55−/− islets in a TNF-α–independent fashion. If this were true, T cell proliferation in vivo might differ between mice harboring only p55−/− islets and those carrying only wild-type islets. This was tested by monitoring the degree of in vivo proliferation of the BDC2.5 T cells in the efferent lymph of mice harboring one or the other islet grafts under the left kidney using the decay of the integral membrane dye, CSFE, as an indicator of cell division (43, 56, 57). As depicted in Fig. 7, draining lymph nodes from mice engrafted with p55−/− islets were devoid of reactivated BDC2.5 T cells, whereas the renal lymph nodes from mice engrafted with wild-type islets contained T cells that clearly had undergone several rounds of replication. Therefore, we concluded that the most likely explanation for the infiltration and subsequent destruction of p55−/− islets in both the mixed and twin-kidney grafts was due in part to the activation of the BDC2.5 T cells in response to wild-type islets either proximal or distal to the p55−/− islets. This process required a TNF-α response on the part of the target islet cells but the subsequent destruction of the islet tissue was TNF-α–independent. The nature of the TNF-α response on the part of islets remains unknown, but could be an increase in antigen delivery either in direct response to TNF-α or as a result of islet cell death. In either case, this leads to the subsequent activation of our infiltrating islet-reactive BDC2.5 T cells, which then act to mediate the destruction of islet β cells in a process that does not require TNF-α.
Figure 7.

T cells from the efferent lymph of p55−/− islet engrafted mice have not undergone cell division, whereas those from wild-type islet engrafted mice have. Streptozotocin-treated NOD.scid mice were engrafted with either p55-deficient (p55−/−) or p55-sufficient islets (WT) under the left kidney capsule. Several weeks later, 107 CSFE-labeled BDC2.5 T cells were injected intravenously in each group of mice. On days 4 and 5 after transfer, peri-renal lymph nodes were collected and BDC2.5 T cell proliferation was assessed as measured by diminution of CSFE label on a flow cytometer. Lymph nodes from two to four animals were pooled per group.
Using the same BDC2.5 TCR transgenic model, André et al. have proposed two checkpoints in the progression of diabetes, the first the formation of a benign infiltrate and the second the transition to destructive insulitis (58). We would propose that the transition through checkpoint two is dependent on the active response of islets to TNF-α. Moreover, our results are consistent with the hypothesis that early and local production of TNF-α in the islet acts to enhance the islet's antigenicity and the subsequent activation of disease-causing lymphocytes (16, 17, 53).
In conclusion, these data, taken together, demonstrate that Fas, IFN-γ, and iNOS do not play an obligatory role in the apoptotic destruction of pancreatic β cells induced by a diabetogenic CD4+ T cell population, but that TNF-α plays a critical role in the transformation of islet-reactive CD4+ T cells from a benign state of β cell indifference to an activated state of β cell reactivity. Moreover, these results suggest, for the first time, that the islet cells themselves play an active and TNF-dependent role in facilitating their own death by providing an environment capable of perpetuating T cell–mediated insulitis.
Acknowledgments
We would like to thank Ms. Olga Strots and Mr. Larry McClendon for their excellent technical assistance and animal care. We would also like to thank Drs. Robert D. Schreiber, John H. Russell, Osami Kanagawa, John Mudgett, Werner Lesslauer, and Mark Moore for gifts of reagents, mice, and critical technical advice, and Dr. Stacey Smith for use of her cryostat. We would also like to thank Dr. Paul E. Lacy for his advice on transplantation experiments and critical review of the manuscript.
Abbreviations used in this paper
- CFSE
5,6-carboxy-succinimidyl-fluorescein-ester
- IDDM
insulin-dependent diabetes mellitus
- iNOS
inducible NO synthase
- NO
nitric oxide
- NOD
nonobese diabetic
Footnotes
We particularly wish to thank Dr. Charles Kilo and his Kilo Diabetes and Vascular Research Foundation for their generous financial support. In addition, this work was supported by grants to J.D. Katz from the Juvenile Diabetes Foundation International (JDFI; No. 197030), the Washington University Diabetes Research and Training Center, the National Institutes of Health (R01 AI44416), and a joint NIH/JDFI program project grant (P01 AI39676/995012; Dr. E.R. Unanue, program director). J.D. Katz is a recipient of an American Diabetes Association Career Development Award.
References
- 1.Bach JF. Insulin-dependent diabetes mellitus as an autoimmune disease. Endocr Rev. 1994;15:516–542. doi: 10.1210/edrv-15-4-516. [DOI] [PubMed] [Google Scholar]
- 2.Atkinson MA, MacLaren NK. The pathogenesis of insulin-dependent diabetes mellitus. N Engl J Med. 1994;331:1428–1436. doi: 10.1056/NEJM199411243312107. [DOI] [PubMed] [Google Scholar]
- 3.Tisch R, McDevitt H. Insulin-dependent diabetes mellitus. Cell. 1996;85:291–297. doi: 10.1016/s0092-8674(00)81106-x. [DOI] [PubMed] [Google Scholar]
- 4.Charlton B, Bacelj A, Mandel TE. Administration of silica particles or anti-Lyt2 antibody prevents β-cell destruction in NOD mice given cyclophosphamide. Diabetes. 1988;37:930–935. doi: 10.2337/diab.37.7.930. [DOI] [PubMed] [Google Scholar]
- 5.Miller BJ, Appel MC, O'Neil J, Wicker LS. Both the LYT-2+ and L3T4+T cell subsets are required for transfer of diabetes in nonobese diabetic mice. J Immunol. 1988;140:52–58. [PubMed] [Google Scholar]
- 6.Kurrer MO, Pakala SV, Hanson HL, Katz JD. Beta cell apoptosis in T cell-mediated autoimmune diabetes. Proc Natl Acad Sci USA. 1997;94:213–218. doi: 10.1073/pnas.94.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O'Brien BA, Harmon BV, Cameron DP, Allan DJ. Apoptosis is the mode of beta-cell death responsible for the development of IDDM in the nonobese diabetic (NOD) mouse. Diabetes. 1997;46:750–757. doi: 10.2337/diab.46.5.750. [DOI] [PubMed] [Google Scholar]
- 8.O'Brien BA, Harmon BV, Cameron DP, Allan DJ. Beta-cell apoptosis is responsible for the development of IDDM in the multiple low-dose streptozotocin model. J Pathol. 1996;178:176–181. doi: 10.1002/(SICI)1096-9896(199602)178:2<176::AID-PATH433>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 9.Pober JS, Cotran RS. Cytokines and endothelial cell biology. Physiol Rev. 1990;70:427–451. doi: 10.1152/physrev.1990.70.2.427. [DOI] [PubMed] [Google Scholar]
- 10.Pujol-Borrell R, Todd I, Doshi M, Bottazzo GF, Sutton R, Gray D, Adolf G, Feldmann M. HLA class II induction on human islets by interferon-gamma plus tumor necrosis factor or lymphotoxin. Nature. 1987;326:304–306. doi: 10.1038/326304a0. [DOI] [PubMed] [Google Scholar]
- 11.Feldmann M, Brennan FM, Maini R. Role of cytokines in rheumatoid arthritis. Annu Rev Immunol. 1996;14:397–440. doi: 10.1146/annurev.immunol.14.1.397. [DOI] [PubMed] [Google Scholar]
- 12.Old LJ. Tumor necrosis factor (TNF) Science. 1985;230:630–632. doi: 10.1126/science.2413547. [DOI] [PubMed] [Google Scholar]
- 13.Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383:787–793. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- 14.Satoh J, Seino H, Abo T, Tanaka S, Shintani S, Ohta S, Tamura K, Sawai T, Nobunaga T, Oteki T, et al. Recombinant human tumor necrosis factor α suppresses autoimmune diabetes in nonobese diabetic mice. J Clin Invest. 1989;84:1345–1348. doi: 10.1172/JCI114304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacob CO, Aiso S, Michie SA, McDevitt HO, Acha-Orbea H. Prevention of diabetes in nonobese diabetic mice by tumor necrosis factor (TNF): similarities between TNF-α and interleukin 1. Proc Natl Acad Sci USA. 1990;87:968–972. doi: 10.1073/pnas.87.3.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang XD, Tisch R, Singer SM, Cao ZA, Liblau RS, Schreiber RD, McDevitt HO. Effect of tumor necrosis factor α on insulin-dependent diabetes mellitus in NOD mice. I. The early development of autoimmunity and the diabetogenic process. J Exp Med. 1994;180:995–1004. doi: 10.1084/jem.180.3.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Picarella DE, Kratz A, Li CB, Ruddle NH, Flavell RA. Transgenic tumor necrosis factor (TNF)-alpha production in pancreatic islets leads to insulitis, not diabetes. Distinct patterns of inflammation in TNF-alpha and TNF-beta transgenic mice. J Immunol. 1993;150:4136–4150. [PubMed] [Google Scholar]
- 18.Higuchi Y, Herrera P, Muniesa P, Huarte J, Belin D, Ohashi P, Aichele P, Orci L, Vassalli JD, Vassalli P. Expression of a tumor necrosis factor alpha transgene in murine pancreatic β cells results in severe and permanent insulitis without evolution towards diabetes. J Exp Med. 1992;176:1719–1731. doi: 10.1084/jem.176.6.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grewal IS, Grewal KD, Wong FS, Picarella DE, Janeway CA, Jr, Flavell RA. Local expression of transgene encoded TNF-α in islets prevents autoimmune diabetes in nonobese diabetic (NOD) mice by preventing the development of auto-reactive islet-specific T cells. J Exp Med. 1996;184:1963–1974. doi: 10.1084/jem.184.5.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cope AP, Liblau RS, Yang XD, Congia M, Laudanna C, Schreiber RD, Probert L, Kollias G, McDevitt HO. Chronic tumor necrosis factor alters T cell responses by attenuating T cell receptor signaling. J Exp Med. 1997;185:1573–1584. doi: 10.1084/jem.185.9.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang B, Andre I, Gonzalez A, Katz JD, Aguet M, Benoist C, Mathis D. Interferon gamma impacts at multiple points during the progression of autoimmune diabetes. Proc Natl Acad Sci USA. 1997;94:13844–13849. doi: 10.1073/pnas.94.25.13844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.von Herrath MG, Oldstone MB. Interferon γ is essential for destruction of β cells and development of insulin-dependent diabetes mellitus. J Exp Med. 1997;185:531–539. doi: 10.1084/jem.185.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hultgren B, Huang X, Dybdal N, Stewart TA. Genetic absence of gamma-interferon delays but does not prevent diabetes in NOD mice. Diabetes. 1996;45:812–817. doi: 10.2337/diab.45.6.812. [DOI] [PubMed] [Google Scholar]
- 24.Boehm U, Klamp T, Groot M, Howard J. Cellular responses to interferon-gamma. Annu Rev Immunol. 1997;15:749–795. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- 25.Deiss L, Galinka H, Berissi H, Cohen O, Kimichi A. Cathepsin-D protease mediates programmed cell death induced by interferon-gamma, Fas/APO-1 and TNF-α. EMBO (Eur Mol Biol Organ) J. 1996;15:3861–3870. [PMC free article] [PubMed] [Google Scholar]
- 26.Chaudhary P, Eby M, Jasmin A, Bookwalter A, Murray J, Hood L. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-κB pathway. Immunity. 1997;7:821–830. doi: 10.1016/s1074-7613(00)80400-8. [DOI] [PubMed] [Google Scholar]
- 27.Ankarcrona M, Dypbukt JM, Brune B, Nicotera P. Interleukin-1 beta-induced nitric oxide production activates apoptosis in pancreatic RINm5F cells. Exp Cell Res. 1994;213:172–177. doi: 10.1006/excr.1994.1187. [DOI] [PubMed] [Google Scholar]
- 28.Cailleau C, Diu-Hercend A, Ruuth E, Westwood R, Carnaud C. Treatment with neutralizing antibodies specific for IL-1β prevents cyclophosphamide-induced diabetes in nonobese diabetic mice. Diabetes. 1997;46:937–940. doi: 10.2337/diab.46.6.937. [DOI] [PubMed] [Google Scholar]
- 29.Corbett JA, Wang JL, Sweetland MA, Lancaster JR, Jr, McDaniel ML. Interleukin 1β induces the formation of nitric oxide by β cells purified from rodent islets of Langerhans. Evidence for the β-cell as a source and site of action of nitric oxide. J Clin Invest. 1992;90:2384–2391. doi: 10.1172/JCI116129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Itoh N, Imagawa A, Hanafusa T, Waguri M, Yamamoto K, Iwahashi H, Moriwaki M, Nakajima H, Miyagawa J, Namba M, et al. Requirement of Fas for the development of autoimmune diabetes in nonobese diabetic mice. J Exp Med. 1997;186:613–618. doi: 10.1084/jem.186.4.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chervonsky AV, Wang Y, Wong FS, Visintin I, Flavell RA, Janeway CA, Jr, Matis LA. The role of Fas in autoimmune diabetes. Cell. 1997;89:17–24. doi: 10.1016/s0092-8674(00)80178-6. [DOI] [PubMed] [Google Scholar]
- 32.Katz JD, Wang B, Haskins K, Benoist C, Mathis D. Following a diabetogenic T cell from genesis through pathogenesis. Cell. 1993;74:1089–1100. doi: 10.1016/0092-8674(93)90730-e. [DOI] [PubMed] [Google Scholar]
- 33.Huang S, Hendriks W, Althage A, Hemmi S, Bluethmann H, Kamijo R, Vilcek J, Zinkernagel RM, Aguet M. Immune response in mice that lack the interferon-gamma receptor. Science. 1993;259:1742–1745. doi: 10.1126/science.8456301. [DOI] [PubMed] [Google Scholar]
- 34.Rothe J, Lesslauer W, Lotscher H, Lang Y, Koebel P, Kontgen F, Althage A, Zinkernagel R, Steinmetz M, Bluethmann H. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. . Nature. 1993;364:798–802. doi: 10.1038/364798a0. [DOI] [PubMed] [Google Scholar]
- 35.Erickson SL, de Sauvage FJ, Kikly K, Carver-Moore K, Pitts-Meek S, Gillett N, Sheehan KC, Schreiber RD, Goeddel DV, Moore MW. Decreased sensitivity to tumour-necrosis factor but normal T-cell development in TNF receptor-2-deficient mice. Nature. 1994;372:560–563. doi: 10.1038/372560a0. [DOI] [PubMed] [Google Scholar]
- 36.MacMicking, J.D., C. Nathan, G. Hom, N. Chartrain, D.S. Fletcher, M. Trumbauer, K. Stevens, Q.W. Xie, K. Sokol, N. Hutchinson, et al. 1995. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. 81:641–650. (See published erratum 81: following 1170.) [DOI] [PubMed]
- 37.Tomonari K, Lovering E, Spence S. Correlation between the Vβ4+ CD8+ T cell population and the H-2dhaplotype. Immunogenetics. 1990;31:333–339. doi: 10.1007/BF02115007. [DOI] [PubMed] [Google Scholar]
- 38.Lacy PE, Kostianovsky M. Method for the isolation of intact islets of Langerhans from the rat pancreas. Diabetes. 1967;16:35–39. doi: 10.2337/diab.16.1.35. [DOI] [PubMed] [Google Scholar]
- 39.Sullivan FP, Ricordi C, Hauptfeld V, Lacy PE. Effect of low temperature culture and site of transplantation on hamster islet xenograft survival (hamster to mouse) Transplantation. 1987;44:465–468. doi: 10.1097/00007890-198710000-00001. [DOI] [PubMed] [Google Scholar]
- 40.Leenen PJ, de Bruijn MF, Voerman JS, Campbell PA, van Ewijk W. Markers of mouse macrophage development detected by monoclonal antibodies. J Immunol Methods. 1994;174:5–19. doi: 10.1016/0022-1759(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 41.Streeter PR, Rouse BT, Butcher EC. Immunohistologic and functional characterization of a vascular addressin involved in lymphocyte homing into peripheral lymph nodes. J Cell Biol. 1988;107:1853–1862. doi: 10.1083/jcb.107.5.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pakala SV, Kurrer MO, Katz JD. T helper 2 (Th2) T cells induce acute pancreatitis and diabetes in immune-compromised nonobese diabetic (NOD) mice. J Exp Med. 1997;186:299–306. doi: 10.1084/jem.186.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lyons AB, Parish CR. Determination of lymphocyte division by flow cytometry. J Immunol Methods. 1994;171:131–137. doi: 10.1016/0022-1759(94)90236-4. [DOI] [PubMed] [Google Scholar]
- 44.Shinkai Y, Koyasu S, Nakayama K, Murphy KM, Loh DY, Reinherz EL, Alt FW. Restoration of T cell development in RAG-2-deficient mice by functional TCR transgenes. Science. 1993;259:822–825. doi: 10.1126/science.8430336. [DOI] [PubMed] [Google Scholar]
- 45.Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269:1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- 46.Gianani R, Sarvetnick N. Viruses, cytokines, antigens, and autoimmunity. Proc Natl Acad Sci USA. 1996;93:2257–2259. doi: 10.1073/pnas.93.6.2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haskins K, McDuffie M. Acceleration of diabetes in young NOD mice with a CD4+islet-specific T cell clone. Science. 1990;249:1433–1436. doi: 10.1126/science.2205920. [DOI] [PubMed] [Google Scholar]
- 48.Haskins K, Portas M, Bradley B, Wedmann D, Lafferty KJ. T-lymphocyte clone specific for pancreatic islet antigen. Diabetes. 1988;37:1444–1448. doi: 10.2337/diab.37.10.1444. [DOI] [PubMed] [Google Scholar]
- 49.Haskins K, Portas M, Bergman B, Lafferty K, Bradley B. Pancreatic islet-specific T-cell clones from nonobese diabetic mice. Proc Natl Acad Sci USA. 1989;86:8000–8004. doi: 10.1073/pnas.86.20.8000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nagata S, Golstein P. The Fas death factor. Science. 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 51.Stassi G, Maria RD, Trucco G, Rudert W, Testi R, Galluzzo A, Giordano C, Trucco M. Nitric oxide primes pancreatic β cells for Fas-mediated destruction in insulin-dependent diabetes mellitus. J Exp Med. 1997;186:1193–1200. doi: 10.1084/jem.186.8.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McDaniel ML, Kwon G, Hill JR, Marshall CA, Corbett JA. Cytokines and nitric oxide in islet inflammation and diabetes. Proc Soc Exp Biol Med. 1996;211:24–32. doi: 10.3181/00379727-211-43950d. [DOI] [PubMed] [Google Scholar]
- 53.Jacob CO, Aiso S, Schreiber RD, McDevitt HO. Monoclonal anti-tumor necrosis factor antibody renders non-obese diabetic mice hypersensitive to irradiation and enhances insulitis development. Int Immunol. 1992;4:611–614. doi: 10.1093/intimm/4.5.611. [DOI] [PubMed] [Google Scholar]
- 54.Green EA, Eynon EE, Flavell RA. Local expression of TNF-α in neonatal NOD mice promotes diabetes by enhancing presentation of islet antigens. Immunity. 1998;9:733–743. doi: 10.1016/s1074-7613(00)80670-6. [DOI] [PubMed] [Google Scholar]
- 55.Picarella DE, Kratz A, Li CB, Ruddle NH, Flavell RA. Insulitis in transgenic mice expressing tumor necrosis factor beta (lymphotoxin) in the pancreas. Proc Natl Acad Sci USA. 1992;89:10036–10040. doi: 10.1073/pnas.89.21.10036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kurts C, Heath WR, Carbone FR, Allison J, Miller JF, Kosaka H. Constitutive class I–restricted exogenous presentation of self antigens in vivo. J Exp Med. 1996;184:923–930. doi: 10.1084/jem.184.3.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kurts C, Kosaka H, Carbone FR, Miller JF, Heath WR. Class I–restricted cross-presentation of exogenous self-antigens leads to deletion of autoreactive CD8+T cells. J Exp Med. 1997;186:239–245. doi: 10.1084/jem.186.2.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.André I, Gonzalez A, Wang B, Katz J, Benoist C, Mathis D. Checkpoints in the progression of autoimmune disease: lessons from diabetes models. Proc Natl Acad Sci USA. 1996;93:2260–2263. doi: 10.1073/pnas.93.6.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]