

Abstract
Variable (V) region gene replacement was recently implicated in B cell repertoire diversification, but the contribution of this mechanism to antibody responses is still unknown. To investigate the role of V gene replacements in the generation of antigen-specific antibodies, we analyzed antiviral immunoglobulin responses of “quasimonoclonal” (QM) mice. The B cells of QM mice are genetically committed to exclusively express the anti-(4-hydroxy-3-nitrophenyl) acetyl specificity. However, ∼20% of the peripheral B cells of QM mice undergo secondary rearrangements and thereby potentially acquire new specificities. QM mice infected with vesicular stomatitis virus (VSV), lymphocytic choriomeningitis virus, or poliovirus mounted virus-specific neutralizing antibody responses. In general, kinetics of the antiviral immunoglobulin responses were delayed in QM mice; however, titers similar to control animals were eventually produced that were sufficient to protect against VSV-induced lethal disease. VSV neutralizing single-chain Fv fragments isolated from phage display libraries constructed from QM mice showed VH gene replacements and extensive hypermutation. Thus, our data demonstrate that secondary rearrangements and hypermutation can generate sufficient B cell diversity in QM mice to mount protective antiviral antibody responses, suggesting that these mechanisms might also contribute to the diversification of the B cell repertoire of normal mice.
Keywords: B cell gene rearrangement, vesicular stomatitis virus, mutation, viral antibodies, antibody diversity
The immune system of vertebrates has evolved complex mechanisms to generate sufficient lymphocyte receptor diversity to protect the organism against a wide range of pathogens. Ab diversity is the result of several distinct mechanisms, the major one being the rearrangement of V(D)J gene segments to generate the coding sequences of the V regions of the heavy and light chains of a functional Ab molecule (1). Secondary rearrangements have mostly been implicated in the elimination of autoreactive Abs in a process called receptor editing (2–9). However, recent experiments have suggested a role of receptor editing in the generation of peripheral B cell diversity (8, 10–13). Despite these observations, the actual contribution of secondary rearrangements to the shaping of the primary B cell repertoire is unclear because these rearrangements are often indistinguishable from the canonical V(D)J rearrangements. Fortunately, gene-targeted mice expressing a very limited B cell repertoire have been generated, one of which is the “quasimonoclonal” (QM)1 mouse (14), which should allow assessment of the role of VH gene replacement in the generation of protective antiviral Ig responses.
The QM mouse was generated by gene-targeted replacement of the endogenous JH elements by the rearranged 17.2.25 VH(D)JH region isolated from a (4-hydroxy-3-nitrophenyl) acetyl (NP)-specific hybridoma, whereas the other allele was rendered nonfunctional by the targeted deletion of all JH segments. In addition, QM mice have no functional κ light chain allele; therefore, the transgenic heavy chain can only pair with endogenously rearranged λ chains. When paired with a λ light chain, the targeted heavy chain allows binding to NP-hapten, rendering the QM primary B cell repertoire theoretically monospecific. However, despite this genetic predisposition, 20% of the peripheral B cells of QM mice do not bind NP (15). In the absence of immunization, VH gene replacement intermediates were observed in bone marrow and splenic B cells of QM mice, and recombinase-activating gene (RAG)-1 transcripts were detected in the spleen (16). To assess the extent of diversification generated through secondary rearrangements, we infected QM mice with different viruses and monitored their specific antiviral responses. Our results show that VH replacements together with hypermutation can generate protective antiviral Ab specificities in QM mice and indicate that both mechanisms could contribute to diversify the B cell repertoire of normal mice.
Materials and Methods
Mice and Viruses.
The generation of the QM mouse has been previously described (14). C57BL/6 mice were obtained from the breeding colony of the Institut für Labortierkunde, Faculty of Veterinary Medicine, University Zürich-Irchel, Zürich, Switzerland. Experiments were carried out with age- and sex-matched animals kept under specific pathogen–free conditions. All animals were 8–16 wk old unless indicated otherwise.
VSV-IND (vesicular stomatitis virus Indiana serotype; Mudd-Summers isolate) was originally obtained from D. Kolakofsky (University of Geneva, Switzerland), and was grown on BHK cells in MEM with 5% FCS to virus stocks containing 109 PFU/ml. Poliovirus (PV) stock solutions of serotypes I, II, and III were obtained from the Swiss Serum and Vaccine Institute (Bern, Switzerland). Lymphocytic choriomeningitis virus (LCMV)-WE was originally provided by F. Lehmann-Grube (Heinrich Pette Institut für Experimentelle Virologie und Immunologie, University of Hamburg, Hamburg, Germany), and was grown on L-929 cells for 48 h in MEM with 5% FCS after infection with an initial multiplicity of infection (moi) of 0.01.
Immunizations.
Mice were immunized intravenously on day 0 with a standard dose of 2 × 106 PFU of VSV-IND or 200 PFU of LCMV-WE as indicated. For PV experiments, 0.5 ml of PV vaccine, Salk (minimal content [min.] 30 D-antigen U type 1, min. 6 D-antigen U type 2, min. 24 D-antigen U type 3, phenoxyethanol; Poliomyelitis-Impstoff Berna, Swiss Serum and Vaccine Institute) was injected intravenously on days 0 and 14. Blood was collected at various time points as indicated.
VSV and PV Neutralization Assay.
Serial twofold dilutions of serum samples (previously diluted 1:40) were mixed with equal volumes of VSV or PV (PV serotype II was used) containing 500 PFU/ml, and the mixtures were incubated for 90 min at 37°C in an atmosphere containing 5% CO2. 100 μl of the mixture was transferred onto Vero cell monolayers in 96-well plates and incubated for 1 h at 37°C. Monolayers were overlaid with 100 μl of DMEM containing 1% methylcellulose, incubated for 24 h (up to 48 h for PV) at 37°C, then the overlay was removed and the monolayer was fixed and stained with 0.5% crystal violet dissolved in 5% formaldehyde, 50% ethanol, 4.25% NaCl. The dilution reducing the number of plaques by 50% was taken as titer (17). To determine IgG titers, undiluted serum was treated with an equal volume of 0.1 M 2-ME in MEM tissue culture medium for 1 h at room temperature.
LCMV Neutralization Assay: Infectious Focus Reduction Assay.
LCMV neutralization in vitro was determined as described previously (18). In brief, serial 2-fold dilutions of 10-fold prediluted sera were incubated with LCMV for 90 min at 37°C in a 96-well plate. MC57G mouse fibroblasts were added, and incubated for 4 h to allow cells to settle and be infected by nonneutralized virus; cells were then overlaid with 1% methylcellulose in DMEM. After 48 h, cell monolayers were fixed with 4% formalin and infectious foci were detected by intracellular LCMV staining of infected cells with the rat anti-LCMV mAb VL-4. For LCMV neutralization by total Ig, sera were tested under nonreducing conditions. For LCMV neutralization by IgG, sera were first reduced by addition of 2-ME (final concentration 0.05 M) for 60 min. All samples were heat inactivated at 56°C for 30 min to destroy complement.
Detection of Abs by ELISA.
To detect LCMV nucleoprotein– specific Abs, 96-well plates were coated overnight at 4°C with recombinant baculovirus–derived LCMV nucleoprotein (19, 20), washed three to five times with PBS 0.05%/Tween 20, and blocked with 1% BSA in PBS for 2–3 h at room temperature. Sera diluted in PBS containing 0.1% BSA were added and incubated for 90 min. Horseradish peroxidase–labeled specific goat anti–mouse IgG Ab (1:1,000; Zymed Laboratories) was added for 1 h, and the reaction was developed by addition of 0.4 mM ABTS (Boehringer Mannheim), in NaH2PO4, pH 4.0, 0.01% H2O2. The OD was read at 405 nm using a microplate reader (model 3550; Bio-Rad Laboratories). Positive titers were defined as 3 SD above the mean value of negative controls. To measure the different IgG isotype-specific anti-VSV Abs, ELISA plates were coated with 10 μg/ml of polyethylene glycol (PEG)-precipitated VSV-IND, and the assay was performed as described above.
Phage Display Library Construction.
Phage display libraries were constructed as described previously (21). In brief, mRNA was isolated from 107 spleen cells using the QuickPrep mRNA purification kit (Amersham Pharmacia Biotech) and reverse transcribed using random hexamer primers according to the manufacturer's instructions (first strand synthesis for RT-PCR; Amersham Life Science). The V region of the heavy chain (VH) was amplified using the VHback and VHfor mixes of degenerate primers, and the light chain V region (VL) was amplified using the λ-specific primers LBλ and LFλ as described previously (21). The VH and VL PCR products were purified and assembled in a single-chain Fv (scFv) configuration using splicing by overlap extension PCR. The full-length scFv fragment product was gel-purified, digested with SfiI, and ligated into the phage display vector pAK100 (gift of Dr. Andreas Plückthun, University of Zürich-Irchel, Zürich, Switzerland). The ligation mixes were electroporated into XL1-Blue electrocompetent cells (Stratagene, Inc.) and plated. Colonies were scraped off the plates, and the scFv displaying phages were rescued by infection with the VCS-M13 helper phage (Stratagene, Inc.). Phage particles were PEG precipitated twice and resuspended in PBS. VSV-binding phages were selected by panning using immunotubes (Maxisorp; Nunc, Inc.) coated with 100 μg/ml PEG-precipitated VSV-IND particles by overnight incubation at room temperature. Tubes were blocked with 4% dried skim milk powder in PBS and washed 20 times with PBS containing 0.1% Tween and 20 times with PBS. Bound phages were eluted with 0.1 M glycine/HCl, pH 2.2, for 10 min and immediately neutralized with 2 M Tris/HCl, pH 8.5. Eluted phages were used to infect XL1-Blue cells that were plated, rescued as described above, and subjected to another round of panning before isolated clones were analyzed by ELISA. Isolated colonies from the second round of panning were grown in 96-well plates and rescued with VCS-M13 helper phage. After overnight incubation, 100 μl of the phage-containing supernatant was added to VSV-coated plates (10 μg/ml) and incubated for 90 min. The wells were washed five times with PBS containing 0.1% Tween, and peroxidase-labeled anti-M13 antiserum (Amersham Pharmacia Biotech) was then added and the plates incubated for another 90 min. The plates were again washed five times with PBS/0.1% Tween, and the bound peroxidase was revealed by incubation with O-phenylenediamine (Sigma Chemical Co.) and hydrogen peroxide. The reaction was stopped with 1 N HCl, and the absorbance was read at 490 nm using a Bio-Rad 3550 microplate reader.
Results and Discussion
Induction of Neutralizing Abs against VSV-IND in QM Mice.
VSV is a cytopathic RNA negative strand virus of the Rhabdoviridae family closely related to rabies virus (22). After VSV infection, virus-neutralizing Abs mediate protection against a progressive paralytic disease (23, 24). Most VSV-IND–specific mAbs are directed against overlapping subsites clustered within one major antigenic site of the viral glycoprotein (25–27). The early IgM response (days 3–5) can be induced in the absence of T cell help (28, 29), whereas the switch to IgG (days 6–8) is T cell help dependent (30).
QM and C57BL/6 mice were immunized with 2 × 106 PFU of VSV-IND intravenously, and neutralizing serum Abs were monitored for 150 d. VSV total Ig neutralizing titers were measurable in QM mice 4 d after infection, but these titers were ∼30-fold lower than in C57BL/6 control mice (Fig. 1 a). The switch to the IgG subclass was delayed by ∼6 d in QM mice: VSV-neutralizing IgG Abs appeared ∼12 d after infection, whereas in control mice they were first detected at about day 6 (Fig. 1 a). About 40 d after infection, total anti-VSV neutralizing Abs reached similar levels in QM and control mice, and remained elevated for the duration of the 150-d observation period (Fig. 1 a). Similar kinetics were observed after infection with 102 or 104 PFU of VSV-IND (data not shown). Despite the delayed kinetics of Ab production, QM mice were able to control the viral infection and survived. However, the LD50 was decreased 10-fold from 2 × 108 in C57BL/6 control mice to 2 × 107 in QM mice (data not shown).
Figure 1.
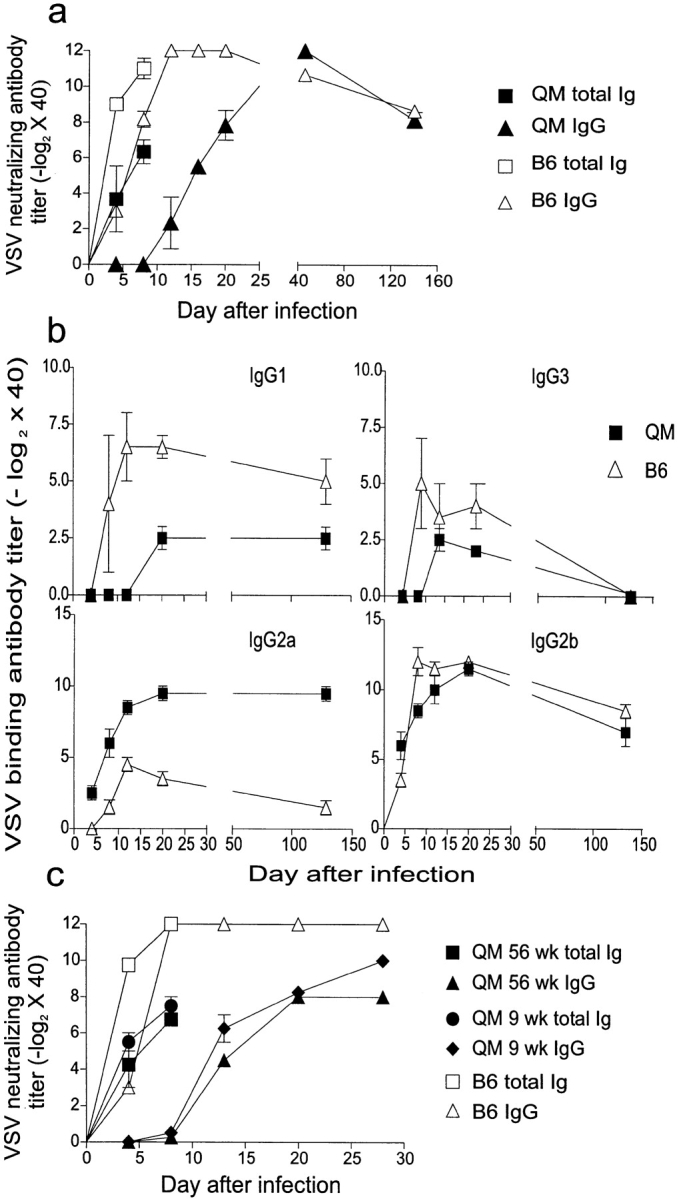
QM mice mount VSV-specific Ig responses. Groups of three QM and C57BL/6 mice were immunized with 2 × 106 PFU of VSV-IND intravenously. Blood was taken at the indicated time points, sera were separated and prediluted 40-fold, and the Ig responses were analyzed. (a) Kinetics of VSV-neutralizing Ab responses in QM and C57BL/6 mice. (b) Titration of the different VSV-specific Ig isotypes in infected QM and C57BL/6 mice. Titers were defined as 3 SD above mean values of negative controls. (c) Kinetics of VSV-neutralizing responses of 9- and 56-wk-old QM mice. C57BL/6 mice were used as controls.
Determination of the isotypes of the VSV-specific IgG Abs revealed that IgG2a was more abundant in infected QM mice than in control mice (Fig. 1 b). In contrast, IgG1 titers were higher in the control animals than in QM mice, while IgG2b and IgG3 Ab titers were approximately the same (Fig. 1 b). Similarly to infected QM mice, naive QM mice also show elevated IgG2a and IgG2b levels, which may indicate a certain bias towards Th1 cell development in QM mice (14).
The delayed kinetics of the VSV-neutralizing Ig response in QM mice was not due to the absence of kappa light chains because Cκ-deficient mice (31) mounted virtually normal anti-VSV Ig responses (data not shown). The delayed kinetics observed in QM mice is likely to be attributable to the time required for the small number of anti-VSV B cell precursors present in naive QM mice to be activated or expanded and to produce sufficient serum Abs to be detected in the neutralization assay. It has previously been reported that the amount of NP-specific serum IgG1 decreased from 13.1% of total IgG1 in 4–5-wk-old QM mice to 2.8% in 20–24-wk-old mice, which is consistent with the idea that B cells with variant receptors are preferentially expanded as QM mice age (15). This could lead to increased numbers of VSV-specific B cell precursors and an accelerated VSV-neutralizing Ab response in older mice. To test this hypothesis, 9- and 56-wk-old QM mice were infected with 2 × 106 PFU of VSV-IND. The anti-VSV neutralizing Ab response in both 9- and 56-wk-old QM mice displayed similar kinetics, suggesting that there was no significant increase in the number of VSV-specific B cell precursors in QM mice older than 9 wk (Fig. 1 c).
Analysis of VSV-specific scFv Fragments Isolated from QM Mice.
To analyze V region sequences of VSV-specific neutralizing Abs generated in QM mice, phage display libraries were constructed from splenic mRNA isolated from naive QM mice and from QM mice after primary and secondary VSV-IND infections. In a screening ELISA, 37% of the phages isolated from the naive library and ∼57% of the phages isolated from QM spleens after primary or secondary VSV infections bound VSV. Thus, it can be concluded from this result that VSV-specific B cells were present in QM mice before infection. The serum of the mouse used to generate the naive library was tested for the presence of VSV-specific Abs and was found to be negative. In addition, it must be emphasized that mice kept in our facilities for prolonged periods of time have been tested repetitively during the last 10 yr, and in no circumstances have we found the presence of VSV-IND–specific Abs, which could indicate accidental spreading of virus. Individual clones showing strong positive signals in the screening ELISA were concentrated by PEG precipitation and tested for their relative binding potential to VSV in a second ELISA. As shown in Fig. 2 a, scFv-displaying phages showed a specific dose-dependent binding to VSV-coated plates. To identify clones specific for the viral glycoprotein (VSV-G), binding of phages to VSV-infected EL4 cells expressing VSV-G on their surface was analyzed cytofluorometrically. From 48 tested clones, 15 showed binding to VSV-infected EL4 cells (data not shown). These 15 glycoprotein-specific clones were tested in a VSV-neutralization assay, which revealed that 12 out of 15 clones neutralized viral infectivity in vitro. The neutralizing titers of six independent scFv clones (see below) are shown in Fig. 2 b.
Figure 2.

Binding, neutralizing capacity, and sequence analysis of VSV-specific scFv fragments isolated from phage display libraries. (a) Phage clones displaying VSV-specific scFv fragments isolated from a naive QM mouse library (0B1 and 0G6) and libraries generated from QM mice after primary (1A10 and 1E12) or secondary (2F7 and 2H5) infections were tested for their capacity to bind VSV-coated plates in ELISA. (b) VSV-neutralizing titers of PEG-precipitated phage particles. (c) The nucleotide sequence of six VSV-specific clones is compared with the 17.2.25 transgene sequence (reference 16) shown in bold. Mutations in the DSP2.10 and JH4 segments are underlined. The remaining 3′ sequence of the JH4 segments did not contain somatic hypermutations. The sequences of the complete scFv fragments are available from EMBL/GenBank/DDBJ under accession nos. AF127092–AF127097.
The V regions of the 12 VSV-neutralizing scFv Ab fragments were sequenced and aligned to the 17.2.25 VH sequence expressed in QM mice. From the 12 clones analyzed, 6 independent sequences were identified, 2 from each of the three libraries (Fig. 2 c). Clones 0B1 and 0G6 were isolated from the naive library, clones 1A10 and 1E12 from the primary library, and clones 2F7 and 2H5 from the secondary library. The other six clones were multiple isolates of clone 0G6 (four copies) and of clone 1A10 (two copies); such repeated isolation is often the consequence of the amplification of binding clones during the panning procedure. All six independent clones used a VL fragment belonging to the λ1 family. In addition, they all showed evidence for VH gene replacement. Instead of the original 17.2.25 VH gene segment, all clones used a different VH gene segment belonging to the VH1 (J558) family that has previously been shown to play a role in secondary and hyperimmune responses against VSV in BALB/c mice (32). All six analyzed clones underwent secondary rearrangements by recombining a rearranged VHD segment with the D element of the rearranged VHDJH segment of the QM mouse (Fig. 2 c). This type of VHD to VHDJH rearrangement has previously been observed in naive QM mice in the majority of idiotype-negative sorted B cells (14, 15). However, unlike the events described here, a small proportion of idiotype-negative B cells did show signs of canonical VH to VHDJH secondary rearrangements (14, 15). Therefore, it is possible that such VHD to VHDJH rearrangements are preferred to the canonical secondary rearrangements because they generate greater diversity by drastically varying the length of CDR3, which might in turn increase the chances of generating new specificities. It is unclear whether the heptamer motif embedded at the 3′ end of the VH gene segment of the 17.2.25 V region and most other VH gene segments was involved in these rearrangements because the recombination breakpoint cannot be unambiguously determined. In addition, N regions flanking the inserted D elements were present in all analyzed clones (Fig. 2 c), which argues for the involvement of the normal rearrangement machinery including terminal deoxynucleotidyl transferase (TdT), which is expressed in bone marrow B cells and has recently been shown to be also reexpressed in germinal center B cells (13). Furthermore, the 6 VSV-specific clones sequenced showed extensive hypermutation in the remaining DSP2.10 and JH4 segments of clone 17.2.25 with a high frequency of 3.8% (15 mutations in 393 bp cumulated from the 6 scFv sequences). Interestingly, mutations were present in clones isolated from the naive library, confirming the results obtained in B cells isolated from the peripheral blood of unimmunized QM mice that also showed a high frequency of somatic mutations (14).
Induction of LCMV- and PV-specific Abs in QM Mice.
Since germline Abs can already efficiently neutralize VSV infection in vitro and a relatively restricted B cell repertoire can mount an efficient early anti-VSV response (27), it is possible that by chance the right VH/VL pairing in QM mice could give rise to VSV-specific but not other virus-specific Abs. Therefore, in addition to the VSV-specific Ig response, the responses of QM mice to LCMV and PV were analyzed.
LCMV is a noncytopathic ambisense RNA virus of the Arenaviridae family for which the mouse is the natural host. Acute LCMV infection is predominantly controlled by CTLs in a perforin-dependent manner (33, 34). Early after infection, at about day 8, a strong Ab response specific for the LCMV nucleoprotein is mounted (20) that does not exhibit virus-neutralizing capacity (35, 36). Late after infection, between days 30 and 60, LCMV glycoprotein–specific neutralizing Abs develop (20, 37, 38) that have been shown to play an important role in protection against reinfection (39–41).
QM and C57BL/6 mice were immunized intravenously with 102 PFU of LCMV-WE, and the LCMV nucleoprotein–specific ELISA Ab and the LCMV-neutralizing Ab responses were monitored. QM mice mounted a delayed LCMV nucleoprotein–specific Ab response at about day 12 that was initially reduced by ∼10-fold in comparison with control mice, but which eventually reached similar levels by day 32 (Fig. 3 a).
Figure 3.
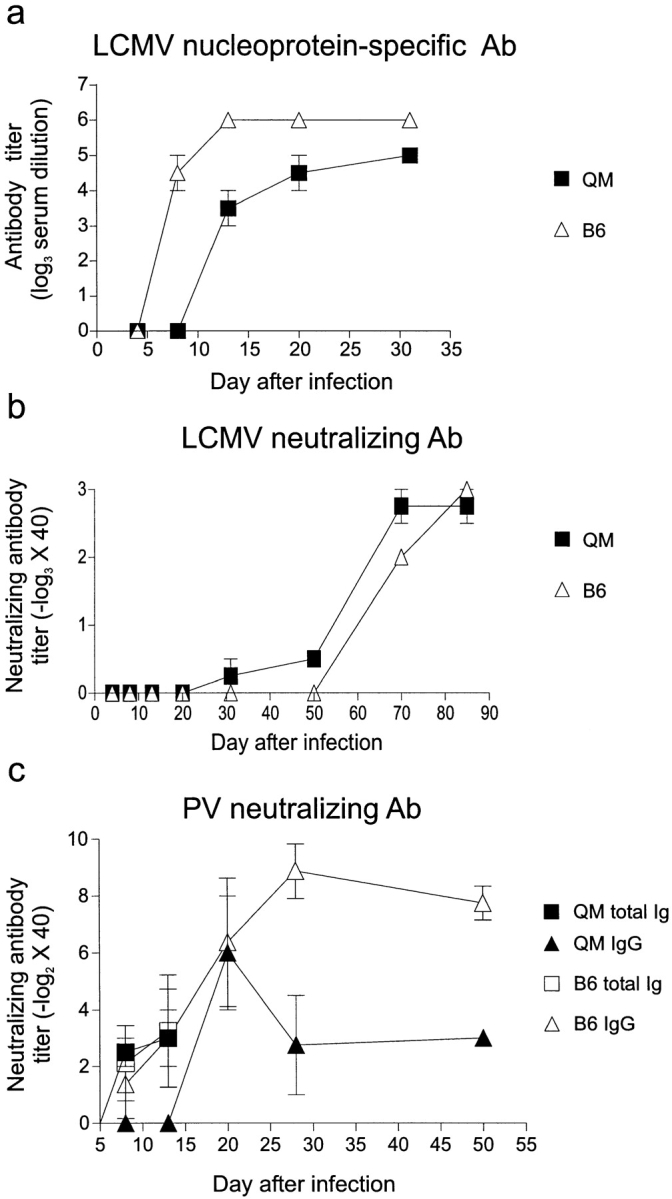
QM mice mount neutralizing Ig responses against LCMV and PV. Groups of three mice were immunized with 200 PFU of LCMV-WE or 0.5 ml of PV (Salk vaccine) intravenously. (a) LCMV nucleoprotein–specific Ab titers were measured by ELISA. (b) LCMV-neutralizing Abs and (c) PV-neutralizing Abs were measured as described in Materials and Methods.
Neutralizing Abs to LCMV normally appearing late after infection (20, 37, 38) followed the same slow kinetics in QM and control mice (Fig. 3 b). Low neutralizing Ab titers were first detected after 30 d of infection, and by day 70 a distinct neutralizing Ab response was measured in both QM and C57BL/6 mice (Fig. 3 b). The long time period required to mount a neutralizing response against LCMV could well have permitted the generation of sufficient LCMV-specific precursors to mount a neutralizing Ab response comparable to control animals. On the other hand, the reason for the long period of time needed to generate LCMV-neutralizing Abs is not well understood and could be dependent on factors other than low B cell precursor frequency (e.g., immunopathology [42]).
PV is a cytopathic positive strand RNA virus of the Picornaviridae family. The viral surface contains four regularly ordered proteins (VP1–4) against which neutralizing Abs are directed (43). In BALB/c mice, PV induces an early T cell–independent IgM response followed by a T cell–dependent IgG response (44). Early after immunization with formalin-inactivated PV (Salk vaccine), total neutralizing Ig titers in QM mice were similar to control animals (Fig. 3 c). The IgG response was initially delayed, but 20 d after infection titers reached the levels observed in C57BL/6 control mice. Interestingly, in the memory phase after 28 d of infection, serum titers in QM mice showed a three- to fourfold reduction compared with controls (Fig. 3 c). This could reflect differences in the numbers or in the expansion potential of the PV-specific B cell clones generated in QM mice. In addition, the inactivated PV vaccine used in this experiment might have provided a weaker stimulus for the selection and expansion of PV-specific QM B cells.
Taken together, our experiments have shown that VH replacement and hypermutation generated the VSV-specific immune response observed in QM mice and demonstrated the surprisingly great diversification potential of the QM B cell repertoire, establishing that this phenomenon is not restricted to one particular viral antigen.
Where was VH replacement taking place in QM mice, and what triggered it? The current model for the role of receptor editing in B cell development incorporates two different events (45). During the early phase of B cell development in the bone marrow, editing is induced by the interaction between a self-antigen and the receptor of a developing B cell (2–4). At a later phase, weak interaction between antigen and the B cell receptor of a mature peripheral B cell would induce editing, whereas strong binding turns it off (12, 13). Evidence for the occurrence of VH replacement in both bone marrow and spleen of nonimmunized QM mice was recently reported (16). However, a slight accumulation of idiotype-negative cells in the spleen compared with the bone marrow suggests that the spleen might be a privilege site for ongoing secondary rearrangement (16). In addition, a higher proportion of idiotype-negative B cells was also observed in B cells isolated from the peritoneum when compared with peripheral blood (15). Thus, it would appear that there is a constant need for diversification of the QM mouse B cell repertoire throughout B cell development and in different tissues.
The molecular events involved in the induction of secondary rearrangements are still not clearly defined, but different triggering mechanisms could be envisaged. First, B cells could have a certain intrinsic capacity to undergo receptor editing in an antigen-independent fashion through help from T cells, cytokines, or other stimuli. Although this scenario cannot be ruled out, there is enough evidence supporting the notion that B cell receptor triggering is required for induction of secondary rearrangements both in the bone marrow (7, 8) and in the periphery (10–12) to make this unlikely. The B cell receptor in QM mice would not be expected to bind any other antigen than NP with high affinity. However, it could be argued that weak cross-reactive interactions between NP-specific B cells and different antigens could induce V gene replacement and generate virus-specific B cells. Second, it was proposed that environmental antigenic pressure could be the driving force behind the diversification of the V gene repertoire of naive QM mice (15). Recent data suggested that secondary rearrangements in peritoneal B-1 cells might contribute to the development of autoreactive Abs (46). In this study, the authors proposed that frequent exposure of B-1 cells to LPS may lower the threshold for activation of secondary rearrangements resulting in a rapid shift in the Ab repertoire. In view of our findings, this mechanism could generate not only autoreactive B cells but also potentially useful pathogen-specific Abs. On the other hand, de novo VH replacement could have been triggered by cross-reactive binding of the NP-specific B cell receptor to the viral antigens used in this study. Although viral antigens might play a role in the expansion and maintenance of the virus-specific B cells in the infected mice, these interactions may be excluded as initial triggering events because VSV-specific scFv Abs were isolated from a phage display library constructed from a naive QM mouse. Therefore, virus-specific clones were generated in QM mice even before the introduction of viral antigens. In addition, nonimmunized QM mice undergo frequent VH replacement (14, 15).
It is difficult to address the role of these events in the complexity of a normal Ab response. However, new evidence is emerging in favor of the frequent involvement of secondary rearrangements during normal B cell development (47). Moreover, sequence analysis of human heavy and light chain V domains suggested that receptor editing occurs in human peripheral B cells (48). Taken together with our results, these experiments suggest that, together with hypermutation, secondary rearrangement could participate in the shaping of the natural B cell repertoire. In conclusion, our data illustrate the potential of VH gene replacements in the diversification of a restricted repertoire and definitely show that this expanded repertoire is functional.
Acknowledgments
We are grateful to K.J. Maloy, T. Fehr, and K. McCoy for reviewing the manuscript, and to N. Jeanguenat for excellent technical support. We would also like to thank Dr. Andreas Plückthun for providing the pAK100 phage display vector.
This work was supported by the Kanton of Zürich, by a grant from the Swiss National Foundation (31-50884.97), by National Institutes of Health grant R01 AI41570, by a Howard Hughes Institute Transgenic Mouse grant, and by grants from the Junta Nacional de Investigação Científica e Tecnológica (Praxis XXI, BD 3763/94; to M. Cascalho) and Heuber Stifftung (to C. López-Macías). A. Lamarre acknowledges fellowship support from the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Medical Research Council of Canada (MRCC). C. López-Macías is a recipient of a Bundesstipendium from the Eidgenössiche Stipendien Kommission, Bern, Switzerland, and acknowledges the support received from Fundación Universidad Nacional Autónoma de México (UNAM) and Consejo Nacional de Ciencia y Tecnología (CONACYT), Mexico.
Abbreviations used in this paper
- LCMV
lymphocytic choriomeningitis virus
- NP
(4-hydroxy-3-nitrophenyl) acetyl
- PEG
polyethylene glycol
- PV
poliovirus
- QM
“quasimonoclonal”
- VSV
vesicular stomatitis virus
- VSV-IND
VSV Indiana serotype
- VSV-G
VSV glycoprotein
- scFv
single-chain Fv
Footnotes
U. Kalinke's present address is EMBL Mouse Biology Programme, Adriano Buzzati-Traverso Campus, Via E. Ramarini 32, Monterotondo Scalo, I-00016 Rome, Italy.
References
- 1.Tonegawa S. Somatic generation of antibody diversity. Nature. 1983;302:575–581. doi: 10.1038/302575a0. [DOI] [PubMed] [Google Scholar]
- 2.Gay D, Saunders T, Camper S, Weigert M. Receptor editing: an approach by autoreactive B cells to escape tolerance. J Exp Med. 1993;177:999–1008. doi: 10.1084/jem.177.4.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Radic MZ, Erikson J, Litwin S, Weigert M. B lymphocytes may escape tolerance by revising their antigen receptors. J Exp Med. 1993;177:1165–1173. doi: 10.1084/jem.177.4.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tiegs SL, Russell DM, Nemazee D. Receptor editing in self-reactive bone marrow B cells. J Exp Med. 1993;177:1009–1020. doi: 10.1084/jem.177.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen C, Nagy Z, Prak EL, Weigert M. Immunoglobulin heavy chain gene replacement: a mechanism of receptor editing. Immunity. 1995;3:747–755. doi: 10.1016/1074-7613(95)90064-0. [DOI] [PubMed] [Google Scholar]
- 6.Lang J, Jackson M, Teyton L, Brunmark A, Kane K, Nemazee D. B cells are exquisitely sensitive to central tolerance and receptor editing induced by ultralow affinity, membrane-bound antigen. J Exp Med. 1996;184:1685–1697. doi: 10.1084/jem.184.5.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen C, Prak EL, Weigert M. Editing disease-associated autoantibodies. Immunity. 1997;6:97–105. doi: 10.1016/s1074-7613(00)80673-1. [DOI] [PubMed] [Google Scholar]
- 8.Pelanda R, Schwers S, Sonoda E, Torres RM, Nemazee D, Rajewsky K. Receptor editing in a transgenic mouse model: site, efficiency, and role in B cell tolerance and antibody diversification. Immunity. 1997;7:765–775. doi: 10.1016/s1074-7613(00)80395-7. [DOI] [PubMed] [Google Scholar]
- 9.Fang W, Weintraub BC, Dunlap B, Garside P, Pape KA, Jenkins MK, Goodnow CC, Mueller DL, Behrens TW. Self-reactive B lymphocytes overexpressing Bcl-xL escape negative selection and are tolerized by clonal anergy and receptor editing. Immunity. 1998;9:35–45. doi: 10.1016/s1074-7613(00)80586-5. [DOI] [PubMed] [Google Scholar]
- 10.Han S, Dillon SR, Zheng B, Shimoda M, Schlissel MS, Kelsoe G. V(D)J recombinase activity in a subset of germinal center B lymphocytes. Science. 1997;278:301–305. doi: 10.1126/science.278.5336.301. [DOI] [PubMed] [Google Scholar]
- 11.Papavasiliou F, Casellas R, Suh H, Qin XF, Besmer E, Pelanda R, Nemazee D, Rajewsky K, Nussenzweig MC. V(D)J recombination in mature B cells: a mechanism for altering antibody responses. Science. 1997;278:298–301. doi: 10.1126/science.278.5336.298. [DOI] [PubMed] [Google Scholar]
- 12.Hertz M, Kouskoff V, Nakamura T, Nemazee D. V(D)J recombinase induction in splenic B lymphocytes is inhibited by antigen-receptor signalling. Nature. 1998;394:292–295. doi: 10.1038/28419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meffre E, Papavasiliou F, Cohen P, de Bouteiller O, Bell D, Karasuyama H, Schiff C, Banchereau J, Liu YJ, Nussenzweig MC. Antigen receptor engagement turns off the V(D)J recombination machinery in human tonsil B cells. J Exp Med. 1998;188:765–772. doi: 10.1084/jem.188.4.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cascalho M, Ma A, Lee S, Masat L, Wabl M. A quasi-monoclonal mouse. Science. 1996;272:1649–1652. doi: 10.1126/science.272.5268.1649. [DOI] [PubMed] [Google Scholar]
- 15.Cascalho M, Wong J, Wabl M. VH gene replacement in hyperselected B cells of the quasimonoclonal mouse. J Immunol. 1997;159:5795–5801. [PubMed] [Google Scholar]
- 16.Bertrand FE, Golub R, Wu GE. V(H) gene replacement occurs in the spleen and bone marrow of non-autoimmune quasi-monoclonal mice. Eur J Immunol. 1998;28:3362–3370. doi: 10.1002/(SICI)1521-4141(199810)28:10<3362::AID-IMMU3362>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 17.Charan S, Zinkernagel RM. Antibody mediated suppression of secondary IgM response in nude mice against vesicular stomatitis virus. J Immunol. 1986;136:3057–3061. [PubMed] [Google Scholar]
- 18.Battegay M, Cooper S, Althage A, Baenziger J, Hengartner H, Zinkernagel RM. Quantification of lymphocytic choriomeningitis virus with an immunological focus assay in 24- or 96-well plates. J Virol Methods. 1991;33:191–198. doi: 10.1016/0166-0934(91)90018-u. [DOI] [PubMed] [Google Scholar]
- 19.Matsuura Y, Possee RD, Overton HA, Bishop DH. Baculovirus expression vectors: the requirements for high level expression of proteins, including glycoproteins. J Gen Virol. 1987;68:1233–1250. doi: 10.1099/0022-1317-68-5-1233. [DOI] [PubMed] [Google Scholar]
- 20.Battegay M, Moskophidis D, Waldner H, Brundler MA, Fung-Leung W-P, Mak TW, Hengartner H, Zinkernagel RM. Impairment and delay of neutralizing antiviral antibody responses by virus-specific cytotoxic T cells. J Immunol. 1993;151:5408–5415. [PubMed] [Google Scholar]
- 21.Krebber A, Bornhauser S, Burmester J, Honegger A, Willuda J, Bosshard HR, Plückthun A. Reliable cloning of functional antibody variable domains from hybridomas and spleen cell repertoires employing a reengineered phage display system. J Immunol Methods. 1997;201:35–55. doi: 10.1016/s0022-1759(96)00208-6. [DOI] [PubMed] [Google Scholar]
- 22.Wagner, R.R. 1987. The Rhabdoviruses. Plenum Press, New York. 544 pp.
- 23.Lefrancois L. Protection against lethal viral infection by neutralizing and nonneutralizing monoclonal antibodies: distinct mechanisms of action in vivo. J Virol. 1984;51:208–214. doi: 10.1128/jvi.51.1.208-214.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bachmann MF, Kündig TM, Kalberer CP, Hengartner H, Zinkernagel RM. How many specific B cells are needed to protect against a virus? . J Immunol. 1994;152:4235–4241. [PubMed] [Google Scholar]
- 25.Roost H-P, Bachmann MF, Haag A, Kalinke U, Pliska V, Hengartner H, Zinkernagel RM. Early high-affinity neutralizing anti-viral IgG responses without further overall improvements of affinity. Proc Natl Acad Sci USA. 1995;92:1257–1261. doi: 10.1073/pnas.92.5.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roost H-P, Haag A, Burkhart C, Zinkernagel RM, Hengartner H. Mapping of the dominant neutralizing antigenic site of a virus using infected cells. J Immunol Methods. 1996;189:233–242. doi: 10.1016/0022-1759(95)00252-9. [DOI] [PubMed] [Google Scholar]
- 27.Kalinke U, Krebber A, Krebber C, Bucher E, Plückthun A, Zinkernagel RM, Hengartner H. Monovalent single-chain Fv fragments and bivalent miniantibodies bound to vesicular stomatitis virus (VSV) protect against lethal infection. Eur J Immunol. 1996;26:2801–2806. doi: 10.1002/eji.1830261202. [DOI] [PubMed] [Google Scholar]
- 28.Burns W, Billups LC, Notkins AL. Thymus dependence of viral antigens. Nature. 1975;256:654–656. doi: 10.1038/256654a0. [DOI] [PubMed] [Google Scholar]
- 29.Charan S, Hengartner H, Zinkernagel RM. Antibodies against the two serotypes of vesicular stomatitis virus measured by enzyme-linked immunosorbent assay: immunodominance of serotype-specific determinants and induction of asymmetrically cross-reactive antibodies. J Virol. 1987;61:2509–2514. doi: 10.1128/jvi.61.8.2509-2514.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leist TP, Cobbold SP, Waldmann H, Aguet M, Zinkernagel RM. Functional analysis of T lymphocyte subsets in antiviral host defense. J Immunol. 1987;138:2278–2281. [PubMed] [Google Scholar]
- 31.Takeda S, Zou YR, Bluethmann H, Kitamura D, Muller U, Rajewsky K. Deletion of the immunoglobulin kappa chain intron enhancer abolishes kappa chain gene rearrangement in cis but not lambda chain gene rearrangement in trans. EMBO (Eur Mol Biol Organ) J. 1993;12:2329–2336. doi: 10.1002/j.1460-2075.1993.tb05887.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalinke U, Bucher EM, Oxenius A, Ernst B, Roost H-P, Geley S, Kofler R, Zinkernagel RM, Hengartner H. The role of somatic mutation in the generation of the protective humoral immune response against vesicular stomatitis virus (VSV) Immunity. 1996;5:639–652. doi: 10.1016/s1074-7613(00)80277-0. [DOI] [PubMed] [Google Scholar]
- 33.Kägi D, Ledermann B, Bürki K, Seiler P, Odermatt B, Olsen KJ, Podack ER, Zinkernagel RM, Hengartner H. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature. 1994;369:31–37. doi: 10.1038/369031a0. [DOI] [PubMed] [Google Scholar]
- 34.Zinkernagel RM, Leist T, Hengartner H, Althage A. Susceptibility to lymphocytic choriomeningitis virus isolates correlates directly with early and high cytotoxic T cell activity, as well as with footpad swelling reaction, and all three are regulated by H-2D. J Exp Med. 1985;162:2125–2141. doi: 10.1084/jem.162.6.2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kimmig B, Lehmann-Grube F. The immune response of the mouse to lymphocytic choriomeningitis virus. I. Circulating antibodies. J Gen Virol. 1979;45:703–710. doi: 10.1099/0022-1317-45-3-703. [DOI] [PubMed] [Google Scholar]
- 36.Moskophidis D, Cobbold SP, Waldmann H, Lehmann-Grube F. Mechanism of recovery from acute virus infection: treatment of lymphocytic choriomeningitis virus-infected mice with monoclonal antibodies reveals that Lyt-2+ T lymphocytes mediate clearance of virus and regulate the antiviral antibody response. J Virol. 1987;61:1867–1874. doi: 10.1128/jvi.61.6.1867-1874.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bruns M, Cihak J, Muller G, Lehmann-Grube F. Lymphocytic choriomeningitis virus. VI. Isolation of a glycoprotein mediating neutralization. Virology. 1983;130:247–251. doi: 10.1016/0042-6822(83)90135-6. [DOI] [PubMed] [Google Scholar]
- 38.Buchmeier MJ, Lewicki HA, Tomori O, Oldstone MBA. Monoclonal antibodies to lymphocytic choriomeningitis and pichinde viruses: generation, characterization, and cross-reactivity with other arenaviruses. Virology. 1981;113:73–85. doi: 10.1016/0042-6822(81)90137-9. [DOI] [PubMed] [Google Scholar]
- 39.Baldridge JR, Buchmeier MJ. Mechanisms of antibody-mediated protection against lymphocytic choriomeningitis virus infection: mother-to-baby transfer of humoral protection. J Virol. 1992;66:4252–4257. doi: 10.1128/jvi.66.7.4252-4257.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seiler P, Brundler MA, Zimmermann C, Weibel D, Bruns M, Hengartner H, Zinkernagel RM. Induction of protective cytotoxic T cell responses in the presence of high titers of virus-neutralizing antibodies: implications for passive and active immunization. J Exp Med. 1998;187:649–654. doi: 10.1084/jem.187.4.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wright KE, Buchmeier MJ. Antiviral antibodies attenuate T-cell-mediated immunopathology following acute lymphocytic choriomeningitis virus infection. J Virol. 1991;65:3001–3006. doi: 10.1128/jvi.65.6.3001-3006.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Planz O, Seiler P, Hengartner H, Zinkernagel RM. Specific cytotoxic T cells eliminate cells producing neutralizing antibodies. Nature. 1996;382:726–729. doi: 10.1038/382726a0. [DOI] [PubMed] [Google Scholar]
- 43.Rueckert, R.R. 1990. Picornaviridae and their replication. In Virology. B.N. Fields and D.M. Knipe, editors. Raven Press, New York. 507–548.
- 44.Fehr T, Naim HY, Bachmann MF, Ochsenbein AF, Spielhofer P, Bucher E, Hengartner H, Billeter MA, Zinkernagel RM. T-cell independent IgM and enduring protective IgG antibodies induced by chimeric measles viruses. Nat Med. 1998;4:945–948. doi: 10.1038/nm0898-945. [DOI] [PubMed] [Google Scholar]
- 45.Nussenzweig MC. Immune receptor editing: revise and select. Cell. 1998;95:875–878. doi: 10.1016/s0092-8674(00)81711-0. [DOI] [PubMed] [Google Scholar]
- 46.Qin XF, Schwers S, Yu W, Papavasiliou F, Suh H, Nussenzweig A, Rajewsky K, Nussenzweig MC. Secondary V(D)J recombination in B-1 cells. Nature. 1999;397:355–359. doi: 10.1038/16933. [DOI] [PubMed] [Google Scholar]
- 47.Retter MW, Nemazee D. Receptor editing occurs frequently during normal B cell development. J Exp Med. 1998;188:1231–1238. doi: 10.1084/jem.188.7.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Wildt RMT, Hoet RMA, van Venrooij WJ, Tomlinson IM, Winter G. Analysis of heavy and light chain pairings indicates that receptor editing shapes the human antibody repertoire. J Mol Biol. 1999;285:895–901. doi: 10.1006/jmbi.1998.2396. [DOI] [PubMed] [Google Scholar]