

Abstract
In studying the subdominant status of two cysteine-containing influenza virus nuclear protein (NP) determinants (NP39–47 and NP218–226) restricted by H-2Kd, we found that the antigenicity of synthetic peptides was enhanced 10–100-fold by treatment with reducing agents, despite the fact that the affinity for Kd was not enhanced. Reducing agents also markedly enhanced the immunogenicity of cysteine-containing peptides, as measured by propagation of long-term T cell lines in vitro. Similar enhancing effects were obtained by substituting cysteine with alanine or serine in the synthetic peptides, demonstrating that sulfhydryl modification of cysteine is responsible for the impaired antigenicity and immunogenicity of NP39–47 and NP218–226. We found similar effects for two widely studied, cysteine-containing peptides from lymphocytic choriomeningitis virus. The major modifications of cysteine-containing synthetic peptides are cysteinylation and dimerization occurring through cysteine residues. We demonstrate that both of these modifications occur in cells synthesizing a cytosolic NP218–226 minigene product and, further, that T cells specific for cysteinylated NP218–226 are induced by influenza virus infection in mice, demonstrating that this modification occurs in vivo. These findings demonstrate that posttranslational modifications affect the immunogenicity and antigenicity of cysteine-containing viral peptides and that this must be considered in studying the status of such peptides in immunodominance hierarchies.
Keywords: antigen processing, cysteine/immunology, major histocompatibility complex/immunology, viral vaccines/immunology, peptides/immunology
CD8+ T cells (TCD8+) recognize peptides, usually of 8–10 residues, complexed to MHC class I molecules. The peptide binding site is formed by class I heavy chains, which are encoded by genes that are highly polymorphic in most species. Most of the variability between heavy chain alleles resides in residues located in the peptide binding site. As a result, each class I allele presents a unique spectrum of peptides to the immune system (1). Although the rules that govern peptide binding to different alleles are far from simple, most peptides recovered from a given allele conform to a simple motif dictated largely by the nature of two to three “pockets” present at the bottom of the binding groove (2). Using these motifs, it is possible to identify upwards of 90% of peptides in a given sequence that could potentially bind to a given allele with biologically significant affinity. Synthetic peptides corresponding to these sequences can be tested for immunogenicity, antigenicity, and binding to class I molecules.
Although this method is capable of identifying antigenic regions of proteins, it remains a considerable challenge to establish the precise structure of the naturally processed determinant. As a first step, the chromatographic properties of synthetic and naturally processed peptides recovered from class I molecules can be compared by HPLC. Coelution indicates a close structural relationship, but the exact structure of the naturally processed peptide can only be established by mass spectroscopy (3). This is sufficiently difficult to preclude publication of mass spectroscopic analysis of viral peptides to date.
The possibilities for surprises in the structure of naturally processed peptides have been amply documented by Meadows et al. (4), who have identified peptides with a number of posttranslational modifications. Of direct relevance to the present study, naturally processed peptides with cysteinylated cysteine residue were identified, as were TCD8+ that required this modification for activation (4).
Due to its free SH group, cysteine is the most chemically reactive of the 20 common amino acids under physiological conditions. Cysteine is the second least frequent residue in proteins, representing 1.7% of the residues present in eukaryotic proteins. Its occurrence in antigenic peptides is consistent with random usage, being present in 14% of 384 class I ligands (1) (predicted frequency in nonamers is 90.83 = 14%). The frequency of cysteine-containing determinants is expected to be even higher in cysteine-rich proteins, such as Her2/Neu, a promising target for tumor immunotherapy (5). In this study, we show that modification of cysteine residues in naturally processed and synthetic peptides has profound negative and positive effects on the antigenicity and immunogenicity of viral determinants.
Materials and Methods
Cell Culture.
All media, including cystine-containing and cystine/methionine-free DMEM, were purchased from Biofluids, Inc. The mastocytoma cell line P815 (H-2d) and the thymoma cell line EL-4 (H-2b) were maintained in DMEM containing 10% fetal bovine serum (FBS),1 5 × 10−5 M β-ME, antibiotics, and 2 mM glutamine (DME-10). Kd-transfected T2 (T2-Kd) and RMA-S (RMA-S/Kd) cells were cultured in RPMI 1640 containing 10% FBS and the above supplements (RP-10). CTL stimulation and maintenance were performed in RP-10 medium containing 10 U/ml of recombinant human IL-2. In some assays, cells were incubated with IMDM supplemented with 10% FBS (I-10).
CTL Priming In Vivo and Assay.
8–10-wk-old female BALB/c mice or C57BL/6 mice were injected intraperitoneally with 1 ml of a 1:10 dilution of chicken egg allantoic fluid containing influenza A virus Puerto Rico/8/34 (PR8) or intravenously with 107 PFU of lymphocytic choriomeningitis virus (LCMV) WE strain. Splenocytes were stimulated with peptide-pulsed APCs for 7 d in vitro at least 3 wk after virus priming. Cells were stimulated in RP-10 with 10 U/ml IL-2 in 6-well plates. In brief, 3 × 107 splenocytes were stimulated with 6 × 105 virus-infected or peptide-pulsed (1 nM) APC, which were irradiated with 200 Gy before addition to cultures. After 4 d, live cells were recovered via a Ficoll–Hypaque gradient and recultured with fresh IL-2–containing medium. CTL activities were tested after 7 d in standard 51Cr-release microcytotoxicity assays. In some cases, short-term CTL lines were used, which were restimulated weekly for up to 5–6 wk.
Virus Infections.
For PR8 infection, log-phase cells were harvested and washed in serum-free Autopow MEM (Life Technologies), adjusted to pH 6.6, and resuspended in 200 μl of the same medium containing 100 μl PR8-containing allantoic fluid per 106 cells. For vaccinia virus (VV) infection, cells were infected at a multiplicity of infection of 10 in basal salt solution supplemented with 0.1% BSA (wt/vol) at a concentration of 5 × 106 cells/ml. Cells were incubated for 1 h at 37°C in a water bath before 2 ml of prewarmed complete medium was added. Cells were then further incubated for 1–2 h before 51Cr labeling, unless indicated otherwise.
Peptides, Binding Assays, and Flow Cytometry.
All peptides were synthesized, HPLC-purified, and analyzed by mass spectrometry by the Biologic Resource Branch, National Institute of Allergy and Infectious Diseases. Peptides were dissolved at 1 mM in DMSO and stored at −20°C unless otherwise indicated. For analytical purposes, peptide masses were determined by matrix assisted laser desorption ionization with time of flight detection (MALDI-TOF) using a Hewlett Packard mass spectrometer (model G2025A) and cyano-4-hydroxycinnamic acid as the matrix. Peptide binding to live cells was determined by protection of class I molecules to melting (6). In brief, T2-Kd cells were cultured for 14–16 h at 26°C. Synthetic peptides, diluted in FBS-free DME in the presence or absence of tris (2-carboxyethyl) phosphine hydrochloride (TCEP; Pierce Chemical Co.) as indicated, were added to cells, which were incubated at 37°C for 2 h to denature Kd molecules not stabilized by peptide binding. Cells were then washed and stained with fluorescein-conjugated SF1-1.1 (PharMingen). Live cells were gated based on scattering and exclusion of ethidium homodimer (Molecular Probes, Inc.) present at 5 μg/ml for 5 min before the last wash. For each histogram, 10,000–20,000 cells were counted on a Becton Dickinson FACScan™, and live cells were analyzed using CELLQuest™ software (Becton Dickinson).
Extraction of Cellular Peptides and Fractionation by Reversed-phase HPLC (RP-HPLC).
Natural peptides were recovered and analyzed as previously described (7). In brief, cultures of P815 cells were expanded in roller bottles. 5 × 108–109 cells were infected as described above and incubated for 6 h at 37°C before being pelleted, washed twice in PBS, lysed with ice-cold 0.33% TFA/ H2O, and further disrupted using a TenBroek tissue homogenizer on ice. At this stage, synthetic peptides were added to uninfected P815 cells as control. Lysates were sonicated and centrifuged at 10,000 g for 30 min, and the supernatants were passed through a 3K cutoff filter (Macrosep™ filtron 3K; Pall Filtron Corp.). Samples were dried to a volume >400 μl using a SpeedVac (Savant Instruments, Inc.) and fractionated on a C18 column (Deltapack; Waters) at 1 ml/min on TFA/acetonitrile gradient (7). Either 0.25- or 1-ml fractions were collected.
Microcytotoxicity Assay.
Generally, 106 target cells were labeled with 100 μCi of Na51CrO4 (Dupont) in minimum volume of medium at 37°C for 60 min. For some experiments, RMA-S/Kd cells that had been cultured for 12–14 h at 26°C were labeled at 26°C for the same time period. After two washes, 104 cells were aliquoted into round-bottom, 96-well plates containing serial dilutions of effector TCD8+. For testing HPLC fractions, target cells in 50 μl of either PBS or FCS-free medium were exposed to 5 μl of fractions for 30 min at 26°C before TCD8+ were added. In some experiments, TCEP was freshly dissolved in H2O and used at 200 μM, both at peptide-pulsing and microcytotoxicity assay stages. The radioactivity in supernatants collected after 4–6-h incubation at 37°C was determined using a gamma counter. The percent specific release was then determined as: % specific release = (CTL-induced release − spontaneous release)/(release by detergent − spontaneous release) × 100.
Results
TCD8+ Specific for Cysteine-containing Determinants Apparently Require More Peptide–Class I Complexes.
We previously reported that in Kd-restricted responses to PR8 influenza virus nuclear protein (NP), NP147–155 is the immunodominant determinant, with NP39–47 and NP218–226 exhibiting subdominant status (8). This hierarchy is not accounted for by peptide affinity, as NP147–155 binds to Kd with the lowest efficiency, as determined by a Kd “melting” assay performed either with RMA-S cells expressing Kd from a transfected gene (data not shown) or T2 cells (8). We initially focused on TCD8+ avidity to explain the immunodominance of NP147–155, as 10-fold less synthetic NP147–155 was usually required to sensitize target cells for lysis by TCD8+ lines raised to the individual peptides under conditions similar to those used for the peptide binding assay (Fig. 1 B). This was observed using either short- or long-term lines stimulated in vitro by synthetic peptides derived from animals immunized either with PR8 or rVV expressing NP or cytosolic or endoplasmic reticulum (ER)-targeted minigene product versions of the determinants. Taking into account the lower efficiency of NP147–155 binding to Kd, the data in Fig. 1 suggest that <10% of Kd-NP147–155 complexes are required for TCD8+ triggering relative to Kd complexed to either of the subdominant determinants.
Figure 1.

Antigenicity of synthetic peptides corresponding to dominant and subdominant determinants. (A) T2-Kd cells were cultured for 14 h at 26°C and added to the wells containing synthetic peptides at the indicated concentrations. The samples were immediately shifted to 37°C and incubated for 2 h to denature Kd molecules lacking peptides and then stained with a fluorescein-conjugated anti-Kd mAb. The mean channel fluorescence (MCF) of viable cells was determined by flow cytometry. (B) Splenocytes from PR8-primed animals stimulated in vitro for 7 d with synthetic peptides corresponding to NP39–47, NP147–155, or NP218–226 were tested in a microcytotoxicity assay for their ability to lyse 51Cr- labeled P815 target cells incubated in I-10 with synthetic peptides at the indicated concentrations.
Several findings, however, suggested that matters might be a bit more complicated. Unlike NP147–155, the dose–response curves of NP39–47 and NP218–226 varied considerably between experiments, depending in part on the manner in which the assay was executed. We also experienced difficulties in stimulating and maintaining TCD8+ lines to these subdominant determinants, often observing slower growth after restimulation and morphological abnormalities of the cells, which were frequently larger than TCD8+ specific for NP147–155. This was not strictly related to the subdominant status of these determinants, as TCD8+ specific for other subdominant determinants behaved similarly to NP147–155-specific TCD8+.
A property shared by NP218–226 and NP39–47 is the presence of cysteine (Table I). The report by Meadows et al. (4) demonstrating the dramatic effects of sulfhydryl modification of cysteine-containing residues on TCD8+ recognition prompted us to examine possible effects of cysteine modification on NP39–47 and NP218–226 binding and antigenicity. We first studied the properties of synthetic peptides in which cysteine is replaced by serine or alanine (the most conservative substitutions). For NP39–47, the cysteine→ serine substitution had no significant effect on peptide binding, whereas the cysteine→ alanine substitution increased peptide potency by ∼10-fold (Fig. 2 A). For NP218–226, either substitution reduced peptide potency in stabilizing Kd molecules by ∼10-fold (Fig. 2 B). Each of the substitutions resulted in large increases in antigenicity relative to the wild-type peptides (Fig. 2, C and D). For NP218–226, the substituted peptides were 1,000–10,000-fold more antigenic on a per-complex basis (assuming that complex formation at the endpoint of peptide titrations is proportional to peptide binding efficiency determined by the melting assay).
Table I.
Properties of Peptides Used in This Study
| Peptide | Sequence | HPLC elution time | ||
|---|---|---|---|---|
| min | ||||
| NP39–47 | FYIQMCTEL | |||
| NP147–155 | TYQRTRALV | |||
| NP218–226 | AYERMCNIL | 25 | ||
| CysNP218–226 | AYERMCNIL | |||
|
: C |
27 | |||
| dimNP218–226 | AYERMCNIL:
|
|||
| AYERMCNIL | 29 | |||
| GP33–41 | KAVYNFATC | |||
| GP276–286 | SGVENPGGYCL | |||
| NP396–404 | FQPQNGQFI | |||
| GP, glycoprotein; :, disulfide bond. | ||||
Figure 2.

Binding and antigenicity of synthetic peptides. A and B show data from the Kd-melting experiment displayed in Fig. 1 A. In this case, data for wild-type NP39–47 and NP218–226 are replotted with the substituted peptides. In C and D, TCD8+ specific for NP39–47 or NP218–226 were tested in a microcytotoxicity assay for their ability to lyse 51Cr-labeled P815 target cells incubated in I-10 in the presence of the synthetic peptides at the indicated concentrations.
These findings prompted us to study the effects of sulfhydryl modification on the antigenicity of NP39–47 and NP218–226. In most experiments described below, the two peptides were studied in parallel. Because the results were highly similar, only results with NP218–226 are shown for the sake of clarity and simplicity.
Reduction of Cysteine Enhances the Antigenicity of Cysteine-containing Viral Peptides.
Cysteine readily forms disulfide bonds at neutral or slightly basic pH in the presence of O2 at atmospheric tension, and oxidation to the disulfide is stimulated by trace amounts of iron salts that are present in tissue culture media. The disulfide can be reduced to the original thiol form by exposure to reducing agents. To determine whether disulfide formation affected peptide antigenicity, synthetic NP218–226 was added to cells in the presence of dithiothreitol or TCEP, and cells were tested for lysis by NP218–226-specific TCD8+. Either of these reducing agents increased peptide potency by ∼10-fold (Fig. 3 A). Reducing agents did not affect the potency of noncysteine-containing peptides, including NP147–155 (Fig. 3 B), an LCMV peptide (described below), or the cysteine→ serine- or cysteine→ alanine-substituted NP39–47 peptides (not shown). Enhancement of cysteine peptide recognition is, as expected, dependent on the concentration of reducing agent, with TCEP being more effective on a molar basis than dithiothreitol (not shown). The optimal concentration for TCEP was 200 μM (used in additional experiments), as higher concentrations (1 mM) sometimes increased spontaneous release values in 51Cr-release assays. The effect of TCEP on NP218–226 antigenicity is particularly impressive when considered in view of the 100–1,000-fold decrease in peptide binding to Kd in the presence of TCEP (described below).
Figure 3.

Effects of reducing agents on the antigenicity and Kd binding of synthetic NP218–226. Synthetic peptides were diluted in I-10 with or without 200 μM of the indicated reducing agents and added to assay wells containing 51Cr-labeled target cells and TCD8+ specific for NP218–226 (A) or NP147–155 (B), and lysis was determined by microcytotoxicity assay.
In additional experiments, we found that inclusion of reducing agents in the media used to stimulate and propagate TCD8+ specific for NP218–226 or NP39–47 greatly enhanced their growth and altered their appearance, to the extent that these TCD8+ were indistinguishable from TCD8+ raised to NP147–155 or other immunodominant determinants.
These findings indicated that sulfhydryl modification can have major effects on the antigenicity and immunogenicity of synthetic peptides, effects that can lead to erroneous conclusions regarding the nature of their interactions with class I molecules, the affinity of TCD8+ specific for the peptide, and the growth characteristics of the cells. To broaden these findings, we examined the effect of reducing agents on the in vitro antigenicity of three oft-studied, Db-restricted determinants from LCMV: two containing cysteine (GP33–41 and GP276–284) and a control determinant lacking cysteine (NP396–404) (Table I). As with the influenza virus determinants, the cysteine-containing peptides were recognized 10–100-fold less efficiently than the cysteine-free peptide in the absence of reducing agent (Fig. 4, A and B), whereas TCEP had no affect on the antigenicity of NP396–404 (Fig. 4 C).
Figure 4.

The antigenicity of Db-restricted synthetic peptides corresponding to LCMV determinants is enhanced by reducing agents. Splenocytes from C57BL/6 mice infected 3 wk previously with LCMV were stimulated in vitro by incubation for 7 d with synthetic peptide–pulsed EL-4 cells and restimulated in the same manner for an additional 5 d. Synthetic peptides were diluted in PBS in the presence or absence of 200 μM TCEP and added to assay wells containing LCMV-specific TCD8+ and 51Cr-labeled EL-4 cells, and lysis was determined by microcytotoxicity assay in I-10.
Based on these findings, it is clear that sulfhydryl modification of cysteine-containing synthetic peptides can have major effects on TCD8+ growth and target cell recognition that must be taken into consideration in investigating the biological properties of the corresponding naturally produced determinants and the TCD8+ they induce.
Nature of Thiol-modifying Agents Affects NP218–226.
We turned our attention to why reducing agents enhance the antigenicity of synthetic NP218–226. The major possibilities were reduction of disulfide-linked peptide dimers and reduction of disulfide-bound species derived from culture media. The most abundant sulfhydryl-containing compound present in DMEM is cystine (cysteine–cysteine dimers). To determine whether NP218–226 becomes cysteinylated in DMEM, freshly dissolved NP218–226 was incubated in normal or cystine-free DMEM for 2 h. Peptides present in media were then separated by RP-HPLC, and the masses in peptide-containing fractions were determined by mass spectroscopy (Fig. 5). After incubation in cysteine-free media, the only modification detected was a small amount of dimerization (Fig. 5 B). By contrast, in cysteine-containing media, most of the monomer was converted to a separate eluting form representing cysteinylated peptide (Fig. 5 C). This fraction also contained a minor species with an additional mass of 16 daltons that probably represents oxidation of the neighboring methionine residue in the peptide.
Figure 5.

RP-HPLC mass spectrometric characterization of synthetic NP218–226. 10 μmol of NP218–226, either a freshly made stock in PBS (A) or 1-yr stock in DMSO (D), were added to 300 μl of cystine-free or cystine-containing DME, incubated for 2 h at room temperature, and analyzed by RP-HPLC. The masses of material in eluting peaks were determined by MALDI-TOF, revealing that cysteinylated NP218–226 elutes at 25 min, unmodified NP218–226 at 27 min, and dimeric NP218–226 at 29 min. A, in PBS + TCEP; B, monomer in cystine-free medium; C, monomer in cystine-containing medium; D, in DMSO; E, dimer in cystine-free medium; F, dimer in cystine-containing medium.
The effect of cysteinylation on NP218–226 antigenicity was examined by measuring the Kd binding and antigenicity of an unmodified preparation of NP218–226 incubated for 2 h in cystine-containing DMEM with or without TCEP before addition to cells. As seen in Fig. 6 A, the resulting cysteinylation was associated with an ∼10-fold increase in rescuing Kd molecules from melting and an ∼10,000-fold decrease in capacity to sensitize target cells for lysis by TCD8+ induced by APCs pulsed with unmodified NP218–226.
Figure 6.

Binding and antigenicity of cysteine-modified NP218–226. (A) NP218–226 peptide freshly dissolved in PBS was diluted as indicated in 200 μl of cystine-containing DMEM and incubated for 2 h to produce CysNP218–226 or incubated in cystine-free DMEM with TCEP as a noncysteinylated control. Alternatively, an old stock consisting primarily of dimeric NP218–226 was treated in cystine-free DMEM in the same manner in the presence or absence of TCEP. Diluted peptides were then incubated for 1 h at 26°C with T2-Kd cells previously cultured for 14 h at 26°C and shifted to 37°C for 2 h to melt Kd molecules. The cells were then analyzed by flow cytometry after staining with fluorescein-conjugated SF1.1.1 mAb. (B) TCD8+ specific for unmodified NP218–226 were tested in a microcytotoxicity assay for their ability to lyse 51Cr-labeled P815 target cells incubated in the presence of peptides treated as above.
We also examined the effect of NP218–226 dimerization on antigenicity and Kd binding. Analysis of various stocks by RP-HPLC in conjunction with mass spectrometry revealed that a 1-yr-old stock of peptide in DMSO was >95% dimerized (Fig. 5 D). Using this stock as an NP218–226 dimer source, we investigated the effect of 2-h incubation at room temperature in cystine-free or cystine-containing DMEM. NP218–226 dimers were stable under these conditions (Fig. 5, E and F). Having identified a source of dimers and demonstrated the stability of dimers in DMEM, we could examine the Kd binding and antigenicity of dimers (Fig. 6), which revealed that dimers behaved similarly to cysteinylated NP218–226.
We draw two conclusions from these findings. First, cysteinylation and dimerization of NP218–226 is associated with enhanced Kd binding yet greatly reduced antigenicity, using TCD8+ restimulated by the reduced peptide. Second, in normal DMEM, cysteinylation occurs preferentially to peptide dimerization, even when NP218–226 is present at relatively high concentrations. As a second-order reaction, dimerization should be greatly disfavored at decreasing peptide concentrations, whereas cysteinylation continues at a first-order reaction rate. Therefore, cysteinylation is probably the major process for modifying cysteine-containing peptides at the concentrations used in Kd-binding and 51Cr-release assays.
Cysteinylated NP218–226 Is Produced by Virus-infected Cells.
Given the potential for cysteine modification in vitro, we examined whether NP218–226 produced by PR8-infected cells was modified in vivo. Low Mr peptides present in acid extracts from whole cells were fractionated by RP-HPLC and tested for their abilities to sensitize target cells for lysis by TCD8+ raised to reduced NP218–226. TCEP was added to the fractions to reveal the presence of SH-modified forms of peptides rendered nonantigenic by the modification. As shown in Fig. 7 A, antigenic peptides were recovered in fractions eluting from 25–27 min, matching the elution times of cysteinylated (25 min) and unmodified (27 min) NP218–226. No activity was present in the 29-min fraction, where dimeric NP218–226 elutes. The amounts of peptide recovered were well below that required for saturation (peak lysis of column fraction of 40% versus 70% with a saturating amount of synthetic peptide; Fig. 7 A, top right), suggesting that a considerable fraction of NP218–226 recovered from PR8-infected cells is cysteinylated.
Figure 7.
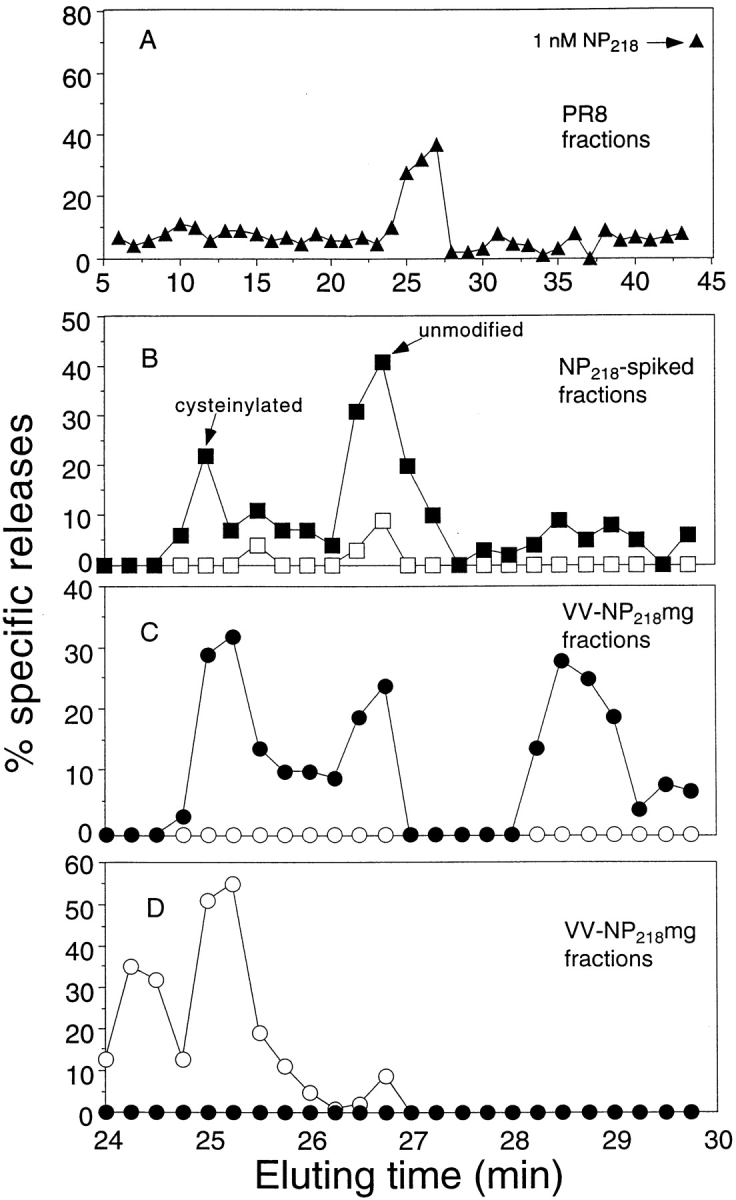
Cysteinylated and dimerized NP218–226 species can be recovered from virus-infected cells. Low-molecular mass, TFA-soluble material present in lysates from 109 PR8-infected P815 cells (A), 5 × 108 P815 cells spiked with synthetic unmodified NP218–226 (B), and 5 × 108 VV-NP218–226–infected P815 cells (C and D) were fractionated by RP-HPLC, and 5 μl of each fraction (1 ml in A; 0.25 ml in B–D) was then added to low temperature–induced, 51Cr-labeled RMA-S/Kd cells in the presence (filled symbols) or absence (open symbols) of 200 μM TCEP and tested for lysis in a microcytotoxicity assay, using TCD8+ lines raised to TCEP-treated (A–C) or cysteinylated NP218–226. TCD8+ lines were derived by four rounds of stimulation of splenocytes from PR8-infected mice with APCs pulsed with HPLC-purified, unmodified or cysteinylated NP218–226. Note that B–D show only fractions collected from 24–30 min.
Cysteinylation of NP218–226 might have occurred artefactually during the extraction process. To examine this possibility, cell homogenates were doped with synthetic, unmodified NP218–226 and then processed identically to virus-infected cells (note that in this and subsequent experiments, to increase the chromatographic resolution, fraction size was reduced from 1 ml to 0.25 ml) (Fig. 7 B). In this case, <1% of the antigenic activity (as determined by titrating fractions; data not shown) was recovered in the cysteinylated form. In this experiment, peptides were tested in the presence or absence of TCEP. Even unmodified NP218–226 required TCEP treatment, which we attribute to rapid peptide cysteinylation during target cell sensitization.
To examine themaximal potential for posttranslational modification of NP218–226, we infected cells with an rVV (VV-NP218–226) that expresses the peptide in the cytosol as a minigene product (with NH2-terminal methionine to enable translation initiation). As reported previously (7–9), this greatly enhances the number of peptide–class I complexes generated by cells. RP-HPLC fractionation from minigene-expressing cells revealed the presence of material coeluting with unmodified NP218–226, as well as cysteinylated peptide (Fig. 7 C). For the first time, dimeric peptide was also detected. These activities cannot be attributed to methionine-extended NP218–226, which, for each form, elutes slightly later than NP218–226 (data not shown). Titration of the antigenic activities in the fractions (not shown) revealed a ratio of unmodified/cysteinylated/dimeric forms of ∼6:3:1.
In an additional experiment (data not shown), we examined the Kd dependence of NP218–226 recovery in HPLC fractions after VV-NP218–226 by using L929 cells and L929 cells expressing Kd from a transgene. Peptides corresponding to cysteinylated and unmodified NP218–226 were recovered from L929-Kd cells but not L929 cells. Dilution of peak fractions revealed that expression of Kd resulted in at least a 25-fold increase in the recovery of NP218–226. This confirms numerous prior studies demonstrating the MHC dependence of antigenic peptide recovery (1).
Cysteinylated NP218–226 Is Presented In Vivo.
The ultimate demonstration of the biological relevance of cysteinylated NP218–226 is that specific TCD8+ are elicited in PR8-infected mice. We could show this by stimulating splenocytes derived from PR8-infected mice with RP-HPLC–purified, cysteinylated peptide. After three to four rounds of stimulation, we obtained TCD8+ that preferentially recognize cysteinylated peptide (Fig. 8 A). In the same assay, the noncysteinylated peptide is preferentially recognized by TCD8+ induced in the standard manner (Fig. 8 B). The recovery of TCD8+ specific for the cysteinylated peptide could not be attributed to in vitro stimulation of naive TCD8+, as we failed to obtain any activity using splenocytes from nonimmunized mice (data not shown).
Figure 8.

Cysteinylated NP218–226 is presented in vivo. TCD8+ were produced from PR8-infected mice by stimulation with HPLC-purified, cysteinylated (A) or unmodified (B) NP218–226. Synthetic NP218–226 was cysteinylated by dilution into serum-free DME and incubation for 2 h at room temperature or maintained in unmodified form by addition of 200 μM TCEP. Diluted peptides were then added into assay wells containing TCD8+ and 51Cr-labeled P815 targets, and lysis was determined by microcytotoxicity assay.
Using TCD8+ stimulated by the cysteinylated NP218–226, it was possible to formally demonstrate that the 25-min fraction derived from minigene-expressing cells contained cysteinylated peptide (Fig. 7 D). Indeed, now antigenicity was destroyed by TCEP exposure, in contrast to the enhancing activity observed with other peptides. We also detected an additional peak at 24.5 min. This probably represents the methionine-oxidized form of cysteinylated NP218–226, which, based on experience with other peptides, often elutes slightly faster than the nonoxidized form, providing evidence that this modification occurs in cells.
Discussion
In this paper, we confirm and extend the findings of Meadows et al. (4) that modification of the SH group of cysteine-containing peptides has important positive and negative effects on their antigenicity and immunogenicity in vitro and in vivo. Failure to consider these effects can have disastrous consequences for the accurate interpretation of several different types of experiments.
In our own case, the use of synthetic peptides in the absence of reducing agents led us to erroneously favor the idea that the subdominant status of two cysteine-containing peptides was due to a greater number of complexes required for TCD8+ recognition and correlated with the atypical growth of TCD8+ in vitro. In other experiments (our unpublished results), it also led us to the incorrect quantitation of peptides recovered from virus-infected cells. A further potential methodological pitfall is that autooxidation of reducing agents can cause additional artifacts. This can be minimized by inclusion of a chelating agent, such as DTPA (diethylenetriaminepentaacetic acid), with the reducing agent.
These errors are probably widespread. We demonstrate that two cysteine-containing LCMV determinants restricted by a different class I molecule are also modified in vitro through their sulfhydryl groups, with a resulting 10–100-fold loss in antigenicity. These determinants were the subject of a recent study (10) focused on the factors involved in the immunodominance hierarchy of the determinants. The failure to add reducing agents during peptide titration probably led to erroneous calculations of peptides present in virus-infected cells, particularly because the peptide titration curves were nearly identical to those we obtained in the absence of reducing agents. Results obtained in this study with in vivo transfer of TCD8+ lines must also be questioned, as the cells were propagated in vitro with synthetic peptides in the absence of reducing agents. In another recent study of the fine specificity of a TCD8+ clone for a cysteine-containing peptide, amino acids were substituted for cysteine, many of which enhanced the antigenicity of the peptide (11). Based on our findings, we would predict that simple reduction of the wild-type peptide would have a similar (or greater) effect.
In addition to reducing antigenicity, modification of cysteine can result in the generation of TCD8+ specific for the modified determinant. We show that PR8-infected mice generate TCD8+ that prefer cysteinylated NP218–226. We also demonstrate that PR8-infected cells generate an SH-modified peptide that coelutes with cysteinylated NP218–226 and is recognized by TCD8+ specific for cysteinylated NP218–226. This species almost certainly represents cysteinylated NP218–226, although definitive evidence requires mass spectroscopy. The recognition of posttranslationally modified peptides by TCD8+ adds to the already formidable challenge of understanding in vivo TCD8+ responses but can be ignored only at the peril of the investigator.
We can only speculate where NP218–226 is cysteinylated during its processing and presentation by virus-infected cells. It is theoretically possible that the cystine derives naturally from a disulfide bond present in NP. It is difficult, though admittedly not impossible, to imagine the proteolytic liberation of cysteinylated NP218–226. Given the highly reducing environment of the cytosol and nucleus, it also seems unlikely that cysteinylation would occur before peptide translocation into the ER. The ER provides a much more oxidizing environment and possesses resident proteins that catalyze thiol–disulfide interchange, including protein disulfide isomerase (12). There is evidence that exogenous homocysteine is added to HLA class I molecules in an early secretory compartment (13). If cysteinylation occurs in the ER, it may occur before peptide loading onto class I molecules. Alternatively, cysteinylation could occur after peptide binding, particularly in the case of NP218–226, as the SH must be directed away from the groove (so as to accommodate dimer binding). In this case, it could occur anywhere from the ER to the cell surface. Cysteine is thought to be the major reductant in the endosomal pathway, and although there is no evidence that functional Kd molecules visit these compartments, this remains a possibility.
It was previously reported that disulfide-linked homodimeric peptides could bind to class I molecules (14). We provide another example of this and further demonstrate that this can occur naturally in cells. Recovery of dimers required expression of NP218–226 as a cytosolic minigene, which results in at least 10–1,000-fold overproduction of peptide–class I complexes relative to expression of peptide in its natural context (7, 9), and the extent to which overexpression of peptide is required remains to be determined. As peptide cross-linking is expected to be a second-order reaction, it implies that NP218–226 is present at a very high concentration intracellularly. It is hard to imagine this occurring outside of the ER, and it may occur in the vicinity of TAP, which is required for transport of NP218–226 into the ER (our unpublished results).
These findings have important clinical implications. First, for synthetic peptide vaccines (or other exogenous antigen vaccine preparations with vulnerable cysteine residues in antigenic peptides), modification of the cysteine in vitro or in vivo can obviously have major negative effects on immunogenicity. This can be avoided by modifying the side chain to a nonreactive form. For NP218–226 and NP39–47, this is achieved simply by substitution with alanine or serine, which did not detrimentally affect peptide binding or TCD8+ triggering. This strategy will probably work for most peptides. For others, it is possible that chemical modification of the SH group (e.g., treatment with an alkylating agent or a heavy metal) will do the job. Second, if increased cysteinylation is associated with a disease process, this could lead to autoimmune recognition of cysteinylated self peptides.
Abbreviations used in this paper
- ER
endoplasmic reticulum
- FBS
fetal bovine serum
- LCMV
lymphocytic choriomeningitis virus
- NP
nuclear protein
- RP-HPLC
reversed-phase HPLC
- VV
vaccinia virus
Footnotes
Bethany Buschling provided outstanding technical assistance.
W. Chen was supported by a C.J. Martin Fellowship (967036) from the Australian National Health and Medical Research Council.
References
- 1.Rammensee, H.-G., J. Bachmann, and S. Stevanovic. 1997. MHC Ligands and Peptide Motifs. Landes Bioscience, Austin, Texas.
- 2.Madden DR. The three-dimensional structure of peptide-MHC complexes. Annu Rev Immunol. 1995;13:587–622. doi: 10.1146/annurev.iy.13.040195.003103. [DOI] [PubMed] [Google Scholar]
- 3.Engelhard VH. Structure of peptides associated with MHC class I molecules. Curr Opin Immunol. 1994;6:13–23. doi: 10.1016/0952-7915(94)90028-0. [DOI] [PubMed] [Google Scholar]
- 4.Meadows L, Wang W, den Haan JM, Blokland E, Reinhardus C, Drijfhout JW, Shabanowitz J, Pierce R, Agulnik AI, Bishop CE, et al. The HLA-A*0201-restricted H-Y antigen contains a posttranslationally modified cysteine that significantly affects T cell recognition. Immunity. 1997;6:273–281. doi: 10.1016/s1074-7613(00)80330-1. [DOI] [PubMed] [Google Scholar]
- 5.Fisk B, Blevins TL, Wharton JT, Ioannides CG. Identification of an immunodominant peptide of HER-2/neu protooncogene recognized by ovarian tumor–specific cytotoxic T lymphocyte lines. J Exp Med. 1995;181:2109–2117. doi: 10.1084/jem.181.6.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen W, Khilko S, Fecondo J, Margulies DH, McCluskey J. Determinant selection of major histocompatibility complex class I–restricted antigenic peptides is explained by class I–peptide affinity and is strongly influenced by nondominant anchor residues. J Exp Med. 1994;180:1471–1483. doi: 10.1084/jem.180.4.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Antón LC, Yewdell JW, Bennink JR. MHC class I-associated peptides produced from endogenous gene products with vastly different efficiencies. J Immunol. 1997;158:2535–2542. [PubMed] [Google Scholar]
- 8.Deng Y, Yewdell JW, Eisenlohr LC, Bennink JR. MHC affinity, peptide liberation, T cell repertoire, and immunodominance all contribute to the paucity of MHC class I-restricted peptides recognized by antiviral CTL. J Immunol. 1997;158:1507–1515. [PubMed] [Google Scholar]
- 9.Porgador A, Yewdell JW, Deng Y, Bennink JR, Germain RN. Localization, quantitation, and in situ detection of specific peptide-MHC class I complexes using a monoclonal antibody. Immunity. 1997;6:715–726. doi: 10.1016/s1074-7613(00)80447-1. [DOI] [PubMed] [Google Scholar]
- 10.Gallimore A, Dumrese T, Hengartner H, Zinkernagel RM, Rammensee HG. Protective immunity does not correlate with the hierarchy of virus-specific cytotoxic T cell responses to naturally processed peptides. J Exp Med. 1998;187:1647–1657. doi: 10.1084/jem.187.10.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manning TC, Schodin BA, Kranz DM. A strategy for the synthesis and screening of thiol-modified peptide variants recognized by T cells. J Immunol Methods. 1996;192:125–132. doi: 10.1016/0022-1759(96)00048-8. [DOI] [PubMed] [Google Scholar]
- 12.Freedman RB. Protein disulfide isomerase: multiple roles in the modification of nascent secretory protein. Cell. 1989;57:1069–1072. doi: 10.1016/0092-8674(89)90043-3. [DOI] [PubMed] [Google Scholar]
- 13.Gao XM, Wordsworth P, McMichael AJ, Kyaw MM, Seifert M, Rees D, Dougan G. Homocysteine modification of HLA antigens and its immunological consequences. Eur J Immunol. 1996;26:1443–1450. doi: 10.1002/eji.1830260707. [DOI] [PubMed] [Google Scholar]
- 14.Di Modugno F, Mammi C, Rosano L, Rubiu O, Nistico P, Chersi A. MHC-peptide binding: dimers of cysteine-containing nonapeptides bind with high affinity to HLA-A2.1 class I molecules. J Immunother. 1997;20:431–436. [PubMed] [Google Scholar]