

Abstract
In extravasation of T cells, little is known about the mechanisms of transendothelial migration subsequent to the T cells' tight adhesion to endothelium. To investigate these mechanisms, we developed a monoclonal antibody (mAb), termed anti-4C8, that blocks transmigration but not adhesion in a culture system in which high CD26–expressing (CD26hi) T cells preferentially migrate through human umbilical vein endothelial cell (HUVEC) monolayers cultured on collagen gels. Anti-4C8 reacted with all CD3+ T cells and monocytes but not neutrophils or HUVECs. The structure defined by this antibody was an 80-kD molecule. The mAb at 1 μg/ml inhibited 80–90% of migration of CD3+ T cells through unstimulated and interferon γ–stimulated HUVEC monolayers without interfering with adhesion and cell motility. When added to the cultures after the adhesion, anti-4C8 completely blocked subsequent transmigration of adherent T cells. Phase–contrast and electron microscopy revealed that T cells are arrested at the intercellular junctions of HUVECs in the presence of anti-4C8. Anti-4C8 exhibited agonistic effects on resting T cells without other stimuli under culture conditions in which anti-4C8 can stimulate T cells. First, in the checkerboard assay using collagen gels, the antibody promoted chemokinetic migration of the cells in a dose-dependent manner from 0.1 to 10 μg/ml. The predominant population of T cells that migrated into collagen gels with impregnated anti-4C8 were CD26hi. Second, solid-phase–immobilized anti-4C8 induced adhesion of T cells to the substrate, often with polarizations in cell shape and large pseudopods rich in filamentous (F-) actin. Third, soluble anti-4C8 augmented F-actin content preferentially in CD26hi T cells when added to T cells at a high dose of 10 μg/ml. Finally, both anti-4C8–induced chemokinetic migration and transendothelial migration were inhibited by pretreatment of T cells with pertussis toxin. These findings suggest that stimulation via the 4C8 antigen increases cell motility of CD26hi cells with profound cytoskeletal changes through signaling pathways including G proteins. The 4C8 antigen may be involved in preferential transmigration of CD26hi cells adherent to HUVECs.
Keywords: lymphocyte, transmigration, mechanism, cell motility, actin polymerization
Adhesion of leukocytes to the luminal surface of vascular endothelium is the first step for their extravasation, which is essential for immune surveillance and inflammatory reactions (1, 2). The adhesion process is thought to be a multistep process. Initially, leukocytes roll along endothelium by selectin-mediated interactions. The leukocyte integrins are activated during this contact with the endothelial surface, which facilitates tight adhesion and spreading of the leukocytes to the endothelial cell (EC)1 surface. The second step is transmigration of the adherent leukocytes across the vessel wall. In general, this step consists of leukocyte diapedesis and penetration of the subendothelial basement membrane; the former involves locomotion of the adherent cells to and then through nearby endothelial cell-to-cell junctions.
Little is known about the mechanisms of the transmigration process. However, it is becoming evident that leukocyte–EC interactions at the junctional level play an important role in the process (3–6). A candidate for supporting these interactions is CD31–platelet–endothelial cell adhesion molecule-1 (PECAM-1), which is an immunoglobulin superfamily expressed by platelets and leukocytes and localized at the EC junctions (7, 8). Several in vitro and in vivo studies suggest that homophilic PECAM-1–CD31 adhesion between leukocytes and ECs mediates the transmigration process of leukocytes, including neutrophils, monocytes, and NK cells (9–14). Although the contribution of CD31 to T cell transmigration is controversial (4, 15), it has been demonstrated that naive type T cells (CD45RA+) migrate through CD31–PECAM-1–transfected murine fibroblast monolayers (8).
With an in vitro system using human umbilical vein EC (HUVEC) monolayers, we and other investigators have shown that memory type T cells (CD45RO+) expressing high CD26 (CD26hi) predominantly migrate through HUVEC monolayers without a chemokine gradient (16–18). In contrast, although CD26-negative (CD26−) T cells also adhere to the monolayers, most of them do not transmigrate. This finding indicates that there are mechanisms other than CD31–PECAM-1–mediated transmigration that promote transmigration of adherent CD26hi T cells but not CD26− cells. Transmigration is directional movement of leukocytes from the apical surface of ECs to the subendothelial space. It is therefore assumed that molecules involved in transmigration mechanisms selectively stimulate cell motility of CD26hi T cells. Here we report that a mAb, anti-4C8, inhibits transmigration of CD26hi T cells subsequent to their adhesion to HUVEC monolayers and induces cell movement similar to chemokinesis as well as an increase in filamentous (F-) actin content in CD26hi cells. This is the first report suggesting that the 4C8 antigen is involved in the process of postadhesive transendothelial migration of CD26hi T cells.
Materials and Methods
Reagents.
Type I collagen solution extracted from porcine skin (Cellmatrix I-A) was purchased from Nitta Gelatin Co. EC growth supplement (ECGS) and porcine heparin were purchased from Collaborative Research and Nakarai Chemical Co., respectively. Recombinant human IFN-γ was provided by Shionogi Pharmaceutical Co. Monocyte chemoattractant protein-1 (MCP-1) was provided by T. Kasahara (Kyoritsu College of Pharmacy, Tokyo, Japan; 19). Rhodamine-conjugated anti-CD26 mAb and phycoerythrin-Cy5–conjugated anti-CD3 mAb were obtained from Coulter Corporation. Anti-CD11a mAb and purified mouse IgG3 were obtained from Becton Dickinson and Zymed Laboratories, Inc., respectively. FCS was purchased from Cell Culture Laboratories. BSA, Hepes buffer, gelatin, diisopropyl fluorophosphate (DFP), papain, L-cysteine, and collagenase (type 1-A) were obtained from Sigma Chemical Co. Pertussis toxin (PT) and M199 were obtained from Seikagaku Corporation and GIBCO BRL, respectively.
Preparation of Cells.
PBMCs were prepared from heparinized healthy human venous blood by Ficoll-Conray density gradient centrifugation as described previously (16). The T cell–enriched fraction was obtained by passing the mononuclear cells through a nylon wool column. CD3+ cells were negatively selected by inclusion of the fraction with magnetic anti-CD16 mAb (Advanced Magnetics, Inc.) The selected cells contained >96% CD3+ cells, as determined by flow cytometry. Neutrophils were prepared by dextran sedimentation, centrifugation with Ficoll-Conray, and hypotonic lysis of contaminating erythrocytes (20). Neutrophil fractions contained >95% neutrophils. Endothelial cells were obtained from human umbilical cord veins treated with 0.1% collagenase as described previously (16). Cells were grown on gelatin-precoated dishes in M199 containing 20% heat-inactivated FCS, 60 μg/ml ECGS, 100 μg/ml heparin, 1% penicillin and streptomycin solution, and 15 mM Hepes buffer. Culture medium was changed every 3 d. These experiments used cells in passages 2 and 3 only.
Production of Anti-4C8 mAb.
The anti-4C8 mAb was produced by standard techniques after immunization of BALB/c mice with PBMCs cocultured with HUVEC monolayers. In brief, after removal of nonadherent PBMCs from the cocultures, the cocultured adherent cells containing at least 107 lymphocytes were intraperitoneally injected five times at 2–3 wk intervals. The final immunization was performed by intravenous injection of 7 × 106 transmigrated T cells isolated by using an in vitro vessel model as previously described (16). 3 d later, the spleen was removed and cells were fused with NS-1 cell line. Hybridoma cultures producing antibodies that inhibited T cell migration across but not adhesion to HUVEC monolayers were selected, cloned, and recloned by limiting dilution methods in the presence of IL-6. Malignant ascites were then developed and further purified by an IgG purification kit (Pierce Chemical Co.). The anti-4C8 mAb was shown to be of the IgG3 subclass by an ELISA method for determining subclasses of mouse IgG. Fab fragments were produced by incubating purified IgG with 10 μg/ml papain, 5 mM L-cysteine, and 2 mM EDTA and then purified by passing over DEAE-cellulose. SDS-PAGE performed under nonreducing conditions proved that the fragments were properly cut and that no extraneous bands were present on Coomassie blue stain. Fluorescein-conjugated anti-4C8 mAb was prepared by using a fluorescein labeling kit (Sigma Chemical Co.).
Adhesion and Transmigration Assays.
For the adhesion and transmigration assays, we modified the original system that was described elsewhere (16). HUVEC monolayers were grown to confluence on collagen gels (50 μl/well) in 96-well flat bottom plates (Becton Dickinson), followed by treatment for 48 h with or without IFN-γ (500 U/ml) before assay. Freshly isolated CD3+ T cells suspended in M199–0.1% BSA were or were not pretreated with mAbs for 20 min on ice. The cells were added to the wells without washing (3 × 105 cells/100 μl/well). The plate was centrifuged for 1 min at 50 g and incubated for 3–4 h at 37°C in a 5% CO2-humidified incubator. In the adhesion assay, unbound T cells were gently washed out, and then adherent cells were immediately fixed with 1% paraformaldehyde in PBS. The transmigration assay was performed simultaneously with the adhesion assay. To count migrated cells, adherent T cells and HUVECs were removed from the surface of collagen gels by 0.4% EDTA treatment. In some experiments, mAbs were added after unbound cells had been washed from the cultures. Adherent cells on the apical surfaces of HUVECs or cells that had transmigrated into the collagen gels were counted by phase–contrast microscopy in a blinded manner. The cells in a field of 0.25 mm2 were counted at a magnification of 100. Adhesion or migration index (%) was calculated as follows: the number of cells with antibody/the number of cells without antibody × 100. All experiments were performed in triplicate.
Release of Adherent and Transmigrated T Cells.
After T cells (2 × 107 cells) were cultured for 5 h with a confluent HUVEC monolayer on 2 ml of collagen gels (60 mm dish), adherent and transmigrated cells were collected as described (16). In brief, after unbound T cells were removed, T cells bound to HUVECs were incubated for 20 min with 0.4% EDTA. Almost all adherent T cells could be obtained by this treatment. The HUVEC monolayer was then removed from the surface of collagen gels by the EDTA treatment for another 30 min. The collagen gels containing transmigrated T cells were incubated with 0.05% collagenase in PBS for 3 min to release the cells. This collagenase treatment was repeated twice. No changes in the expression of surface proteins of T cells were found following the treatment.
Chemotaxis and Checkerboard Assays Using Collagen Gels.
We modified a chemotaxis assay using collagen gels as described by others (21). Resting and activated T cells were prepared by culturing freshly isolated T cells for 2 d in RPMI 1640 containing 10% FCS and for 6 d on anti-CD3–coated dishes (0.4 μg/ml) in medium with 100 U/ml of IL-2, respectively. The cells were washed, resuspended in M199 plus 0.1% BSA, and added directly onto collagen gels (50 μl/well), with or without impregnated MCP-1 (100 ng/ml), in 96-multiwell plates (1–4 × 105 cells/ well). After 1.5–2 h, unbound cells and cells attached on the surfaces of the gels were washed out with 0.4% EDTA in PBS. Cells that migrated into the gels were counted under a phase–contrast microscope at 200× as described above. In the checkerboard assay, mAbs were impregnated into collagen gels and/or added directly to freshly isolated T cells (4 × 105 cells/well) above the gels at varying concentrations in 96-multiwell plates. T cells were incubated for 4 h under these conditions. In these experiments, to reduce spontaneous migration of T cells, the collagen gels were prepared with collagen solution at a concentration of 3 mg/ ml, which is three times higher than in the transmigration assays. Migrated cells in the gels were carefully counted and the migration index was calculated as described above. All experiments were performed in triplicate.
Flow Cytometric Analysis.
Cells were treated for 20 min with saturating amounts of fluorescein-conjugated mAb and washed three times with PBS containing 0.1% BSA and 0.01% sodium azide. The stained cells (10,000 cells) were analyzed on a FACScan® flow cytometer (Becton Dickinson) with gating on the lymphocyte, monocyte, or neutrophil population. All staining procedures were performed at 4°C. Lysis II software (Becton Dickinson) was used to analyze the data obtained.
Western Blotting.
PBMCs or neutrophils (107 cells) were incubated for 30 min with intermittent agitation with or without DFP (1 mM ) on ice. The pellets of the cells were resuspended for 60 min in 100 μl of ice cold extraction buffer containing 50 mM Hepes (pH 7.4), 2 mM sodium orthovanadate, 100 mM sodium fluoride, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 1 mM PMSF, 100 μg/ml aprotinin, and 10 μg/ml leupeptin. Cells were treated for 30 min with 1 mM DFP on ice before cell lysis. After centrifugation, the supernatant was mixed 1:1 with 2× sample buffer (4% SDS, 20% glycerol, 10% mercaptoethanol, and a trace amount of bromophenol blue dye in 125 mM Tris-HCl, pH 6.8), heated at 100°C for 5 min, and loaded onto an 8% SDS–polyacrylamide gel. After electrophoresis, proteins were transferred onto a nitrocellulose membrane (Pierce Chemical Co.). Residual binding sites on the membrane were blocked by incubating the membrane in Tris-buffered saline (pH 7.6) containing 0.1% Tween-20 and 5% nonfat dry milk for 2 h at room temperature. The membranes were incubated with anti-4C8 mAb and then with biotin-conjugated anti–mouse IgG antibody. After incubation, enzymatic development was performed by using peroxidase-conjugated streptoavidin (GIBCO BRL) and the ECL system (Amersham).
Cell Morphology Assay and F-actin Staining.
The anti-4C8– induced changes in cell shape and F-actin formation were visualized by staining with TRITC-labeled phalloidin (Sigma Chemical Co.) as described previously by others (22). Glass slides (uncoated eight-well CultureSlide; Becton Dickinson) were coated with anti-4C8 mAb (10 μg/ml, 250 μl/well) or control IgG3 overnight. T cells (5 × 105 cells/well in M199 with 0.1% BSA) were added to the slides and incubated for 2 h at 37°C. After incubation, attached cells were fixed with 3.7% paraformaldehyde, permeabilized with 0.1% Triton X-100/PBS, and stained for F-actin with TRITC-labeled phalloidin. Microscopic analysis was performed using an Olympus microscope equipped with fluorescence accessories and photographed with an ×100 oil immersion objective. In some experiments, T cells (5 × 105 cells/500 μl of 0.1% BSA–M199/Eppendorf tube) were incubated for 3 h with anti-4C8 mAb or control IgG3 at 37°C. After fixation and permeabilization, the cells were double-stained with FITC-conjugated phalloidin and rhodamine-conjugated anti-CD26 mAb. The stained cells were then analyzed by a flow cytometer.
Scanning Electron Microscopy.
The transmigration assay was performed in the presence of anti-4C8 (1 μg/ml) in 24-multiwell plastic plates (Falcon Labware). After a 5-h incubation, cultures were fixed overnight in 2% electron microscopy–grade glutaraldehyde and 5% sucrose in 0.1 M sodium cacodylate buffer (pH 7.4), followed by postfixation with 1% OsO4. The fixed cells and collagen gels were removed from the plate and processed for scanning electron microscopy by critical-point drying and gold coating.
Statistical Analysis.
All values are represented as means ± SD. When comparing two groups, P values were calculated by Student's t test. P values <0.05 were considered to indicate a significant difference.
Results
Expression of the 4C8 Antigen on Human PBLs and HUVECs and Its Structure.
We first examined immunofluorescence profiles of 4C8 expression on PBLs and HUVECs by a flow cytometer. As shown in Fig. 1, the 4C8 antigen was expressed intensely on CD3+ T cells and to a lesser extent on CD3− cells (largely CD16+ cells). Staining of the cells gated on the monocyte population was positive and two peaks were seen. In contrast, anti-4C8 did not react with neutrophils or unstimulated HUVECs. The negative expression of neutrophils was unaffected by stimulation with LPS or TNF-α (data not shown). Immunofluorescence microscopy also showed no significant staining of confluent monolayers of HUVECs (not shown). To address the molecular weight of the 4C8 antigen, Western blotting analysis was performed using lysates from PBMCs and neutrophils (Fig. 2). Anti-4C8 reacted with a single band of 80 kD in lysates from PBMCs but not neutrophils. It is possible that the 4C8 antigen was cleaved by numerous proteolytic enzymes released from neutrophils during the procedure of cell lysis. However, anti-4C8 did not react with the lysates in the presence of DFP, a serine protease inhibitor. There were no differences between blots from gels electrophoresed under reducing and nonreducing conditions (data not shown). In addition, immunoprecipitation failed to detect the 4C8 antigen by a standard method (not shown). This finding is consistent with fluorescence profiles of the 4C8 antigen on these cells and further suggests that the antigen is not present in the intracellular contents of neutrophils.
Figure 1.

Immunofluorescence profiles of the 4C8 antigen on human PBLs and HUVECs. PBMC and neutrophils from a healthy donor were stained with FITC-conjugated anti-4C8 and phycoerythrin-Cy5–conjugated anti-CD3 mAb or control FITC-conjugated IgG3. The stained leukocytes as well as HUVECs (10,000 cells) were analyzed by a FACScan® (Becton Dickinson) flow cytometer with gating on the lymphocyte, monocyte, and neutrophil populations.
Figure 2.

Western blot analysis of the 4C8 antigen. SDS–gel electrophoresis of lysates from PBMC and neutrophils (5.5 × 106 cells/lane) was performed under reducing conditions. DFP, a serine protease inhibitor, was used to prevent proteolytic activity in the neutrophil lysates. Control Ab was purified mouse IgG3 of the same isotype as anti-4C8. See Materials and Methods for details.
Anti-4C8 mAb Inhibits T Cell Transmigration Subsequent to LFA-1–mediated Adhesion to a HUVEC Monolayer.
In our system, 20–30% of total added CD3+ T cells adhered to unstimulated HUVEC monolayers after 3–5 h of incubation, and 10–20% of the adherent cells transmigrated during this period (16, 17). IFN-γ augments the expression of intercellular adhesion molecule-1 (ICAM-1), the ligand for CD11a/CD18 (LFA-1 α/β), on HUVEC. Stimulation of HUVEC with IFN-γ increased T cell adhesion and transmigration to 1.5- and 3-fold the base line values, respectively (Fig. 3). We then assessed the changes in adhesion and transmigration in the presence of antibodies. Anti-CD11a mAb (10 μg/ml) inhibited T cell adhesion to and transmigration through unstimulated and IFN-γ–stimulated HUVEC monolayers by 50–60 and 80%, respectively. This indicates that T cell transmigration is largely dependent upon LFA-1–mediated adhesion to HUVECs. On the other hand, anti-4C8 mAb (1 μg/ml) did not inhibit T cell adhesion but rather induced a small increase in adhesion to IFN-γ–stimulated HUVEC monolayers (Fig. 3 A). The small increase was in accord with the number of T cells that were retained on HUVECs by the transmigration-blocking effect of anti-4C8. However, transmigration was strikingly inhibited: 79 and 87% with unstimulated and IFN-γ–stimulated HUVEC monolayers, respectively (Fig. 3 B). Control IgG3 (the same isotype as anti-4C8) showed neither inhibition of T cell adhesion nor transmigration. To determine even more directly whether anti-4C8 mAb acts subsequently to LFA-1–mediated adhesion, the antibody blocking study was performed following removal of unbound T cells 1 h after coculturing T cells and IFN-γ–stimulated HUVEC monolayers. As shown in Fig. 4, both anti-4C8 IgG (1 μg/ml) and Fab fragments (10 μg/ml), but not anti-CD11a or control IgG3, completely blocked subsequent migration of adherent T cells during a 5-h incubation. The blocking effect was not due to detachment of the adherent cells from the apical surfaces of HUVECs (data not shown). However, it is possible that the blockage is caused by a direct suppressive effect of anti-4C8 on cell motility. To examine this, we next performed chemotaxis assays using a three-dimensional collagen matrix (collagen gels), with or without impregnated MCP-1. Resting and anti-CD3–activated T cells were added to the gels and incubated for 1.5–2 h in the presence or absence of anti-4C8 at 1 μg/ml. Although spontaneous migration of both resting and activated T cells into the gels was enhanced two to three times by MCP-1, anti-4C8 had no effect on spontaneous or MCP-1–induced migration (Fig. 5). Taken together, the data suggest that anti-4C8 mAb inhibits postadhesive transmigration of T cells without affecting adhesion or suppressing cell motility.
Figure 3.


Anti-4C8 mAb inhibits T cell migration through, but not adhesion to, HUVEC monolayers. Freshly isolated CD3+ T cells were incubated with unstimulated and IFN-γ–stimulated (500 U/ml, 48 h) HUVEC monolayers cultured on collagen gels in the continuous presence of anti-4C8 (1 μg/ml), anti-CD11a (10 μg/ml), or control IgG3 (10 μg/ml). The numbers of adherent cells and migrated cells were determined as described in Materials and Methods. Results of T cell adhesion (A) and migration (B) are expressed as the adhesion and the migration index, respectively, and represent the mean ± SD of five independent experiments. The index (%) is calculated as follows: the number of adherent or migrated T cells with antibody/the number of adherent or migrated T cells without antibody × 100. The numbers of the background adhesion and migration with unstimulated HUVEC are 376 ± 84 and 131 ± 4, respectively.
Figure 4.

Anti-4C8 mAb inhibits postadhesive transmigration of T cells. T cells were incubated for 1 h with IFN-γ–stimulated HUVEC monolayers. After nonadherent T cells were washed out, the cultures were further incubated with anti-4C8 IgG (1 μg/ml), anti-4C8 Fab fragments (3 and 10 μg/ml), anti-CD11a (10 μg/ml), or control IgG3 (10 μg/ml). Results are expressed as the migration index calculated as described in the Fig. 3 legend and represent the mean ± SD of four independent experiments.
Figure 5.
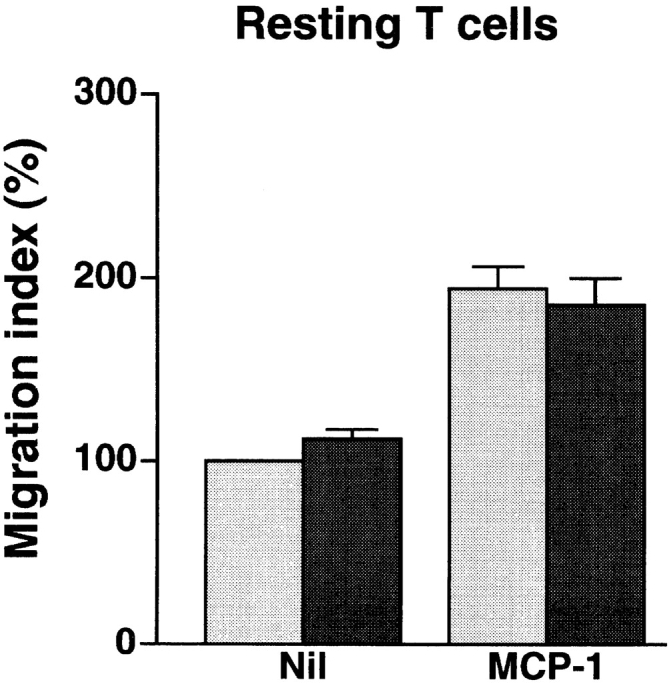
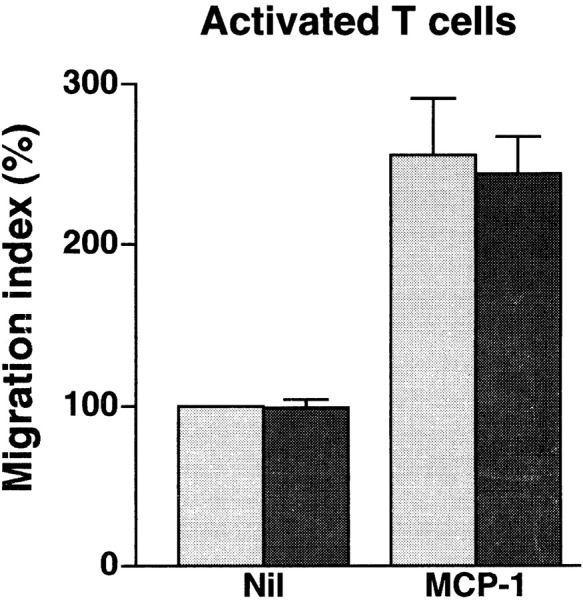
Anti-4C8 mAb has no effect on spontaneous or chemotactic migration of resting or activated T cells into collagen gels. Resting and activated T cells were prepared by incubation for 2 d without stimuli and for 6 d on anti-CD3–coated dishes with IL-2, respectively. The cells were placed on collagen gels with or without 100 ng/ml of impregnated MCP-1 in 96-multiwell plates and incubated for 1.5–2 h in the presence or absence of 1 μg/ml of anti-4C8 in medium. After incubation, T cells that had migrated into the gels were counted. Results are expressed as the migration index (calculated as described in the Fig. 3 legend), and represent the mean ± SD of three independent experiments. Gray bar, Nil; black bar, +Anti-4C8.
Anti-4C8 mAb Inhibits T Cell Transmigration at the Intercellular Junctions of HUVECs.
It has been reported that monocytes treated with anti-CD31 remained bound to the apical surface of HUVEC monolayers at the intercellular junctions (10). We therefore examined where the blockage of T cell transmigration by anti-4C8 occurs on the apical surface of IFN-γ–treated HUVEC monolayers. Although numerous T cells migrated across the monolayer into collagen gels below in the presence of control IgG3, the migration was strongly inhibited in the presence of anti-4C8 at 1 μg/ml (Fig. 6). Compared to the controls, T cells that remained on the apical surface of the monolayer increased in number, and most of them appeared to be arrested at the intercellular junctions of HUVECs. Scanning electron microscopy revealed that these T cells were firmly attached and flattened on the EC surface, with pseudopods extending into the junction (Fig. 7).
Figure 6.
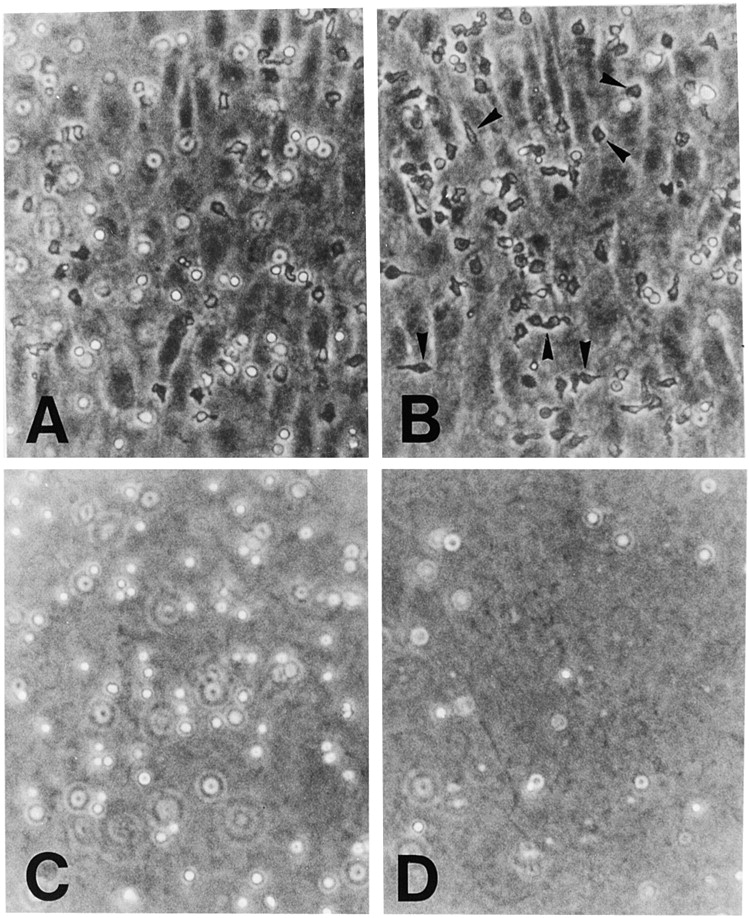
Anti-4C8 mAb inhibits T cell transmigration at the intercellular junctions of HUVECs. T cells were incubated on IFN-γ–stimulated (500 U/ml, 48 h) HUVEC monolayers cultured on collagen gels in the presence of anti-4C8 (1 μg/ml; B and D) or control IgG3 (1 μg/ml; A and C). After 4 h, monolayers were washed to remove nonadherent cells and fixed with 1% paraformaldehyde in PBS or further treated with 0.4% EDTA to remove monolayers from the surface of collagen gels. Cells were photographed under a phase–contrast microscope (×100). In the control Ab sample, although a number of T cells still adhered to the apical surface of the HUVEC monolayer (A), numerous cells that migrated into the collagen gel below could be seen (C). However, in the anti-4C8– treated sample, migration was strongly inhibited (D), whereas the number of adherent cells was increased (B) compared to the control. Most adherent cells appear to be arrested at the EC junctions (B). The black arrows indicate these T cells.
Figure 7.
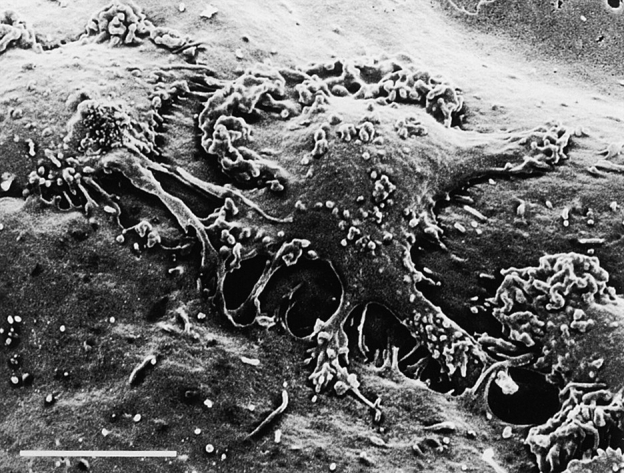
Anti-4C8 mAb blocks transmigration of T cells at the junctional level. T cells were incubated for 4 h on an IFN-γ–stimulated HUVEC monolayer in the presence of anti-4C8 (1 μg/ml). The culture was washed, fixed, and prepared for scanning electron microscopy. Tightly adherent cells extending pseudopods into the junction could be seen. Bar, 5 μm.
Anti-4C8 mAb Impregnated into Collagen Gels Predominantly Stimulates Chemokinetic Migration of CD26hi T Cells.
Activation of cell motility is a critical event in the transmigration process of T cells through the EC junctions. If the 4C8 antigen plays an essential role in the process, stimulation of T cells via the antigen should promote cell motility and migration. To examine this hypothesis, we used collagen gels in which anti-4C8 had been impregnated to substitute for the 4C8 ligand. The impregnated anti-4C8 IgG increased T cell migration in a dose-dependent manner, whereas anti-4C8 Fab fragments, as well as control IgG3 and anti-CD11a, had no such effect (Table I). In the checkerboard analysis, when soluble anti-4C8 was directly added to T cells above plain gels, migration was significantly enhanced only at the high dose of 10 μg/ml but not at doses ≤1 μg/ml. When the dose of the antibody was the same above and in the gels, the degree of migration was almost equivalent to that induced by anti-4C8 impregnated in the gels, irrespective of the presence of a gradient. Thus, stimulation via the 4C8 antigen appeared to induce migration with increased random motility resembling chemokinesis but not chemotaxis. In our transmigration assay system using HUVEC monolayers, most T cells that do not adhere to HUVECs express low CD26 (CD26lo), whereas adherent (but not migrating) cells are predominantly CD26− and migrating cells are CD26hi (Fig. 8 A). If the 4C8 antigen is involved in the transmigration, its chemokinetic action should result in migration of CD26hi cells. We therefore examined whether anti-4C8 stimulation actually causes selective migration of CD26hi cells. After T cells were incubated for 5 h on collagen gels, with or without impregnated anti-4C8 (10 μg/ml), cells that migrated were isolated and analyzed for CD26 expression. The CD26 profiles of migrated T cells with or without anti-4C8 were different from profiles of the initial T cells (Fig. 8 B). The proportion of CD26− cells increased among cells that had spontaneously migrated without anti-4C8, whereas the proportion of CD26hi cells substantially increased among cells that migrated after stimulation with anti-4C8. The CD26 profile of cells that migrated in response to anti-4C8 stimulation was similar to the profile of cells that migrated through HUVEC monolayers.
Table I.
Checkerboard Analysis of Anti-4C8 mAb
| Concentration of antibody in collagen gels (μg/ml) | Concentration of antibody above collagen gels (μg/ml) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Control IgG3 | Anti-CD11a | Anti-4C8 Fab fragments | Anti-4C8 IgG | |||||||||||
| 0 | 0 | 0 | 0 | 0.1 | 1.0 | 10.0 | ||||||||
| 0 | 100 | 100 | 100 | 100 | 98 ± 14 | 106 ± 14 | 192 ± 36* | |||||||
| 0.1 | ND | ND | ND | 101 ± 5 | 99 ± 13 | ND | ND | |||||||
| 1.0 | 103 ± 10 | 100 ± 9 | 103 ± 11 | 140 ± 16 | ND | 161 ± 35* | ND | |||||||
| 10.0 | 109 ± 10 | 101 ± 14 | 103 ± 5 | 300 ± 73* | ND | ND | 288 ± 70* | |||||||
The checkerboard assays were performed by varying concentrations of antibody both in and above collagen gels. Collagen gels were prepared and impregnated with antibodies as described in Materials and Methods. T cells were suspended in media with or without antibodies and placed on the surface of collagen gels. After a 4-h culture, T cells that had migrated into the gels were counted by phase–contrast microscopy. All experiments were performed in triplicate. Results are expressed as percent of migration under control media and represent the mean ± SD of three independent experiments.
P < 0.02, compared with control, paired Student's t test.
Figure 8.

The profile of CD26 expression on T cells that migrated through HUVEC monolayers (A) and into collagen gels with impregnated anti-4C8 mAb (B). (A) After T cells were incubated for 5 h with resting HUVEC monolayers, unbound, adherent (but not migrating), and migrated T cells were isolated as described in Materials and Methods. (B) T cells were incubated for 5 h on collagen gels with impregnated anti-4C8 (10 μg/ml) or control IgG3 (10 μg/ml). After unbound cells and cells adhering to the apical surface of collagen gels were removed with EDTA treatment, migrated cells were released from the gels by treatment with collagenase. The isolated cells were stained with fluorescein-conjugated anti-CD26 and anti-CD3 mAbs. The CD26 expression of the cells was analyzed by flow cytometry with gating on CD3+ cells.
Anti-4C8 Stimulation Induces Formation of Pseudopods Rich in F-actin and Increases F-actin Content in CD26hi T Cells.
Polarization of F-actin content and changes in cell shape are essential events in cell movement (23–25). Therefore, these changes should occur selectively in CD26hi cells with cell movement induced by anti-4C8. When T cells were incubated for 1 h on the substrate with immobilized anti-4C8, the majority of T cells attached by the mAb displayed cell flattening and irregularities in cell shape. With fluorescence microscopy using TRITC-conjugated phalloidin staining, ∼15% of the cells revealed extreme cell polarization with lamellipodia or filopodia rich in F-actin, whereas with immobilized control IgG3, almost all T cells maintained spherical morphologies (Fig. 9). We determined whether the anti-4C8–induced increase in F-actin content is characteristic of CD26hi T cells. After stimulation for 3 h with antibodies in solution, T cells were fixed and double-stained with FITC-conjugated phalloidin and rhodamine-conjugated anti-CD26 mAb. Flow cytometry revealed that a definite increase in F-actin content was induced in CD26hi cells, and to a lesser extent in CD26lo cells, by 10 μg/ml of anti-4C8 but not by control IgG3 at the same dose (Fig. 10). The small increase of F-actin in CD26lo cells was consistent with the observation that anti-4C8 stimulation also induced minimal migration of CD26lo cells as described above. However, 1 μg/ml of anti-4C8, which strongly blocked transmigration, had no such effect on CD26hi cells. These data suggest that signaling via the 4C8 antigen induces an increase in F-actin content and morphologic polarization most strongly in CD26hi T cells, resulting in activated motility and migration of the subset.
Figure 9.


Morphologic changes of T cells and redistribution of F-actin induced by immobilized anti-4C8 mAb. T cells were incubated for 2 h on glass slides precoated with anti-4C8 (10 μg/ml) or control IgG3 (10 μg/ml). After staining with TRITC-conjugated phalloidin, microscopic observation was performed (×1,000). Extreme cell polarization and large pseudopods rich in F-actin were noted with immobilized anti-4C8 (B) but not IgG3 (A).
Figure 10.
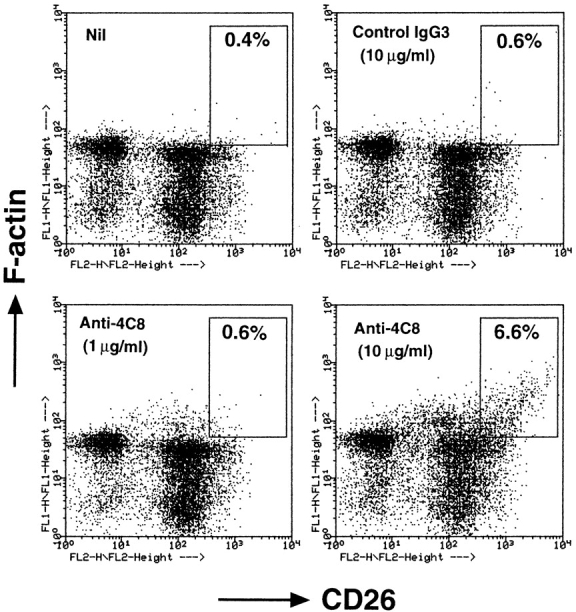
Soluble anti-4C8 mAb augments F-actin content in CD26hi T cells. T cells were incubated for 3 h with anti-4C8 or control IgG3. The cells were fixed, permeabilized, and stained with FITC-conjugated phalloidin and rhodamine-conjugated anti-CD26 mAb. The stained cells were analyzed by a flow cytometer. Increased F-actin content was observed in CD26hi T cells at 10 μg/ml, but not 1 μg/ml, of anti-4C8. The staining shown is representative of three independent experiments.
G proteins Are Involved in Postadhesive T Cell Transmigration and Migration Induced by Anti-4C8 Impregnated in Collagen Gels.
In general, chemoattractant receptor signaling is G protein linked and can be inhibited by PT (26, 27). G proteins may mediate the signal via the 4C8 antigen, as anti-4C8 stimulation induced chemokinetic migration into collagen gels. Finally, we determined that PT-sensitive G proteins are involved in T cell migration, as well as adhesion and transmigration with IFN-γ–stimulated HUVEC monolayers. The anti-4C8–induced migration and the transendothelial migration were strongly inhibited by PT pretreatment of T cells (Fig. 11). In contrast, spontaneous T cell migration and adhesion to the monolayers were insensitive to PT. The inhibitory effect of PT suggests that both anti-4C8–induced chemokinetic migration and transendothelial migration are mediated by signaling pathways that include G proteins.
Figure 11.


Inhibitory effect of PT on T cell transmigration through HUVEC monolayers and migration into collagen gels impregnated with anti-4C8 mAb. T cells were pretreated for 1 h with PT (100 ng/ml), washed three times, and cultured for 2 h with IFN-γ–stimulated HUVEC monolayers (A) or for 3 h on collagen gels with or without impregnated anti-4C8 (10 μg/ml; B). Adhesion and migration are expressed as the adhesion and the migration index, respectively. The data are presented as the mean ± SD of three independent experiments.
Discussion
In our culture system, a restricted subset, ∼6%, of the added T cells migrate through resting HUVEC monolayers. However, the migrating subset mainly consists of CD26hi cells, despite adhesion of both CD26− and CD26hi cells to the HUVECs (16; Fig. 8 A). This strongly suggests that there are molecules (distinct from those mediating adhesion) that selectively stimulate the motility of CD26hi cells, causing them to move toward the subendothelial space. The present data indicate that the antigen recognized by anti-4C8 mAb is one of the molecules, due to the following lines of evidence: first, anti-4C8 blocked postadhesive transmigration of T cells without interfering with adhesion and cell motility; second, anti-4C8 impregnated in collagen gels induced chemokinetic migration of T cells, predominantly CD26hi cells; and third, anti-4C8 stimulation induced formation of large pseudopods rich in F-actin and increased F-actin content selectively in CD26hi cells. These results suggest that the cellular interaction via the 4C8 antigen between T cells and HUVECs predominantly stimulates cell motility and transmigration of CD26hi cells.
Previous studies by others have suggested the possibility that adhesion molecules, such as integrins or ICAMs, participate in transendothelial migration of adherent lymphocytes (28–31). For example, ICAM-2 peptide or cross-linking of LFA-1 (CD11a/CD18) receptors induces migration of NK cells and actin polymerization (28). In the present study, we have shown that anti-CD11a mAb blocked T cell transmigration. However, this blockage is due to the inhibitory effect of anti-CD11a on T cell adhesion, because the mAb did not inhibit subsequent transmigration of T cells after tight adhesion to HUVEC monolayers (Fig. 4). Based on this finding, although stimulation of LFA-1 may be capable of triggering cell locomotion, the molecule seems unlikely to directly mediate T cell transmigration in our system. This conclusion is in line with a recent report showing that activated T cells can not migrate through ICAM-1–transfected monolayers in the absence of a chemotactic gradient (8). The authors speculated that the diffuse distribution of ICAM-1 on the apical cell surface may not support directed locomotion of T cells across the monolayers. In this regard, the concentrated distribution of CD31 at the EC junctions may be an appropriate stimulus for transmigration of leukocytes, as it has been reported that pretreatment of the EC junctions with anti-CD31 blocks transmigration of monocytes (10). A hypothesis is now proposed that homophilic interactions of leukocyte and EC CD31 mediate transmigration of adherent leukocytes, including T cells, monocytes, neutrophils, and NK cells (3, 8–13). However, it should be noted that CD31+ T cells display a naive phenotype characterized by CD45RA+ expression, whereas a majority of T cells that migrate through HUVEC monolayers express a memory type phenotype characterized by CD45RO+ expression (15–18). Indeed, CD31−CD45RO+ cells have been shown to be the predominant transmigrated cells (15, 18, and our unpublished data). Thus, CD31 appears not to be required for transmigration of memory T cells across HUVEC monolayers.
Soluble anti-4C8 inhibited T cell transmigration up to 90% at a dose as low as 1 μg/ml. In contrast, it also promoted cell migration into collagen gels when impregnated in the gels at doses from 1 to 10 μg/ml or when used in solution at 10 μg/ml (Table I). Since Fab fragments of anti-4C8 did not stimulate migration into collagen gels, it seems likely that anti-4C8, at the high dose of 10 μg/ml, cross-links the 4C8 antigen existing on T cells at high density and generates a signal to increase F-actin content, consequently activating cell movement. Thus, the blockage of transmigration might be due to random movement stimulated by anti-4C8, which is sufficient to overcome the force to transmigrate generated on HUVEC monolayers. However, soluble anti-4C8 at 1 μg/ml affected neither F-actin content in T cells nor the spontaneous and chemotactic motility of resting and activated T cells. Rather, these findings suggest that anti-4C8 at the low dose blocked the cellular interaction through the 4C8 antigen (which mediates T cell transmigration) between T cells and HUVEC monolayers without cross-linking of the antigen. Complete inhibition of postadhesive transmigration by Fab fragments strongly supports this notion. Microscopic observations imply that the interaction occurs at the EC junction and that the 4C8 ligand might be concentrated in the junctions like endothelial CD31. More studies are needed to elucidate the existence and the nature of the ligand.
CD26hi T cells actively transmigrate, but CD26− T cells remain adherent to HUVEC. This finding strongly suggests that additional cytoskeletal changes should occur to support active cell movement of adherent CD26hi cells, but not CD26− cells, directed toward the subendothelial space. To migrate, cells must acquire the redistribution of actin-based cytoskeleton from symmetry around the cell rim to concentration in a particular region, followed by morphological polarization characterized by extension of lamellipodia and filopodia (23, 24, and 32). Interestingly, anti-4C8 caused these changes in resting T cells under culture conditions in which the mAb can stimulate T cells. Solid-phase– immobilized anti-4C8 induced changes in cell shape, including extreme morphological polarization and formation of lamellipodia or filopodia rich in F-actin (Fig. 9). When impregnated in collagen gels, anti-4C8 promotes migration of T cells into the gels. The checkerboard analysis suggests that although a gradient is established with the mAb, the migration appears to depend upon the concentration of the antibody itself but not the gradient. Moreover, these effects were observed preferentially in CD26hi cells (Figs. 8 and 10). The 4C8 antigen thus appears to transduce a signal preferentially in CD26hi cells for cell polarization, with protrusion of the membrane that is tightly coupled to polymerization of F-actin. A similar cellular polarization induced by mAbs other than anti-4C8 has been reported with use of T lymphoblasts (33–36). Those reports showed that, similar to chemokines, engagement of ICAM-3 or CD43 with specific mAbs activates the integrin-mediated adhesion and causes the development of a cytoplasmic projection at the tailing edge, termed the uropod, and also induces the redistribution of ICAM-1, -3, CD43, and CD44 to the uropod. These morphological and functional changes may contribute to lymphocyte locomotion and recruitment. However, the 4C8 antigen is definitely distinct from ICAM-3 and CD43 in structural characteristics and tissue distribution. Whether the cellular events caused by anti-4C8 are similar to those caused by anti-ICAM-3 and anti-CD43 mAbs remains to be studied.
It is unclear why CD26hi T cells are preferentially stimulated by anti-4C8, despite the intense expression of the 4C8 antigen on all CD3+ T cells. CD26 is a widely distributed cell surface glycoprotein of 110 kD with multiple functions (37, 38). On human T cells, its expression is preferentially restricted to the CD4+ memory subset and strongly upregulated following cell activation. Recent studies indicate that CD26 has dipeptidyl–peptidase IV activity, is not only a functional receptor for collagen but also a receptor for adenosine deaminase, and acts as a costimulatory molecule for T cell activation. More recently, the chemokine RANTES (regulated on activation, normal T cell expressed and secreted) has been shown to be a natural substrate for CD26 (39, 40). These findings suggest that CD26 plays an important role in immune system events such as T cell activation and migration. However, two different anti-CD26 mAbs failed to inhibit T cell transmigration (our unpublished data), suggesting that CD26 is not directly involved in the transmigration process. The avidity of LFA-1 for ICAM-1 is enhanced by a conformational change in the integrin upon activation (41). Similarly, the activation state of T cells as related to CD26 expression might regulate the affinity of the 4C8 antigen.
We have shown that PT pretreatment of T cells inhibited both anti-4C8–induced chemokinetic migration and postadhesive transmigration. Thus, both of these types of cell motility are mediated via PT-sensitive, G protein–coupled pathways. This is consistent with the idea that the 4C8 antigen is involved in the process of transmigration after adhesion of T cells. The result also raises the question of whether the 4C8 antigen belongs to the chemokine receptor family, because chemoattractant signaling is generally mediated by G proteins and inhibited by PT (26, 27). CC chemokines have been shown to attract a subset of CD26hi CD45RO+ T cells (42–44). More recently, it has been reported that transendothelial migration can be triggered by an agonistic mAb against the CC chemokine receptor 2 (CCR2) via a PT-sensitive signaling pathway (45). Therefore, the 4C8 antigen might be a receptor for these chemokines. However, the 4C8 appears to be different from chemokine receptors identified to date in the following ways: the molecular mass of the 4C8 antigen is 80 kD, whereas the molecular mass of a chemokine receptor was estimated to be ∼35 kD (45, 46); in contrast to the 4C8 antigen, chemokine receptor levels on T cells are generally much lower than those on neutrophils and eosinophils (43, 47); and the CCR2 expression is low on the CD26hi T cells and undetectable on other T cells. Moreover, a novel membrane-bound chemokine with a CX3C motif that was recently identified has potent adhesive and chemoattractant activity for unstimulated lymphocytes, particularly CD16+ NK cells (48, 49). The chemokine is induced on activated endothelial cells and thus presumed to regulate leukocyte trafficking. However, its receptor is expressed on only a small population (up to 14%) of CD3+ T cells (49). Thus, although chemokine receptors and the 4C8 antigen show functional similarities, they appear to be different molecules based on the differences described above. To our knowledge, the 4C8 antigen is a previously unknown molecule. Gene cloning and identification will define the structural nature of this molecule in the near future.
Finally, we propose herein a hypothesis that the interaction via the 4C8 antigen between a T cell and a HUVEC transduces a signal to stimulate crawling locomotion of CD26hi cells with profound changes in cell morphology and cytoskeletal assembly, resulting in preferential migration of this T cell subset.
Acknowledgments
We thank Dr. M. Miyasaka (Osaka University, Osaka, Japan) for helpful comments on the manuscript, Dr. Y. Terano and Mr. A. Shimada (Gifu Research Laboratory, Immunology Division, JBC Inc., Gifu, Japan) for preparing Fab fragments of anti-4C8 mAb, and Ms. Mamiko Semba for her technical assistance.
This work was supported by grants from the Japanese Ministry of Education and Ministry of Welfare.
Abbreviations used in this paper
- DFP
diisopropyl fluorophosphate
- EC
endothelial cell
- ECGS
EC growth supplement
- F-actin
filamentous actin
- HUVEC
human umbilical vein EC
- ICAM-1
intercellular adhesion molecule-1
- MCP-1
monocyte chemoattractant protein-1
- PECAM-1
platelet–endothelial cell adhesion molecule-1
- PT
pertussis toxin
References
- 1.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 2.Butcher EC, Picker LJ. Lymphocyte homing and homeostasis. Science. 1996;272:60–66. doi: 10.1126/science.272.5258.60. [DOI] [PubMed] [Google Scholar]
- 3.Bianchi E, Bender JR, Blasi F, Pardi R. Through and beyond the wall: late steps in leukocyte transendothelial migration. Immunol Today. 1997;18:586–591. doi: 10.1016/s0167-5699(97)01162-6. [DOI] [PubMed] [Google Scholar]
- 4.Muller WA. The role of PECAM-1 (CD31) in leukocyte emigration: studies in vitro and in vivo. J Leukoc Biol. 1995;57:523–528. doi: 10.1002/jlb.57.4.523. [DOI] [PubMed] [Google Scholar]
- 5.Allport JR, Ding H, Collins T, Gerristen ME, Luscinskas FW. Endothelial-dependent mechanisms regulate leukocyte transmigration: a process involving the proteasome and disruption of the vascular endothelial-cadherin complex at endothelial cell-to-cell junctions. J Exp Med. 1997;186:517–527. doi: 10.1084/jem.186.4.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Del Maschio A, Zanetti A, Corada M, Rival Y, Ruco L, Lampugnani MG, Dejana E. Polymorphonuclear leukocyte adhesion triggers the disorganization of endothelial cell-to-cell adherens junctions. J Cell Biol. 1996;135:497–510. doi: 10.1083/jcb.135.2.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muller WA, Ratti CM, McDonnell SL, Cohn ZA. A human endothelial cell–restricted, externally disposed plasmalemmal protein enriched in intercellular junctions. J Exp Med. 1989;170:399–414. doi: 10.1084/jem.170.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zocchi MR, Ferrero E, Leone BE, Rovere P, Bianchi E, Toninelli E, Pardi R. CD31/PECAM-1-driven chemokine-independent transmigration of human T lymphocytes. Eur J Immunol. 1996;26:759–767. doi: 10.1002/eji.1830260406. [DOI] [PubMed] [Google Scholar]
- 9.Muller WA, Weigl SA. Monocyte-selective transendothelial migration: dissection of the binding and transmigration phases by an in vitro assay. J Exp Med. 1992;176:819–828. doi: 10.1084/jem.176.3.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muller WA, Weigl SA, Deng X, Phillips DM. PECAM-1 is required for transendothelial migration of leukocytes. J Exp Med. 1993;178:449–460. doi: 10.1084/jem.178.2.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berman ME, Xie Y, Muller WA. Roles of platelet/endothelial cell adhesion molecule-1 (PECAM-1, CD31) in natural killer cell transendothelial migration and β2 integrin activation. J Immunol. 1996;156:1515–1524. [PubMed] [Google Scholar]
- 12.Liao F, Ali J, Greene T, Muller WA. Soluble domain 1 of platelet–endothelial cell adhesion molecule (PECAM) is sufficient to block transendothelial migration in vitro and in vivo. J Exp Med. 1997;185:1349–1357. doi: 10.1084/jem.185.7.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vaporciyan AA, DeLisser HM, Yan H-C, Mendiguren II, Thom SR, Jones ML, Ward PA, Albelda SM. Involvement of platelet-endothelial cell adhesion molecule-1 in neutrophil recruitment in vivo. Science. 1993;262:1580–1582. doi: 10.1126/science.8248808. [DOI] [PubMed] [Google Scholar]
- 14.Wakelin MW, Sanz M-J, Dewar A, Albelda SM, Larkin SW, Boughton-Smith N, Williams TJ, Nourshargh S. An anti-platelet–endothelial cell adhesion molecule-1 antibody inhibits leukocyte extravasation from mesenteric microvessels in vivo by blocking the passage through the basement membrane. J Exp Med. 1996;184:229–239. doi: 10.1084/jem.184.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bird IN, Spragg JH, Ager A, Matthews N. Studies of lymphocyte transendothelial migration: analysis of migrated cell phenotypes with regard to CD31 (PECAM-1), CD45RA and CD45RO. Immunology. 1993;80:553–560. [PMC free article] [PubMed] [Google Scholar]
- 16.Masuyama J, Berman JS, Cruikshank WW, Morimoto C, Center DM. Evidence for recent as well as long term activation of T cells migrating through endothelial cell monolayers in vitro. J Immunol. 1992;148:1367–1374. [PubMed] [Google Scholar]
- 17.Berman JS, Mahorney K, Saukkonen JJ, Masuyama J. Migration of distinct subsets of CD8+ blood T cells through endothelial cell monolayers in vitro. J Leukoc Biol. 1995;58:317–324. doi: 10.1002/jlb.58.3.317. [DOI] [PubMed] [Google Scholar]
- 18.Brezinschek RI, Lipsky PE, Galea P, Vita R, Oppenheimer-Marks N. Phenotypic characterization of CD4+ T cells that exhibit a transendothelial migratory capacity. J Immunol. 1995;154:3062–3077. [PubMed] [Google Scholar]
- 19.Takahashi M, Masuyama J, Ikeda U, Kasahara T, Takahashi Y, Kiragawa S, Takahashi Y, Shimada K, Kano S. Induction of monocyte chemoattractant protein-1 synthesis in human monocytes during transendothelial migration in vitro. Circ Res. 1995;76:750–757. doi: 10.1161/01.res.76.5.750. [DOI] [PubMed] [Google Scholar]
- 20.Takahashi M, Kitagawa S, Masuyama J, Ikeda U, Kasahara T, Takahashi Y, Furukawa Y, Kano S, Shimada K. Human monocyte-endothelial cell interaction induces synthesis of granulocyte-macrophage colony-stimulating factor. Circulation. 1996;93:1185–1193. doi: 10.1161/01.cir.93.6.1185. [DOI] [PubMed] [Google Scholar]
- 21.Wilkinson PC. A visual study of chemotaxis of human lymphocyte using a collagen gel assay. J Immunol Methods. 1984;76:105–120. doi: 10.1016/s0022-1759(85)80004-1. [DOI] [PubMed] [Google Scholar]
- 22.Parsey MV, Lewis GK. Actin polymerization and pseudopod reorganization accompany anti-CD3-induced growth arrest in Jurkat T cells. J Immunol. 1993;151:1881–1893. [PubMed] [Google Scholar]
- 23.Gumbiner BM. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell. 1996;84:345–357. doi: 10.1016/s0092-8674(00)81279-9. [DOI] [PubMed] [Google Scholar]
- 24.Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- 25.Howard TH, Meyer WH. Chemokine peptide modulation of actin assembly and locomotion in neutrophils. J Cell Biol. 1984;98:1265–1271. doi: 10.1083/jcb.98.4.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bokoch GM. Chemoattractant signaling and leukocyte activation. Blood. 1995;86:1649–1660. [PubMed] [Google Scholar]
- 27.Premack BA, Schall TJ. Chemokine receptors: gateways to inflammation and infection. Nat Med. 1996;2:1174–1178. doi: 10.1038/nm1196-1174. [DOI] [PubMed] [Google Scholar]
- 28.Somersalo K, Carpen O, Saksela E, Gahmberg CG, Nortamo P, Timonen T. Activation of natural killer cell migration by leukocyte integrin-binding peptide from intracellular adhesion molecule-2 (ICAM-2) J Biol Chem. 1995;270:8629–8636. doi: 10.1074/jbc.270.15.8629. [DOI] [PubMed] [Google Scholar]
- 29.Van Epps DE, Potter J, Vachula M, Smith CW, Anderson DA. Suppression of human lymphocyte chemotaxis and transendothelial migration by anti-LFA-1 antibody. J Immunol. 1989;143:3207–3210. [PubMed] [Google Scholar]
- 30.Dustin ML, Carpen O, Springer TA. Regulation of locomotion and cell-cell contact area by the LFA-1 and ICAM-1 adhesion receptors. J Immunol. 1992;148:2654–2663. [PubMed] [Google Scholar]
- 31.Poggi A, Costa P, Zocchi MR, Moretta L. Phenotypic and functional analysis of CD4+ NKRP1A+ human T lymphocytes. Direct evidence that the molecule is involved in transendothelial migration. Eur J Immunol. 1997;27:2345–2350. doi: 10.1002/eji.1830270932. [DOI] [PubMed] [Google Scholar]
- 32.Haston WS, Shield JM, Wilkinson PC. Lymphocyte locomotion and attachment on two-dimensional surface and in three-dimensional matrices. J Cell Biol. 1982;92:747–752. doi: 10.1083/jcb.92.3.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.del Pozo MA, Sánchez-Mateos P, Nieto M, Sánchez-Madrid F. Chemokines regulate cellular polarization and adhesion receptor redistribution during lymphocyte interaction with endothelium and extracellular matrix. Involvement of cAMP signaling pathway. J Cell Biol. 1995;131:495–508. doi: 10.1083/jcb.131.2.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Campanero MR, Sánchez-Mateos P, del Pozo MA, Sánchez-Madrid F. ICAM-3 regulates lymphocyte morphology and integrin-mediated T cell interaction with endothelial cell and extracellular matrix ligands. J Cell Biol. 1994;127:867–878. doi: 10.1083/jcb.127.3.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.del Pozo MA, Cabanas C, Montoya MC, Ager A, Sánchez-Mateos P, Sánchez-Madrid F. ICAMs redistributed by chemokines to cellular uropods as a mechanism for recruitment of T lymphocytes. J Cell Biol. 1997;137:493–508. doi: 10.1083/jcb.137.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sánchez-Mateos P, Campanero MR, del Pozo MA, Sánchez-Madrid F. Regulatory role of CD43 leukosialin on integrin-mediated T-cell adhesion to endothelial and extracellular ligands and its polar redistribution to a cellular uropod. Blood. 1995;86:2228–2239. [PubMed] [Google Scholar]
- 37.Morimoto C, Schlossman SF. The structure and function of CD26 in the T-cell immune response. Immunol Rev. 1998;161:55–70. doi: 10.1111/j.1600-065x.1998.tb01571.x. [DOI] [PubMed] [Google Scholar]
- 38.von Bonin A, Huhn J, Fleischer B. Dipeptidyl-peptidase IV/CD26 on T cells: analysis of an alternative T-cell activation pathway. Immunol Rev. 1998;161:43–53. doi: 10.1111/j.1600-065x.1998.tb01570.x. [DOI] [PubMed] [Google Scholar]
- 39.Oravecz T, Pall M, Roderiquez G, Gorrell MD, Ditto M, Nguyen NY, Boykins R, Unsworth E, Norcross MA. Regulation of the receptor specificity and function of the chemokine RANTES (regulated on activation, normal T cell expressed and secreted) by dipeptidyl peptidase IV (CD26)-mediated cleavage. J Exp Med. 1997;186:1865–1872. doi: 10.1084/jem.186.11.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Proost P, De Meester I, Schols D, Struyf S, Lambeir AM, Wuyts A, Opdenakker G, De Clercq E, Scharpe S, Van Damme J. Amino-terminal truncation of chemokines by CD26/dipeptidyl-peptidase IV. Conversion of RANTES into a potent inhibitor of monocyte chemotaxis and HIV-1-infection. J Biol Chem. 1998;273:7222–7227. doi: 10.1074/jbc.273.13.7222. [DOI] [PubMed] [Google Scholar]
- 41.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 42.Roth SJ, Carr MW, Springer TA. C-C chemokines, but not the C-X-C chemokines interleukin-8 and interferon-γ inducible protein-10, stimulate transendothelial chemotaxis of T lymphocytes. Eur J Immunol. 1995;25:3482–3488. doi: 10.1002/eji.1830251241. [DOI] [PubMed] [Google Scholar]
- 43.Qin S, LaRosa G, Campbell JJ, Smith-Heath H, Kassam N, Shi X, Zeng L, Butcher EC, Mackay CR. Expression of monocyte chemoattractant protein-1 and interleukin-8 receptors on subsets of T cells: correlation with transendothelial chemotactic potential. Eur J Immunol. 1996;26:640–647. doi: 10.1002/eji.1830260320. [DOI] [PubMed] [Google Scholar]
- 44.Bleul CC, Wu L, Hoxie JA, Springer TA, Mackay CR. The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc Natl Acad Sci USA. 1997;94:1925–1930. doi: 10.1073/pnas.94.5.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frade JMR, Mellado M, del Real G, Gutierrez-Ramos JC, Lind P, Martinez C -A. Characterization of the CCR2 chemokine receptor: functional CCR2 receptor expression in B cells. J Immunol. 1997;159:5576–5584. [PubMed] [Google Scholar]
- 46.Murphy PM. The molecular biology of leukocyte chemoattractant receptors. Annu Rev Immunol. 1994;12:593–633. doi: 10.1146/annurev.iy.12.040194.003113. [DOI] [PubMed] [Google Scholar]
- 47.Mackay CR. Chemokine receptors and T cell chemotaxis. J Exp Med. 1996;184:799–802. doi: 10.1084/jem.184.3.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bazan JF, Bacon KB, Hardiman G, Wang W, Soo K, Rossi D, Greaves DR, Zlotnik A, Schall T. A new class of membrane-bound chemokine with a CX3C motif. Nature. 1997;385:640–644. doi: 10.1038/385640a0. [DOI] [PubMed] [Google Scholar]
- 49.Imai T, Hieshima K, Haskell C, Baba M, Nagira M, Nishimura M, Kakizaki M, Takagi S, Nomiyama H, Schall TJ, et al. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell. 1997;91:521–530. doi: 10.1016/s0092-8674(00)80438-9. [DOI] [PubMed] [Google Scholar]