

Abstract
Staphylococcus epidermidis releases factors that activate the HIV-1 long terminal repeat, induce cytokine release, and activate nuclear factor κB in cells of macrophage lineage. The active material had a mass of 34,500 daltons, was inactivated by proteases and partitioned into the phenol layer on hot aqueous phenol extraction, and thus was termed phenol-soluble modulin (PSM). High performance liquid chromatography (HPLC) of crude PSM yielded two peaks of activity designated PSM peak 1 and peak 2. MALDI-TOF (matrix-assisted laser desorption ionization-time of flight) mass spectroscopy indicated the presence of two components in peak 1, which were designated PSMα and PSMβ. Peak 2 contained a single component, designated PSMγ. Separation of PSMα and PSMβ in peak 1 could be achieved by a second HPLC procedure. The structure of each component was determined by amino acid sequence analysis and identification and sequencing of their genes. PSMα, PSMβ, and PSMγ were 22-, 44-, and 25-amino acid, respectively, strongly hydrophobic polypeptides. PSMγ was identified as Staphylococcus epidermidis delta toxin, whereas PSMα and PSMβ exhibited more distant homology to previously described staphylococcal toxins. They appeared to exist as a complex or aggregate with activity greater than the component parts. The properties of the S. epidermidis PSMs suggest that they may contribute to the systemic manifestations of Gram-positive sepsis.
Keywords: Staphylococcus epidermidis, HIV-1 long terminal repeat, phenol-soluble modulin, inflammatory polypeptide, nuclear factor κB
The systemic manifestations of sepsis are, in large part, the result of cytokine production by host cells in response to microbial products. The general term modulin has been coined to describe these virulence factors (1, 2). They include a variety of compounds of diverse structure (2). The most studied and best characterized modulin is LPS, which has been implicated in the development of the shock-like state in Gram-negative sepsis through the induction of cytokine production. However, the microbial factors responsible for the induction of Gram-positive sepsis have not been well characterized. Cell wall components such as lipoteichoic acid (LTA)1 and peptidoglycan, as well as certain exotoxins, have been implicated in some instances. However, a more generally relevant, potent Gram-positive modulin has yet to be identified (2).
We have recently reported that a factor or factors were released by staphylococci that activated the HIV-1 LTR in the transfected monocytic cell line THP-1 as measured by the induction of luciferase synthesis (3). All of the 31 strains of staphylococci tested (9 S. aureus, 11 S. epidermidis, 2 S. hemolyticus, 4 S. hominis, 2 S. capitis, 2 S. warneri, and 1 S. saprophyticus) were active, although considerable variation among strains was observed. The active components were readily released by the staphylococci as indicated by their presence in the overnight culture supernatant and in the extracellular fluid after vigorous mixing of intact bacteria in aqueous medium.
The HIV-1 LTR contains the promoter and enhancer sequences required for the initiation of HIV-1 gene transcription, including two binding sites for the transcription factor nuclear factor κB (NF-κB). NF-κB is released from a cytoplasmic complex with an inhibitory protein IκB in response to a variety of extracellular signals, and rapidly translocates as homo- or heterodimers into the nucleus where it activates a number of host defense–related genes by binding to the DNA κB consensus sequence (4, 5). We report here that the staphylococcal product also activates NF-κB in THP-1 cells and induces cytokine synthesis in THP-1 cells and monocytes. This suggests that the induction of luciferase synthesis in the transfected THP-1 cells serves not only as a marker of the activation of the HIV-1 LTR in a cell of macrophage lineage, but also, in a broader sense, as a marker of the activation of other host defense– related genes dependent on NF-κB, such as genes involved in cytokine production. The possibility that the staphylococcal products may be important in the induction of the systemic effects of Gram-positive sepsis prompted the purification of the factors released by S. epidermidis and their structural and functional characterization.
Materials and Methods
Special Reagents.
TAQ polymerase was from Boehringer-Mannheim, radiolabeled γ-[32P]ATP was from New England Nuclear, PMA, dimethylacetamide, DMSO, and polymyxin B sulfate were from Sigma Chemical Co., human recombinant TNF-α was provided by Genentech, and neutralizing polyclonal rabbit anti–human TNF-α was from Genzyme Corp. LPS from Escherichia coli 055:B5 was obtained from List Biological Labs., Inc., and LTA from S. aureus was obtained from Sigma Chemical Co. According to the suppliers, both the LPS and LTA were prepared by hot, aqueous phenol extraction using modifications of the methods of Westphal and Jann (6) and Fischer et al. (7), respectively. The LPS preparation was reported by the manufacturer to contain <1.7% protein.
Cells.
A stable transfectant of THP-1 cells (THP-1 LTRluc) containing the luciferase reporter gene under the control of the HIV-1 LTR was prepared as previously described (3). IG5, a Jurkat T cell–derived cell line containing a stably integrated HIV-1 luciferase construct (Jurkat LTRluc) (8) was obtained through the Research and Reference Reagent Program, Division of AIDS, NIAID, National Institutes of Health, Bethesda, MD. The HL-60 and U937 cell lines were obtained from the American Type Culture Collection. The cells were maintained in RPMI-1640 containing 10 mM Hepes buffer, 2 mM l-glutamine, 50 U/ml penicillin, 50 mg/ml streptomycin sulfate, and 10% heat-inactivated FBS (RF-10) (BioWhittaker). Human monocytes were isolated from peripheral blood as previously described (9). Cell viability was >98% as determined by trypan blue exclusion. Monocytes (5 × 105) were added to each well of 24-well plates (Falcon 3847 Primaria; Becton Dickinson) and allowed to adhere for 2 h. Nonadherent cells were removed by washing with PBS and the adherent cells overlaid with RF-10 plus 10% heat-inactivated human AB serum (BioWhittaker). The monolayer was incubated overnight at 37°C in a CO2 incubator and just before use was washed twice with RPMI without serum.
Activation of the HIV-1 LTR.
The components of the reaction mixture were incubated at 37°C in a CO2 incubator (5% CO2/95% air) for 6 h, and the luciferase activity was determined as previously described (3) and designated as relative light units (RLU). For studies with large numbers of samples, for example column fractions with serial dilutions of each fraction, the reaction was conducted in 96-well filter plates (MultiScreen GV; Millipore) with the stimulus added to 2.5 × 105 THP-1 LTRluc in a final volume of 250 μl RPMI. The cells were harvested, rinsed, and lysed in the wells before determination of luciferase activity as described above.
Isolation of Crude Phenol-soluble Modulin.
S. epidermidis UW-3 (University of Washington hospital strain 3; reference 3) was grown overnight at 37°C with shaking in 10 liters of IMDM with Hepes and l-glutamine (BioWhittaker) that had been brought to 1% glucose and filter-sterilized (0.22 μm Zap Cap; Schleicher and Schuell). All subsequent steps were conducted at 4°C except as noted. The bacteria were removed by centrifugation (6000 g, 30 min) and filtration, and the supernatant was concentrated by tangential (Prep/Scale CDUF 02.5LB; Millipore) and centrifugal (Centriprep 10; Amicon) ultrafiltration to ∼20 ml. The preparation was dialyzed extensively against 0.4 M NaCl in 25 kD mol wt cutoff (MWCO) tubing (Spectra/Por CE; Spectrum) and then against distilled water in 12 kD MWCO tubing (Sigma Chemical Co.). 50 ml of buffer-saturated phenol (GIBCO BRL) and enough 1 M sodium acetate (pH 4.7) was added to the retentate to bring the aqueous portion to 0.1 M. The mixture was maintained as a single phase by raising the temperature to 65°C and agitated for 1 h. After cooling and centrifugation for 15 min, the phenol layer was removed and the aqueous layer was extracted twice more with 25 ml of phenol. The pooled phenol layers were extensively dialyzed against distilled water at 4°C in 12 kD MWCO tubing. A fine precipitate formed which was vigorously mixed into the aqueous retentate. This material, designated phenol-soluble modulin (PSM), was lyophilized and stored at −20°C.
Chromatography.
Gel chromatography was performed to determine size using a fast protein liquid chromatography apparatus and an HR 10/30 Superose 12 column (Pharmacia). After equilibrating the column in PBS, 100 μl of concentrated bacterial supernatant or 100 μg of phenol-extracted material was injected and subsequently eluted with PBS at 0.4 ml/min. 2-ml fractions were collected and assayed. Molecular weight was estimated by comparing the elution time of standard proteins to the elution time of active fractions, as per manufacturer's instructions.
HPLC was performed on an LKB (now Pharmacia) 2150 instrument equipped with a Vydak 0.46 × 15 cm C4 column and a flow cell detector measuring absorbance at 214 nm. For the rapid (10 min) HPLC procedure, a 10 min gradient was run from 36 to 100% 1-propanol, and the aqueous portion was buffered with 2.5 mM ammonium acetate, pH 6.7. 100 μg of dialyzed, lyophilized PSM was injected in 20 μl 20% 1-propanol, and fractions were collected, lyophilized, and frozen. Slow (57 min) HPLC was done on pooled peak 1 material from several rapid HPLCs with a gradient from 28 to 48% 1-propanol, buffered as above. Slow (57 min) HPLC of crude material was done similarly except that the gradient was from 25 to 45% 1-propanol. For assay, the lyophilized material was dissolved in PBS with vigorous vortexing, and dilutions of these fractions were compared with a standard curve made from material that was similarly processed (in terms of lyophilization and solvent exposure) but not subjected to the HPLC separation.
Chemical Analysis of PSMα, PSMβ, and PSMγ.
Endotoxin was measured by a Limulus Amebocyte Lysate kit (BioWhittaker). Total phosphorous content after ashing of the specimen was determined by the ascorbic acid/ammonium molybdate method (10). Amino acid composition was done by AAA Laboratory on a Beckman 7300 Analyzer after 20 h of hydrolysis in 6 N HCl, 0.05% mercaptoethanol, and 0.02% phenol at 115°C. Mass spectrometry and NH2-terminal sequencing was performed by the University of Washington Biochemistry Core Facility. Mass spectral ionization was via MALDI-TOF (matrix-assisted laser desorption ionization–time of flight) on a Voyager Elite (PerSeptive Biosystems) instrument. Sequencing was done by Edman degradation on an Applied Biosystems 470A sequencer with on-line 120A phenol-thio-hydantoin analyzer. COOH-terminal sequencing was performed by partial digestion with carboxypeptidase Y and subsequent mass spectrometry, as previously described (11). Peak 1 from the rapid HPLC (see above) was rechromatographed using acetonitrile/0.05% TFA in a second, rapid HPLC, and the single peak was submitted to the Harvard Microchemistry Facility (Cambridge, MA) for tryptic digestion, separation of the fragments by HPLC, and sequencing of the fragments by Edman degradation. Fragment sequences were confirmed by mass spectrometry using a Finnigan TSQ Triple Quadrupole Mass Spectrometer. Sequence alignment was performed using the Basic Local Alignment Search Tool (BLAST).
SDS-PAGE was performed using a 12.5% gel and stained for protein with silver stain and Coomassie blue and for polysaccharide with Alcian blue–silver (12). High percentage tricine gels (16%) used the method of Schagger and von Jagow (13) and had a stacking gel of 12% and a resolving gel of 16%. Gels were fixed with 20% TCA.
Proteinase digestion of PSM was performed using proteinases immobilized on beaded agarose (proteinase K, Streptomyces griseus protease, trypsin [Sigma Chemical Co.]). Beads were washed three times with PBS and resuspended in 1 ml PBS containing 20 μg PSM. After incubation for 3 h at 37°C with agitation, the proteinases were removed by low speed (325 g) centrifugation and the supernatant was tested for its ability to activate the HIV-1 LTR in THP-1 cells.
Cytokine Production.
HL-60, U937, THP-1 (THP-1 LTRluc), and human monocytes were incubated with LPS, LTA, and the PSM preparations, as described in the figure legends, for 6 h, and the supernatant fluid collected and frozen at −20°C. At intervals the samples were thawed and TNF-α, IL-1β, and IL-6 levels were determined using commercially available ELISA kits (TNF-α, Endogen; IL-1β, Genzyme; IL-6, Immunotech).
Electrophoretic Mobility Shift Assay.
The nuclear extraction protocol is essentially that of Dignam et al. (14) as modified by Brophy et al. (15). For the preparation of nuclear extracts, 107 THP-1 LTRluc were incubated with 100 ng/ml PSM, PSM peak 1, or PSM peak 2 in RPMI at a final volume of 2.0 ml for 1 h at 37°C in 5% CO2/95% air. The protein concentration of the extracts was determined using the Bradford assay (Bio-Rad). The extracts were stored in aliquots at −70°C until use. The double stranded κB consensus oligonucleotide (5′-AGT TGA GGG GAC TTT CCC AGG C-3′), obtained from Promega, was end-labeled with γ-[32P]ATP and T4 polynucleotide kinase using the Promega Gel Shift Assay kit. For supershifts, 1 μl of NF-κB p65 rabbit antiserum (provided by Dr. Karol Bomsztyk, University of Washington, Seattle, WA), 4 μl (4 μg) of NF-κB p50 rabbit polyclonal IgG antibody (Santa Cruz Biotechnology), or 2 μl (2 μg) of NF-κB c-Rel rabbit polyclonal IgG antibody (Santa Cruz Biotechnology) were preincubated with the nuclear extracts in binding buffer for 30 min before addition of the labeled probe. For studies with cold probe, 50× cold probe was added at the time of addition of the labeled probe. Electrophoresis of the DNA-protein complexes was performed with 6% nondenaturing polyacrylamide gels as described in the Promega Gel Shift Assay System protocol.
Genetic Analysis.
S. epidermidis UW-3 DNA was obtained by incubating the bacteria with 50 μg/ml lysostaphin (Sigma Chemical Co.) for 2 h at 37°C and then using the standard isolation procedure for Gram-negative bacteria (16). Southern blots of genomic DNA digested with an array of restriction endonucleases were probed with radiolabeled, degenerate oligonucleotides based on Edman degradation data (17). Plasmids were isolated using a Quiagen kit, and sequencing was done by fluorescent dye termination on an ABI Prism Model 377 at the University of Washington Biochemistry Facility using ABI (Perkin-Elmer) dRhodamine or BigDye kits. The genetic sequence downstream of each PSM gene was obtained by anchor PCR (18, 19). PCR was performed using established procedures (20) with an Eppendorf MicroCycler. All PCR was done with a 94°C presoak for 4 min and a final extension step at 55°C for 9.5 min, and, except where noted, used 35 cycles of 15 s at 94°C, 15 s at 55°C, and 90 s at 72°C. A “doubled” PCR is one in which 1 μl from a completed PCR reaction is used as template in an identical PCR reaction.
The anchor PCR for PSMα incorporated a single-strand extension and Exonuclease III digestion as previously described (21) except that the final PCR was doubled. Sequence obtained in this manner generated the primers for direct PCR of PSMα (upstream: 5′-CGA ATA ATA CTA TTA ATA TAT TTT AAA TGA GCA AGA GTG TCA ATG G) (downstream: 5′-CAT GGT TGT AAA ACA TAT AAA AAT GCT AAT GAG TGT GAC AAT AAT TGA TGA TAA ACT GG).
Anchor PCR for PSMβ revealed that the gene occurred as two copies with ∼50 bases in between. A size-selected (5 kB) library of EcoRV cut genomic DNA in pBS was screened via colony hybridization to an oligomer specific for this PSMβ intergenic region (5′-TCT TAG TTT TTT AAA ATA TAA ATT TAA ATA ATT AAT TAG GGA GAG ATA) as well as two degenerate oligomers based on the amino acid sequence. Positive colonies were picked, grown out, and sequenced.
Anchor PCR for PSMγ yielded ∼500 bases of downstream sequence. From this sequence was generated reverse (5′-TGC TTC TCA CTT GCT TAG TTT ATA TTA GTA AAT TAT TAA GTT GGG ATG GCT CAA CAA CTC) and forward (5′-TTT GCT AGT AAC TGT AGT TTC CTT GGA CTC AGT GTT ACG TAT TAT TCT TAG CTA CCT TAA) primers for inverse PCR (22) on a Sau 3AI digested template. This inverse PCR provided sequence for the creation of upstream (5′-GAT ATT TTA CCA TAT TTA GTT TTA CAG TTG AGT ACT AAA TAT TGC TAT) and downstream (5′-CCA CAT CTT TAT AAA TAG CAT AGT TAA AGC CGT GAG C) primers for directly amplifying PSMγ.
To compensate for errors introduced by TAQ, the sequences of several direct PCR products using primers for PSMα and for PSMγ were compared, and at least three identical assignments were found for each nucleotide in the sequence shown. The gene for PSMβ was isolated as two identical, 5-kB recombinant clones sequenced in their entirety.
Statistical Analyses.
The results are presented as the mean ± SEM. The Mann-Whitney U rank-sum test (unpaired, two-tailed) was used to analyze differences for significance unless otherwise indicated. P > 0.05 was considered not significant.
Results
S. epidermidis PSM.
S. epidermidis strain UW-3 was grown overnight in glucose-enriched IMDM with Hepes and l-glutamine. This medium supported the rapid growth of the organism and, being a defined medium lacking large mol wt components (>3.5 kD), could be subsequently removed by dialysis. The culture medium, free of microorganisms, was concentrated ∼500-fold and dialyzed. About half the activity was lost if dialysis was carried out with 25 kD MWCO tubing against distilled water; these losses were prevented by dialysis against 0.4 M NaCl. A two-step dialysis program was therefore used, with 25 kD MWCO tubing in 0.4 M NaCl followed by 12 kD MWCO tubing in distilled water. Dialysis suggested a mol wt for the active material of >25,000 daltons and this was supported by gel chromatography using a Superose 12 column which indicated an effective mass for the concentrated and dialyzed material of 34,500 ± 5,900 daltons (mean ± SD, n = 3) Hot aqueous phenol extraction was performed as used in the purification of LPS (6) and LTA (7). In contrast to LPS and LTA, which partition into the aqueous layer, the S. epidermidis factor was found to be entirely in the phenol layer, thus the term phenol-soluble modulin (PSM). The mass of the active material, as measured by Superose 12 chromatography, remained ∼35 kD, allowing the phenol to be removed by dialysis. However, MALDI-TOF mass spectroscopy of the PSM did not reveal any component in the 35 kD region, but rather indicated the presence of four low mol wt components of 2,489, 2,648, 2,849, and 4,668 daltons (M + I) (Fig. 1). The recovery of activity in two large scale purifications, beginning with 10 liters of culture medium and proceeding through concentration, dialysis, and three phenol extractions was 190% (wt 74.8 mg) for preparation A and 87% (wt 230 mg) for preparation B. In five smaller scale purifications using 500 ml of culture medium and a single phenol extraction, recovery was 71 ± 29% (mean ± SD). The total amount of material was determined by weight after dialysis and lyophilization, and the recovery was determined by comparing the activity of a weighed sample of the product to that of the starting material.
Figure 1.

MALDI-TOF mass spectroscopy of S. epidermidis PSM. The large box has a range of 500–50,500 m/z and the insert is a magnification of the 2,000–5,000 m/z range. The designated masses are M+1 (H).
The S. epidermidis PSM had a small but significant activating effect on the HIV-1 LTR in THP-1 cells at 1 ng/ml with the activation increasing to a maximum at 30 ng/ml and remaining high as the PSM concentration was increased to 1 μg/ml (Fig. 2). PSM was routinely used at 100 ng/ml. In contrast to PSM, LPS did not activate the HIV-1 LTR in THP-1 cells at comparable concentrations; however, a small but significant activation was observed at 10 and 100 μg/ml. LTA from S. aureus was ineffective over the entire concentration range used. In contrast to the LTR in THP cells, the HIV-1 LTR introduced into Jurkat T cells in association with the luciferase reporter gene was unaffected by PSM as well as by LPS and LTA under comparable conditions. The background luciferase activity (29,379 ± 6,538, n = 4) remained unchanged on the addition of 0.001–1 μg/ml PSM or 0.01–100 μg/ml LPS or LTA.
Figure 2.

Activation of the HIV-1 LTR in THP-1 cells by PSM, LPS, and LTA. The reaction mixture contained 2 × 106 THP-1 LTRluc and PSM, LPS, or LTA at the concentrations indicated in RPMI at a final volume of 2.0 ml. The y-axis was broken to allow for an increased scale in the lower range to illustrate the significant effect of high concentrations of LPS. The results are the mean ± SEM of 4–13 experiments, with the asterisk indicating a significant difference from no stimulant (P < 0.05).
The activation of the HIV-1 LTR in THP-1 cells by the S. epidermidis PSM was not due to the release of TNF-α, since blocking antibody to TNF-α (diluted 1:2,000), although it inhibited activation via exogenously added TNF-α (100 U/ml), did not affect activation by PSM (100 ng/ml) (background, 1,189 ± 509 [RLU ± SEM]; PSM, 363,987 ± 95,583; PSM + anti–TNF-α antibody, 429,532 ± 105,532, NS versus PSM alone; TNF-α, 15,048 ± 5,585; TNF-α + anti–TNF-α antibody, 2,278 ± 332, P < 0.05 versus TNF-α alone; n = 4). The low activity of TNF-α as compared with PSM further supported the absence of an association.
The S. epidermidis PSM was heat resistant, requiring 15 min at 100°C for a partial loss of activity and 60–120 min for complete loss of activity (Fig. 3 A). Exposure to three different proteinases (proteinase K, Streptomyces griseus protease, trypsin) immobilized on agarose beads resulted in a loss of activity (Fig. 3 B), suggesting that the active material is proteinaceous.
Figure 3.

Effect of heat and proteinases on PSM activity. (A) PSM at 10 μg/ml in RPMI was heated in a boiling water bath for the periods indicated. For determination of luciferase synthesis, the reaction mixtures contained 2 × 106 THP-1 LTRluc, either unheated or heated PSM at 100 ng/ml, and RPMI at a final volume of 2.0 ml. The results are the mean (− background) ± SEM of four experiments. The asterisk indicates a significant difference from unheated PSM. (B) PSM was preincubated with immobilized proteinase (proteinase K, Streptomyces griseus protease, trypsin) as described under Materials and Methods and activation of the LTR compared with that produced by untreated PSM or PSM preincubated with agarose beads without bound proteinase. The results are the mean ± SEM of three experiments. The asterisk indicates a significant difference from PSM alone (P < 0.05, unpaired Student's t test).
S. epidermidis PSM Peak 1 and Peak 2.
Separation of PSM by HPLC with a C4 column, a 36–100% 1-propanol gradient, an elution rate of 1 ml/min, and an elution time of 10 min yielded two peaks of activity in the THP-1 LTRluc assay, which were designated PSM peak 1 and peak 2 (Fig. 4 A). Peak 1 had a lower absorbance at 214 nm but considerably more activity than peak 2. Approximately 25% of the activity applied to the column was recovered. Neither active peak contained phosphorous as assessed by a modified Fiske-Subborow assay. SDS-PAGE of each peak revealed a band that ran at the dye front even with high percentage tricine gels that could resolve down to 6 kD. These low mol wt bands stained positively for protein with Coomassie blue and silver stains. Silver staining was not intensified by Alcian blue pretreatment, suggesting the absence of polysaccharide.
Figure 4.
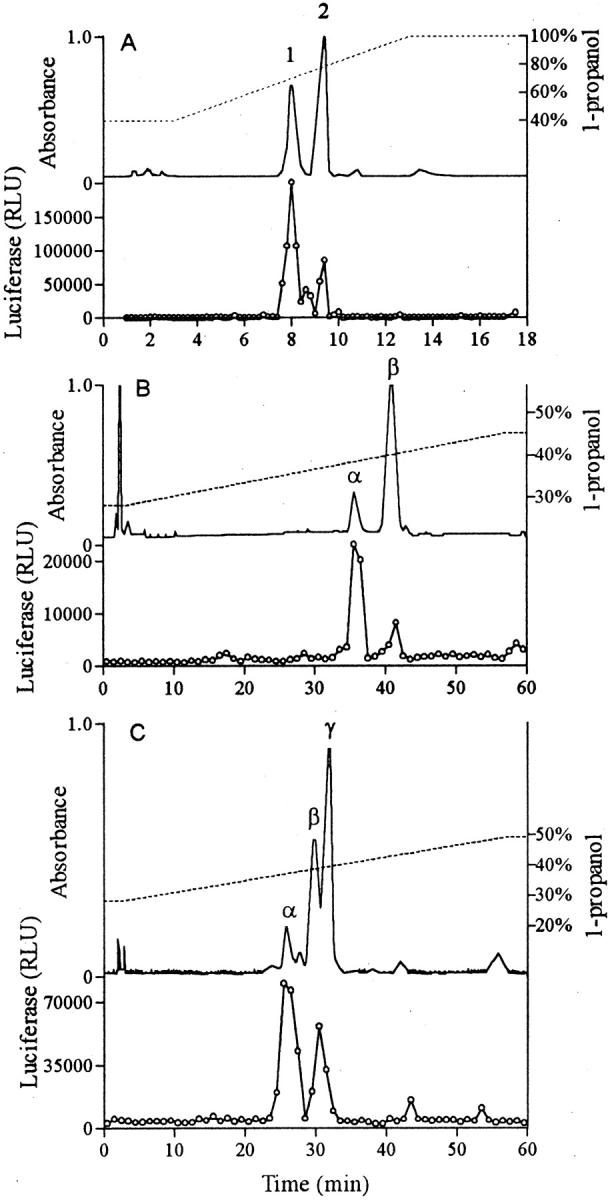
HPLC separation. The upper section of each segment indicates the absorbance at 214 nm (solid line) and the 1-propanol gradient (broken line). The lower section indicates the activity of the fractions in the THP-1 LTRluc assay. (A) Rapid HPLC of crude PSM: C4 column; 36–100%, 10 min, 1-propanol gradient; elution rate 1 ml/min. (B) HPLC of peak 1 from A above: C4 column; 25–45%, 57 min, 1-propanol gradient; elution rate 1 ml/min. (C) Slow HPLC of crude PSM: C4 column; 28–48%, 57 min, 1-propanol gradient; elution rate 1 ml/min.
The effect of PSM, PSM peak 1, and PSM peak 2 on THP-1 cells was not limited to the activation of the HIV-1 LTR. Fig. 5 demonstrates the stimulatory effect of 100 ng/ml PSM, PSM peak 1, and, to a lesser degree, PSM peak 2 on TNF-α and IL-1β production by THP-1 cells. Little IL-6 was produced by untreated THP-1 cells, with PSM producing a small but significant increase by paired analysis. As with activation of the HIV-1 LTR, LPS or LTA had no effect on the production of these cytokines by THP-1 cells. In contrast to its effect on THP-1 cells, PSM as well as LPS and LTA did not induce TNF-α production by HL-60 (cells alone, 32 ± 23; LPS, 17 ± 10; LTA, 17 ± 10; PSM, 15 ± 15 pg/ml; n = 4, NS) or U937 cells (cells alone, 14 ± 8; LPS, 11 ± 7; LTA, 11 ± 6; PSM, 9 ± 9 pg/ml; n = 4, NS).
Figure 5.
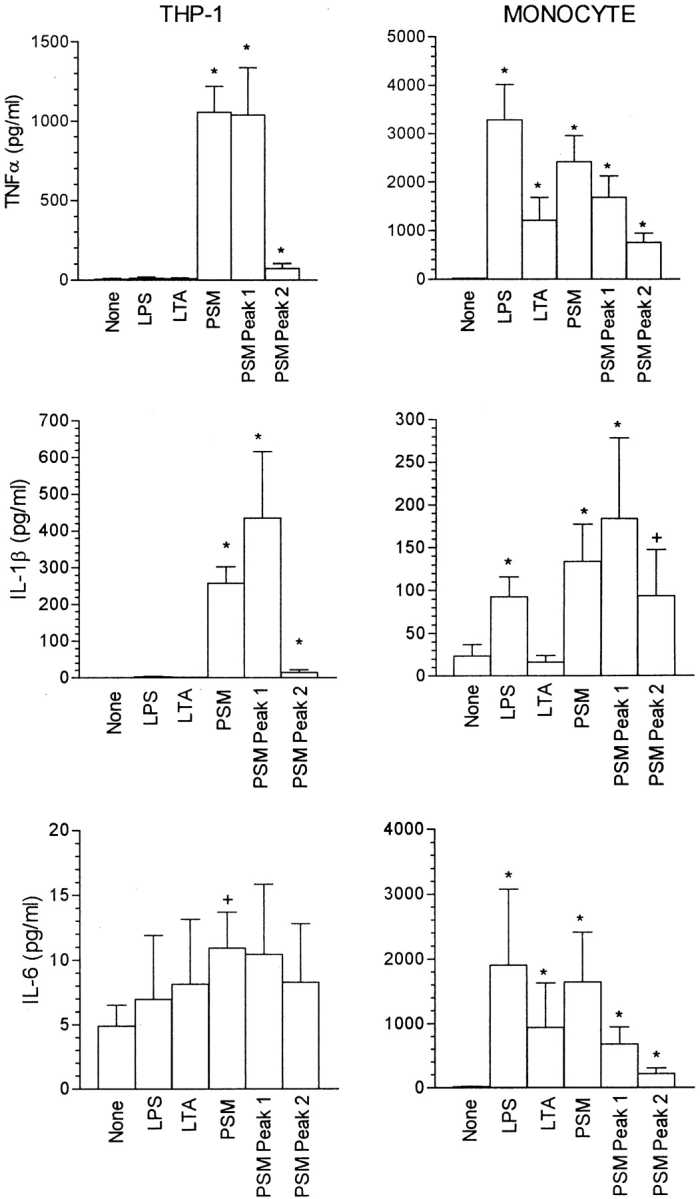
Effect of LPS, LTA, PSM, PSM peak 1, and PSM peak 2 on cytokine production by THP-1 cells and human monocytes. With THP-1 cells, the reaction mixture contained 2 × 106 THP-1 cells, and where indicated 100 ng/ml LPS, LTA, PSM, PSM peak 1, or PSM peak 2 in RPMI at a final volume of 2.0 ml. With monocytes, 100 ng/ml LPS, LTA, PSM, PSM peak 1, or PSM peak 2 were added to a monolayer of 5 × 105 monocytes in 1.0 ml of RPMI. TNF-α, IL-1β, and IL-6 levels were determined as described in Materials and Methods. The results are the mean ± SEM of four to six experiments. The asterisk and plus sign indicate a significant difference from no stimulant (none) by the unpaired and paired Mann-Whitney U tests, respectively.
TNF-α, IL-1β, and IL-6 production by human monocytes also was stimulated by PSM, PSM peak 1, and, to a lesser degree, PSM peak 2 (Fig. 5). In contrast to THP-1 cells, LPS and LTA also induced cytokine production by monocytes, although LTA did not affect monocytic production of IL-1β. PSM was less effective than LPS, but more effective than LTA as a stimulant of TNF-α production by monocytes (Fig. 6). A significant effect of LPS was observed at concentrations down to 0.1 ng/ml with the effect at 0.01 ng/ml just not significant by the Mann-Whitney U test (P = 0.07), and just significant by the Student's t test (P = 0.04). PSM was effective at concentrations down to 10 ng/ml, whereas LTA was stimulatory only at 100 ng/ml.
Figure 6.

Dose response of PSM, LPS, and LTA on TNF-α production by human blood monocytes. We added PSM, LPS, and LTA at the concentrations indicated, and RPMI at a final volume of 1.0 ml, to monolayers containing 5 × 105 monocytes. After incubation for 6 h, the supernatant fluid was removed for the determination of TNF-α levels. The results are the mean ± SEM of three to eight experiments. The asterisk indicates a significant difference from background (P < 0.05).
The passage of PSM into the phenol layer upon hot phenol extraction and the low activity of LPS or LTA suggests that the activation of the HIV-LTR in THP-1 cells by PSM is not due to the presence of LPS or LTA. Further evidence against LPS or LTA involvement was the absence of inhibition by polymyxin B. Polymyxin B alone at 1 μg/ml had no effect on the LTR in THP-1 cells (background, 1,088 ± 154 [RLU ± SEM]; polymyxin B, 1,388 ± 320; n = 4, NS), nor did it decrease the activation by 100 ng/ml PSM (PSM, 243,012 ± 29,691; PSM + polymyxin B, 259,048 ± 27,766; n = 4, NS). Polymyxin B at 1 μg/ml also had no effect on TNF-α production by monocytes induced by 100 ng/ml PSM, whereas strongly inhibiting production by 100 ng/ml LPS or LTA (PSM without polymyxin B, 2,781 ± 997 pg/ml; with polymyxin B, 2,070 ± 779; n = 5-6, NS; LPS without polymyxin B, 3,160 ± 1,074; with polymyxin B, 358 ± 140; n = 5-6, P < 0.005; LTA without polymyxin B, 1,088 ± 421; with polymyxin B, 88 ± 71; n = 5-6, P < 0.05). The endotoxin level of a reaction mixture containing 100 ng/ml of PSM in RPMI was below the level of detection (<1 pg/ml) by the Limulus Amebocyte Lysate assay.
Both the HIV-1 LTR and the promoter region of the cytokine genes contain binding sites for the transcription factor NF-κB. The activation of NF-κB in THP-1 cells by S. epidermidis PSM, PSM peak 1, and PSM peak 2, as determined by the electrophoretic mobility shift assay, is shown in Fig. 7. Nuclear extracts obtained from THP-1 cells pretreated with PSM, PSM peak 1, and PSM peak 2 produced a retardation in the migration of the 32P-labeled kB consensus oligonucleotide, which was not seen when extracts from untreated THP-1 cells were used. Antibodies to the NF-κB components p65 and p50 caused a further retardation in migration (supershift) whereas antibody to c-Rel had no effect. The specificity of the mobility shift assay for NF-κB was further supported by competition studies in which unlabeled NF-κB oligonucleotide prevented the appearance of the retarded band.
Figure 7.

Activation of NF-κB in THP-1 cells by PSM. The lanes from left to right are: 1, probe alone; 2, nuclear extract from untreated THP-1 cells; 3, nuclear extract from PSM-treated THP-1 cells alone; 4, nuclear extract from PSM-treated THP-1 cells with anti-p65 antibody; 5, nuclear extract from PSM-treated THP-1 cells with anti-p50 antibody; 6, nuclear extract from PSM-treated THP-1 cells with anti–c-Rel antibody; 7, nuclear extract from PSM-treated THP-1 cells with 10× cold probe; 8, nuclear extract from peak 1–treated THP-1 cells; and 9, nuclear extract from peak 2–treated THP-1 cells.
S. epidermidis PSMα, PSMβ, and PSMγ.
MALDI-TOF mass spectroscopy of PSM peak 1 revealed two components, a minor component with a mass of 2,503.8 daltons (designated PSMα) and a major component of 4,683.8 daltons (designated PSMβ), whereas mass spectroscopy of PSM peak 2 indicated a single component with a mass of 2,864.8 daltons (designated PSMγ). The two components in PSM peak 1 could be separated by a second, slow HPLC procedure using the same column and solvents but a longer, shallower gradient (Fig. 4 B). The composition of these peaks was verified by mass spectrometry. It was observed that the second slow HPLC of peak 1 resulted in a considerable loss of activity; <5% of the activity applied would typically be recovered. In every instance, PSMα was more active than PSMβ and in some instances PSMβ was inactive. A single HPLC separation of the S. epidermidis PSM using a slow HPLC yielded three major peaks of 214 nm-absorbing material (Fig. 4 C). The first peak, PSMα by mass spectrometry, contained most of the activity. The two later peaks were not completely separated from each other. Mass spectroscopy indicated that the peak eluting at 29–31 min contained PSMβ as its major component and that the peak eluting at 31–33 min was largely PSMγ. The luciferase assay indicated a single peak of activity that overlapped both of these later peaks.
Amino Acid Sequence Analyses.
PSMα derived from the single, slow (57 min) HPLC separation using the 28–48% 1-propanol gradient was subjected to Edman degradation, which revealed the NH2-terminal 11 amino acids. Edman degradation of peak 1 provided the NH2-terminal 24 amino acids of PSMβ. PSMα was present in peak 1 in relatively small amounts that did not interfere with the sequencing of PSMβ. The complete sequence of PSMα and PSMβ was determined by NH2-terminal Edman degradation of tryptic digests. No fragments were found that did not correspond to PSMα or PSMβ. Verification of the COOH-terminal 8 amino acids of PSMβ was obtained by carboxypeptidase Y digestion and sequential mass spectroscopy. PSM peak 2 was used for the derivation of the NH2-terminal 16 amino acids of PSMγ.
Genetic Characterization of PSMα, PSMβ, and PSMγ.
The amino acid sequence enabled the creation of degenerate oligonucleotides and the isolation of the genes for the three PSMs. Anchor PCR allowed for the determination of sequence downstream from each gene. The isolation of the entire gene resulted from additional anchor PCR (for PSMα), colony hybridization (for PSMβ), and inverse PCR (for PSMγ). Although PSMβ could be readily cloned in E. coli, PSMα and PSMγ could not, so the gene sequence for these two was determined via several directly sequenced PCRs. All coding regions were preceded by a putative Shine-Delgarno sequence and followed by a stop codon. The deduced amino acid sequence was in agreement with that found by Edman degradation and was consistent with amino acid analysis.
PSMα is a 22-amino acid polypeptide without a high degree of homology to any known protein, although some similarity to delta toxin is evident (Fig. 8). PSMβ is a 44-amino acid polypeptide (Fig. 9) whose gene was found to exist in two copies with an intervening 58 nucleotides. The deduced amino acid sequence for the two copies was identical, although slight codon variation was noted. Homology to S. hemolyticus antigonococcal polypeptides 1, 2, and 3, and S. lugdunensis SLUSH polypeptides A, B, and C is shown. PSMγ is a 25-amino acid polypeptide with the sequence MAADIISTIGDLVKWIIDTVNKFKK, which is identical to that of S. epidermidis delta toxin (23). The gene sequence of PSMγ with 100 flanking bases was identical to that reported by Otto et al. (24) except for two base differences in the flanking regions (Fig. 10).
Figure 8.

PSMα. (A) The complete amino acid and genomic squence with 100 flanking bases is shown. (B) Alignment of PSMα and delta toxin (PSMγ). Identical residues are designated as dots, and each polypeptide has a single gap to facilitate alignment.
Figure 9.

PSMβ. (A) The complete amino acid and gene sequence for both copies is shown along with the intergenic sequence and 100 bases of flanking sequence. The deduced amino acid sequence is the same for the two genes and there is a high degree of homology in the genetic sequence 30 bases upstream of each copy. (B) Homology between PSMβ and S. hemolyticus antigonococcal proteins 1, 2, and 3 and S. lugdunensis SLUSH proteins A, B, and C.
Figure 10.

PSMγ. The complete amino acid and genomic sequence with 100 flanking bases is shown. Base differences from the published sequence (24) are underlined.
The mass of PSMα, PSMβ, and PSMγ calculated from the amino acid sequence was 2,460.0, 4,639.3, and 2,820.4, respectively. Mass spectrometry of PSM before HPLC indicated the presence of components of mass 2,487.9, 4,667.2, and 2,848.5, which exceeded the calculated mass of PSMα, PSMβ, and PSMγ by ∼28 daltons in each instance, a difference compatible with formylation of the polypeptides. After HPLC, the mass of each PSM was further increased by 16 daltons, consistent with the oxidation of methionine. A fourth component of 2648.0 daltons found at low levels in the postphenol extraction PSM was not detected on the subsequent HPLC separations.
The gene sequence data, including 100 bases of flanking sequence, for PSMα, PSMβ, and PSMγ, are available from EMBL/GenBank/DDBJ under accession numbers AF068632, AF068633, and AF068634, respectively.
Discussion
We describe here modulins from S. epidermidis that partition into the phenol layer on hot aqueous phenol extraction. A number of lines of evidence indicate that the active components were neither LPS nor LTA. Both LPS and LTA partition into the aqueous layer on hot phenol extraction, whereas PSM partitioned into the phenol layer. LTA from S. aureus did not activate the HIV-1 LTR in THP-1 cells at concentrations up to 100 μg/ml and List E. coli 055:B5 LPS was only weakly active requiring concentrations of 10–100 μg/ml. In contrast, PSM was active at concentrations down to 1 ng/ml. Furthermore, polymyxin B, which binds to and inactivates LPS and LTA, had no effect on the activation of the HIV-1 LTR in THP-1 cells nor did it inhibit TNF-α production in monocytes induced by PSM. In contrast, polymyxin B strongly inhibited LPS- or LTA-induced production of TNF-α by monocytes. Active material also was devoid of sugars and phosphorous, and proteinase inactivation suggested that it was a protein or polypeptide. Finally, LPS could not be detected in the PSM preparation.
The S. epidermidis PSM could be separated into three active polypeptides, designated PSMα, PSMβ, and PSMγ. PSMα is a 22-amino acid polypeptide whose amino acid sequence shows 40% identity to S. epidermidis delta toxin when a single residue gap is placed in each polypeptide (Fig. 8 B). PSMβ is a 44-amino acid polypeptide with considerable structural homology to previously described gonococcal inhibitor 1, 2, and 3 from S. hemolyticus (25) and to a lesser degree to three structurally related peptides from S. lugdunensis with synergistic hemolytic activity (SLUSH A, B, and C) (26) (Fig. 9 B). Closest homology was to S. hemolyticus gonococcal inhibitor 2 (73% identity). The polypeptides from the three staphylococcal strains had in common similar molecular weights (S. epidermidis and S. hemolyticus, 44 amino acids; S. lugdunensis, 43 amino acids), strong hydrophobicity, and the absence of arginine, cysteine, histidine, proline, and tyrosine. It has been proposed on the basis of its primary structure that the S. hemolyticus antigonococcal proteins are signal sequences for secreted or membrane-associated proteins (25). Antibody to the S. hemolyticus polypeptides recognized a 51-kD protein in the cytoplasmic fraction of S. hemolyticus, and it was proposed that this protein may contain a signal peptide with homology to the S. hemolyticus antigonococcal proteins (27). As all three S. epidermidis PSMs have a stop codon immediately after their COOH terminus, such a role is unlikely for these molecules. Two copies of the gene for PSMβ were detected. PSMγ is identical to delta toxin of S. epidermidis (23). Delta toxin from S. epidermidis consists of 25 amino acids that share a high degree of homology with delta toxin from S. aureus. The latter consists of 26 amino acids with the substitution of glutamine for alanine at position 3 and the presence of threonine at position 24 which was absent in the S. epidermidis toxin (28). S. aureus delta toxin (delta hemolysin) is an amphipathic, helical peptide which can insert into hydrophobic membranes to form cation-selective ion channels with each channel consisting of six delta toxin alpha helices oriented with their hydrophilic face lining a central aqueous pore (29–33). Under some experimental conditions, lysis of the cell can occur. Helical wheel analysis of PSMα also suggests its orientation as an amphipathic alpha helix, with hydrophobic amino acids on one face and hydrophilic amino acids on the other.
PSMα, PSMβ, and PSMγ are all strongly hydrophobic polypeptides that lack arginine, cysteine, histidine, proline, and tyrosine. Two (PSMα and PSMγ) have some structural homology to each other. Based upon their absorption at 214 nm and normalized for molecular weight, the three components occur in a molar ratio of ∼1:2:5 PSMα/ PSMβ/PSMγ. Their low molecular weight confirms the findings with SDS-PAGE and MALDI-TOF mass spectroscopy which indicated mol wt <5 kD. However, sephadex gel chromatography either of the crude extract or the S. epidermidis PSM suggested an active product with mol wt ∼35 kD, which is consistent with its retention during dialysis and ultrafiltration. Both S. aureus delta toxin (34, 35) and S. hemolyticus gonococcal growth inhibitor (36) have been reported to form complexes or aggregates of various sizes and similar aggregation or complexing of S. epidermidis PSM appears to occur as well. It is not clear whether the polypeptides assemble into a highly ordered complex or whether they aggregate due to hydrophobic interactions with little or no order. We do not know the degree to which each polypeptide participates in the aggregate nor whether other components, proteinaceous or not, also are involved.
Although our routine assay for the S. epidermidis PSMs is the activation of the HIV-1 LTR in THP-1 cells, their effects are not limited to this reaction or to this cell type. Cytokine production (TNF-α, IL-1β, IL-6) by either THP-1 cells or peripheral blood monocytes also is increased, as is the activation of NF-κB in THP-1 cells. Both the HIV-1 LTR and the cytokine genes contain NF-κB binding sites in their promoter region, suggesting that activation of NF-κB by the S. epidermidis PSMs may be central to the other effects observed. Cell-free supernatants of Gram-positive bacteria have been reported to stimulate TNF-α production from whole human blood and, of 63 strains tested, supernatants from coagulase-negative staphylococci were the most effective (37). The S. epidermidis PSMs did not activate the HIV-1 LTR in Jurkat T cells, indicating some selectivity for cells of macrophage lineage.
We have concentrated here on the release from S. epidermidis of three small hydrophobic polypeptides which can induce gene activation through an effect on the transcription faction NF-κB. S. epidermidis can induce septic shock in humans (38–46) and a similar shock-like state in animals (47). Although most attention has focused on the role of lipoteichoic acid and peptidoglycan as the inducers of cytokine release and shock in this condition, evidence for the involvement of other factors has been presented. Thus, S. epidermidis has been reported to be a significant pathogen in neonatal necrotizing enterocolitis (NEC) (48, 49) and to be a common pathogen in a milder form of enterocolitis in infants (50). In one study (48), >90% of the S. epidermidis isolates from the stools of patients with NEC produced a toxin resembling delta toxin, and delta toxin was detected by ELISA in the stools of 11 out of 35 patients with NEC and toxin-positive staphylococci. Delta toxin was shown to be enteropathic in infant rats, producing mucosal necrosis and hemorrhage (48). These findings suggest that coagulase-negative staphylococci may be enteropathic in humans through the formation of delta toxin. Delta toxin is produced by most clinical isolates of coagulase-negative staphylococci, including strains of S. epidermidis, S. saprophyticus, and S. hemolyticus (51, 52), and a PSMβ-like molecule is produced by S. epidermidis, S. hemolyticus, and S. lugdunensis (25, 26).
Strains of S. aureus also release factors that activate the HIV-1 LTR in THP-1 cells (3) and induce the release of TNF-α and IL-1β by human monocytes (53). Essentially, all strains of S. aureus produce delta toxin; however, it is not yet known whether a PSMα- or PSMβ-equivalent molecule is produced by this organism. An S. aureus component that binds CD14 and induces IL-6 production by U373 cells and human PBMCs has been described (54, 55). This factor differs from S. epidermidis PSM in that the active material distributes into the aqueous phase on hot phenol extraction. Delta toxin has been shown to induce the release of TNF-α from human monocytes (56).
In summary, the pathogenesis of Gram-positive septic shock remains unclear. In 1996, Henderson et al. (2) described the issue as follows: “Gram-positive bacteria can produce a septic shock-like condition and as many people die each year from Gram-positive as from Gram-negative sepsis. LPS is obviously a very potent inducer of cytokine synthesis. Peptidoglycans and teichoic acids are believed to be the equivalent components, inducing cytokine-induced shock in patients with Gram-positive sepsis. However, given the relatively weak cytokine-inducing activity of peptidoglycan and teichoic acids in the studies described above, perhaps we have to look at other Gram-positive bacterial components as the inducers of the shock-like state.” The staphylococcal PSM is of interest in this regard in that it is more effective than lipoteichoic acid as an inducer of cytokine release by THP-1 cells or human monocytes. Further, it is normally shed or secreted by the bacteria as indicated by its release into the culture medium on overnight growth and into the extracellular fluid by vortexing of intact organisms. These properties make it a prime candidate for the initiation of a systemic effect contributing to septic shock in vivo.
Acknowledgments
We thank Peggy Sue O'Brien for the preparation of the manuscript.
This study was supported in part by National Institutes of Health grant AI07763.
Abbreviations used in this paper
- LTA
lipoteichoic acid
- MALDI-TOF
matrix-assisted laser desorption ionization-time of flight
- MWCO
molecular weight cut off
- NEC
neonatal necrotizing enterocolitis
- PSM
phenol-soluble modulin
- RLU
relative light units
References
- 1.Henderson B, Wilson M. Modulins: a new class of cytokine-inducing, pro-inflammatory bacterial virulence factor. Inflamm Res. 1995;44:187–197. doi: 10.1007/BF01782257. [DOI] [PubMed] [Google Scholar]
- 2.Henderson B, Poole S, Wilson M. Bacterial modulins: a novel class of virulence factors which cause host tissue pathology by inducing cytokine synthesis. Microbiol Rev. 1996;60:316–341. doi: 10.1128/mr.60.2.316-341.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klebanoff SJ, Kazazi F, Van Voorhis WC, Schlechte KG. Activation of the human immunodeficiency virus long terminal repeat in THP-1 cells by a staphylococcal extracellular product. Proc Natl Acad Sci USA. 1994;91:10615–10619. doi: 10.1073/pnas.91.22.10615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baeuerle PA, Henkel T. Function and activation of NF-κB in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 5.Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-κB. Annu Rev Cell Biol. 1994;10:405–455. doi: 10.1146/annurev.cb.10.110194.002201. [DOI] [PubMed] [Google Scholar]
- 6.Westphal O, Jann K. Bacterial lipopolysaccharides. Extraction with phenol-water and further applications of the procedure. Methods Carbohydr Chem. 1965;5:83–91. [Google Scholar]
- 7.Fischer W, Koch HU, Haas R. Improved preparation of lipoteichoic acids. Eur J Biochem. 1983;133:523–530. doi: 10.1111/j.1432-1033.1983.tb07495.x. [DOI] [PubMed] [Google Scholar]
- 8.Aguilar-Cordova E, Chinen J, Donehower L, Lewis DE, Belmont JW. A sensitive reporter cell line for HIV-1 tatactivity, HIV-1 inhibitors, and T cell activation effects. AIDS Res Hum Retrovir. 1994;10:295–301. doi: 10.1089/aid.1994.10.295. [DOI] [PubMed] [Google Scholar]
- 9.Neill MA, Henderson WR, Klebanoff SJ. Oxidative degradation of leukotriene C4 by human monocytes and monocyte-derived macrophages. J Exp Med. 1985;162:1634–1644. doi: 10.1084/jem.162.5.1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen PS, Jr, Toribara TY, Warner H. Microdetermination of phosphorus. Anal Chem. 1956;28:1756–1758. [Google Scholar]
- 11.Patterson DH, Tarr GE, Regnier FE, Martin SA. C-terminal ladder sequencing via matrix-assisted laser desorption mass spectrometry coupled with carboxypeptidase Y time-dependent and concentration-dependent digestions. Anal Chem. 1995;67:3971–3978. doi: 10.1021/ac00117a024. [DOI] [PubMed] [Google Scholar]
- 12.Corzo J, Perez-Galdona R, Leon-Barrios M, Gutierrez-Navarro AM. Alcian blue fixation allows silver staining of the isolated polysaccharide component of bacterial lipopolysaccharides in polyacrylamide gels. Electrophoresis. 1991;12:439–441. doi: 10.1002/elps.1150120611. [DOI] [PubMed] [Google Scholar]
- 13.Schagger H, von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 14.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brophy VH, Sibley CH. Expression of CD14 corrects the slow response to lipopolysaccharide in the 1B8 mutant of the B cell lymphoma 70Z/3. Immunogenetics. 1998;47:196–205. doi: 10.1007/s002510050348. [DOI] [PubMed] [Google Scholar]
- 16.Ausubel, F.M., R. Brent, R.E. Kingston, D.D. Moore, J.G. Seidman, J.A. Smith, and K. Struhl. 1989. Current Protocols in Molecular Biology. John Wiley & Sons, New York.
- 17.Ruef BJ, Hecht JH, Manning JE. A method of identifying and isolating a unique member of a multigene family: application to a trypanosome surface antigen gene. Nucleic Acids Res. 1991;19:1811–1815. doi: 10.1093/nar/19.8.1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roux KH, Dhanarajan P. A strategy for single site PCR amplification of dsDNA: priming digested cloned or genomic DNA from an anchor-modified restriction site and a short internal sequence. Biotechniques. 1990;8:48–57. [PubMed] [Google Scholar]
- 19.Templeton NS, Urcelay E, Safer B. Reducing artifact and increasing the yield of specific DNA target fragments during PCR-RACE or anchor PCR. Biotechniques. 1993;15:48–52. [PubMed] [Google Scholar]
- 20.Sambrook, J., E.F. Fritsche, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. Cold Spring Laboratory Press, Cold Spring Harbor, New York.
- 21.Willems H. Adaptor PCR for the specific amplification of unknown DNA fragments. Biotechniques. 1998;24:26–28. doi: 10.2144/98241bm03. [DOI] [PubMed] [Google Scholar]
- 22.Ochman H, Gerber AS, Hartl DL. Genetic applications of an inverse polymerase chain reaction. Genetics. 1988;120:621–623. doi: 10.1093/genetics/120.3.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKevitt AI, Bjornson GL, Mauracher CA, Scheifele DW. Amino acid sequence of a deltalike toxin from Staphylococcus epidermidis. . Infect Immun. 1990;58:1473–1475. doi: 10.1128/iai.58.5.1473-1475.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Otto M, Submuth R, Jung G, Gotz F. Structure of the pheromone peptide of the Staphylococcus epidermidis agrsystem. FEBS Lett. 1998;424:89–94. doi: 10.1016/s0014-5793(98)00145-8. [DOI] [PubMed] [Google Scholar]
- 25.Watson DC, Yaguchi M, Bisaillon J-G, Beaudet R, Morosoli R. The amino acid sequence of a gonococcal growth inhibitor from Staphylococcus haemolyticus. . Biochem J. 1988;252:87–93. doi: 10.1042/bj2520087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Donvito B, Etienne J, Denoroy L, Greenland T, Benito Y, Vandenesch F. Synergistic hemolytic activity of Staphylococcus lugdunensis is mediated by three peptides encoded by a non-agrgenetic locus. Infect Immun. 1997;65:95–100. doi: 10.1128/iai.65.1.95-100.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sevigny G, Beaudet R, McSween G, Morosoli R, Bisaillon J-G. Detection and purification of a potential precursor protein or a prohaemolysin produced by Staphylococcus haemolyticus. . Microbios. 1992;71:203–215. [PubMed] [Google Scholar]
- 28.Fitton JE, Dell A, Shaw WV. The amino acid sequence of the delta haemolysin of Staphylococcus aureus. . FEBS Lett. 1980;115:209–212. doi: 10.1016/0014-5793(80)81170-7. [DOI] [PubMed] [Google Scholar]
- 29.Freer JH, Birkbeck TH. Possible conformation of delta-lysin, a membrane-damaging peptide of Staphylococcus aureus. . J Theor Biol. 1982;94:535–540. doi: 10.1016/0022-5193(82)90299-5. [DOI] [PubMed] [Google Scholar]
- 30.Mellor IR, Thomas DH, Sansom MSP. Properties of ion channels formed by Staphylococcus aureusδ-toxin. Biochim Biophys Acta. 1988;942:280–294. doi: 10.1016/0005-2736(88)90030-2. [DOI] [PubMed] [Google Scholar]
- 31.Sansom MSP. The biophysics of peptide models of ion channels. Prog Biophys Mol Biol. 1991;55:139–235. doi: 10.1016/0079-6107(91)90004-c. [DOI] [PubMed] [Google Scholar]
- 32.Kerr ID, Dufourcq J, Rice JA, Fredkin DR, Sansom MSP. Ion channel formation by synthetic analogues of staphylococcal δ-toxin. Biochim Biophys Acta. 1995;1236:219–227. doi: 10.1016/0005-2736(95)00051-4. [DOI] [PubMed] [Google Scholar]
- 33.Kerr ID, Doak DG, Sankararamakrishnan R, Breed J, Sansom MSP. Molecular modelling of staphylococcal δ-toxin ion channels by restrained molecular dynamics. Protein Engineering. 1996;9:161–171. doi: 10.1093/protein/9.2.161. [DOI] [PubMed] [Google Scholar]
- 34.Kantor HS, Temples B, Shaw WV. Staphylococcal delta hemolysin: purification and characterization. Arch Biochem Biophys. 1972;151:142–156. doi: 10.1016/0003-9861(72)90483-3. [DOI] [PubMed] [Google Scholar]
- 35.Fitton JE. Physiochemical studies on delta haemolysin, a staphylococcal cytolytic polypeptide. FEBS Lett. 1981;130:257–260. doi: 10.1016/0014-5793(81)81133-7. [DOI] [PubMed] [Google Scholar]
- 36.Loyer M, Beaudet R, Bisaillon J-G. Comparative study of hemolytic substances produced by coagulase-negative Staphylococcusstrains. Infect Immun. 1990;58:2144–2148. doi: 10.1128/iai.58.7.2144-2148.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bayston K, Tomlinson M, Cohen J. In-vitro stimulation of TNF-α from human whole blood by cell-free supernatants of Gram-positive bacteria. Cytokine. 1992;4:397–402. doi: 10.1016/1043-4666(92)90084-5. [DOI] [PubMed] [Google Scholar]
- 38.Smith, I.M. 1981. Staphylococcus epidermidis septicemia: 1974– 1978 compared to 1949–1958. In Staphylococci and staphylococcal infections. J. Jeljaszewicz, editor. Gustav Fischer Verlag, Stuttgart, Germany. 765–772.
- 39.Christensen GD, Bisno AL, Parisi JT, McLaughlin B, Hester MG, Luther RW. Nosocomial septicemia due to a multiply antibiotic-resistant Staphylococcus epidermidis. . Ann Intern Med. 1982;96:1–10. doi: 10.7326/0003-4819-96-1-1. [DOI] [PubMed] [Google Scholar]
- 40.Burchard KW, Minor LB, Slotman GJ, Gann DS. Staphylococcus epidermidissepsis in surgical patients. Arch Surg. 1984;119:96–100. doi: 10.1001/archsurg.1984.01390130078014. [DOI] [PubMed] [Google Scholar]
- 41.Ponce de Leon S, Wenzel RP. Hospital- acquired bloodstream infections with Staphylococcus epidermidis. . Am J Med. 1984;77:639–644. doi: 10.1016/0002-9343(84)90354-1. [DOI] [PubMed] [Google Scholar]
- 42.Martin MA, Pfaller MA, Wenzel RP. Coagulase-negative staphylococcal bacteremia. Ann Intern Med. 1989;110:9–16. doi: 10.7326/0003-4819-110-1-9. [DOI] [PubMed] [Google Scholar]
- 43.Nafziger DA, Wenzel RP. Coagulase-negative staphylococci. Epidemiology, evaluation and therapy. Infect Dis Clin North Am. 1989;3:915–929. [PubMed] [Google Scholar]
- 44.Lina G, Fleer A, Etienne J, Greenland TB, Vandenesch F. Coagulase-negative staphylococci isolated from two cases of toxic shock syndrome lack superantigenic activity, but induce cytokine production. FEMS Immunol Med Microbiol. 1996;13:81–86. doi: 10.1111/j.1574-695X.1996.tb00219.x. [DOI] [PubMed] [Google Scholar]
- 45.Johnson-Robbins LA, El-Mohandes AE, Simmens SJ, Keiser JF. Staphylococcus epidermidissepsis in the intensive care nursery: a characterization of risk association in infants <1,000 g. Biol Neonat. 1996;69:249–256. doi: 10.1159/000244318. [DOI] [PubMed] [Google Scholar]
- 46.Fidalgo S, Vazquez F, Mendoza MC, Perez F, Mendez FJ. Bacteremia due to Staphylococcus epidermidis: microbiologic, epidemiologic, clinical, and prognostic features. Rev Infect Dis. 1990;12:520–528. doi: 10.1093/clinids/12.3.520. [DOI] [PubMed] [Google Scholar]
- 47.Wakabayashi G, Gelfand JA, Jung WK, Connolly RJ, Burke JF, Dinarello CA. Staphylococcus epidermidis induces complement activation, tumor necrosis factor and interleukin-1, a shock-like state and tissue injury in rabbits without endotoxemia. Comparison to Escherichia coli. . J Clin Invest. 1991;87:1925–1935. doi: 10.1172/JCI115218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scheifele DW, Bjornson GL, Dyer RA, Dimmick JE. Delta-like toxin produced by coagulase-negative staphylococci is associated with neonatal necrotizing enterocolitis. Infect Immun. 1987;55:2268–2273. doi: 10.1128/iai.55.9.2268-2273.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Molitt DL, Tepas JJ, Talbert JL. The role of coagulase-negative Staphylococcusin neonatal necrotizing enterocolitis. J Pediatr Surg. 1988;23:60–63. doi: 10.1016/s0022-3468(88)80542-6. [DOI] [PubMed] [Google Scholar]
- 50.Gruskay JA, Abbasi S, Anday E, Baumgart S, Gerdes J. Staphylococcus epidermidis-associated enterocolitis. J Pediatr. 1986;109:520–524. doi: 10.1016/s0022-3476(86)80135-4. [DOI] [PubMed] [Google Scholar]
- 51.Kleck JL, Donahue JA. Production of thermostable hemolysin by cultures of Staphylococcus epidermidis. . J Infect Dis. 1968;118:317–323. doi: 10.1093/infdis/118.3.317. [DOI] [PubMed] [Google Scholar]
- 52.Gemmell CG, Thelestam M. Toxinogenicity of clinical isolates of coagulase-negative staphylococci towards various animal cells. Acta Path Microbiol Immunol Scand Sect B. 1981;89:417–421. [PubMed] [Google Scholar]
- 53.Veldkamp KE, Van Kessel KPM, Verhoef J, Van Strijp JAG. Staphylococcal culture supernates stimulate human phagocytes. Inflammation. 1997;21:541–551. doi: 10.1023/a:1027315814817. [DOI] [PubMed] [Google Scholar]
- 54.Kusunoki T, Hailman E, Juan TS-C, Lichenstein HS, Wright SD. Molecules from Staphylococcus aureusthat bind CD14 and stimulate innate immune response. J Exp Med. 1995;182:1673–1682. doi: 10.1084/jem.182.6.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kusunoki T, Wright SD. Chemical characteristics of Staphylococcus aureusmolecules that have CD14-dependent cell-stimulating activity. J Immunol. 1996;157:5112–5117. [PubMed] [Google Scholar]
- 56.Schmitz F-J, Veldkamp K-E, Van Kessel KPM, Verhoef J, Van Strijp JAG. Delta-toxin from Staphylococcus aureusas a costimulator of human neutrophil oxidative burst. J Infect Dis. 1997;176:1531–1537. doi: 10.1086/514152. [DOI] [PubMed] [Google Scholar]