

Abstract
The aim of this work was to decipher how graft-versus-host disease (GVHD) affects T cell production and homeostasis. In GVHD+ mice, thymic output was decreased fourfold relative to normal mice, but was sufficient to maintain a T cell repertoire with normal diversity in terms of Vβ usage. Lymphoid hypoplasia in GVHD+ mice was caused mainly by a lessened expansion of the peripheral postthymic T cell compartment. In 5-bromo-2′-deoxyuridine pulse-chase experiments, resident T cells in the spleen of GVHD+ mice showed a normal turnover rate (proliferation and half-life). When transferred into thymectomized GVHD− secondary hosts, T cells from GVHD+ mice expanded normally. In contrast, normal T cells failed to expand when injected into GVHD+ mice. Thus, the reduced size of the postthymic compartment in GVHD+ mice was not due to an intrinsic lymphocyte defect, but to an extrinsic microenvironment abnormality. We suggest that this extrinsic anomaly is consistent with a reduced number of functional peripheral T cell niches. Therefore, our results show that GVHD-associated T cell hypoplasia is largely caused by a perturbed homeostasis of the peripheral compartment. Furthermore, they suggest that damage to the microenvironment of secondary lymphoid organs may represent an heretofore unrecognized cause of acquired T cell hypoplasia.
Keywords: adoptive transfer, bone marrow transplantation, cell survival, thymus gland, T lymphocyte subsets
Although GVHD causes damage to many recipient organs, one of the most important hallmarks of this disease is a profound defect in the development of donor- derived T cells that is responsible for a long-lasting immunodeficiency state (1–7). GVHD is initiated by host-reactive T cells that produce copious amounts of cytokines and cause direct cytotoxicity via Fas-, TNF-, and perforin-mediated pathways (8–12). This antihost allogeneic attack, commonly referred to as acute GVHD, occurs during the first few days/weeks after transplantation and is of limited time duration (13) since donor-derived antihost T cells undergo massive activation-induced cell death (Brochu, S., B. Rioux-Massé, D.C. Roy, and C. Perreault, manuscript submitted for publication). After the acute phase of GVHD has abated, most recipients are left with a chronic immunodeficiency state characterized by frequent opportunistic infections and autoimmune manifestations; i.e., the so-called chronic phase of GVHD (4, 13, 14). The immune deficiency found during chronic GVHD mainly involves T cells, and is characterized by a severe lymphoid atrophy and inadequate responses to both recall antigens and new epitopes (15–20). It remains unclear how a time-limited antihost T cell response can induce long lasting (commonly permanent) disturbances in the differentiation and homeostasis of donor-derived T cells.
During the last decade, studies using transgenic mice and transplant chimeras have shed light on the intricacies of T cell differentiation and homeostasis. It was shown that T cell production can proceed along both thymic and extrathymic pathways (21–23). This has been well ascertained by investigations using mice transplanted with congeneic histocompatible hematopoietic stem cell grafts. In young euthymic recipients, T cell reconstitution is carried out by donor hematopoietic progenitor cells that differentiate into the host's thymus (21, 23). In contrast, in athymic recipients, T cell reconstitution entirely depends on extrathymic pathways and is influenced by the presence/absence of post thymic T cells in the graft. When T lymphocytes are present in the graft, their progeny is responsible for the repopulation of host secondary lymphoid organs (21, 23). This underscores the fact that mature T cells have a considerable proliferation potential (up to 8 × 1015-fold) and can survive long term in the absence of competing recent thymic emigrants (24–27). However, when the graft does not contain T lymphocytes, reconstitution of peripheral T cell compartments can only proceed through extrathymic maturation of donor hematopoietic progenitors (22, 23, 28). In various mouse models, extrathymic differentiation of hematopoietic stem cells has been detected in the bone marrow (28, 29), the intestinal cryptopatches (30), the liver (22), and the lymph nodes (31). The ability of these organs to replenish and maintain lymph node and spleen T cell compartments is considered to be much inferior to that of the thymus.
The number of peripheral CD4+ and CD8+ T lymphocytes is tightly regulated by poorly defined homeostatic mechanisms that do not rely only on the cellular input into the peripheral compartment (24, 32, 33). In support of this, thymic output is not influenced by downstream alterations in peripheral T cell pool size or CD4/CD8 ratio (34, 35) and, furthermore, the size of peripheral T cell compartments shows relatively modest variations when confronted with a low/absent thymic output or with a major increase in thymic output (e.g., hyperthymic mice and some transgenic models) (34–40). It thus seems that the number of available T cell niches in secondary lymphoid organs determines the size of peripheral T cell compartments (38, 41). The term niche designates an environment that provides local conditions (such as expression of specific chemokines, cytokines, and MHC molecules) required for T cells to seed and survive long-term in the peripheral compartment (41, 42). Recently, a number of evidences have been presented suggesting that resident dendritic cells represent fundamental constituents of the peripheral T cell niches (43– 46). Because of their abundant expression of MHC class I and II molecules and their specific chemokine and cytokine expression profile, dendritic cells seem to have a unique ability to control the homing of postthymic T cells and to provide the continuous TCR ligation required for the survival of naïve and memory T cells in the periphery (33, 44, 47).
The goal of this work was to decipher the mechanisms responsible for the GVHD-associated hypoplasia of peripheral T cell pools. Therefore, we addressed two questions. Firstly, how does GVHD influence thymic and extrathymic generation of donor-derived T lymphocytes? Knowing that GVHD per se causes thymic dysfunction (17, 48, 49), we wanted to quantify the impact of thymic lesions on thymic output and determine whether GVHD also affected extrathymic T cell production. Secondly, how does GVHD influence T cell production, proliferation, and survival? In otherwise normal subjects, the size of peripheral T cell compartments can be maintained even in the presence of a severely decreased thymic output. Why, therefore, is chronic GVHD associated with persistent lymphoid hypoplasia? Establishing how GVHD affects the size and repertoire of peripheral T cell compartments should be instrumental in understanding why patients with chronic GVHD present more infections, autoimmune diseases, and neoplasia (14, 50–52). Our results demonstrate that GVHD not only impairs thymic production of new T cells, but also abrogates expansion of mature T cell pools in secondary lymphoid organs. Furthermore, they indicate that the defective expansion of the peripheral postthymic T cell compartment in GVHD+ mice is not due to an intrinsic T cell proliferative defect, but rather to an extrinsic anomaly consistent with a restriction in the number of functional peripheral T cell niches. These findings allow for the development of a model in which GVHD-associated lymphoid hypoplasia would represent a disease of soil (environment) rather than seed (T cell). Accordingly, damage to the stroma of the thymus and of secondary lymphoid organs inflicted during the acute phase of GVHD would be responsible for a prolonged impairment of the development of donor-derived T cells.
Materials and Methods
Mice.
The following strains of mice were used: C57BL/6J (H-2b) (Thy1.2+, Ly5.2+), B6.PL-Thy1a/Cy (B6.PL; Thy1.1+, Ly5.2+), B6.SJL-PtprcaPep3b/BoyJ (Ly5a) (B6.SJL; Thy1.2+, Ly5.1+), and A.BY-H2b H2-T18b/SnJ (ABY; Thy1.2+, Ly5.2+). Mice, originally purchased from The Jackson Laboratory, were bred and housed in specific pathogen-free conditions at the Guy-Bernier Research Center according to the standards of the Canadian Committee for Animal Protection. All mice used as primary cell donors or irradiated recipients were between 8 and 20 wk of age. For 5-bromo-2′-deoxyuridine (BrdU)1 incorporation studies, mice were given sterile drinking water containing 0.8 mg/ml BrdU (Sigma Chemical Co.), which was made fresh, changed daily, and protected from light.
Thymectomy.
At 4–8 wk of age, mice were anesthetized by intraperitoneal injection of 75 mg/kg sodium pentobarbital (Somnotol; MTC Pharmaceuticals). Thymectomy was performed with a suction cannula introduced over the suprasternal notch. Completeness of thymectomy was verified in each animal by visual inspection at the time of killing. Cell transplantation was performed at least 2 wk after surgery.
Cell Transplantation and T Cell Depletion.
Bone marrow cells were T cell depleted and transplanted as previously described (23). Recipient mice received 10 Gy total body irradiation on day 0, the day of transplant. Bone marrow cells were obtained from the tibias and femurs of donor mice and T cell depleted with a specific anti-Thy1.2 mAb (5a-8; mouse IgG) or with a rabbit anti–mouse T cells (Thy1) antiserum, both obtained from Cedarlane Labs., and rabbit serum (Low-Tox-M rabbit complement; Cedarlane Labs.) as a source of complement. Efficacy of depletion was assessed by flow cytometry. Spleen or axillary, cervical, and inguinal LN cells were harvested and washed. The number of T cells injected was determined by flow cytometry using an anti-Thy1.1 or anti-Thy1.2 Ab. Bone marrow and spleen or LN cells were given as a single intravenous injection, via the tail vein, in a volume of 0.5 ml.
mAbs.
The following Abs were obtained from PharMingen: biotinylated anti-CD8α (53-6.7; rat IgG2a) detected with Cy-chrome™–streptavidin, FITC-conjugated anti–TCR-α/β (H57-597; hamster IgG), anti-Vβ3 (KJ25; hamster IgG), anti-Vβ5.1,2 (MR9-4; mouse IgG1), anti-Vβ6 (RR4-7; rat IgG2b), anti-Vβ7 (TR310; rat IgG2b), anti-Vβ8.1,2 (MR5-2; mouse IgG2a), anti-Vβ9 (MR10-2; mouse IgG1), anti-Vβ10b (B21.5; rat IgG2a), anti-Vβ11 (RR3-15; rat IgG2b), anti-Vβ13 (MR12-3; mouse IgG1), anti-Vβ14 (14-2; rat IgM), anti-Vβ17a (KJ23; mouse IgG2a), anti-CD45.1 (Ly5.1; 104; mouse IgG2a), anti-CD45.2 (Ly5.2; A20; mouse IgG2a); PE-conjugated anti-Thy1.2 (30-H12; rat IgG2b), anti-Thy1.1 (OX-7, mouse IgG1,k), anti-CD62L (MEL-14; rat IgG2a,k), anti–TCR-γ/δ (GL3; hamster IgG), and specific Cy-chrome™-conjugated anti-CD4 (RM4-5; rat IgG2a), anti-CD8α (53-6.7; rat IgG2a) Abs, and their isotypic controls. PE-conjugated anti-CD4 (YTS 191.1, rat IgG2b), anti-CD8α (YTS 169.4, rat IgG2b) Abs and their isotypic controls were purchased from Cedarlane Labs., and the FITC-conjugated anti-BrdU Ab was purchased from Becton Dickinson.
Flow Cytometry.
Cell surface staining was performed as previously described (23). BrdU labeling was performed as described by Tough and Sprent (27). In brief, after surface staining, cells were resuspended in cold 0.15 M NaCl, fixed by dropwise addition of cold 95% ethanol, incubated for 30 min on ice, and washed with PBS. The cells were then incubated with PBS containing 1% paraformaldehyde and 0.01% Tween 20 for 30 min, pelleted, and then incubated for 30 min with 50 KU of DNase I (Sigma Chemical Co.) in 0.15 M NaCl and 4.2 mM MgCl2, pH 5. After washing, cells were incubated with FITC-conjugated anti-BrdU for 30 min and washed. Cells were analyzed on a FACScalibur® using the CellQuest program or on a FACScan® using the Lysis II program (all from Becton Dickinson).
Results and Discussion
The Influence of GVHD on T Cell Development in Allogeneic Chimeras.
Irradiated euthymic or thymectomized A.BY (Thy1.2, Ly5.2) recipients received a graft containing B6.PL (Thy1.1, Ly5.2) bone marrow cells (as a source of hematopoietic progenitors) with or without mature T cells harvested from the LNs of B6.SJL donors (Thy1.2, Ly5.1). The origin of recipient T cells was determined according to their Thy1/ Ly5 phenotype. Recipients were studied on day 100 after transplant; i.e., after the allogeneic acute phase of GVHD was terminated. We found that two factors affected the level of T cell reconstitution: the presence/absence of a host thymus and the presence/absence of GVHD (Fig. 1). Indeed, based on the number of both CD4+ and CD8+ T cells found in the spleen of day 100 chimeras, the following hierarchy was observed (Fig. 1): thymus+ GVHD− (group A) > thymus− GVHD− (group B) > thymus+ GVHD+ (group C) > thymus− GVHD+ (group D). These results confirm that, in thymectomized recipients of a T cell–depleted graft, some extrathymic differentiation of donor hematopoietic progenitors may take place, but that this cannot compensate for the absence of the classical thymic differentiation pathway (group B versus A) (22, 23, 28, 29, 53). More importantly, they show that, in terms of T cell reconstitution of secondary lymphoid organs, the impact of GVHD (groups C and D) is even more deleterious than that caused by the mere absence of thymus (group B).
Figure 1.

The influence of GVHD on T cell development in allogeneic chimeras. Irradiated euthymic (A and C) or thymectomized (B and D) A.BY (Thy1.2, Ly5.2) recipients received a graft containing 107 T cell–depleted B6.PL (Thy1.1, Ly5.2) bone marrow cells (as a source of hematopoietic progenitors) with or without mature T cells (0.4 or 1.6 × 106) harvested from the LNs of B6.SJL donors (Thy1.2, Ly5.1). Chimeras' spleen cells were analyzed by three-color staining on day 100 ± 5 posttransplant. The origin of recipient T cells was determined according to their Thy1/Ly5 phenotype. Results are presented as the mean ± SD (three to four mice per group). Note that the ordinate scale is different in A and B versus C and D. nd, not determined.
In GVHD+ euthymic recipients grafted with low or high numbers of donor T cells (group C), the number of spleen T cells was decreased 6–12-fold relative to recipients without GVHD (group A). T cell hypoplasia was slightly more severe when the number of grafted T cells was increased from 0.4 to 1.6 × 106. Interestingly, most T cells found in euthymic GVHD+ recipients (group C) did not derive from expansion of grafted postthymic T cells, but rather from the donor hematopoietic stem cells. We could ascertain that the vast majority of these T lymphocytes originated from donor hematopoietic progenitors that had differentiated in the GVHD+ thymus and not in extrathymic sites, because their numbers were decreased eightfold in athymic GVHD+ recipients (group D versus C).
In athymic hosts, the occurrence of GVHD (group D) caused an extremely severe T cell hypoplasia with T cell numbers decreased by fivefold compared with athymic GVHD− recipients (group B). Among the few T cells that were found in athymic GVHD+ mice, ∼55% were derived from grafted mature T cells, while ∼45% had the phenotype of donor hematopoietic stem cells and were likely derived from the extrathymic differentiation of these progenitors. T cells derived from grafted postthymic T cells were much less abundant (∼14-fold) in athymic GVHD+ hosts (group D) than what we previously observed in similarly treated syngeneic (GVHD−) recipients (23). Two main conclusions can be drawn from these results. First, GVHD not only impairs reconstitution via the thymic pathway, but also abrogates expansion of grafted postthymic T cells. Second, although thymus function is impaired in GVHD+ chimeras, intra-thymic maturation still represents the most effective pathway for T cell reconstitution in these mice.
The TCR Vβ Repertoire of GVHD+ Mice.
Results from Fig. 1 indicate that in GVHD+ mice, the ability of thymic-independent pathways to restore peripheral T cell pools is extremely poor and much inferior to that of the classical thymic pathway. Besides these quantitative considerations, we were interested in determining whether the different origin of T lymphocytes in athymic versus euthymic GVHD+ chimeras would have any impact on their Vβ profile. This question was addressed because a continuous thymic output can contribute to the maintenance of T cell diversity, whereas the repertoire of T cell pools that rely solely on the expansion of postthymic T cells is more prone to skewing after stochastic encounter with antigens (38, 54). Thus, we evaluated by flow cytometry the TCR Vβ profile of T cells (Thy1.1+) that differentiated in the thymus of nonthymectomized GVHD+ recipients, of (Thy1.2+) cells that originated from the expansion of grafted mature T cells in athymic GVHD+ recipients, and of age-matched A.BY controls.
In both controls and euthymic GVHD+ mice, the proportion of CD4+ and CD8+ cells bearing various Vβ elements was remarkably constant and, except for Vβ11+ cells, showed very little variation from mouse to mouse (Fig. 2). In contrast, considerable variability in the usage of Vβ chains was found in athymic GVHD+ mice (Fig. 2). Thus, among CD8+ lymphocytes, the percentage of cells expressing Vβ5 or Vβ6 elements was 14–16 and 7–7.5%, respectively, in euthymic GVHD+ mice, but 4–20 and 2–14.5%, respectively, in athymic GVHD+ mice. Analyses based on size heterogeneity or on sequence of the CDR3 region will be required to assess more precisely the diversity and clonality of these T cell populations (55, 56). Nevertheless, our results indicate that the TCR repertoire of T cells derived from the expansion of grafted mature T cells is subject to dramatic skewing in athymic GVHD+ mice. The absence of Vβ skewing in euthymic GVHD+ mice suggest that, even though their thymus is damaged, it has the ability to maintain a Vβ profile that is similar to that of age-matched controls.
Figure 2.

Skewing of Vβ usage by CD4+ and CD8+ T cells in athymic GVHD+ chimeras. Three-color staining was performed using Abs against CD4 or CD8, Thy1.1 or Thy1.2, and specific Vβ elements. Chimeras were studied on day 100 ± 5. Results are presented as the mean ± SD (three to four mice per group).
Survival of Euthymic Versus Thymectomized GVHD+ Recipients.
To address the biological significance of the residual thymus function detected in nonthymectomized GVHD+ mice, we followed up to day 95 posttransplant the survival of mice from experimental groups presented in Fig. 1 (Fig. 3). No deaths were observed either in thymectomized or nonthymectomized GVHD− recipients. In contrast, the presence/absence of the thymus significantly influenced the survival of GVHD+ mice. Indeed, during the 100-d observation period, death rates were greater in thymectomized than in nonthymectomized hosts: 25 vs. 0% and 65 vs. 10% for thymectomized versus nonthymectomized recipients grafted with 0.4 and 1.6 × 106 donor T cells, respectively. Thus, at least in mice housed under specific pathogen-free conditions, the limited T cell reconstitution afforded by the GVHD+ thymus has a major impact on posttransplant survival.
Figure 3.
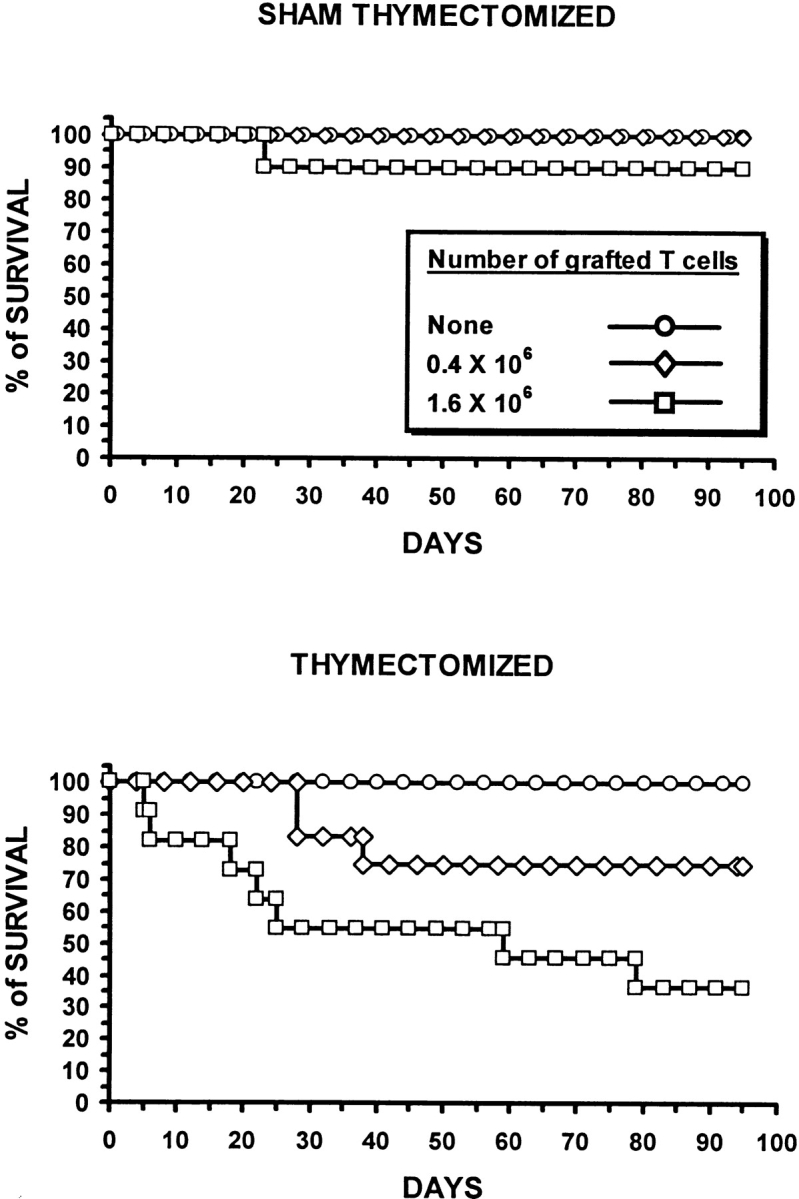
The influence of GVHD on survival of euthymic and thymectomized recipients. Irradiated thymectomized or sham thymectomized A.BY recipients received a graft (C57BL/6) containing 107 bone marrow cells with either no, 0.4 × 106, or 1.6 × 106 LN T cells (10–12 mice per group). Survival was assessed daily up to day 95. In GVHD+ mice transplanted with 0.4 or 1.6 × 106 donor T cells, the survival of thymectomized recipients was inferior to that of nonthymectomized recipients (P < 0.05, Student's t test).
Thymus Output in GVHD+ Mice.
Having found that practically all T cells found in day 100 GVHD+ mice derived from intra-thymic differentiation of donor hematopoietic stem cells, we wanted to quantitatively assess the thymic function of GVHD+ mice. Relative to normal mice, thymus cellularity was decreased by 33–50% in GVHD+ mice transplanted with a low or high number of T cells (Fig. 4 A). The proportion of immature double- versus single-positive CD4+ and CD8+ thymocytes was similar in GVHD+ versus normal thymi (Fig. 4 B). To get a better appraisal of thymus function, we measured the rate of production of recent thymic emigrants after in vivo BrdU labeling (27). Euthymic A.BY recipients of B6.PL bone marrow cells with or without 0.4 × 106 B6.SJL LN T cells were given BrdU-supplemented drinking water for 21 d beginning on day 70 posttransplant, and three-color flow cytometry analyses were performed selectively on CD62L+ T cells. For both the CD4+ and the CD8+ subsets, recent thymic emigrants were defined as CD62L+BrdUlo cells. The CD62L− T cell subset contains antigen-experienced activated/memory T cells, while the CD62L+ subset contains practically all naïve T cells and some “revertant” antigen-experienced T cells (27, 57). The intensity of BrdU labeling provides a convenient way to distinguish recent thymic emigrants (BrdUlo) from “older” peripheral T cells (BrdUhi) that divide during the BrdU-labeling period. Low BrdU labeling of recent thymic emigrants results from cold target competition in the thymus (27). Indeed, high levels of apoptosis with local breakdown of DNA, as found in the thymus but not in secondary lymphoid organs, leads to cold target competition for BrdU incorporation.
Figure 4.

The effect of GVHD on thymic cellularity and thymic output. (A) Thymic cellularity and (B) proportion of double- and single-positive thymocytes in euthymic A.BY recipients of a B6.PL bone marrow graft supplemented with no, 0.4 × 106, or 1.6 × 106 B6.SJL LN T cells. Three mice per group. (C) Accumulation of recent thymic emigrants (CD62L+/BrdUlo cells) in the spleen of normal A.BY mice, GVHD− recipients, and GVHD+ hosts. All mice received BrdU-supplemented drinking water for 21 d, beginning on day 70 posttransplant in the case of GVHD− and GVHD+ mice. GVHD− and GVHD+ mice were euthymic A.BY recipients transplanted with 107 B6.PL bone marrow cells supplemented with no (GVHD−) or 0.4 × 106 (GVHD+) B6.SJL LN T cells. The data show the values obtained for two mice per time point. (D) Percentage of CD62L+ elements that were BrdUlo in the three groups presented in C.
After administration of BrdU for 21 d, the total number of recent thymic emigrants (CD4+/CD62L+/BrdUlo and CD8+/CD62L+/BrdUlo) per spleen was fourfold lower in GVHD+ hosts relative to normal mice and GVHD− recipients (Fig. 4 C). Thus, thymus output in GVHD+ mice was significantly decreased but not absent. Interestingly, for both CD4+ and CD8+ T cells, the percentage of CD62L+ elements that were BrdUlo was similar in mice with or without GVHD (Fig. 4 D). This suggests that the reduced size of the CD62L+ compartment, which encompasses naïve and revertant T cells, cannot be wholly ascribed to a decreased input of recent thymic emigrants. If this were the case, as in senescent individuals, one would expect not only the absolute number but also the proportion of BrdUlo elements among CD62L+ cells to be decreased (58).
Results presented in Fig. 1 showed that the mere absence of thymus (group B) does not entail the severe level of lymphoid hypoplasia found in GVHD+ mice (groups C and D). In addition, we found that the thymus of GVHD+ mice was still functional with a thymocyte production equivalent to 25% that of 8-wk-old controls or 18-wk-old GVHD− recipients (Fig. 4, C and D). Such a level of thymus export has been shown to be sufficient to sustain the size of T cell compartments in secondary lymphoid organs of otherwise normal senescent mice (39, 40). Thus, thymus hypoplasia cannot be held solely responsible for GVHD- associated T cell hypoplasia. These data point to the existence of other factors that perturb peripheral homeostasis of T cell compartments in GVHD+ mice.
Kinetics of BrdU Labeling and Disappearance of BrdU-labeled Peripheral T Cells in GVHD+ Mice.
To evaluate the rate of division of peripheral T cells, BrdU labeling was performed for 21 d, as in the previous experiment. Results for CD62L+ and CD62L− subsets were analyzed separately because it has been shown that CD62L− cells divide more rapidly than CD62L+ cells (27), and the proportion of CD62L+ versus CD62L− T cells were different in our experimental groups (Fig. 5 A). Strikingly, the kinetics of BrdU labeling was similar in GVHD− recipients, GVHD+ hosts and normal controls (Fig. 5 B). This suggests that the reduced size of the CD62L+ and CD62L− peripheral compartments is not due to a proliferative defect of resident T cells, otherwise the proportion of dividing (BrdU+) T cells would have been decreased.
Figure 5.
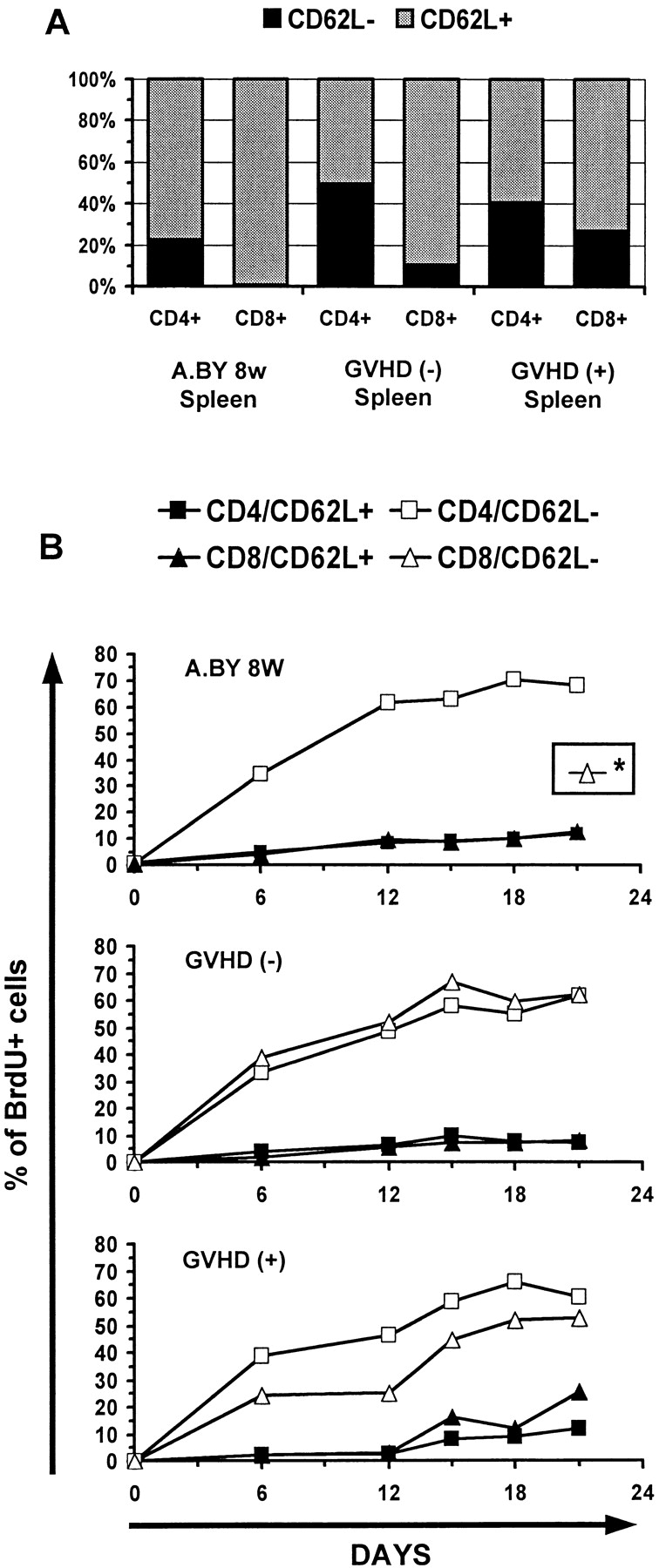
Peripheral T cells from GVHD+ mice have a normal rate of cell division (BrdU incorporation). (A) Proportion of CD62L+ and CD62L− cells in the spleen of normal A.BY mice, GVHD− recipients, and GVHD+ hosts. (B) Kinetics of BrdU labeling of T cells in the three experimental groups. Mice received BrdU-supplemented drinking water for 21 d as in Fig. 4. Results are presented as the percentage of BrdU-labeled (BrdUlo and BrdUhi) elements in four cell subsets: CD4/CD62L+, CD4/CD62L−, CD8/CD62L+, and CD8/ CD62L−. *BrdU labeling of CD8/CD62L− elements could not be studied in the A.BY control group because this cell subset is practically undetectable in 8-wk-old A.BY mice (A). Two to three mice per point. The intensity of BrdU labeling of the various T cell subsets on day 21 is presented in Fig. 6.
Since BrdU is not reused, it was possible to perform pulse–chase experiments. Thus, after being placed on BrdU water for 21 d, mice were transferred to normal water to examine the rate of decay of BrdU-labeled cells up to day 75. This approach has been used notably to show that the peripheral T cell lymphopenia of BB rats is caused by a shortened survival of T cells associated with an increased apoptotic death rate relative to WF rat controls (59). The intensity of BrdU labeling in T cells from GVHD+ and control mice on days 21 and 75 is depicted in Fig. 6. The rate of disappearance of BrdU-labeled T cells was similar in the three experimental groups so that, by day 75, the remaining proportion of BrdU-labeled cells was ∼20% in normal mice, GVHD− recipients, and GVHD+ hosts (Fig. 7). We deducted from the above BrdU-labeling experiments that the failure of peripheral homeostatic mechanisms to compensate for the moderate decrease in thymic output (Fig. 4) and to prevent the severe T cell depletion in GVHD+ mice (Fig. 1) could neither be ascribed to an impairment of T cell proliferative activity (Fig. 5), nor to a shortened half-life of BrdU-labeled cells (Fig. 7). These findings suggest that the deficient expansion of the postthymic T cell compartment in GVHD+ mice was not due to an intrinsic lymphocyte defect but to an extrinsic microenvironment abnormality. This led us to raise the intriguing possibility that the number of peripheral T cell niches might be decreased in GVHD+ mice. This premise, according to which T cell hypoplasia would represent a problem of soil (environment) rather than seed (lymphocytes), entails two crucial corollaries: (a) T cells from GVHD+ mice should expand normally in normal mice, and (b) lymphoid hypoplasia of GVHD+ mice should not be corrected by infusion of (host-tolerant) T cells from normal mice.
Figure 6.
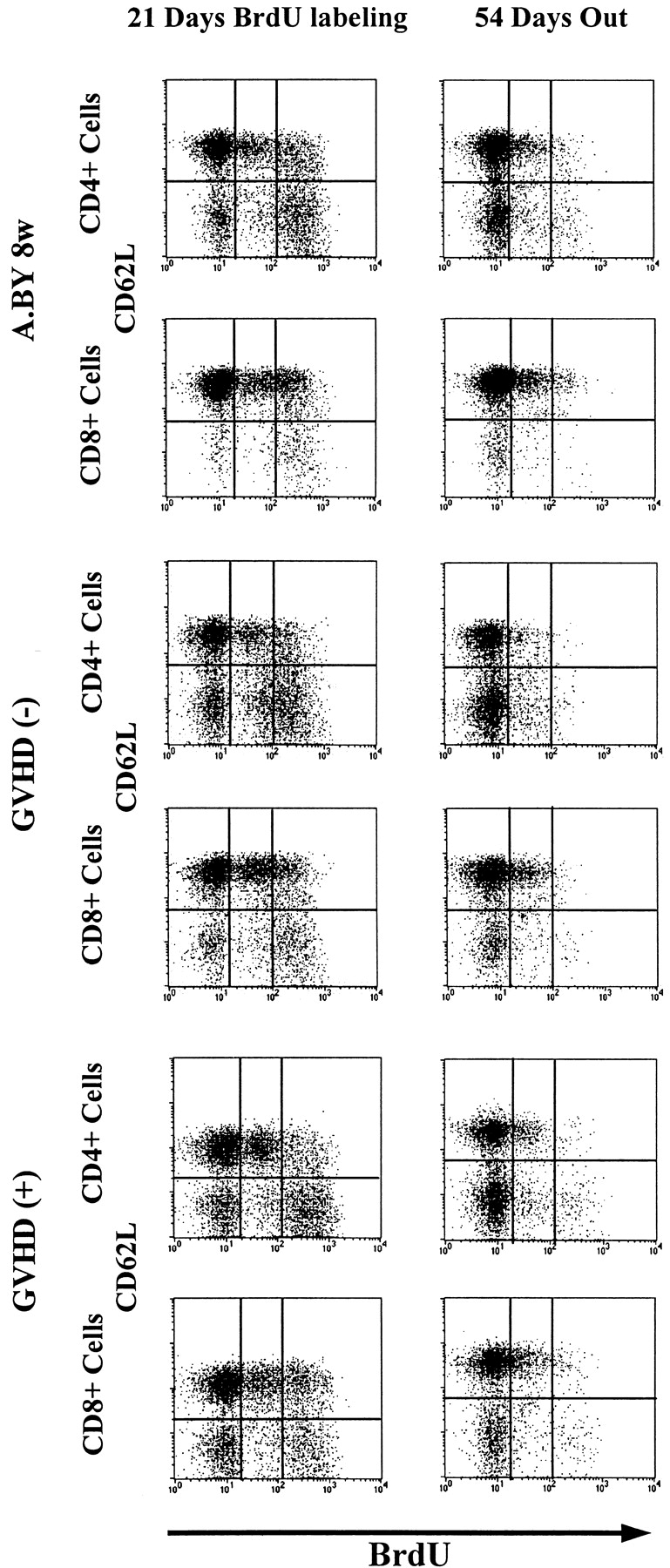
Intensity of BrdU labeling of T cells from mice placed on BrdU water for 21 d, and then on normal water for a further 54 d. The intensity of BrdU labeling (negative, low, high) is presented as a function of CD62L expression (negative or positive) for both the CD4+ and CD8+ T cell subsets. Each result is representative of three experiments.
Figure 7.
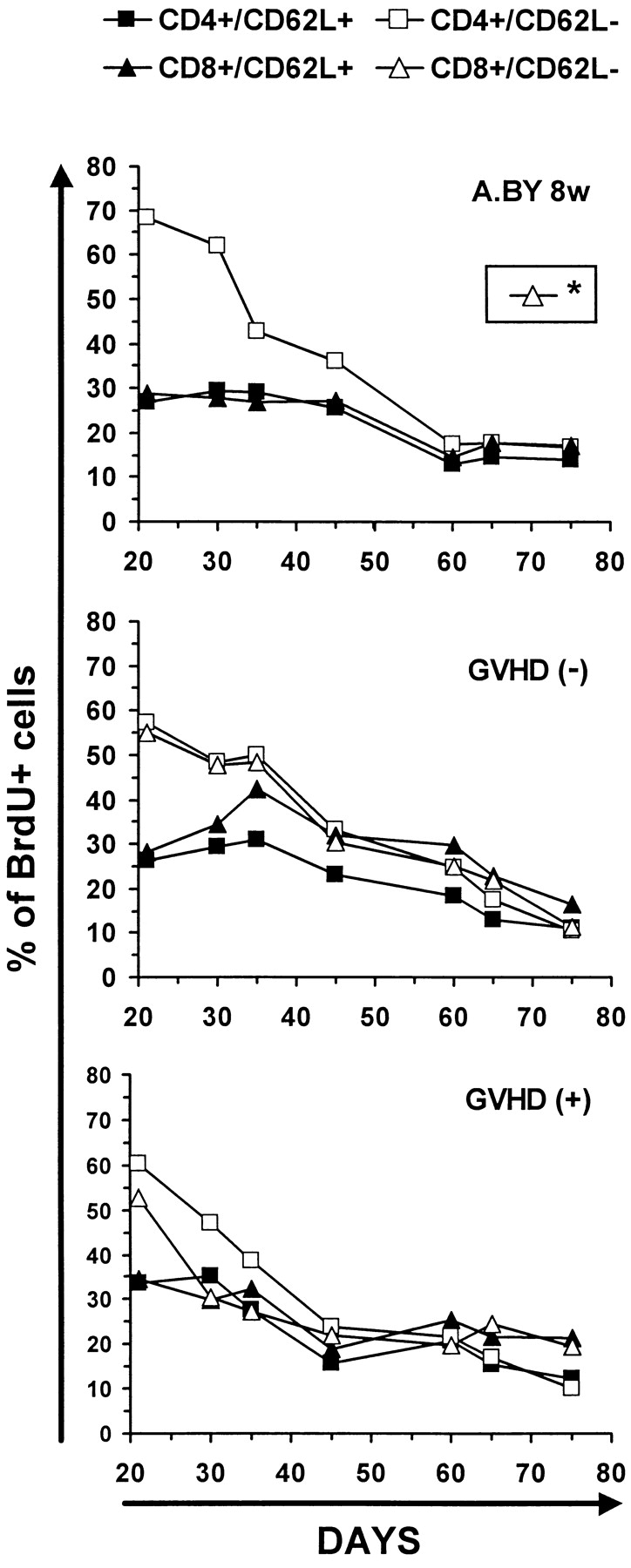
Kinetics of disappearance of BrdU-labeled T cells in GVHD− and GVHD+ mice. Decline in percent BrdU labeling of T cells after transferring mice to normal water for 54 d. The intensity of BrdU labeling on day 75 is presented in Fig. 6. Two to three mice per point. *The fate of CD8/CD62L− elements could not be studied in the A.BY control group because this cell subset is practically undetectable in 8-wk-old A.BY mice (compare Fig. 5 A).
Normal Expansion of T Cells from GVHD+ Mice after Adoptive Transfer into Normal Hosts.
To determine whether T cells from GVHD+ mice would expand in normal mice, we compared the expansion of T cells from GVHD+ mice with that of normal T cells after injection in normal thymectomized/irradiated recipients (with no defect in peripheral niches). Thus, thymectomized/irradiated A.BY or B6.SJL recipients were transplanted with 2 × 106 Thy1.1+ Ly5.2+ T cells harvested from the spleen of nonthymectomized GVHD+ mice (compare Fig. 1 C) and 107 T cell– depleted C57BL/6 bone marrow cells. The expansion of T cells from GVHD+ mice was compared with that of normal T cells 30 d after injection (together with T cell–depleted bone marrow cells) into thymectomized/irradiated syngeneic recipients (Fig. 8). In all recipients, the origin of T lymphocytes could be determined according to their Thy1/Ly5 phenotype. Strikingly, the level of T cell reconstitution found in syngeneic (B6.SJL) or allogeneic (A.BY) secondary recipients of GVHD+ T cells was similar to that of hosts repopulated with normal T cells (Fig. 8). The numbers of T cells harvested from secondary recipients were increased twofold relative to those in day 100 GVHD+ mice. This difference is impressive when one considers that (a) the T cell reconstitution of normal secondary hosts was achieved after only 30 d (versus 100 d in GVHD+ mice), and (b) GVHD+ mice had a measurable thymic output, whereas reconstitution of thymectomized secondary hosts was achieved strictly by expansion of transplanted postthymic T cells. Hence, T cells from GVHD+ mice expanded normally when transferred to normal hosts. As suggested by the studies presented in Fig. 5, this shows that the failure of T cells to expand in GVHD+ mice is not due to an intrinsic T cell–proliferative defect.
Figure 8.

Expansion of T cells from GVHD+ mice after adoptive transfer into normal hosts. Three-color staining was performed on spleen cell suspensions using Abs specific for CD4 or CD8, Ly5.1 or Ly5.2, and Thy1.1 or Thy1.2. (A) Numbers of CD4+ and CD8+ T cells of B6.PL origin (Thy1.1, Ly5.2) found in the spleen of day 100 nonthymectomized GVHD+ recipients (negative controls). (B) Numbers of CD4+ and CD8+ T cells of B6.PL origin (Thy1.1, Ly5.2) found in the spleen of irradiated/thymectomized B6.SJL or A.BY secondary hosts, on day 30 after injection of 107 T cell–depleted C57BL/6 bone marrow cells + 2 × 106 T cells harvested from the spleen of day 70 GVHD+ mice (test group). (C) Numbers of Thy1.2+Ly5.2+ T cells found in the spleen of irradiated/thymectomized B6.SJL or A.BY hosts, on day 30 after injection of 107 T cell–depleted B6.PL bone marrow cells + 2 × 106 T cells harvested from the spleen of normal syngeneic donors (A.BY for A.BY secondary recipients, C57BL/6 for B6.SJL secondary recipients) (positive controls). GVHD+ mice were prepared as in Fig. 1 C (irradiation followed by transplantation of 107 B6.PL bone marrow cells + 0.4 × 106 B6.SJL LN T cells). Results are presented as the mean ± SD (three to four mice per group). *P < 0.05, **P < 0.005 relative with numbers of CD4+ or CD8+ T cells in GVHD+ mice (Student's t test). No significant difference was found between T cell numbers presented in B and C.
Interestingly, the fact that normal reconstitution was achieved not only in syngeneic (B6.SJL) but also in allogeneic (A.BY) secondary hosts indicates that T cells found in the spleen of day 70 GVHD+ mice could not elicit GVHD, and thus were purged of functional host-reactive T cells. As these T cells had differentiated in the thymus of the GVHD+ mice, this observation supports the concept that, at least after the acute phase of GVHD, the thymus of GVHD+ mice efficiently performs negative selection of host-reactive thymocytes (60). Furthermore, the lack of antihost alloreactive T cells in day 70 GVHD+ mice argues against the possibility that a persistent GVHD activity per se could be responsible, notably via production of some cytokine(s), for the persistent T cell hypoplasia found in long-term (day 100) chimeras.
Failure of Normal Postthymic T Cells to Restore the Size of the Peripheral Pool in GVHD+ Mice.
We next evaluated whether adoptive transfer of normal postthymic T cells would correct the T cell lymphopenia of GVHD+ mice. The fate of normal T cells after passive transfer to histocompatible recipients depends on the quantity of available T cell niches. When the size of the peripheral T cell compartment is normal, such that few T cell niches are available, most donor T cells disappear soon after transfer (61). In contrast, transfer of T cells to “T-less” recipients, in which numerous empty niches are available, is followed by the persistence of a large proportion of donor-derived T cells and a considerable antigen-driven expansion of these cells, which results in the restoration of the size of the peripheral compartment (26, 62).
To obtain T cells of B6 origin that were tolerant to A.BY antigens, normal irradiated A.BY mice were transplanted with 107 T cell–depleted C57BL/6 bone marrow cells. 60 d later, a spleen cell suspension from these chimeras containing 5 × 106 Thy1.2+Ly5.2+ T cells (of C57BL/6 origin) was injected into day 60 GVHD+ secondary hosts, and the fate of Thy1.2+Ly5.2+ T cells was assessed 40 d after transfer. Before transfer, no Thy1.2+Ly5.2+ cells were present in GVHD+ secondary hosts that had been constructed by injection of 107 B6.PL bone marrow cells + 0.4 × 106 B6.SJL LN T cells into irradiated nonthymectomized A.BY hosts. As a positive control, the proliferative potential of postthymic T cells transferred into secondary hosts was evaluated in a group of irradiated/thymectomized GVHD− recipients. When transferred into GVHD− secondary hosts, postthymic T cells showed a major expansion (Fig. 9 C), whose magnitude was similar to what has been reported after transfer into irradiated/thymectomized syngeneic recipients (23). In contrast, transfer of host-tolerant C57BL/6 T cells did not increase the size of the peripheral T cell pool of GVHD+ recipients (Fig. 9 B). Indeed, 40 d after their transfer into GVHD+ secondary hosts, only trace amounts of Thy1.2+Ly5.2+ T cells were recovered. Thus, supply of postthymic T cells that had differentiated in a normal GVHD− thymus and possessed a normal proliferation potential had no influence on the size of the splenic T cell pool in GVHD+ mice. Together with those presented in Fig. 8, these results demonstrate that the perturbed homeostasis of the peripheral T cell pool in GVHD+ mice is not due to a lymphocyte intrinsic anomaly, but rather to a failure of the peripheral environment to support the seeding and/or expansion of postthymic T cells.
Figure 9.
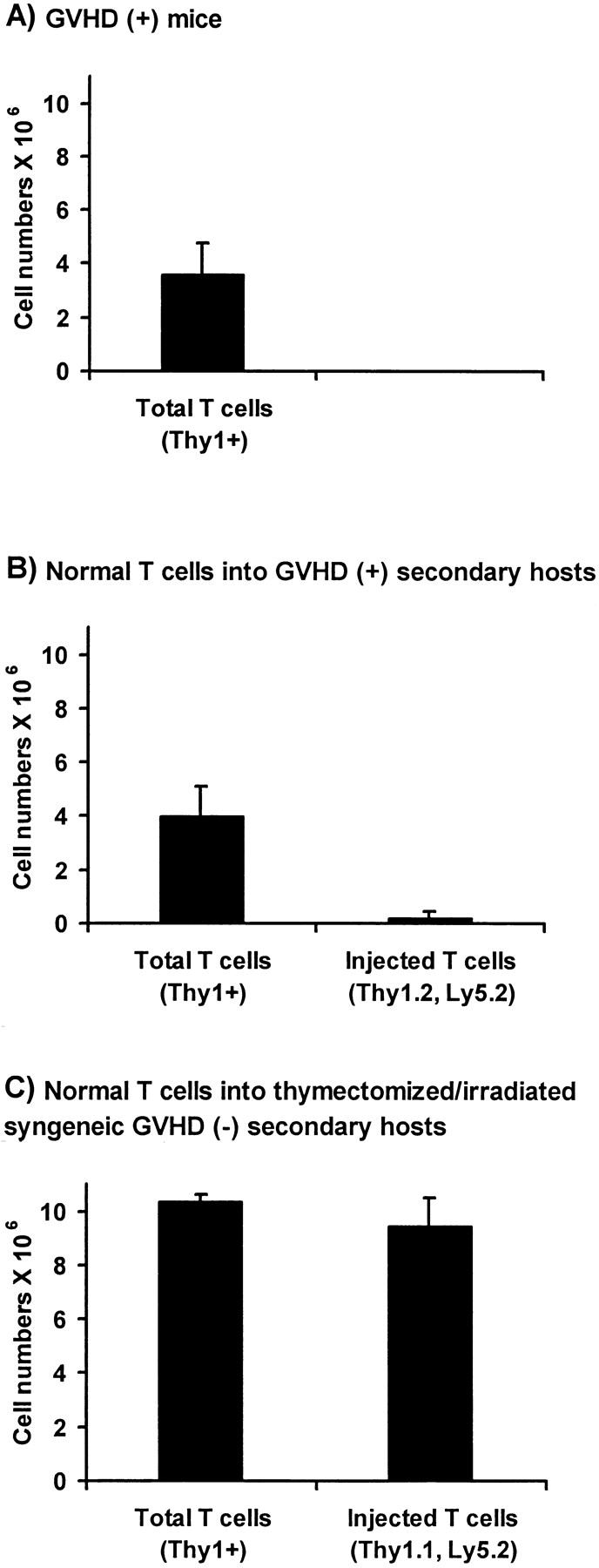
Adoptive transfer of normal host (A.BY)-tolerant T cells into nonthymectomized GVHD+ mice and thymectomized GVHD− recipients. (A) Number of T cells in the spleen of day 100 GVHD+ mice. GVHD+ mice were prepared as in Fig. 1 C (irradiation followed by transplantation of 107 B6.PL bone marrow cells + 0.4 × 106 B6.SJL T cells). (B) Number of T cells in the spleen of day 100 GVHD+ mice previously injected, on day 60, with 5 × 106 host-tolerant T cells of C57BL/6 (Thy1.2,Ly5.2) origin. (C) Number of T cells in the spleen of thymectomized A.BY mice 40 d after irradiation and injection of 107 T cell–depleted B6.SJL bone marrow cells + 5 × 106 host-tolerant B6.PL T cells. Host (A.BY)-tolerant T cells used in B and C experiments were obtained from the spleen of nonthymectomized A.BY hosts, 60 d after irradiation and injection of T cell–depleted C57BL/6 or B6.PL bone marrow cells. Results are presented as the mean ± SD (three to four mice per group).
In conclusion, the results presented in this paper show that both central and peripheral mechanisms contribute to the long lasting T cell hypoplasia found in GVHD+ mice. GVHD+ mice have a decreased thymic output, but this defect cannot be held solely responsible for peripheral T cell hypoplasia. The lack of reconstitution of normal peripheral T cell compartments is largely accounted for by an inability of postthymic T cells to expand in the secondary lymphoid organs of GVHD+ hosts. Collectively, our results provide convincing evidence that the failure of mature T cells to expand in GVHD+ mice is not due to an intrinsic lymphocyte defect, but to an undefined abnormality of the lymphoid microenvironment: (a) postthymic T cells from GVHD+ mice expanded normally in syngeneic or allogeneic secondary hosts, and (b) normal T cells failed to expand in GVHD+ mice.
Until it is better defined, we think that this microenvironment defect is most consistent with a decrease in the number of functional peripheral T cell niches. As mentioned in the introduction, resident dendritic cells may represent the most crucial elements of these niches. Thus, the possible influence of quantitative and/or qualitative (e.g., expression of MHC molecules, chemokines, cytokines) dendritic cell defects on T cell homeostasis in GVHD deserves further investigation. To our knowledge, these questions have not been addressed in mouse models of GVHD. However, according to the limited information available from human studies, it is noteworthy that chronic GVHD appears to be associated with a major decrease in the numbers of dendritic cells in the skin and secondary lymphoid organs (63–65). Alternatively, we cannot discard the possibility that the microenvironment defect epitomized herein as a reduction of functional T cell niches could be related to the presence of a toxic and/or absence of supportive soluble factor produced by cells other than dendritic cells. Nevertheless, two elements argue against some involvement of an inhibitory T cell–derived cytokine: no GVHD-inducing T cells were detected in our day 100 GVHD+ mice, and the defect was not transferred to secondary recipients of T cells from GVHD+ mice (Fig. 8).
These considerations emphasize the need for a precise definition of the peripheral T cell niches, whose number likely dictates the size of peripheral T cell pools. Furthermore, it will be important to determine whether a similar microenvironment defect could represent an unrecognized cause of acquired immunodeficiency in other settings. For example, damage to peripheral T cell niches could provide a plausible explanation for the prolonged hypoplasia of secondary lymphoid organs observed after massive T cell responses (66, 67). Importantly, a loss of peripheral T cell niches must not be considered irreversible a priori, and may possibly be amenable to therapy. Indeed, recent studies in RIP-LT transgenic mice have demonstrated that engineered local release of lymphotoxin can trigger lymphoid neogenesis characterized by the formation of well organized and functional “tertiary” lymphoid tissue (68, 69).
Acknowledgments
We are grateful to Drs. Guy Sauvageau and Stan Vukmanovic for their thoughtful review of the manuscript.
This work was supported by a grant from the National Cancer Institute of Canada to C. Perrault. D.-C. Roy is a senior scholar of the Fonds de la Recherche en Santé du Québec.
Abbreviation used in this paper
- BrdU
5-bromo-2′-deoxyuridine
References
- 1.Noel DR, Witherspoon RP, Storb R, Atkinson K, Doney K, Mickelson EM, Ochs HD, Warren RP, Weiden PL, Thomas ED. Does graft-versus-host disease influence the tempo of immunologic recovery after allogeneic human marrow transplantation? An observation on 56 long-term survivors. Blood. 1978;51:1087–1105. [PubMed] [Google Scholar]
- 2.Atkinson K, Storb R, Prentice RL, Weiden PL, Witherspoon RP, Sullivan K, Noel D, Thomas ED. Analysis of late infections in 89 long-term survivors of bone marrow transplantation. Blood. 1979;53:720–731. [PubMed] [Google Scholar]
- 3.Rozans MK, Smith BR, Burakoff SJ, Miller RA. Long-lasting deficit of functional T cell precursors in human bone marrow transplant recipients revealed by limiting dilution methods. J Immunol. 1986;136:4040–4048. [PubMed] [Google Scholar]
- 4.Via CS, Shearer GM. T-cell interactions in autoimmunity: insights from a murine model of graft-versus-host disease. Immunol Today. 1988;9:207–213. doi: 10.1016/0167-5699(88)91215-7. [DOI] [PubMed] [Google Scholar]
- 5.Storek J, Witherspoon RP, Storb R. T cell reconstitution after bone marrow transplantation into adult patients does not resemble T cell development in early life. Bone Marrow Transplant. 1995;16:413–425. [PubMed] [Google Scholar]
- 6.Kook H, Goldman F, Padley D, Giller R, Rumelhart S, Holida M, Lee N, Peters C, Comito M, Huling D, Trigg M. Reconstruction of the immune system after unrelated or partially matched T-cell–depleted bone marrow transplantation in children: immunophenotypic analysis and factors affecting the speed of recovery. Blood. 1996;88:1089–1097. [PubMed] [Google Scholar]
- 7.Hakim, F.T., and C.L. Mackall. 1997. The immune system: effector and target of graft-versus-host disease. In Graft-vs.-Host Disease. J.L.M. Ferrara, H.J. Degg, and S.J. Burakoff, editors. Marcel Dekker, Inc., New York. 257–289.
- 8.Korngold R, Sprent J. Lethal GVHD across minor histocompatibility barriers: nature of the effector cells and role of the H-2 complex. Immunol Rev. 1983;71:5–29. doi: 10.1111/j.1600-065x.1983.tb01066.x. [DOI] [PubMed] [Google Scholar]
- 9.Antin JH, Ferrara JL. Cytokine dysregulation and acute graft-versus-host disease. Blood. 1992;80:2964–2968. [PubMed] [Google Scholar]
- 10.Braun MY, Lowin B, French L, Acha-Orbea H, Tschopp J. Cytotoxic T cells deficient in both functional fas ligand and perforin show residual cytolytic activity yet lose their capacity to induce lethal acute graft-versus-host disease. J Exp Med. 1996;183:657–661. doi: 10.1084/jem.183.2.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baker MB, Altman NH, Podack ER, Levy RB. The role of cell-mediated cytotoxicity in acute GVHD after MHC-matched allogeneic bone marrow transplantation in mice. J Exp Med. 1996;183:2645–2656. doi: 10.1084/jem.183.6.2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Speiser DE, Bachmann MF, Frick TW, McKall-Faienza K, Griffiths E, Pfeffer K, Mak TW, Ohashi PS. TNF receptor p55 controls early acute graft-versus-host disease. J Immunol. 1997;158:5185–5190. [PubMed] [Google Scholar]
- 13.Parkman R. Clonal analysis of murine graft-vs-host disease. I. Phenotypic and functional analysis of T lymphocyte clones. J Immunol. 1986;136:3543–3548. [PubMed] [Google Scholar]
- 14.Siadak M, Sullivan KM. The management of chronic graft-versus-host disease. Blood Rev. 1994;8:154–160. doi: 10.1016/0268-960x(94)90076-t. [DOI] [PubMed] [Google Scholar]
- 15.Tsoi MS, Dobbs S, Brkic S, Ramberg R, Thomas ED, Storb R. Cellular interactions in marrow-grafted patients. II. Normal monocyte antigen-presenting and defective T-cell–proliferative functions early after grafting and during chronic graft-versus-host disease. Transplantation (Baltimore) 1984;37:556–561. [PubMed] [Google Scholar]
- 16.Wall DA, Hamberg SD, Reynolds DS, Burakoff SJ, Abbas AK, Ferrara JL. Immunodeficiency in graft-versus-host disease. I. Mechanism of immune suppression. J Immunol. 1988;140:2970–2976. [PubMed] [Google Scholar]
- 17.Lapp WS, Ghayur T, Mendes M, Seddik M, Seemayer TA. The functional and histological basis for graft-versus-host–induced immunosuppression. Immunol Rev. 1985;88:107–133. doi: 10.1111/j.1600-065x.1985.tb01155.x. [DOI] [PubMed] [Google Scholar]
- 18.Atkinson K. Reconstruction of the haemopoietic and immune systems after marrow transplantation. Bone Marrow Transplant. 1990;5:209–226. [PubMed] [Google Scholar]
- 19.Baker MB, Riley RL, Podack ER, Levy RB. Graft-versus-host-disease–associated lymphoid hypoplasia and B cell dysfunction is dependent upon donor T cell–mediated Fas-ligand function, but not perforin function. Proc Natl Acad Sci USA. 1997;94:1366–1371. doi: 10.1073/pnas.94.4.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pignata C, Sanghera JS, Soiffer RJ, Chartier S, Eder M, Pelech SL, Ritz J. Defective activation of mitogen-activated protein kinase after allogeneic bone marrow transplantation. Blood. 1996;88:2334–2341. [PubMed] [Google Scholar]
- 21.Mackall CL, Granger L, Sheard MA, Cepeda R, Gress RE. T-cell regeneration after bone marrow transplantation: differential CD45 isoform expression on thymic-derived versus thymic-independent progeny. Blood. 1993;82:2585–2594. [PubMed] [Google Scholar]
- 22.Sato K, Ohtsuka K, Hasegawa K, Yamagiwa S, Watanabe H, Asakura H, Abo T. Evidence for extrathymic generation of intermediate T cell receptor cells in the liver revealed in thymectomized, irradiated mice subjected to bone marrow transplantation. J Exp Med. 1995;182:759–767. doi: 10.1084/jem.182.3.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dulude G, Brochu S, Fontaine P, Baron C, Gyger M, Roy DC, Perreault C. Thymic and extrathymic differentiation and expansion of T lymphocytes following bone marrow transplantation in irradiated recipients. Exp Hematol. 1997;25:992–1004. [PubMed] [Google Scholar]
- 24.Rocha B, Dautigny N, Pereira P. Peripheral T lymphocytes: expansion potential and homeostatic regulation of pool sizes and CD4/CD8 ratios in vivo. Eur J Immunol. 1989;19:905–911. doi: 10.1002/eji.1830190518. [DOI] [PubMed] [Google Scholar]
- 25.Rocha B, Penit C, Baron C, Vasseur F, Dautigny N, Freitas AA. Accumulation of bromodeoxyuridine- labeled cells in central and peripheral lymphoid organs: minimal estimates of production and turnover rates of mature lymphocytes. Eur J Immunol. 1990;20:1697–1708. doi: 10.1002/eji.1830200812. [DOI] [PubMed] [Google Scholar]
- 26.Sprent J, Schaefer M, Hurd M, Surh CD, Ron Y. Mature murine B and T cells transferred to SCID mice can survive indefinitely and many maintain a virgin phenotype. J Exp Med. 1991;174:717–728. doi: 10.1084/jem.174.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tough DF, Sprent J. Turnover of naive- and memory-phenotype T cells. J Exp Med. 1994;179:1127–1135. doi: 10.1084/jem.179.4.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dejbakhsh-Jones S, Jerabek L, Weissman IL, Strober S. Extrathymic maturation of αβ T cells from hemopoietic stem cells. J Immunol. 1995;155:3338–3344. [PubMed] [Google Scholar]
- 29.Garcia-Ojeda ME, Dejbakhsh-Jones S, Weissman IL, Strober S. An alternate pathway for T cell development supported by the bone marrow microenvironment: recapitulation of thymic maturation. J Exp Med. 1998;187:1813–1823. doi: 10.1084/jem.187.11.1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saito H, Kanamori Y, Takemori T, Nariuchi H, Kubota E, Takahashi-Iwanaga H, Iwanaga T, Ishikawa H. Generation of intestinal T cells from progenitors residing in gut cryptopatches. Science. 1998;280:275–278. doi: 10.1126/science.280.5361.275. [DOI] [PubMed] [Google Scholar]
- 31.Clegg CH, Rulffes JT, Wallace PM, Haugen HS. Regulation of an extrathymic T-cell development pathway by oncostatin M. Nature. 1996;384:261–263. doi: 10.1038/384261a0. [DOI] [PubMed] [Google Scholar]
- 32.Tanchot C, Rocha B. The peripheral T cell repertoire: independent homeostatic regulation of virgin and activated CD8+ T cell pools. Eur J Immunol. 1995;25:2127–2136. doi: 10.1002/eji.1830250802. [DOI] [PubMed] [Google Scholar]
- 33.Tanchot C, Rocha B. The organization of mature T-cell pools. Immunol Today. 1998;19:575–579. doi: 10.1016/s0167-5699(98)01344-9. [DOI] [PubMed] [Google Scholar]
- 34.Gabor MJ, Scollay R, Godfrey DI. Thymic T cell export is not influenced by the peripheral T cell pool. Eur J Immunol. 1997;27:2986–2993. doi: 10.1002/eji.1830271135. [DOI] [PubMed] [Google Scholar]
- 35.Berzins SP, Boyd RL, Miller JF. The role of the thymus and recent thymic migrants in the maintenance of the adult peripheral lymphocyte pool. J Exp Med. 1998;187:1839–1848. doi: 10.1084/jem.187.11.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kisielow P, Bluthmann H, Staerz UD, Steinmetz M, von Boehmer H. Tolerance in T-cell–receptor transgenic mice involves deletion of nonmature CD4+8+ thymocytes. Nature. 1988;333:742–746. doi: 10.1038/333742a0. [DOI] [PubMed] [Google Scholar]
- 37.Volkmann A, Doffinger R, Ruther U, Kyewski BA. Insertional mutagenesis affecting programmed cell death leads to thymic hyperplasia and altered thymopoiesis. J Immunol. 1996;156:136–145. [PubMed] [Google Scholar]
- 38.Tanchot C, Rosado MM, Agenes F, Freitas AA, Rocha B. Lymphocyte homeostasis. Semin Immunol. 1997;9:331–337. doi: 10.1006/smim.1997.0090. [DOI] [PubMed] [Google Scholar]
- 39.Chen J, Astle CM, Harrison DE. Delayed immune aging in diet-restricted B6CBAT6 F1 mice is associated with preservation of naive T cells. J Gerontol A Biol Sci Med Sci. 1998;53:B330–B337. doi: 10.1093/gerona/53a.5.b330. [DOI] [PubMed] [Google Scholar]
- 40.Mackall CL, Punt JA, Morgan P, Farr AG, Gress RE. Thymic function in young/old chimeras: substantial thymic T cell regenerative capacity despite irreversible age-associated thymic involution. Eur J Immunol. 1998;28:1886–1893. doi: 10.1002/(SICI)1521-4141(199806)28:06<1886::AID-IMMU1886>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 41.Freitas AA, Agenes F, Coutinho GC. Cellular competition modulates survival and selection of CD8+ T cells. Eur J Immunol. 1996;26:2640–2649. doi: 10.1002/eji.1830261115. [DOI] [PubMed] [Google Scholar]
- 42.Teague TK, Marrack P, Kappler JW, Vella AT. IL-6 rescues resting mouse T cells from apoptosis. J Immunol. 1997;158:5791–5796. [PubMed] [Google Scholar]
- 43.Baggiolini M. Chemokines and leukocyte traffic. Nature. 1998;392:565–568. doi: 10.1038/33340. [DOI] [PubMed] [Google Scholar]
- 44.Nesic D, Vukmanovic S. MHC class I is required for peripheral accumulation of CD8+thymic emigrants. J Immunol. 1998;160:3705–3712. [PubMed] [Google Scholar]
- 45.Sallusto F, Schaerli P, Loetscher P, Schaniel C, Lenig D, Mackay CR, Qin S, Lanzavecchia A. Rapid and coordinated switch in chemokine receptor expression during dendritic cell maturation. Eur J Immunol. 1998;28:2760–2769. doi: 10.1002/(SICI)1521-4141(199809)28:09<2760::AID-IMMU2760>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 46.Willimann K, Legler DF, Loetscher M, Stuber R, Roos, Belen M, Delgado, Clark-Lewis I, Bagglioni M, Moser B. The chemokine SLC is expressed in T cell areas of lymph nodes and mucosal lymphoid tissues and attracts activated T cells via CCR7. Eur J Immunol. 1998;28:2025–2034. doi: 10.1002/(SICI)1521-4141(199806)28:06<2025::AID-IMMU2025>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 47.Tanchot C, Lemonnier FA, Perarnau B, Freitas AA, Rocha B. Differential requirements for survival and proliferation of CD8 naive or memory T cells. Science. 1997;276:2057–2062. doi: 10.1126/science.276.5321.2057. [DOI] [PubMed] [Google Scholar]
- 48.Fukushi N, Arase H, Wang B, Ogasawara K, Gotohda T, Good RA, Onoe K. Thymus: a direct target tissue in graft-versus-host reaction after allogeneic bone marrow transplantation that results in abrogation of induction of self-tolerance. Proc Natl Acad Sci USA. 1990;90:6301–6305. doi: 10.1073/pnas.87.16.6301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Desbarats J, Lapp WS. Thymic selection and thymic major histocompatibility complex class II expression are abnormal in mice undergoing graft-versus-host reactions. J Exp Med. 1993;178:805–814. doi: 10.1084/jem.178.3.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Witherspoon RP, Kopecky K, Storb RF, Flournoy N, Sullivan KM, Sosa R, Deeg HJ, Ochs HD, Cheever MA, Fefer A, Thomas ED. Immunological recovery in 48 patients following syngeneic marrow transplantation or hematological malignancy. Transplantation (Baltimore) 1982;33:143–149. doi: 10.1097/00007890-198202000-00008. [DOI] [PubMed] [Google Scholar]
- 51.Shulman HM, Sullivan KM. Graft-versus-host disease: allo- and autoimmunity after bone marrow transplantation. Concepts Immunopathol. 1988;6:141–165. [PubMed] [Google Scholar]
- 52.Deeg HJ, Socie G. Malignancies after hematopoietic stem cell transplantation: many questions, some answers. Blood. 1998;91:1833–1844. [PubMed] [Google Scholar]
- 53.Mackall CL, Gress RE. Pathways of T-cell regeneration in mice and humans: implications for bone marrow transplantation and immunotherapy. Immunol Rev. 1997;157:61–72. doi: 10.1111/j.1600-065x.1997.tb00974.x. [DOI] [PubMed] [Google Scholar]
- 54.Mackall CL, Bare CV, Granger LA, Sharrow SO, Titus JA, Gress RE. Thymic-independent T cell regeneration occurs via antigen-driven expansion of peripheral T cells resulting in a repertoire that is limited in diversity and prone to skewing. J Immunol. 1996;156:4609–4616. [PubMed] [Google Scholar]
- 55.Gapin L, Fukui Y, Kanellopoulos J, Sano T, Casrouge A, Malier V, Beaudoing E, Gautheret D, Claverie JM, Sasazuki T, Kourilsky P. Quantitative analysis of the T cell repertoire selected by a single peptide-major histocompatibility complex. J Exp Med. 1998;187:1871–1883. doi: 10.1084/jem.187.11.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Friedman TM, Gilbert M, Briggs C, Korngold R. Repertoire analysis of CD8+T cell responses to minor histocompatibility antigens involved in graft-versus-host disease. J Immunol. 1998;161:41–48. [PubMed] [Google Scholar]
- 57.Sprent J. Immunological memory. Curr Opin Immunol. 1997;9:371–379. doi: 10.1016/s0952-7915(97)80084-2. [DOI] [PubMed] [Google Scholar]
- 58.Douek DC, McFarland RD, Keiser PH, Gage EA, Massey JM, Haynes BF, Polis MA, Haase AT, Feinberg MB, Sullivan JL, et al. Changes in thymic function with age and during the treatment of HIV infection. Nature. 1998;396:690–695. doi: 10.1038/25374. [DOI] [PubMed] [Google Scholar]
- 59.Ramanathan S, Norwich K, Poussier P. Antigen activation rescues recent thymic emigrants from programmed cell death in the BB rat. J Immunol. 1998;160:5757–5764. [PubMed] [Google Scholar]
- 60.Hakim FT, Payne S, Shearer GM. Recovery of T cell populations after acute graft-vs-host reaction. J Immunol. 1994;152:58–64. [PubMed] [Google Scholar]
- 61.Modigliani Y, Coutinho G, Burlen-Defranoux O, Coutinho A, Bandeira A. Differential contribution of thymic outputs and peripheral expansion in the development of peripheral T cell pools. Eur J Immunol. 1994;24:1223–1227. doi: 10.1002/eji.1830240533. [DOI] [PubMed] [Google Scholar]
- 62.Von BH, Hafen K. The life span of naive αβ T cells in secondary lymphoid organs. J Exp Med. 1993;177:891–896. doi: 10.1084/jem.177.4.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Perreault C, Pelletier M, Landry D, Gyger M. Study of Langerhans cells after allogeneic bone marrow transplantation. Blood. 1984;63:807–811. [PubMed] [Google Scholar]
- 64.Horny HP, Horst HA, Ehninger G, Kaiserling E. Lymph node morphology after allogeneic bone marrow transplantation for chronic myeloid leukemia: a histological and immunohistological study focusing on the phenotype of the recovering lymphoid cells. Blut. 1988;57:31–40. doi: 10.1007/BF00320632. [DOI] [PubMed] [Google Scholar]
- 65.Horny HP, Ruck M, Kaiserling E, Ehninger G. Immunohistology of the human spleen after bone marrow transplantation for leukemia with special reference to the early post-transplantation period. Pathol Res Pract. 1990;186:775–783. doi: 10.1016/S0344-0338(11)80269-4. [DOI] [PubMed] [Google Scholar]
- 66.Aichele P, Brduscha-Riem K, Oehen S, Odermatt B, Zinkernagel RM, Hengartner H, Pircher H. Peptide antigen treatment of naive and virus-immune mice: antigen-specific tolerance versus immunopathology. Immunity. 1997;6:519–529. doi: 10.1016/s1074-7613(00)80340-4. [DOI] [PubMed] [Google Scholar]
- 67.Ehl S, Hombach J, Aichele P, Rulicke T, Odermatt B, Hengartner H, Zinkernagel R, Pircher H. Viral and bacterial infections interfere with peripheral tolerance induction and activate CD8+T cells to cause immunopathology. J Exp Med. 1998;187:763–774. doi: 10.1084/jem.187.5.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kratz A, Campos-Neto A, Hanson MS, Ruddle NH. Chronic inflammation caused by lymphotoxin is lymphoid neogenesis. J Exp Med. 1996;183:1461–1472. doi: 10.1084/jem.183.4.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sacca R, Turley S, Soong L, Mellman I, Ruddle NH. Transgenic expression of lymphotoxin restores lymph nodes to lymphotoxin-α-deficient mice. J Immunol. 1997;159:4252–4260. [PubMed] [Google Scholar]