

Abstract
We have analyzed the presentation of human histocompatability leukocyte antigen-A*0201–associated tumor peptide antigen MAGE-3271–279 by melanoma cells. We show that specific cytotoxic T lymphocyte (CTL)-recognizing cells transfected with a minigene encoding the preprocessed fragment MAGE-3271–279 failed to recognize cells expressing the full length MAGE-3 protein. Digestion of synthetic peptides extended at the NH2 or COOH terminus of MAGE-3271–279 with purified human proteasome revealed that the generation of the COOH terminus of the antigenic peptide was impaired. Surprisingly, addition of lactacystin to purified proteasome, though partially inhibitory, resulted in the generation of the antigenic peptide. Furthermore, treatment of melanoma cells expressing the MAGE-3 protein with lactacystin resulted in efficient lysis by MAGE-3271–279–specific CTL. We therefore postulate that the generation of antigenic peptides by the proteasome in cells can be modulated by the selective inhibition of certain of its enzymatic activities.
Keywords: HLA class I molecule, antigen processing, melanoma cells, ubiquitin, mass spectrometry
The proteasome is a central, nonlysosomal protease localized in the nuclei and cytosol of eukaryotic cells. It has been implicated in the catabolism of numerous intracellular proteins, including transcription factors and regulatory proteins as well as misfolded proteins (1–3). It is composed of a 20S core onto which a cap, formed by at least 15 different protein subunits, associates in an ATP-dependent manner, resulting in a 26S proteasome particle (4). The 26S proteasome has been shown to degrade ubiquitylated protein substrates (3). A second cap, formed by the heterodimeric PA-28 α/β (REG α/β) has also been described (5, 6). This cap increases the catalytic activity of the 20S proteasome toward oligopeptides and, in certain cases, modulates the cleavage activity of the proteasome, at least in vitro (7). This form of the proteasome does not degrade ubiquitylated substrates.
The 20S core proteasome is composed of 14 different subunits arranged in 4 heptameric rings denoted as α7β7β7α7 (8, 9). The catalytic centers of the proteasome are localized in the interior of the β subunits. Studies on the degradation of short fluorogenic peptides by purified proteasome in vitro have demonstrated the presence of at least five different enzymatic activities: a tryptic-like activity, cleaving after basic residues; a chymotryptic-like activity, cleaving after hydrophobic amino acids; a peptidylglutamylpeptide hydrolyzing (PGPH)1 activity, cleaving after acidic residues; a “branched chain amino acid–preferring” (BrAAP) activity; and a “small neutral amino acid–preferring” (SNAAP) activity (10).
The participation of the proteasome in the generation of antigenic peptides presented by MHC class I molecules has been well documented (11–13). After their generation, antigenic peptides are transported across the membrane of the endoplasmic reticulum (ER) by the transporters associated with antigen presentation (TAP). In the ER, newly synthesized MHC class I heavy chain, β2-microglobulin, and peptide form a trimolecular complex that is then transported to the cell surface (14). Purification and biochemical characterization of peptides naturally associated with defined MHC class I alleles led to the discovery that these peptides are of relatively homogenous length (8–10 amino acids) and carry conserved amino acid residues at certain positions. These residues, defined as anchor residues, were shown to form direct contacts with the MHC molecule. Based on this information, the prediction of putative antigenic peptides for defined MHC class I proteins has become possible.
MAGE-3 belongs to the human MAGE multigene family, is characterized by its tumor-restricted expression, and gives rise to several antigenic peptides presented by different MHC class I molecules (15–17). MAGE-3 antigenic peptides have therefore become attractive candidates for the study of antitumor responses by CTL. In an attempt to identify antigenic peptides, a series of 9-mer peptide sequences derived from the MAGE-3 protein and carrying anchor residues for HLA-A*0201 were chemically synthesized and tested for binding to HLA-A*0201. Among the MAGE-3–derived peptides binding to HLA-A*0201, one peptide, MAGE-3271–279, was able to induce a specific CTL response in vitro when added to a culture of PBL from an HLA-A*0201+ healthy individual (17). Subsequently, peptide MAGE-3271–279 was used to derive specific CTL using PBL from melanoma patients (18). Surprisingly, these CTL were able to lyse HLA-A*0201+ melanoma cells pulsed with exogenous peptides but were unable to recognize melanoma cells expressing the MAGE-3 protein endogenously. To elucidate this discrepancy, we have investigated the processing of the MAGE-3 gene product.
Here we show that the lack of presentation of peptide MAGE-3271–279 by HLA-A*0201+ cells is caused by the inaccurate cleavage of the MAGE-3 protein during its processing. Cells were tested for recognition by MAGE-3271–279–specific CTL following intracellular expression of a minigene-encoded MAGE-3271–279 peptide extended either at the NH2 or COOH terminus. In contrast to cells expressing the NH2-terminally extended peptide precursor, cells expressing the COOH-terminally extended peptide precursor were not recognized by specific CTL. Mass spectrometry analysis of the corresponding synthetic peptide precursors incubated with purified human proteasome demonstrated that the antigenic peptide could be generated from the NH2-terminally extended precursor but not from the COOH-terminally extended precursor. Surprisingly, addition of a specific proteasome inhibitor, lactacystin, restored the generation of the antigenic peptide from the COOH-terminally extended precursor. Analysis of the enzymatic activity of the proteasome in the presence of the proteasome inhibitor supported this discrepancy. Finally, treatment of HLA-A*0201+ melanoma cells expressing MAGE-3 with lactacystin or another proteasome inhibitor, N-acetyl-Leu-Leu-norleucinal (LLnL), restored the presentation of MAGE-3271–279. Implication of these findings on the modulatory effect of proteasome inhibitor on the generation of peptides presented by HLA class I is discussed.
Materials and Methods
Cell Lines.
The human melanoma cell lines NA8-MEL (provided by Dr. F. Jotereau, Institut National de la Santé et de la Recherche Médicale, Nantes, France) and SK23-MEL were cultured in RPMI medium (GIBCO BRL) supplemented with 10% FCS, antibiotics, and 20 mM NaHepes (pH 7.3). The CTL line LAU 198 NS (normal stimulation) specific for peptide MAGE-3271–279 was established by stimulating CD8+-enriched cells from a LAU 198 melanoma patient with peptide-pulsed, autologous PBL as described (18). The CTL line was restimulated weekly with irradiated (3,000 rads) autologous PBL. Before irradiation, PBL were incubated for 2 h at 37°C in serum-free medium (X-VIVO 10; BioWhittaker) in the presence of the synthetic peptide MAGE-3271–279 (1 μg/ml) and 3 μg/ml human β2-microglobulin (Sigma Chemical Co.) and then washed extensively to remove unattached peptide. The CTL clone 3C5 used in Fig. 7 was isolated as described previously (19). In brief, PBL were stained with tetramers of HLA-A*0201–β2-microglobulin containing the peptide MAGE-3271–279. After cell sorting by FACS® (Becton Dickinson), positive cells were cloned in the presence of irradiated PBL, 5 μg/ml PHA, and 50 U/ml IL-2 in human serum containing IMDM. The human lymphoblastoid cell lines T2 and LCL721.45 were used as targets in 51Cr release assay. These cells were cultured in DMEM (GIBCO BRL) supplemented with 10% FCS, 0.55 mM arginine, 0.24 mM asparagine, and 1.5 mM glutamine. The TNF-α–sensitive cell line WEHI-164 clone 13 was maintained in RPMI medium (GIBCO BRL) supplemented with 10% FCS, 0.55 mM arginine, 0.24 mM asparagine, and 1.5 mM glutamine.
Figure 7.


(A) Lactacystin restores the presentation of the MAGE-3271–279 in cells expressing virally encoded MAGE-3. When left untreated, the HLA-A*0201+MAGE-3− lymphoblastoid cell line LCL721.45 infected with rec. v.v. encoding MAGE-3 is not recognized by MAGE-3271–279–specific CTL (rec. v.v.-MAGE3). Treatment of the infected cells with lactacystin prior to and during the infection renders them sensitive to the lysis by specific CTL (rec. v.v.-MAGE3 + lactacystin). (B) Effect of proteasome inhibitors on the presentation of peptide MAGE-3271–279 in human melanoma cells. In contrast to the untreated HLA-A*0201+ MAGE-3+ melanoma cell line SK23-MEL, treatment with either 10 μM lactacystin or 100 μM LLnL for 17 h enhances the presentation of peptide MAGE-3271–279 to specific CTL. See Materials and Methods for details.
Construction of Plasmids and Ubiquitin/Protein/Reference (UPR) Technique.
All plasmid constructs described in this study were based on the plasmid pS65T-C1 (Clontech). This vector carries, under the control of the cytomegalovirus immediate early promoter, a mutated version of the green fluorescence protein (GFP), resulting in brighter fluorescence. The stop codon of the GFP moiety has been replaced by a multicloning site allowing COOH-terminal fusions to GFP. A fragment encoding yeast ubiquitin (Ub) carrying a lysine48 to arginine mutation (to preclude that the COOH-terminal Ub moiety could serve as a ubiquitylation/degradation signal) and an influenza hemagglutinin epitope (ha) recognized by a monoclonal antibody was obtained by PCR, using the plasmid pRc/dUb–Met–βgal as template (20). This PCR fragment was digested with KpnI and SmaI and inserted in frame between the KpnI and SmaI sites present in the multicloning site of pS65T-C1, resulting in the vector pGFPAvaI/Ub expressing the fusion GFP–ha–Ub. The ha epitope is located between the GFP and Ub moieties. The plasmid pGFPAvaI/Ub was further digested with XhoI and EcoRI, blunted using Klenow PolI, and religated, thereby eliminating the AvaI site between GFP and Ub and rendering the site AvaI at the 3′ end of Ub unique. This latter construct, termed pGFP/Ub, served as vector for all constructs described below.
Construct I (Fig. 1) was obtained by PCR amplification of a fragment encoding the tumor antigen MAGE-3 (a gift from T. Boon, Ludwig Institute for Cancer Research, Brussels, Belgium). This PCR fragment was digested with SacII and AvaI and inserted in frame between the SacII and AvaI sites of pGFP/Ub, resulting in the plasmid pGFP/Ub–MAGE-31–314. The protein MAGE-3 includes the natural NH2-terminal methionine. Constructs II, III, and IV were obtained by annealing complementary synthetic oligonucleotides encoding the MAGE-3–derived peptide fragments MAGE-3271–279, MAGE-3256–279, and MAGE-3271–285, respectively. The oligonucleotides were designed so as to reconstitute the SacII site at the 5′ end and the AvaI site at the 3′ end of the fragment and included a stop codon immediately upstream of the AvaI site. Upon annealing, the fragments were inserted between the SacII–AvaI sites of pGFP/Ub, resulting in the plasmids pGFP/Ub–MAGE-3271–279 (Fig. 1, construct II), pGFP/Ub– MAGE-3256–279 (construct III), and pGFP/Ub–MAGE-3271–285 (construct IV). The sequences of the critical regions of all constructs were confirmed by DNA sequencing.
Figure 1.


(A) UPR constructs. The plasmid pGFP/Ub–MAGE-31–314 encodes a tripartite fusion protein in which ubiquitin (Ub) is sandwiched between the reference protein GFPha and the full length protein MAGE-3 (construct I). During or shortly after the translation of this fusion, the GFPha–Ub moiety is cleaved at the last residue of Ub by a Ub-specific protease, resulting in equimolar amounts of the reference protein GFPha–Ub and the protein MAGE-3. The plasmid pGFP/Ub–MAGE-3271–279 (construct II) encodes the reference protein GFPha–Ub and a fragment of MAGE-3 corresponding to the antigenic peptide associated with HLA-A*0201. The plasmid pGFP/Ub–MAGE-3256–279 (construct III) encodes the reference protein GFPha–Ub and a fragment of MAGE-3 corresponding to a 15–amino acid NH2-terminal extension of the antigenic peptide MAGE-3271–279. The plasmid pGFP/Ub–MAGE-3271–285 (construct IV) encodes the reference protein GFPha–Ub and a fragment of MAGE-3 corresponding to a six-amino acid COOH-terminal extension of MAGE-3271–279. The residue methionine at the NH2 terminus of the protein GFP results from the translation of the initiation codon ATG. The term ha denotes a peptide sequence derived from the influenza hemagglutinin that is recognized by a specific antibody. The segment corresponding to the antigenic peptide in each construct is indicated as a white box. (B) Synthetic peptides corresponding to the NH2-terminal (MAGE-3256–279) and the COOH-terminal (MAGE-3271–285) extensions of the antigenic 9-mer are described in one-letter code together with their molecular masses in daltons (Da). The antigenic peptide sequence is boxed.
Transfection and Metabolic Labeling.
Cells were transiently transfected using the Lipofectamine reagent (GIBCO BRL) according to the manufacturer's protocol. For metabolic labeling, 3 × 106 cells were transfected with 4 μg plasmid DNA. After 8 h, an equal volume of DMEM containing 10% FCS was added to the cells. After a further 12-h incubation at 37°C, the culture medium was replaced with 1 ml DMEM lacking methionine and cysteine (ICN Biomedicals, Inc.), and the cells were incubated for 1 h at 37°C. Cells were labeled for 90 min at 37°C in the presence of 200 μCi of [35S]Express (New England Nuclear). The cells were then lysed in 1 ml lysis buffer (1% Triton X-100, 0.15 M NaCl, 5 mM EDTA, 20 mM Tris–HCl [pH 7.3], and 0.2 mg/ml phenylmethylsulfonyl fluoride) and maintained on ice. The lysate was cleared by centrifugation at 12,000 g for 10 min. The volumes of supernatant were adjusted to contain equal amount of 10% TCA-insoluble, 35S-labeled material and immunoprecipitated using a mixture of a saturating amount of a monoclonal antibody anti-ha epitope (Berkeley Antibody Co.) and a monoclonal antibody against MAGE-3 (21; a gift from G. Spagnoli, University of Basel, Basel, Switzerland). The samples were incubated with rotary shaking at 4°C for 30 min, followed by the addition of 20 μl protein G–Sepharose and another 30-min incubation at 4°C. The immunoprecipitate was washed three times in lysis buffer containing 0.1% SDS, resuspended in 20 μl SDS–sample buffer (100 mM Tris–HCl, pH 8.8; 1.2 M sucrose; 0.01% bromophenol blue; 2% SDS; and 90 mM dithiothreitol), and boiled at 100°C for 3 min. The samples were subjected to SDS–12% PAGE followed by autoradiography.
TNF-α Release Assay.
Cells were transfected using the same protocol as for the metabolic labeling with the following modifications: 104 cells were transfected in 96-well, round bottom microtiter plates with 200 ng plasmid DNA and 1 μl Lipofectamine in a final volume of 100 μl DMEM. After 6 h at 37°C, 100 μl DMEM containing 10% FCS was added to each well, and the cells were maintained at 37°C for another 14 h. At this point, the transfected cells were tested for their ability to stimulate the release of TNF-α by the MAGE-3271–279–specific CTL line. In brief, CTLs were added at the appropriate effector-to-target cell ratio (E/T) in 100 μl of IMDM supplemented with 10% human serum and 20 U/ml human rIL-2 (Glaxo Wellcome) provided by Dr. M. Nabholz (Swiss Institute for Experimental Cancer Research, Epalinges, Switzerland). After a 24-h incubation at 37°C, supernatants were collected and the TNF-α content was determined in a functional assay using WEHI-164 clone 13 cells (22) as described (23).
Proteasome Purification.
Proteasome was purified from human outdated blood by affinity chromatography using the monoclonal antibody MCP 21 (European Collection of Animal Cell Cultures) immobilized on CnBr-activated Sepharose (6 mg antibody/mg Sepharose; 24). ∼150 ml blood was washed five times with cold PBS, and the cells were pelleted at 2,000 g for 10 min. This pellet was resuspended in 1/5 volume of sterile H2O and mechanically disrupted by douncing in a Dounce homogenator. Immediately after this treatment, sucrose was added to the homogenate to a final concentration of 250 mM. Unsolubilized material was removed by centrifugation. Cleared lysate was incubated for 3 h at 4°C under rotary shaking with the immobilized mAb MCP 21 as a batch preparation. The material was then loaded onto a column and washed extensively with 20 mM Tris–HCl (pH 7.6), and 50 mM NaCl to remove unattached material. 20S proteasome was eluted with 20 mM Tris–HCl (pH 7.6) and 2 M NaCl. The eluate was dialysed for 24 h at 4°C against 20 mM Tris–HCl (pH 7.6) and concentrated to 4 mg/ml. The average yield was ∼1 mg purified proteasome/150 ml blood. Homogeneity of the eluted material was confirmed by analysis of an aliquot by SDS–12% PAGE and Coomassie blue staining of the gel.
Enzymatic Assays.
The activity of the proteasome was assayed using the following fluorogenic peptides: Z–Gly–Gly–Arg–βNA (Z–GGR–βNA; Z = benzyloxycarbonyl and βNA = β-naphtylamide) for tryptic-like activity, Z–Leu–Leu–Glu–βNA (Z–LLE– βNA) for PGPH activity, and Suc–Leu–Leu–Val–Tyr–AMC (Suc–LLVY–AMC; Suc = succinyl, and AMC = 7-amido-4-methylcoumarin) for chymotryptic-like activity. All peptides were purchased from Bachem. Each degradation assay contained 5 μg proteasome plus 100 μM peptides Z–GGR–βNA and Z–LLE– βNA or 10 μM Suc–LLVY–AMC in a final volume of 100 μl. The reaction was incubated at 25°C for 20 min, at which time the reaction was quenched with cold 100% ethanol. Fluorescence emission was measured using a Perkin Elmer fluorometer. Fluorescence excitation/emission wavelengths were 335:410 nm for βNA and 380:440 nm for AMC. Where indicated, the proteasome inhibitor lactacystin (Biomol) was incubated for 15 min with proteasome at 25°C before the addition of the peptide substrates. The final concentration of lactacystin was 50 μM.
Peptide Synthesis.
Peptides were synthesized using standard solid-phase F-moc chemistry on an Applied Biosystems synthesizer. After side chain deprotection and release from the resin, the peptides were purified by reverse-phase preparative HPLC using a Vydac C18 column. Fractions containing the expected product, as judged by mass spectrometry, were pooled and lyophilized. The purified material was then subjected to matrix assisted laser desorption ionization-time of flight (MALDI-TOF) and analytical HPLC analysis. All peptides were >95% pure as indicated by analytical HPLC.
Peptide Digestion and Mass Spectrometry.
For analysis by mass spectrometry, 4 nmol peptide was incubated for 40 min at 37°C with 16 μg purified proteasome in a final volume of 10 μl. Where indicated, lactacystin was added to a final concentration of 50 μM. The samples containing proteasome and lactacystin were incubated for 15 min at 37°C before the addition of peptide. The reaction was terminated by the addition of 5% TFA and immediate immersion in liquid N2. The samples were subsequently lyophilized. The lyophilized samples were dissolved in 20 μl 1% TFA/ H2O. An aliquot of this solution (1.5 μl) was mixed with 24 μl of matrix solution (5 mg/ml α-cyano-4-hydroxyl-cinnamic acid; Sigma Chemical Co.) in 1% TFA/H2O:acetonitrile (1:1, vol/vol) and 1 μl of this mixture was deposited on a gold plated target and vacuum dried before transferring it into the source of the mass spectrometer.
MALDI mass spectra were obtained on a Perseptive Biosystems Voyager RP spectrometer using a 337-nm nitrogen laser, a 25-kV accelerating potential, and a delayed extraction of 150 ns. Each spectrum was the result of an accumulation of 128 single laser shots. External calibration of the MALDI spectra in the linear mode was carried out using a low molecular weight standard (Perseptive Biosystems). The mass determination was based on the mass-to-charge (m/z) ratio. The instrument error ranges between 0.01 and 0.1% of the molecular mass.
Peptide Digestion and 51Cr Release Assay.
Samples used in subsequent CTL assay included 100 pmol peptide and 4 μg purified proteasome. Where indicated, trypsin (1 mg/ml) was added to the peptide and incubated for 20 min at 37°C in 10 mM Tris– HCl, pH 8.0. The reaction performed was identical to the reaction for the mass spectrometry analysis. Target cells were labeled with 51Cr for 1 h at 37°C and washed twice. Labeled target cells (1,000 cells in 50 μl RPMI medium/5% FCS) were added to serial dilutions of the various peptide preparations (100 μl) in v-bottomed microwells for 15 min at room temperature. The CTL were then added at the appropriate E/T in 50 μl RPMI medium/5% FCS, and the release of 51Cr was measured after incubation for 4 h at 37°C. Specific lysis was calculated as follows:
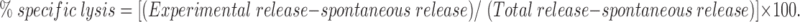 |
Infection with Recombinant Vaccinia Virus and Treatment of Cells with Proteasome Inhibitors.
Approximately 106 LCL721.45 cells (25) were treated with 100 μM lactacystin for 1 h, washed, and incubated overnight in 1 μM lactacystin at 37°C. Cells were then infected with recombinant vaccinia virus (rec. v.v.) expressing the MAGE-31–314 protein at a multiplicity of infection of 10 and simultaneously labeled with 100 μCi of 51Cr for 2 h. The cells were washed and incubated at 37°C for another 4 h before being exposed to the MAGE-3–specific CTL clone 3C5 at the E/T indicated in Fig. 7. Rec. v.v. encoding the MAGE-3 protein was generated by recombination with a pSc11 derivative plasmid as described previously (26). The cDNA coding for MAGE-3 was digested with PstI and XbaI, blunted using Klenow PolI, and inserted into the SmaI site of the vector pSc11.
The HLA-A*0201+ MAGE-3+ melanoma cell line SK23-MEL (18) was treated with 10 μM lactacystin or 100 μM LLnL for 17 h. Subsequently, 106 cells were labeled with 100 μCi of 51Cr for 2 h, washed, and incubated with the MAGE-3–specific CTL clone 3C5 for 8 h at 37°C.
Results
Experimental Strategy.
We have previously derived a MAGE-3271–279–specific CTL line from PBL of a melanoma patient that, although able to readily recognize HLA-A*0201+ cells exposed to exogenously added synthetic MAGE-3271–279 peptide, was unable to lyse cells expressing the MAGE-3 protein (18). To address this discrepancy, we assessed in the present study the recognition of HLA-A*0201+ melanoma cells transfected with cDNA or cDNA fragments encoding the MAGE-3 protein. Expression of the various MAGE-3 gene products in transfected cells was determined by the UPR technique, originally developed to increase the accuracy of protein decay measurements (20).
Ub is a highly conserved protein that, in many instances, serves as a proteasomal degradation signal by forming a multiubiquitin chain on lysine residue(s) of a protein (27). Ub is naturally synthesized as a fused protein—either to itself or to other cellular proteins—and is rapidly cleaved by a member of the Ub-specific proteases. The UPR technique exploits this natural linear arrangement of the Ub gene and relies on a linear fusion with Ub located between a reference protein (here GFP) and a protein of interest (Fig. 1). The rapid cleavage of the fusion protein by Ub-specific proteases after the last residue of Ub yields equimolar amounts of the protein of interest and the reference protein bearing a COOH-terminal Ub moiety. It is noteworthy that the use of Ub fusion also enabled us to bypass the need for a methionine residue at the NH2 termini of the MAGE-3 peptide fragments. Finally, a sequence encoding a peptide tag derived from the influenza ha was inserted between the GFP and Ub moieties to permit the detection and quantitation of GFPha–Ub fusion.
NA8-MEL cells (HLA-A*0201+MAGE-3−) were transiently transfected with the plasmids pGFP/Ub–MAGE-31–314, pGFP/Ub–MAGE-3271–279, pGFP/Ub–MAGE-3256–279, or pGFP/Ub–MAGE-3271–285 coding for the reference GFPha–Ub and either MAGE-31–314, MAGE-3271–279, MAGE-3256–279, or MAGE-3271–285, respectively (Fig. 1, constructs I–IV). The transfected cells were labeled with [35S]methionine/cysteine for 60 min, lysed, immunoprecipitated using saturating amounts of monoclonal anti-ha antibody and monoclonal anti–MAGE-3 antibody, and analyzed by SDS-PAGE and autoradiography. The radioactive band corresponding to the reference GFPha–Ub could be detected in all lanes where cells had been transfected with UPR-based plasmids (Fig. 2, lanes a–d) but not in mock-transfected cells (lane e). The protein MAGE-3 was only detected in cells transfected with the full length MAGE-3 gene (lane a) and migrated as a doublet. The upper band of the doublet is the result of a posttranslational modification other than phosphorylation or N-glycosylation (data not shown). The presence of the reference protein in the other lanes confirmed the expression of the MAGE-3 protein fragments. Using the GFP-based UPR technique, we estimated that the transiently transfected constructs were expressed in ∼2% of the melanoma cells (data not shown).
Figure 2.

The use of UPR technique in assessing the expression of transfected plasmids. NA8-MEL cells were transiently transfected with plasmids pGFP/ Ub–MAGE-31–314 (lane a), pGFP/Ub–MAGE-3271–279 (lane b), pGFP/Ub–MAGE-3256–279 (lane c), pGFP/Ub–MAGE-3271–285 (lane d), or mock transfected (lane e). Transfected cells were metabolically labeled with [35S]methionine/cysteine, followed by immunoprecipitation and SDS-PAGE analysis. The band corresponding to the reference GFPha–Ub is indicated on the left. In lane a, the protein MAGE-31–314 detected by a monoclonal antibody raised against MAGE-3 migrates as a doublet. The upper band of the doublet is the result of a posttranslational modification. The band corresponding to GFPha–Ub is absent in mock-transfected cells (lane e).
CTL Recognition of Cells Expressing MAGE-31–314, MAGE-3271–279, and MAGE-3271–279 Extended Precursors.
NA8-MEL cells transfected with pGFP/Ub–MAGE-31–314 (Fig. 1, construct I) and pGFP/Ub–MAGE-3271–279 (construct II) encoding the reference protein GFPha–Ub and either MAGE-31–314 or MAGE-3271–279, respectively, were incubated with cells from the CTL line specific for MAGE-3271–279 at a lymphocyte-to-target cell ratio of 30:1. After 24 h of coculture, supernatants were assayed for the presence of TNF-α released by activated CTL. The amount of TNF-α produced by CTL incubated with cells expressing MAGE-31–314 was not significantly higher than that produced by CTL incubated with mock-transfected cells (Fig. 3). In contrast, cells expressing MAGE-3271–279 were efficiently recognized by CTL as indicated by the 50-fold increase in the amount of TNF-α produced. As expected, addition of a saturating amount of the corresponding MAGE-3271–279 synthetic peptide to mock-transfected cells resulted in a strong production of TNF-α. Therefore, the impaired presentation of MAGE-3271–279 by NA8-MEL cells transfected with MAGE-31–314 was not caused by an inefficient transport or loading of the peptide on HLA-A*0201 molecules but rather by an inaccurate processing of the protein MAGE-3.
Figure 3.

Recognition of transfected NA8-MEL cells by CTL specific for HLA-A*0201–restricted MAGE-3271–279. Cells transfected with the plasmids pGFP/Ub–MAGE-31–314, pGFP/Ub–MAGE-3271–279, pGFP/ Ub–MAGE-3256–279, and pGFP/Ub–MAGE-3271–285 were cocultured for 24 h with CTL at a lymphocyte to target cell ratio of 30:1. The production of TNF-α was measured by testing the toxicity of the culture supernatant on TNF-sensitive cells. The amount of TNF-α produced by cells expressing MAGE-31–314 or MAGE-3271–285 corresponds to 4 pg/ml and is not significantly different from mock-transfected cells (solid black bar). Cells expressing MAGE-3271–279 or MAGE-3256–279 induced a 50-fold increase in the production of TNF-α. NA8-MEL cells exposed to 1 μg/ml exogenous MAGE-3271–279 peptide (white bar) were efficiently recognized by the CTL.
To locate the region of the protein affecting the generation of the antigenic peptide, we extended the sequence of MAGE-3271–279 at the NH2 terminus by 15 amino acids (Fig. 1, construct III) and at the COOH terminus by 6 amino acids (construct IV). NA8-MEL cells were transfected with the plasmids coding for GFPha–Ub and either MAGE-3256–279 or MAGE-3271–285, respectively. Cells expressing the fragment MAGE-3256–279 were well recognized by CTL (Fig. 3). On the contrary, cells transfected with the plasmid encoding MAGE-3271–285 were not recognized by CTL. The length of the COOH-terminal extension did not influence the recognition of the transfected cells, as cells expressing MAGE-3271–279 with a COOH-terminal extension of 15 amino acids did not lead to a recognition of the cells, either (data not shown). Therefore, the processing protease was unable to generate the MAGE-3271–279 peptide from a COOH-terminally extended precursor but could generate it from a precursor containing a preprocessed COOH terminus as the result of genetic engineering.
Analysis of Peptide Fragments Generated after Digestion with Purified Proteasome In Vitro.
The human proteasome has been implicated in the generation of antigenic peptides presented by HLA class I molecules to CTL (11–13). We tested whether the peptide fragments obtained after exposure of NH2- and COOH-terminally extended MAGE-3271–279 precursors to purified human proteasome correlated with the results obtained using transfected cells. Synthetic peptide MAGE-3271–285 was incubated for 40 min at 37°C in the presence of proteasome purified from blood and was subsequently analyzed by mass spectrometry (Fig. 1 B and Fig. 4, A and B). A major peak corresponding to the original peptide MAGE-3271–285 (theoretical mass 1,766 daltons) was detectable at time 0. Several additional peaks of lower mass appeared after incubation with the proteasome. These degradation products were unambiguously identified as the 11-mer MAGE-3271–281 (1,288 daltons), the 10-mer MAGE-3271–280 (1,187 daltons), the 8-mer MAGE-3271–278 (959 daltons) and the 7-mer MAGE-3272–278 (812 daltons). No peak corresponding to the 9-mer MAGE-3271–279 (1,058 daltons) was detected.
Figure 4.

Fragments generated after digestion of synthetic peptides with purified proteasome in vitro. The synthetic peptide MAGE-3271–285 (A–C) corresponding to a COOH-terminal extension of the antigenic peptide MAGE-3271–279 or MAGE-3256–279 (D–F) corresponding to an NH2-terminal extension of the antigenic peptide were incubated for 0 (A and D) or 40 min (B and E) at 37°C with 15 μg proteasome purified from human blood. A 40-min digestion was also performed in the presence of 50 μM lactacystin (C and F). The samples were then analyzed by mass spectrometry. The identity of the detected peaks was determined based on the specific mass of each peptide fragment and is denoted by the position of the fragment within the precursor peptide sequence. Note the presence of a peak corresponding to the antigenic peptide MAGE-3271–279 in C, E, and F. The sequences of the precursor peptides are indicated, in one-letter code, at the top and the antigenic 9-mer is underlined. Contaminating peaks are not labeled.
A similar analysis was performed using peptide MAGE-3256–279 (Fig. 1 B). The major peak detected at time 0 corresponded to the original peptide (2,767 daltons; Fig. 4 D). After incubation with proteasome, the 9-mer corresponding to MAGE-3271–279 (1,058 daltons) was clearly detected (Fig. 4 E). Moreover, several other peaks were detected and identified as the 10-mer MAGE-3256–265 (1,163 daltons), the 8-mer MAGE-3272–279 (911 daltons), and the 7-mer MAGE-3272–278 (812 daltons). The relative signal intensity of peptide MAGE-3256–279 was reproducibly lower than that of peptide MAGE-3271–285 and may reflect the chemical properties of this peptide. Thus, results obtained with in vitro digestion of synthetic MAGE-3 peptide extensions not only confirmed the results obtained with DNA- encoded peptides expressed intracellularly but also enabled precise identification of the degradation products.
Selective Inhibition of Proteasomal Activity In Vitro Influences the Production of Peptide MAGE-3271–279.
To ascertain the role of the different enzymatic activities of the proteasome in the generation of the proteolytic fragments in vitro, we performed experiments similar to the one described above in the presence of lactacystin, a specific inhibitor of the proteasome. Lactacystin has been shown to irreversibly block the trypsin-like and chymotrypsin-like activities, reversibly inhibit the PGPH activity, and moderately block the BrAAP activity of the proteasome (28, 29).
Purified MAGE-3256–279 and MAGE-3271–285 peptides were incubated for 40 min in the presence of proteasome and 50 μM lactacystin and analyzed by mass spectrometry (Fig. 4, C and F). Digestion of peptide MAGE-3271–285 (C) resulted in the appearance of several peaks with masses similar to those obtained after the digestion without lactacystin (compare with B). Unexpectedly, the peptide species corresponding to the 9-mer MAGE-3271–279 (1,058 daltons) was generated to a detectable level. The peak corresponding to the 7-mer MAGE-3272–278 (812 daltons) was not detectable in the sample incubated with the proteasome inhibitor. Digestion of peptide MAGE-3256–279 in the presence of lactacystin (F) yielded new peaks corresponding to the 12-mer MAGE-3256–267 (1,331 daltons) and the 6-mer MAGE-3256–261 (807 daltons), in addition to the 9-mer MAGE-3271–279 (1,058 daltons) and the 8-mer MAGE-3272–279 (912 daltons) already detected in digestion without lactacystin (E). It is interesting to note that the relative intensity of the peak corresponding to the antigenic 9-mer was reproducibly higher in digestions containing lactacystin than in those performed in the absence of lactacystin (compare E and F).
Lactacystin Inhibits Two of the Three Tested Proteasomal Activities.
To identify the lactacystin-mediated effect on the enzymatic activity of the proteasome, we performed a digestion of the fluorescent substrates Z–GGR–βNA (for trypsin-like activity), Z–LLE–βNA (for PGPH activity), and Suc–LLVY–AMC (for chymotrypsin-like activity) in the presence or absence of 50 μM lactacystin (Fig. 5). Although all fluorescent substrates were cleaved by the proteasome in the absence of lactacystin as detected by increased fluorescence, two of the three activities of the proteasome could be efficiently blocked by the addition of lactacystin. The PGPH activity was only marginally affected by the presence of lactacystin. Further purification of the proteasome by size chromatography did not alter this effect, ruling out the contribution of other low molecular weight proteases present in the purified preparation (data not shown).
Figure 5.

Proteolytic activity of purified proteasome in the absence or presence of proteasome inhibitors. The tryptic-like, PGPH, and chymotryptic-like activities of purified proteasome (5 μg) were tested using 100 μM Z–GGR–βNA (black bar), 100 μM Z–LLE–βNA (hatched bar), or 10 μM Suc–LLVY–AMC (gray bar), respectively. In samples containing the inhibitor lactacystin (50 μM), proteasome was incubated for 15 min at 25°C before the addition of the fluorogenic peptide substrate. The reaction was allowed to proceed for 20 min at 25°C. Peptide hydrolysis was detected by monitoring the increased fluorescence in arbitrary units (A.U.) emitted by the released fluorogenic group βNA (excitation/emission of 335:410 nm) or AMC (excitation/emission of 380:440 nm). In the absence of proteasome (−), only background activity is detected. A representative plot is depicted here. Identical results were obtained using three independent proteasome preparations and two different lots of lactacystin.
Specific CTL Can Recognize Target Cells Pulsed after Digestion of Peptide Precursors.
Since the generation of the 9-mer MAGE-3271–279 from peptides MAGE-3271–285 and MAGE-3256–279 was enhanced in the presence of lactacystin, we tested whether the peptide generated under this condition was able to sensitize target cells for lysis by MAGE-3271–279–specific CTL. Peptides MAGE-3271–285 and MAGE-3256–279 were either added directly to target cells or were first incubated with purified proteasome for 20 min at 37°C in the absence or presence of lactacystin. After 20 min, the reaction was stopped by the addition of 5% TFA and lyophilized. The lyophilized material was resuspended in medium and added in different dilutions to 51Cr-labeled T2 cells. These HLA-A*0201+ cells are TAP deficient but can be efficiently sensitized with exogenous peptides for recognition by specific CTL.
Direct addition of the peptide MAGE-3271–285 resulted in lysis of T2 cells only at very high peptide concentration (Fig. 6 A). This activity was probably caused by the presence of a small amount of peptide MAGE-3271–280 produced by the hydrolysis of peptide MAGE-3271–285. Indeed, the MAGE-3271–279–specific CTL used in the present study was able to recognize the synthetic decapeptide MAGE-3271–280 (50% maximal lysis at 100 nM), although much less efficiently than the 9-mer MAGE-3271–279 (50% maximal lysis at 0.1 nM). No additional activity was observed after digestion of peptide MAGE-3271–285 with proteasome for 20 min (Fig. 6 A). In contrast, incubation of peptide MAGE-3271–285 with proteasome and lactacystin for 20 min resulted in the generation of an antigenic peptide recognized ∼20-fold more efficiently by the MAGE-3271–279– specific CTL (B). Based on the specificity of the CTL and on the degradation products detected by mass spectrometry, we concluded that the antigenic peptide MAGE-3271–279 was indeed produced in the presence of lactacystin. Digestion of peptide MAGE-3271–285 with trypsin completely abrogated recognition of the target cells by specific CTL.
Figure 6.

Lysis of T2 target cells pulsed with proteasome-digested peptide products by MAGE-3271–279–specific CTL. Precursors corresponding to MAGE-3271–285 (A and B) or MAGE-3256–279 (C and D) were incubated with proteasome for 0 or 20 min at 37°C in the absence (A and C) or presence (B and D) of 50 μM lactacystin. In each condition, the precursor was also digested with trypsin or maintained in the absence of proteasome. Upon completion of the incubation, the samples were lyophilized, resuspended in medium, and added in fivefold dilution steps to 51Cr-labeled T2 cells. Cr release was measured after 4 h. The half-maximal lysis of cells pulsed with a saturating amount of the antigenic MAGE-3271–279 peptide was 0.01 nM (not shown).
A similar assay was performed using peptide MAGE-3256–279 as substrate. Again, direct addition of the peptide precursor produced low but detectable lytic activity at high concentration. Digestion of the extended peptide for 20 min in the absence of lactacystin led to a 10-fold increase in lytic activity (Fig. 6 C), an effect compatible with the detection of peptide MAGE-3271–279 in the digestion products analyzed by mass spectrometry (Fig. 4 E). This activity was completely abolished by the addition of trypsin. However, digestion of peptide MAGE-3256–279 in the presence of 50 μM lactacystin resulted in 100-fold–increased recognition (Fig. 6 D). This effect was most likely caused by the increased amount of MAGE-3271–279 generated after proteasomal digestion in the presence of lactacystin as suggested by the increased relative intensity of the MAGE-3271–279 peak detected by mass spectrometry (Fig. 4 F).
Treatment of Cells Expressing MAGE-31–314 with Proteasome Inhibitors Results in Efficient Presentation of MAGE-3271–279.
To ascertain the relevance of the positive effect of lactacystin on the generation of peptide MAGE-3271–279 by the proteasome, we infected HLA-A*0201+ lymphoblastoid cells with rec. v.v. coding for MAGE-31–314 (Fig. 7 A). Infected cells expressing the full length protein MAGE-31–314 were not efficiently recognized by MAGE-3271–279–specific CTL. However, treatment of the same cells with lactacystin prior to and during the infection lead to the efficient lysis of the infected cells. To determine whether the lactacystin-mediated effect observed in lymphoblastoid cells upon infection and high level expression of MAGE-3 could also be observed in uninfected melanoma cells, lysis of HLA-A*0201+MAGE-3+ melanoma cells was measured after treatment with lactacystin. As expected, lysis of untreated target cells was very low (Fig. 7 B). In contrast, treatment with lactacystin led to efficient lysis of the melanoma cells. A similar effect was also observed after treatment of the target cells with the calpain (and proteasome) inhibitor I LLnL. Taken together, these results strongly support the findings obtained after in vitro digestion of peptide precursors by purified proteasome and lead us to conclude that the generation and presentation of peptide MAGE-3271–279 detected after treatment with lactacystin is a direct consequence of the partial inhibition of the proteasome in cells.
Discussion
Two major findings are reported in this work: First, melanoma cells transfected with cDNA encoding either the preprocessed antigenic peptide MAGE-3271–279 (FLWGPRALV) or the same peptide with an NH2-terminal extension were recognized by MAGE-3271–279–specific CTL. In contrast, melanoma cells expressing the full length protein MAGE-31–314 or peptide MAGE-3271–279 with a COOH-terminal extension were not recognized. This discrepancy stems from the inappropriate proteasomal cleavage at the COOH terminus of the antigenic peptide. Second, treatment of MAGE-3+ melanoma cells with lactacystin resulted in the recognition of these normally unrecognized cells by MAGE-3271–279–specific CTL. This effect strongly correlates with the partial inhibition of proteasomal activity in vitro and the lactacystin-induced generation of peptide MAGE-3271–279 from the COOH-terminally extended precursor.
Expression of a minigene product in transfected cells has been difficult to assess by means other than functional assays. However, use of the UPR technique in this study not only allowed us to confirm the presence of the various minigene products via the detection of the reference protein GFP–Ub but also to quantitate the various MAGE-3 gene products, as both the reference protein GFP–Ub and the MAGE-3 protein, or protein fragments, were produced in equimolar amounts. Thus, the observed differences in CTL recognition of target cells transfected with the minigene encoding peptide MAGE-3271–279 or the full length protein MAGE-31–314 could not be attributed to different expression levels of the two products but rather to the generation, or lack thereof, of the antigenic peptide. Cells expressing a short-lived variant of the MAGE-31–314 protein were not recognized by specific CTL, either (data not shown), eliminating the possibility that the lack of recognition was caused by a slow degradation of the MAGE-3 protein leading to a suboptimal concentration of the antigenic peptide. Finally, reciprocal transplantation of the MAGE-3271–279 sequence into unrelated proteins indicated that the impaired generation of the antigenic peptide was not caused by residues flanking peptide MAGE-3271–279 (data not shown). Altogether, these results suggested that the sequence of peptide MAGE-3271–279 itself contributed to the improper processing of the MAGE-3 protein and led us to study the role of the proteasome in the deficient production of the antigenic peptide.
The involvement of the proteasome in the processing of antigenic peptides has been inferred from two sets of data: First, treatment of target cells with specific proteasome inhibitors abolished CTL-mediated recognition of antigenic peptides derived from intracellular proteins without affecting recognition of preprocessed endogenous or exogenous antigenic peptides (13). Second, analysis of proteasome- mediated degradation of synthetic peptide substrates in vitro revealed that the structural features of the digested products were compatible with those of peptides naturally associated with MHC class I molecules (30). However, several reports have recently demonstrated that the presentation of certain antigenic peptides, derived mostly from protein fragments, were insensitive to the effects of proteasome inhibitors (28, 31, 32). Moreover, cell surface expression of assembled MHC class I proteins in cells lacking proteasomal function suggests that other proteases may be involved in the generation of antigenic peptides (33). Therefore, the participation of two complementary and possibly overlapping proteolytic systems in antigen processing could be envisaged and would be compatible with our results obtained after treatment of melanoma cells with lactacystin. To address this question in greater detail, we isolated human proteasome and performed in vitro digestion studies using synthetic peptides. After digestion, the samples were immediately analyzed by mass spectrometry so as to uncover all possible fragments generated by the proteasome. Using this approach, we obtained strong evidence in favor of the involvement of the proteasome in the final proteolytic step leading to the generation of the antigenic peptide MAGE-3271–279, notwithstanding the possibility that distinct cellular proteases may be involved at other stages in the processing of the MAGE-3 protein into precursor peptide fragments.
Five proteolytic activities of the proteasome have been described using short synthetic fluorogenic peptides: a tryptic-like activity, a chymotryptic-like activity, a PGPH activity, a BrAAP activity, and a SNAAP activity (3). Lactacystin has been shown to irreversibly block the trypsin-like and chymotrypsin-like activities and only partially block the PGPH and the BrAAP activities (28, 29, 34). No information is available on the inhibition of the SNAAP activity by lactacystin. In agreement with these results, we also found that the proteasome inhibitor lactacystin did not completely abrogate certain proteasomal activities but resulted in an altered degradation of synthetic peptides. Although previous results indicated that I LLnL blocks all five activities, recent reports have shown only selective inhibition of the chymotryptic-like activity (10, 13, 34).
In the presence of lactacystin, the degradation of MAGE-3 peptide precursors by purified proteasome was only partially and selectively inhibited, even if the tryptic and chymotryptic activities on short fluorogenic peptides were completely abrogated (Figs. 4 and 5). Indeed, we could still detect fragments generated by cleavage after leucine, valine, glutamic acid, alanine, and threonine. Therefore, we envisage two possibilities: First, the remaining PGPH activity not only cleaves at the COOH terminus of glutamic acid but may be more permissive and also cleave after nonacidic amino acid residues, or, second, the catalytic subunits responsible for the BrAAP and SNAAP activities of the proteasome, in addition to those responsible for the PGPH activity, are not affected by lactacystin. In light of recent results obtained after the analysis of peptide fragments generated by purified yeast proteasome (34), we favor the first hypothesis and suggest that the functionally defined PGPH activity is mediated by the same catalytic subunit as are the SNAAP and/or BrAAP activities. It is noteworthy that, in some cases, the PGPH activity of mammalian proteasome has been shown to result in cleavage after aromatic amino acids (35).
Close comparison of the fragments detected by mass spectrometry in the absence or presence of lactacystin suggests that, in the case of peptide MAGE-3271–285, the antigenic fragment MAGE-3271–279 is only detected in the absence of the fragment MAGE-3272–278. The production of the latter appears to be inhibited by lactacystin. Assuming the presence of two competing proteolytic activities of the proteasome, one dominant under normal conditions and producing peptide MAGE-3272–278 and the other resulting in peptide MAGE-3271–279, it is possible that the inhibition of the dominant activity by lactacystin favors the lactacystin-insensitive activity, yielding only the antigenic peptide MAGE-3271–279. Alternatively, the positioning of the peptide in the inner cavity of the proteasome may favor the cleavage, generating peptide MAGE-3272–278 under normal conditions. The covalent binding of lactacystin to the subunit responsible for the chymotryptic activity (29) may reorient the peptide substrate in the cavity and lead to the generation of peptide MAGE-3271–279.
It has been reported that antigenic peptides from the same protein may be processed with different efficiencies (36, 37). In this context, it is noteworthy that the protein MAGE-3 contains, in addition to the poorly presented peptide studied here, other antigenic peptides which are well presented by MAGE-3+ melanoma cells in association with HLA-A1 or HLA-B44 molecules (15, 16). In a survey of antigenic peptides carrying the motif Ala–Leu–Val present at the COOH terminus of peptide MAGE-3271–279, we identified two such peptides. The first one, derived from the influenza nucleoprotein NP147–155 (H-2 Kd–restricted), has been shown to be inefficiently produced in H-2 Kd–positive infected cells (∼30 peptides/cell) in comparison to another antigenic peptide NP50–57 (1,800 peptides/cell; 36). Interestingly, recent results have indicated that the presentation of peptide NP147–155 in infected cells was enhanced by the addition of lactacystin (38). Although it has been shown that amino acids at position 156 could positively influence the generation of the antigenic peptide NP147–155 (39), we did not observe any effect of the amino acid at the COOH terminus of the cleavage site of peptide MAGE-3271–279. Indeed, transplantation of the MAGE-3271–279 sequence into two unrelated proteins (carrying either the residue histidine or isoleucine immediately after the COOH-terminal valine) did not result in the presentation of peptide MAGE-3271–279 (data not shown). The second peptide is derived from the melanosomal TRP-2 protein (TRP-2476-484, HLA-A*0201-restricted). Preliminary results suggest that this peptide is not presented by cells expressing TRP-2 but is efficiently presented when expressed as a minigene (Noppen, C., G. Spagnoli, and F. Lévy, unpublished results). All of these observations can be explained by the presence of unfavorable amino acids surrounding the cleavage site and acting as “negative processing signals.” Elucidation of the exact nature of the postulated negative processing signal and determination of other putative signals is underway and should further refine the current prediction algorithms used to select potential antigenic peptides. More importantly, understanding the proteolytic machinery controlling the generation and presentation of tumor-specific antigenic peptides in tumor cells should facilitate the development of efficient peptide-based vaccines aimed at inducing or enhancing the generation of tumor-reactive CTL in cancer patients. In parallel, demonstration that proteasome inhibitors like lactacystin or LLnL lead to the recognition of a peptide tumor antigen by specific CTL in melanoma cells opens the way to the development of specific proteasome inhibitors able to induce the presentation of antigens recognized by CTL and, hence, broaden the spectrum of CTL-defined tumor antigenic peptides to be considered for cancer vaccines. In this respect, the recent demonstration that the drug ritonavir, originally described as an inhibitor of HIV-1 protease and administered to HIV-1–infected patients, can act as proteasome inhibitor suggests that this drug may also favor the generation of new peptide tumor antigens efficiently recognized by specific CTL (40).
Footnotes
The expert technical assistance of A.-L. Peitrequin, A. Porret, and N. Montandon is gratefully acknowledged. We thank Dr. I. Miconnet and L. Burri for comments on the manuscript.
U. Gileadi, P.R. Dunbar, and V. Cerundolo were supported by the Medical Research Council of the United Kingdom and the Cancer Research Campaign. F. Lévy was supported in part by a grant from the Swiss Cancer League. This work was partly supported by the Federal Office for Education and Science, Switzerland and the European Community (contract BMH4-CT95-1627).
Abbreviations used in this paper: AMC, 7-amido-4-methylcoumarin; BrAAP, branched chain amino acid–preferring; ER, endoplasmic reticulum; E/T, effector-to-target cell ratio; GFP, green fluorescence protein; ha, hemagglutinin epitope; LLnL, N-acetyl-Leu-Leu-norleucinal; MALDI-TOF, matrix assisted laser desorption ionization-time of flight; PGPH, peptidylglutamylpeptide hydrolyzing; rec. v.v., recombinant vaccinia virus; SNAAP, small neutral amino acid–preferring; TAP, transporters associated with antigen presentation; Ub, ubiquitin; UPR, ubiquitin/ protein/reference.
D. Valmori and U. Gileadi contributed equally to this work.
References
- 1.Rivett AJ. Intracellular distribution of proteasomes. Curr Opin Immunol. 1998;10:110–114. doi: 10.1016/s0952-7915(98)80040-x. [DOI] [PubMed] [Google Scholar]
- 2.Baumeister W, Walz J, Zühl F, Seemüller E. The proteasome: paradigm of a self-compartmentalized protease. Cell. 1998;92:367–380. doi: 10.1016/s0092-8674(00)80929-0. [DOI] [PubMed] [Google Scholar]
- 3.Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem. 1996;65:801–847. doi: 10.1146/annurev.bi.65.070196.004101. [DOI] [PubMed] [Google Scholar]
- 4.Ping MC, Vu JH, Proske RJ, Slaughter CA, DeMartino GN. Identification, purification and characterization of a high molecular weight, ATP-dependent activator (PA700) of the 20S proteasome. J Biol Chem. 1994;269:3539–3547. [PubMed] [Google Scholar]
- 5.Ping MC, Slaughter CA, DeMartino GN. Identification, purification and characterization of a protein activator (PA28) of the 20S proteasome (macropain) J Biol Chem. 1992;267:10515–10523. [PubMed] [Google Scholar]
- 6.Dubiel W, Pratt G, Ferrell K, Rechsteiner M. Purification of an 11S regulator of the multicatalytic protease. J Biol Chem. 1992;267:22369–22377. [PubMed] [Google Scholar]
- 7.Dick TP, Ruppert T, Groettrup M, Kloetzel PM, Kuehn L, Koszinowski UH, Stevanovic S, Schild H, Rammensee H-G. Coordinated dual cleavages induced by the proteasome regulator PA28 lead to dominant MHC ligands. Cell. 1996;86:253–262. doi: 10.1016/s0092-8674(00)80097-5. [DOI] [PubMed] [Google Scholar]
- 8.Groll M, Ditzel L, Löwe J, Stock D, Bochtler M, Bartunik HD, Huber R. Structure of 20S proteasome from yeast at 2.4 Å resolution. Nature. 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 9.Kopp F, Hendil KB, Dahlmann B, Kristensen P, Sobek A, Uerkvitz W. Subunit arrangement in the human 20S proteasome. Proc Natl Acad Sci USA. 1997;94:2939–2944. doi: 10.1073/pnas.94.7.2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Orlowski M, Cardozo C, Michaud C. Evidence for the presence of five distinct proteolytic components in the pituitary multicatalytic proteinase complex. Properties of two components cleaving bonds on the carboxyl side of branched chain and small neutral amino acids. Biochemistry. 1993;32:1563–1572. doi: 10.1021/bi00057a022. [DOI] [PubMed] [Google Scholar]
- 11.Svensson K, Lévy F, Sundberg U, Boman H-G, Hendil KB, Kvist S. Proteasomes generate in vitro a natural peptide of influenza-A nucleoprotein functional in HLA–B27 antigen assembly. Int Immunol. 1996;8:467–478. doi: 10.1093/intimm/8.4.467. [DOI] [PubMed] [Google Scholar]
- 12.Groettrup M, Soza A, Kuckelkorn U, Kloetzel PM. Peptide antigen production by the proteasome: complexity provides efficiency. Immunol Today. 1996;17:429–435. doi: 10.1016/0167-5699(96)10051-7. [DOI] [PubMed] [Google Scholar]
- 13.Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, Goldberg AL. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994;78:761–771. doi: 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 14.Pamer E, Cresswell P. Mechanisms of MHC class I–restricted antigen processing. Annu Rev Immunol. 1998;16:323–358. doi: 10.1146/annurev.immunol.16.1.323. [DOI] [PubMed] [Google Scholar]
- 15.Herman J, van der Bruggen P, Luescher IF, Mandruzzato S, Romero P, Thonnard J, Fleischhauer K, Boon T, Coulie P. A peptide encoded by the human MAGE3 gene and presented by HLA–B44 induces cytolytic T lymphocytes that recognize tumor cells expressing MAGE3. . Immunogenetics. 1996;43:377–383. doi: 10.1007/BF02199806. [DOI] [PubMed] [Google Scholar]
- 16.Gaugler B, Van den Eynde B, van der Bruggen P, Romero P, Gaforio JJ, De Plaen E, Lethé B, Brasseur F, Boon T. Human gene MAGE-3codes for an antigen recognized on a melanoma by autologous cytolytic T lymphocytes. J Exp Med. 1994;179:921–930. doi: 10.1084/jem.179.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van der Bruggen P, Bastin J, Gajewski T, Coulie PG, Boel P, De Smet C, Traversari C, Townsend A, Boon T. A peptide encoded by human gene MAGE-3and presented by HLA-A2 induces cytolytic T lymphocytes that recognize tumor cells expressing MAGE-3. Eur J Immunol. 1994;24:3038–3043. doi: 10.1002/eji.1830241218. [DOI] [PubMed] [Google Scholar]
- 18.Valmori D, Liénard D, Waanders G, Rimoldi D, Cerottini J-C, Romero P. Analysis of MAGE-3–specific cytolytic T lymphocytes in human leukocytes antigen–A2 melanoma patients. Cancer Res. 1997;57:735–741. [PubMed] [Google Scholar]
- 19.Dunbar PR, Ogg GS, Chen J, Rust N, van der Bruggen P, Cerundolo V. Direct isolation, phenotyping and cloning of low-frequency antigen-specific cytotoxic T lymphocytes from peripheral blood. Curr Biol. 1998;8:413–416. doi: 10.1016/s0960-9822(98)70161-7. [DOI] [PubMed] [Google Scholar]
- 20.Lévy F, Johnsson N, Rümenapf T, Varshavsky A. Using ubiquitin to follow the metabolic fate of a protein. Proc Natl Acad Sci USA. 1996;93:4907–4912. doi: 10.1073/pnas.93.10.4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kocher T, Schultz-Thater E, Gudat F, Schaefer C, Casorati G, Juretic A, Willimann T, Harder F, Heberer M, Spagnoli GC. Identification and intracellular location of MAGE-3 gene product. Cancer Res. 1995;55:2236–2239. [PubMed] [Google Scholar]
- 22.Espevik T, Nissen-Meyer J. A highly sensitive cell line, WEHI 164 clone 13, for measuring cytotoxic factor/tumor necrosis factor from human monocytes. J Immunol Methods. 1986;95:99–105. doi: 10.1016/0022-1759(86)90322-4. [DOI] [PubMed] [Google Scholar]
- 23.Hansen MB, Nielsen SE, Berg K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J Immunol Methods. 1989;119:203–210. doi: 10.1016/0022-1759(89)90397-9. [DOI] [PubMed] [Google Scholar]
- 24.Ehring B, Meyer TH, Eckerskorn C, Lottspeich F, Tampé R. Effects of major-histocompatibility-complex-encoded subunits on the peptidase and proteolytic activities of human 20S proteasomes. Eur J Biochem. 1996;235:404–415. doi: 10.1111/j.1432-1033.1996.00404.x. [DOI] [PubMed] [Google Scholar]
- 25.DeMars R, Chang CC, Shaw S, Reitnauer PJ, Sondel PM. Homozygous deletions that simultaneously eliminate expressions of class I and class II antigens of EBV-transformed B-lymphoblastoid cells. I. Reduced proliferative responses of autologous and allogeneic T cells to mutant cells that have decreased expression of class II antigens. Human Immunol. 1984;11:77–97. doi: 10.1016/0198-8859(84)90047-8. [DOI] [PubMed] [Google Scholar]
- 26.Cerundolo V, Kelly A, Elliott T, Trowsdale J, Townsend A. Genes encoded in the major histocompatibility complex affecting the generation of peptides for TAP transport. Eur J Immunol. 1995;25:554–562. doi: 10.1002/eji.1830250238. [DOI] [PubMed] [Google Scholar]
- 27.Varshavsky A. The ubiquitin system. Trends Biochem Sci. 1997;22:383–387. doi: 10.1016/s0968-0004(97)01122-5. [DOI] [PubMed] [Google Scholar]
- 28.Vinitsky A, Anton LC, Snyder HL, Orlowski M, Bennink JR, Yewdell JW. The generation of MHC class I-associated peptides is only partially inhibited by proteasome inhibitors. J Immunol. 1997;159:554–564. [PubMed] [Google Scholar]
- 29.Fenteany G, Standaert RF, Lane WS, Choi S, Corey EJ, Schreiber SL. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science. 1995;268:726–731. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]
- 30.Niedermann G, King G, Butz S, Birsner U, Grimm R, Shabanowitz J, Hunt DF, Eichmann K. The proteolytic fragments generated by vertebrate proteasomes: structural relationships to major histocompatibility complex class I binding peptides. Proc Natl Acad Sci USA. 1996;93:8572–8577. doi: 10.1073/pnas.93.16.8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Craiu A, Akopian T, Goldberg A, Rock KL. Two distinct proteolytic processes in the generation of a major histocompatibility complex class I-presented peptide. Proc Natl Acad Sci USA. 1997;94:10850–10855. doi: 10.1073/pnas.94.20.10850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lopez D, Del Val M. Selective involvement of proteasomes and cysteine proteases in MHC class I antigen presentation. J Immunol. 1997;159:5769–5772. [PubMed] [Google Scholar]
- 33.Glas R, Bogyo M, McMaster JS, Gaczynka M, Ploegh HL. A proteolytic system that compensates for loss of proteasome function. Nature. 1998;392:618–622. doi: 10.1038/33443. [DOI] [PubMed] [Google Scholar]
- 34.Dick TP, Nussbaum AK, Deeg M, Heinemeyer W, Groll M, Schirle M, Keilholz W, Stevanovic S, Wolf DH, Huber R, et al. Contribution of proteasomal β-subunits to the cleavage of peptide substrates analyzed with yeast mutants. J Biol Chem. 1998;273:25637–25646. doi: 10.1074/jbc.273.40.25637. [DOI] [PubMed] [Google Scholar]
- 35.Wilk S, Pereira M, Yu B. Probing the specificity of the bovine pituitary multicatalytic proteinase complex by inhibitors, activators, and by chemical modification. Biomed Biochim Acta. 1991;50:471–478. [PubMed] [Google Scholar]
- 36.Anton LC, Yewdell JW, Bennink JR. MHC class I-associated peptides produced from endogenous gene products with vastly different efficiencies. J Immunol. 1997;158:2535–2542. [PubMed] [Google Scholar]
- 37.Sijts AJ, Neisig A, Neefjes J, Pamer EG. Two Listeria monocytogenes CTL epitopes are processed from the same antigen with different efficiencies. J Immunol. 1996;156:685–692. [PubMed] [Google Scholar]
- 38.Anton LC, Snyder HL, Bennink JR, Vinitsky A, Orlowski M, Porgador A, Yewdell JW. Dissociation of proteasomal degradation of biosynthesized viral proteins from generation of MHC class I-associated antigenic peptides. J Immunol. 1998;160:4859–4868. [PubMed] [Google Scholar]
- 39.Yellen-Shaw AJ, Eisenlohr LC. Regulation of class I-restricted epitope processing by local or distal flanking sequence. J Immunol. 1997;158:1727–1733. [PubMed] [Google Scholar]
- 40.André P, Groettrup M, Klenerman P, De Giuli R, Booth BL, Cerundolo V, Bonneville M, Jotereau F, Zinkernagel RM, Lotteau V. An inhibitor of HIV-1 protease modulates proteasome activity, antigen presentation, and T cell responses. Proc Natl Acad Sci USA. 1998;95:13120–13124. doi: 10.1073/pnas.95.22.13120. [DOI] [PMC free article] [PubMed] [Google Scholar]