

Abstract
Chronic inflammation containing CD8+ lymphocytes, neutrophils, and macrophages, and pulmonary emphysema coexist in lungs from patients with chronic obstructive pulmonary disease. Although this inflammatory response is believed to cause the remodeling that is seen in these tissues, the mechanism(s) by which inflammation causes emphysema have not been defined. Here we demonstrate that interferon γ (IFN-γ), a prominent product of CD8+ cells, causes emphysema with alveolar enlargement, enhanced lung volumes, enhanced pulmonary compliance, and macrophage- and neutrophil-rich inflammation when inducibly targeted, in a transgenic fashion, to the adult murine lung. Prominent protease and antiprotease alterations were also noted in these mice. They included the induction and activation of matrix metalloproteinase (MMP)-12 and cathepsins B, H, D, S, and L, the elaboration of MMP-9, and the selective inhibition of secretory leukocyte proteinase inhibitor. IFN-γ causes emphysema and alterations in pulmonary protease/antiprotease balance when expressed in pulmonary tissues.
Keywords: chronic obstructive pulmonary disease, matrix metalloproteinase, cathepsin, neutrophil, secretory leukocyte proteinase inhibitor
Introduction
Chronic obstructive pulmonary disease (COPD) is a generic term covering several clinical syndromes including emphysema and chronic bronchitis. Chronic bronchitis is diagnosed in patients with a history of cough or sputum production on most days for at least 3 mo for two consecutive years without any other explanation 1 2. In contrast, emphysema is defined pathologically as a condition of the lung characterized by abnormal permanent enlargement of the air spaces distal to the terminal bronchiole accompanied by destruction of alveolar walls 1 2. COPD is a pressing clinical problem. In the USA, it affects over 16 million people, accounts for 13% of hospitalizations, and is the fourth leading cause of death 1 3. In addition, 48 million people in the USA smoke cigarettes, the major cause of COPD, and 3,000 people, mostly teenagers, take up the habit each day 1. Worldwide, 1.5 million deaths per year due to COPD are predicted in China alone over the next half century 4. In contrast to its impressive health care importance, COPD is the least funded disease by the USA National Institutes of Health when adjusted for disease burden 5.
Tissue inflammation is noted throughout the bronchial tree and in the parenchyma of lungs from patients with COPD. Neutrophil-, macrophage-, and/or eosinophil-rich tissue and airway responses are seen in these locations 3 6 7 8 9 10 11. Lymphocyte infiltration with the enhanced accumulation of CD8+ cells is also a prominent finding 7 8 9 11. It has long been assumed that this inflammation causes the tissue remodeling seen in COPD 1 12 13. However, the features of this inflammatory response that cause alveolar remodeling and the mediators that are responsible for these changes are poorly understood.
Several lines of evidence suggest that pulmonary lymphocytes contribute in important ways to the pathogenesis of the tissue remodeling responses in COPD. They include studies demonstrating correlations between lymphocyte counts and indices of alveolar destruction 14 and CD8+ cell numbers and the degree of COPD airflow limitation 8 11. These lymphocytes are believed to be predominantly type 1 T cytotoxic (Tc1) cells that produce cytokines like IFN-γ 6 11 15. Surprisingly, the ability of the cytokines that are produced by these cells to generate the alterations seen in pulmonary emphysema have not been defined.
Studies of human α1 antitrypsin (α1-AT) deficiency and experiments with animal models of emphysema induced by papain or other proteases demonstrated that emphysema can be induced by proteolytic injury to the pulmonary matrix, particularly to elastin 1 16. This led to the protease/antiprotease hypothesis, which is still the prevailing concept in the pathogenesis of emphysema. According to this hypothesis, the normal lung is protected by an “antiprotease shield” that neutralizes proteolytic enzymes that are released into the airway or parenchyma. Emphysema is caused by an alteration in this balance with an increase in proteases and/or a reduction in antiproteases 1 3. Ineffective and/or disordered repair of elastic fibers and/or collagen- and oxidant-induced pulmonary injury may also contribute to the generation of these lesions 17 18 19. However, the ability of the inflammatory cells and mediators in COPD tissues to induce protease/antiprotease abnormalities that can contribute to, or activate, pulmonary proteolysis has not been adequately investigated.
In keeping with the belief that CD8+ Tc1 cells play an important role in the pathogenesis of emphysema, we hypothesized that IFN-γ could induce the alveolar destruction characteristic of this disorder. To test this hypothesis, we used an inducible overexpression transgenic modeling system to target IFN-γ to the adult murine lung. These studies demonstrate that IFN-γ causes a phenotype that mirrors human COPD with progressive emphysema, enhanced lung volumes, and macrophage and neutrophilic inflammation. They also define the prominent matrix metalloproteinase (MMP), cathepsin, and antiprotease alterations induced by IFN-γ in this in vivo setting.
Materials and Methods
Transgenic Mice.
Mice and humans are born with premature lungs that contain large sac-like structures. Normal alveolar size and number are acquired only after a growth and septation process that occurs during the first month and years of life, respectively 20. Thus, enlarged alveoli in adult mice or humans can be caused by processes that alter alveolar development or processes that destroy otherwise normal alveolar septae 1 21 22. The emphysema in COPD is caused by the latter 1. Overexpression transgenic modeling in the lung uses either the clara cell 10-kD (CC10) or surfactant apoprotein C promoters most frequently. Both promoters are activated in utero and, when used to drive an appropriate transgene, have been shown to cause developmental abnormalities 22 23 24. The need to differentiate development-dependent and adult onset phenotypes confounds the use of pulmonary overexpression transgenic modeling to study the pathogenesis of COPD. To avoid this confounder, we used a novel, externally regulatable, dual construct overexpression transgenic system developed in our laboratory 22. This transgenic system allows transgene expression to be minimized during phases of lung development and induced after lung development has occurred. The constructs used in this system have been described previously by our laboratory 22. The CC10-rtTA-hGH construct contains the CC10 promoter, the reverse tetracycline transactivator (rtTA), and human growth hormone (hGH) intronic and polyadenylation sequences (Fig. 1). The rtTA is a fusion protein made up of a mutated tetracycline repressor (rtet-R) and the herpes virus VP-16 transactivator. The tet-O-CMV-IFN-γ-hGH construct contains a polymeric tetracycline operator (tet-O), minimal CMV promoter, murine IFN-γ cDNA, and hGH intronic and polyadenylation signals (Fig. 1). In this system, the CC10 promoter directs the expression of rtTA to the lung. In the presence of doxycycline (dox), rtTA is able to bind in trans to the tet-O, and the VP-16 transactivator activates IFN-γ gene transcription. In the absence of dox, rtTA binding occurs at very low levels and only low level gene transcription is noted. The preparation of the CC10-rtTA construct has been described previously 22. The tet-O-CMV-IFN-γ-hGH construct was prepared by replacing the IL-11 cDNA in the construct tet-O-CMV-hIL-11-hGH described previously by our laboratory 22 with the murine IFN-γ cDNA. This construct was checked for correct insert orientation by restriction enzyme digestion and sequencing. Both constructs were purified, linearized, separated by electrophoresis through agarose, and purified as described previously 22. Transgenic mice were prepared in (CBA × C57BL/6)F2 eggs by mixing and simultaneously injecting the constructs into pronuclei as described previously 22.
Figure 1.

Constructs used in the generation of CC10-rtTA-IFN-γ mice.
Documentation of Transgene Status.
The presence or absence of the transgenes was initially evaluated using Southern blot analysis and later by PCR. Southern analysis was performed as described previously using cDNA encoding murine IFN-γ or rtTA 22 25. PCR for rtTA was also performed using protocols described by our laboratory 22 25. PCR for the IFN-γ–containing construct was undertaken using the following primers: upper primer 5′-ACT CAC ATT CAG AAC CCC AAA C-3′, lower primer 5′-CGT CAG ATC GCC TGG AGA C-3′. The cycling conditions were one cycle at 94°C for 4 min, 35 cycles of 94°C for 1 min, followed by 57°C for 1 min and 72°C for 1 min, 1 cycle at 72°C for 5 min, and 1 cycle at 4°C to end. All CC10-rtTA-IFN-γ lineage animals were evaluated for the presence of both the rtTA- and IFN-γ–containing transgenic constructs.
Dox Water Administration.
CC10-rtTA-IFN-γ animals were maintained on normal water until 4–6 wk of age. At that time they were randomized to receive normal water or water containing dox (1.0 mg/ml) as described previously 22.
Bronchoalveolar Lavage and Quantification of IFN-γ Levels.
Mice were killed, the trachea was isolated by blunt dissection, and small caliber tubing was inserted and secured in the airway. Three volumes of 0.75 ml of PBS with 0.1% BSA were then instilled and gently aspirated and pooled. Each bronchoalveolar lavage (BAL) fluid sample was centrifuged and the supernatants were stored at −70°C until used. The levels of IFN-γ were determined immunologically using a commercial ELISA (R&D Systems) as per the manufacturer's instructions.
Lung Volume and Compliance Assessment.
The animals were anesthetized, the trachea was cannulated, and the lungs were ventilated with 100% O2 via a “T” piece attachment. The tracheal cannula was then clamped and oxygen was absorbed in the face of ongoing pulmonary perfusion. At the end of this degassing, the lungs and heart were removed en bloc and inflated to 25 cm pressure with PBS. Static compliance was calculated as the change in volume divided by the change in pressure.
Histologic Analysis.
Animals were killed, a median sternotomy was performed, and right heart perfusion was accomplished with calcium and magnesium–free PBS to clear the pulmonary intravascular space. The heart and lungs were then removed en bloc, inflated to 25 cm with neutral-buffered 10% formalin, fixed overnight in 10% formalin, embedded in paraffin, sectioned at 5 μm, and stained. Hematoxylin and eosin, Congo red, and periodic acid-Schiff with diastase (D-PAS) stains were performed in the Research Histology Laboratory of the Department of Pathology at Yale University School of Medicine.
mRNA Analysis.
mRNA levels were assessed using Northern blot and reverse transcription (RT)-PCR as described previously by our laboratories 25 26. In these experiments, total cellular RNA from lungs or a variety of other mouse tissues were obtained using Trizol reagent (GIBCO BRL) as per the manufacturer's instructions. When Northern blots were being performed, the RNA was fractionated by formaldehyde–agarose gel electrophoresis, transferred to a nylon membrane, and probed with 32P-labeled cDNA encoding the moiety of interest. The cDNA probes were generated using RT-PCR with the primers in Table and whole lung RNA from CC10–IL-13 mice 25. All cDNA was sequenced before utilization. Equity of sample loading and the efficacy of transfer were assessed by stripping and reprobing the membrane with cDNA encoding β-actin.
Table 1.
RT-PCR Primers
| mRNA of interest | Sequences | Product length |
|---|---|---|
| bp | ||
| Gelatinase A (MMP-2) | (UP) 5′-TCT GCG GGT TCT CTG CGT CCT GTG C-3′ (LO) 5′-GTG CCC TGG AAG CGG AAC GGA AAC T-3′ | 861 |
| Matrilysin (MMP-7) | (UP) 5′-ACA TCA GTG GGA ACA GGC TCA G-3′ (LO) 5′-ACA GTA CCG GGA ACA GAA GAG T-3′ | 606 |
| Gelatinase B (MMP-9) | (UP) 5′-CAA AGA CCT GAA AAC CTC CAA CCT C-3′ (LO) 5′-TTC TCC GTT GCC GTG CTC CGT GTA G-3′ | 757 |
| Macrophage metalloelastase (MMP-12) | (UP) 5′-AAG CAA CTG GGC AAC TGG ACA ACT C-3′ (LO) 5′-TGG TGA CAG AAA GTT GAT GGT GGA C-3′ | 631 |
| Collagenase (MMP-13) | (UP) 5′-CTT CTG GCA CAC GCT TTT CCT C-3′ (LO) 5′-CGC AGC GCT CAG TCT CTT CAC C-3′ | 606 |
| MT-MMP1 (MMP-14) | (UP) 5′-GTC TCC TGC TCC CCC TGC TCA C-3 ′ (LO) 5′-CAT GCA CAG CCA CCA AGA AGA T-3′ | 690 |
| TIMP-1 | (UP) 5′-GCA TCT GGC ATC CTC TTG TTG-3 ′ (LO) 5′-GAC AGT GTT CAG GCT TCA GTT TTT C-3′ | 637 |
| TIMP-2 | (UP) 5′-GCA ACA GGC GTT TTG CAA TGC AGA C-3′ (LO) 5′-GCT TTT CAA TTG GCC ACA GGG GCT C-3′ | 598 |
| TIMP-3 | (UP) 5′-CTG GCT TGG GCT TGT CGT GCT CCT GA-3′ (LO) 5′-GGG AAG GAG GTG AGG TGG GGC AGG TC-3′ | 659 |
| TIMP-4 | (UP) 5′-CAC GCC ATT TGA CTC TTC CCT-3′ (LO) 5′-CCA GCA GCC AGT CCG TCC AGA-3′ | 269 |
| Cathepsin B | (UP) 5′-CTT GAT CCT TCT TTC TTG CCT GCT G-3′ (LO) 5′-GAA TCG TAG ACT CCA CCT GAA ACC A-3′ | 513 |
| Cathepsin L | (UP) 5′-CTA CAC AAC GGG GAA TAC AGC AAC G-3′ (LO) 5′-ACT ATA GAA CTG GAG AGA CGG ATG G-3′ | 600 |
| Cathepsin K | (UP) 5′-GGG AGA CAT GAC CAG TGA AGA AGT G-3′ (LO) 5′-TGC TCT CTT CAG GGC TTT CTC GTT C-3′ | 481 |
| Cathepsin H | (UP) 5′-CGA GCT GAC CGT GAA CGC CAT AG-3′ (LO) 5′-AGC TTT TTG GGG GTT GAA TCT GC-3′ | 595 |
| Cathepsin S | (UP) 5′-GGG TTC TTG TGG TGC CTG TTG G-3′ (LO) 5′-CCG TAC AGG AGG GGT CAT CAT A-3′ | 425 |
| SLPI | (UP) 5′-CTG GAC TGT GGA AGG AGG CAA AAA TG-3′ (LO) 5′-AGT AGT TTC CAG AGC ACA CCG AGC AC-3′ | 398 |
| α1-AT | (UP) 5′-TGC CCA TGA TGA CCC TCT C-3′ (LO) 5′-TGG GGC TCT GAG TGT GTT CT-3′ | 498 |
UP, upper; LO, lower; MT-MMPI, membrane-type MMP1.
In the RT-PCR assays, RNA samples were obtained and reverse transcribed. For the proteases and antiproteases, gene-specific primers (Table ) were used to amplify selected regions of each target moiety. When IFN-γ mRNA was being assessed, an upper primer in IFN-γ and a lower primer in hGH were used (upper GAGGAACTGGCAAAAGGATG; lower GATTTTAGGGGCGCTACCT). This yielded a 418-bp reaction product. To verify that equal amounts of undegraded RNA were added in each RT-PCR reaction, β-actin was used as an internal standard. PCR products were quantitated after 20, 25, and 30 cycles of amplification. Amplified PCR products were detected using ethidium bromide gel electrophoresis, quantitated electronically, and confirmed by nucleotide sequencing.
Western Blot Analysis.
Western blot analysis of BAL fluid was performed using antibodies to MMP-12 27 as described previously by our laboratories 26.
Zymography.
Gelatin and casein zymography were performed using BAL fluid as described previously by our laboratories 28.
Active Site Probe Analysis of Cysteine Proteases.
Active site probe analyses were performed as described previously by our laboratories 29. Lung lysates or cell-free BAL, normalized to total protein, were incubated with an 125I-JPM analogue of E-64 and resolved by SDS-PAGE and autoradiography.
Morphometric Analysis.
Alveolar size was estimated from the mean cord length of the airspace as described previously by our laboratories 30. This measurement is similar to the mean linear intercept, a standard measure of air space size, but has the advantage that it is independent of alveolar septal thickness. Sections were prepared as described above. To obtain images at random for analysis, each glass slide was placed on a printed rectangular grid and a series of dots was placed on the coverglass at the intersection of the grid lines, i.e., at ∼1-mm intervals. Fields as close as possible to each dot were acquired by systematically scanning at 2-mm intervals. Fields containing identifiable artifacts or nonalveolated structures such as bronchovascular bundles or pleura were discarded.
A minimum of 10 fields from each mouse lung were acquired into a Macintosh computer through a framegrabber board. Images were acquired in 8 bit grayscale at a final magnification of 1.5 pixels/μm. The images were analyzed on a Macintosh G3 computer using the public domain NIH Image program written by Wayne Rasband at the National Institutes of Health (available at http://rsb.info.nih.gov/nih-image using a custom written macro available from the web site). Images were manually thresholded, then smoothed and inverted. The image was then subject to sequential logical image match “and” operations with a horizontal and then vertical grid. At least 200 measurements per field were made in transgene-positive animals and 400 measurements per field were made in the transgene-negative animals. The length of the lines overlying air space was averaged as the mean chord length. At least four animals were studied at each time point in the presence and absence of dox water. Chord length increases with alveolar enlargement.
Statistical Analysis and Data Presentation.
Values are expressed as means ± SEM. As appropriate, group means were compared by analysis of variance with Scheffe's procedure post hoc analysis or with the Student's two-tailed unpaired t test using the StatView software for the Macintosh (Abacus Concepts, Inc.). The illustrated effects of IFN-γ on the levels of mRNA encoding proteases or antiproteases are representative of the results obtained in evaluations of a minimum of six separate animals.
Results
Generation of Transgenic Mice.
To characterize the effects of IFN-γ in the adult lung, we used a novel, externally regulatable, dual construct overexpression transgenic system developed in our laboratory 22. The constructs required for these transgenics were prepared, purified, and simultaneously microinjected. Tail biopsies were obtained from potential founder animals, DNA was isolated, and the presence or absence of the IFN-γ and rtTA transgenic sequences was determined via Southern blot analysis and PCR. Three dual-positive founder animals were obtained (Fig. 2). They were subsequently backcrossed with C57BL/6 mice to generate transgene-negative and transgene-positive progeny.
Figure 2.

Southern blot analysis of DNA from potential founder mice. The requisite constructs were prepared and microinjected as described in Materials and Methods. Tail biopsies were obtained from potential founder animals, DNA was extracted, and the presence of the rtTA- and IFN-γ–containing constructs was evaluated by Southern blot analysis. The transgenes in three dual transgene-positive founder mice and a transgene-negative littermate control are compared with copy number controls of the CC10-rtTA-hGH and tet-O-CMV-IFN-γ-hGH constructs.
Regulation of IFN-γ Production.
Transgene-negative and transgene-positive mice were kept on normal water until they were 4–6 wk of age. They were then randomized to normal water or water with dox. In the absence of dox administration, BAL IFN-γ levels were <70 pg/ml. As illustrated in Fig. 3, dox administration caused a significant increase in BAL IFN-γ. This increase was noted within 24 h of dox administration, persisted for extended intervals, and returned to baseline values within 4 d of removing the dox from the animal's drinking water. The different transgenic lines had BAL IFN-γ levels between 370 and 1,200 pg/ml after 1 wk of dox administration.
Figure 3.
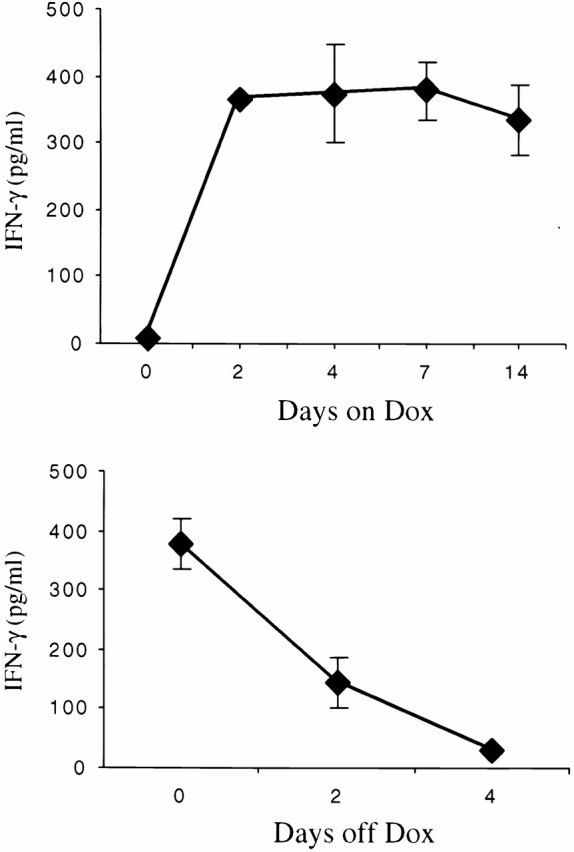
Kinetics of dox regulation of IFN-γ in CC10-rtTA-IFN-γ mice. The kinetics of dox induction of IFN-γ are illustrated in the top panel. In these experiments, BAL was performed on 1-mo-old dual transgene-positive animals (0 d) and identical animals after the noted intervals of dox water exposure. In the bottom panel, the kinetics of IFN-γ elimination are illustrated. In these experiments, 1-mo-old dual transgene-positive animals were placed on dox for 1 wk and then placed back on normal water. The levels of BAL IFN-γ were evaluated at the time the animals went from normal water to dox water (time 0) and at intervals thereafter. The noted values represent the means ± SEM of a minimum of four animals at each time point.
To determine if the IFN-γ that was produced in our transgenic animals was bioactive, studies were undertaken to determine if the IFN-γ–regulated genes IFN-γ–inducible protein 10 (IP-10) and monokine induced by IFN-γ (Mig) were stimulated after dox administration. mRNA encoding either gene could not be appreciated in transgene-negative mice on normal water or dox water (Fig. 4). In contrast, mRNA encoding IP-10 and Mig was weakly detected in transgene-positive mice on normal water, and impressive induction of both genes could be appreciated after dox administration (Fig. 4). These studies clearly demonstrate that the IFN-γ that is produced in the lungs of these transgenic mice is bioactive.
Figure 4.
Demonstration that BAL IFN-γ is bioactive. 1-mo-old transgene-negative and transgene-positive animals were randomized to receive normal water or dox water for up to 14 d. The levels of mRNA encoding IP-10 (A) and Mig (B) were evaluated via Northern blot analysis. Ethidium bromide–stained gels illustrating 28 S and 18 S ribosomal RNA are illustrated below each gel, demonstrating equity of sample loading. Similar results were obtained in three separate experiments.


Organ Specificity of Transgene Expression.
To determine if IFN-γ was appropriately targeted to the lung, RNA was obtained from the lungs and a variety of other tissues from transgene-positive mice that had received dox water for 2 wk. IFN-γ mRNA was then evaluated via RT-PCR analysis. Impressive levels of IFN-γ mRNA could be appreciated in lungs from transgene-positive mice on dox water. In contrast, transgene-induced IFN-γ mRNA was not noted and histologic abnormalities were not appreciated in a variety of visceral tissues from transgene-positive animals (Fig. 5, and data not shown). This demonstrates that our CC10-driven transgenic system, as described previously by this laboratory and others 22 23 31, selectively targets IFN-γ to the lungs of these transgenic animals.
Figure 5.

Representative experiment (of n = 4) illustrating the organ specificity of IFN-γ expression. RNA was isolated from the lungs and a variety of other organs from 6-wk-old dual transgene-positive mice that had received dox water for 2 wk. The levels of mRNA encoding IFN-γ were evaluated by RT-PCR as described in Materials and Methods.
Effect of IFN-γ on BAL Cellularity.
Dual transgene-positive and transgene-negative mice were kept on normal water until they were 4–6 wk of age. They were then placed on normal water or water with dox and maintained on this regimen for an additional month. At the end of this interval they were killed and BAL cellularity was evaluated. The cell recovery and cellular differentials of BAL fluids from transgene-negative mice on normal and dox water were almost identical (Table ). In contrast, IFN-γ increased BAL cell recovery. Modest increases in cellularity were seen in transgene-positive mice on normal water and impressive increases were seen after dox induction. At this time point, BAL fluids from dual transgene-positive mice on dox water had ∼9- and 11.5-fold more cells than the BAL fluids from transgene-negative animals on normal and dox water, respectively (Table ; P < 0.005). The BAL fluids from the transgene-positive mice also had an increased percentage and recovery of neutrophils and lymphocytes and an exaggerated number of macrophages compared with transgene-negative controls (P < 0.05 for all comparisons; Table ). Abnormalities in eosinophil recovery were not noted. These studies demonstrate that IFN-γ increases the pulmonary accumulation of macrophages, lymphocytes, and neutrophils.
Table 2.
IFN-γ Regulation of BAL Cellularity
| Differential | |||||
|---|---|---|---|---|---|
| Transgenestatus | Recovery | M | L | N | E |
| ×104 | % | % | % | % | |
| − | 4.2 ± 0.6 | 97 ± 0.6 | 2.2 ± 0.4 | 0.4 ± 0.3 | 0.3 ± 0.2 |
| −dox | 3.3 ± 0.1 | 97 ± 2.0 | 3.0 ± 1.2 | 0.3 ± 0.1 | 0.1 ± 0.01 |
| + | 10 ± 4.3 | 56 ± 5.1 | 28 ± 5.0 | 15 ± 3.0 | 1 ± 0.1 |
| +dox | 38.1 ± 7.8 | 54 ± 3.6 | 31 ± 3.5 | 15 ± 1.7 | 0.1 ± 0.01 |
Histologic, Physiologic, and Structural Effects of IFN-γ.
Inflammation and mucus metaplasia were not seen in the hematoxylin and eosin and periodic acid-Schiff stains of the lungs from transgene-negative mice on normal water. Similarly, mucus alterations were not noted in the histologic sections from the transgene-positive mice on normal or dox water (data not shown). In contrast, inflammation was noted in lungs from transgene-positive animals. This response contained increased numbers of mononuclear cells and rare neutrophils. It was mild and patchy and, when seen, was noted in peribronchiolar and alveolar parenchymal locations. Abnormalities of the airways in these locations were not appreciated. This response could be appreciated in transgene-positive mice that had received at least 1 mo of dox water but was not consistently detected in transgene-positive mice on normal water (Fig. 6, and data not shown).
Figure 6.

Inflammation in transgenic mice. Transgene-negative, Tg (−), and dual transgene-positive, Tg (+), mice at 1 mo of age were randomized to normal water, dox (−), or dox water, dox (+), for 3 mo. The lungs were then removed, fixed to pressure, sectioned, stained (hematoxylin and eosin), and photographed (original magnification: ×100). These photomicrographs are representative of the results obtained in a minimum of 10 experiments evaluating the histology of transgene-positive and transgene-negative mice on normal water or dox water.
The alveoli in lungs from transgene-negative mice on normal water and dox water were normal in size and appearance (Fig. 7). In contrast, alveolar enlargement was noted in the dual transgene-positive animals. A modest increase in alveolar size was seen in 1-mo-old, dual transgene-positive mice that had received normal water (Fig. 7). Dox-induced IFN-γ caused a further increase in alveolar size. This effect was dose and time dependent. It was most prominent in transgenic lines that produced high levels of IFN-γ after dox administration. In these mice, a marginal increase in alveolar size was noted after 4–6 wk and impressive emphysema was noted after 3 mo of dox administration (Fig. 7, and data not shown). This emphysema manifest as histologic (Fig. 7) and morphometrically obvious (Fig. 8) alveolar enlargement with loss of the orderly appearance of the acinus 32. The morphometric studies revealed a >100% increase in chord length. This is significantly greater than the 25–30% increase in morphometric parameters of alveolar size that we noted in similar mice exposed to cigarette smoke for 6 mo 27.
Figure 7.

Alveolar histology in transgenic mice. Transgene-negative, Tg (−), and dual transgene-positive, Tg (+), mice at 1 mo of age were randomized to normal water, dox (−), or dox water, dox (+), for 3 mo. The lungs were then removed, fixed to pressure, sectioned, stained (hematoxylin and eosin), and photographed (original magnification: ×25). Mild alveolar enlargement is seen in the transgene-positive animals on normal water and further alveolar enlargement is seen in transgene-positive animals receiving dox. These photomicrographs are representative of the results obtained in a minimum of 10 experiments evaluating the alveolar histology of transgene-positive and transgene-negative mice on normal water and dox water.
Figure 8.

Morphometric parameters of alveolar size in CC10-rtTA-IFN-γ mice. Transgene-negative (Neg) and transgene-positive (Pos) mice were randomized to normal water (−Dox) or dox water (+Dox) at 1 mo of age and maintained on this regimen for 3 mo. Lungs were then removed, fixed to pressure, stained, and alveolar chord length was quantitated (*P < 0.01 versus all other conditions). The noted values represent the mean ± SEM of a minimum of five animals.
Enhanced lung volumes and increased pulmonary compliance are characteristic features of human emphysema 1. To determine if this murine model recapitulates these important clinical features of COPD, 1-mo-old transgene-negative and transgene-positive mice were randomized to normal water and dox water for up to 3 mo. Lung volumes were then assessed after pressure fixation. As can be seen in Table and Fig. 9, dox induction of IFN-γ caused a progressive increase in lung volume and an impressive increase in pulmonary compliance compared with dual transgene-positive animals on normal water or transgene-negative animals on normal or dox water. When viewed in combination, these studies demonstrate that IFN-γ causes emphysema with lung volume enlargement and enhanced pulmonary compliance analogous to that seen in human COPD.
Table 3.
Effects of IFN-γ on Lung Volume and Static Compliance
| Transgenestatus | Dox | Lung volume | Compliance |
|---|---|---|---|
| ml | ml/cm | ||
| − | − | 0.9 ± 0.001 | 0.036 ± 0.001 |
| − | + | 0.9 ± 0.04 | 0.036 ± 0.003 |
| + | − | 1.5 ± 0.13 | 0.06 ± 0.001 |
| + | + | 2.4 ± 0.04 | 0.096 ± 0.05 |
Figure 9.

Size of lungs from transgenic mice. 1-mo-old transgene-negative and transgene-positive mice were randomized to receive normal water (Dox −) or dox water (Dox +) for 3 mo. Their lungs were then removed, fixed to pressure, and photographed. The lungs pictured are representative of the results obtained in five separate experiments.
Effect of IFN-γ on MMPs.
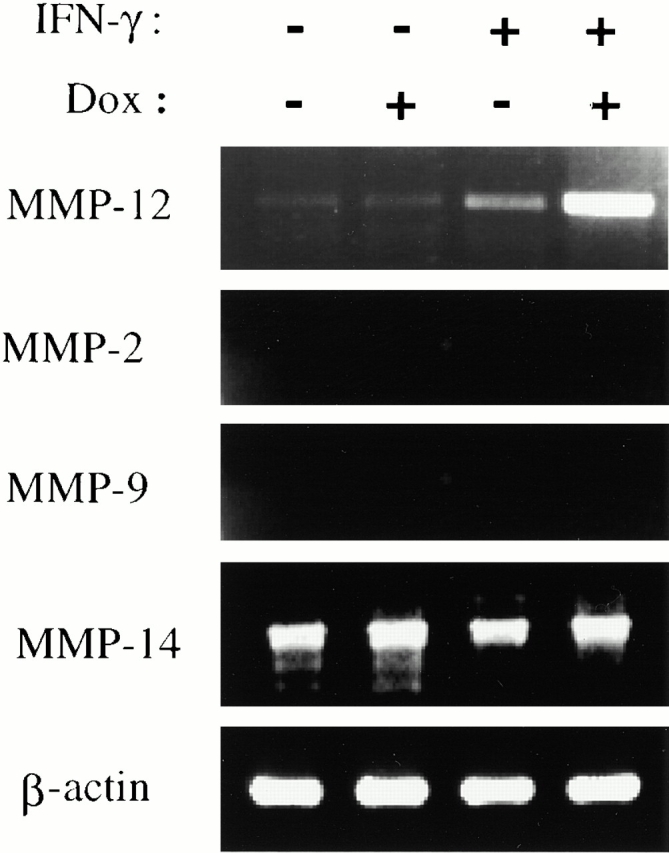
To begin to define the mechanism(s) of IFN-γ–induced alveolar septal destruction, we compared the levels of mRNA encoding a variety of COPD-relevant MMPs in transgene-negative and transgene-positive mice that had been randomized to receive normal water or dox water at 1 mo of age and maintained on this regimen for up to 12 wk. These studies demonstrated that IFN-γ is a potent stimulator of MMP-12 mRNA accumulation (Fig. 10). This effect could be appreciated after as little as 1 wk of dox administration and was readily apparent throughout the 3-mo induction interval (data now shown). MMP-2, MMP-9, and MMP-14 mRNA were not induced in these animals. Western blot analysis confirmed the induction of MMP-12 (Fig. 11). Zymography also demonstrated IFN-γ–induced BAL moieties that comigrated with the unprocessed and/or fully processed forms of MMP-12 (Fig. 11). Interestingly, BAL moieties that comigrated with MMP-9 moieties were also appreciated (Fig. 11). The activities in these bands were inhibited by EDTA, which establishes them as metalloproteinases. Thus, IFN-γ is a potent and selective inducer of MMP-12 and stimulates the release of MMP-9 in vivo.
Figure 10.
Effect of IFN-γ on MMP mRNA. 1-mo-old transgene-negative and dual transgene-positive CC10-rtTA-IFN-γ mice and their littermate controls were randomized to normal water (−) or dox water (+) for 1 mo. RT-PCR was used to compare the levels of mRNA encoding a variety of MMPs in lungs from these animals. The levels of mRNA encoding MMPs are compared with the levels of mRNA encoding β-actin.
Figure 11.
Effect of IFN-γ on MMP protein. 1-mo-old transgene (Tg)-negative and dual transgene-positive CC10-rtTA-IFN-γ mice and their littermate controls were randomized to normal water (−) or dox water (+) for 1 mo. Western blot analysis (A), casein zymography (B), and gelatin zymography (C) were used to demonstrate the presence of MMPs in BAL fluids from these animals.



IFN-γ Regulation of Cysteine Proteinases.
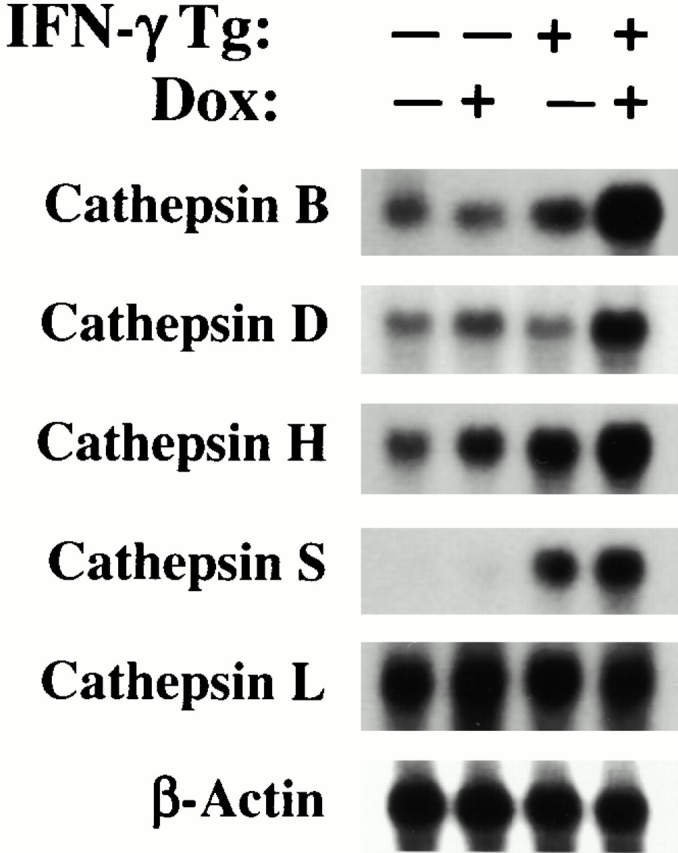
Studies were next undertaken to determine if cysteine proteinases were induced by IFN-γ in our transgenic system. This was done by comparing the levels of mRNA encoding a variety of airway-relevant cysteine proteinases in lungs from dual transgene-positive animals and transgene-negative littermate controls that had been randomized to normal water or dox water at 1 mo of age and maintained on this regimen for 4 wk. RT-PCR and Northern blot evaluations demonstrated that IFN-γ increased the levels of mRNA encoding cathepsins D, H, S, and B. Stimulation of mRNA encoding cathepsins K and L could not be appreciated (Fig. 12, and data not shown). Active site probe analysis, which identifies bioactive cathepsin moieties 29, also demonstrated increased levels of cathepsins H, S, B, and L in lung lysates and free cathepsin B and S in BAL fluids (Fig. 13, and data not shown). Thus, IFN-γ is a potent in vivo inducer of cysteine proteinases in the lung.
Figure 12.
Effect of IFN-γ on respiratory cathepsin mRNA. RT-PCR analysis was used to compare the levels of mRNA encoding the noted cathepsins in lung lysates from 2-mo-old transgene (Tg)-negative and transgene-positive CC10-rtTA-IFN-γ mice that had received normal water or dox water for the 4-wk interval before evaluation.
Figure 13.

Effect of IFN-γ on cathepsin proteins (B, H, S, and L). 1-mo-old transgene-negative and dual transgene-positive mice were randomized to normal water or dox water. 4 wk later, active site probe analysis was used to characterize the levels of cathepsins in lysates from the lungs from these animals.
IFN-γ Regulation of Antiproteases in the Lung.
To fully understand the protease/antiprotease balance in the lungs of IFN-γ transgenic mice, the levels of expression of lung-relevant antiproteases were also evaluated. Studies using RT-PCR and Northern blot analysis demonstrated that IFN-γ did not alter the expression of tissue inhibitor of metalloproteinase (TIMP)-1, TIMP-2, TIMP-3, or α1-AT. However, IFN-γ did significantly inhibit the expression of secretory leukocyte proteinase inhibitor (SLPI; Fig. 14, and data not shown).
Figure 14.
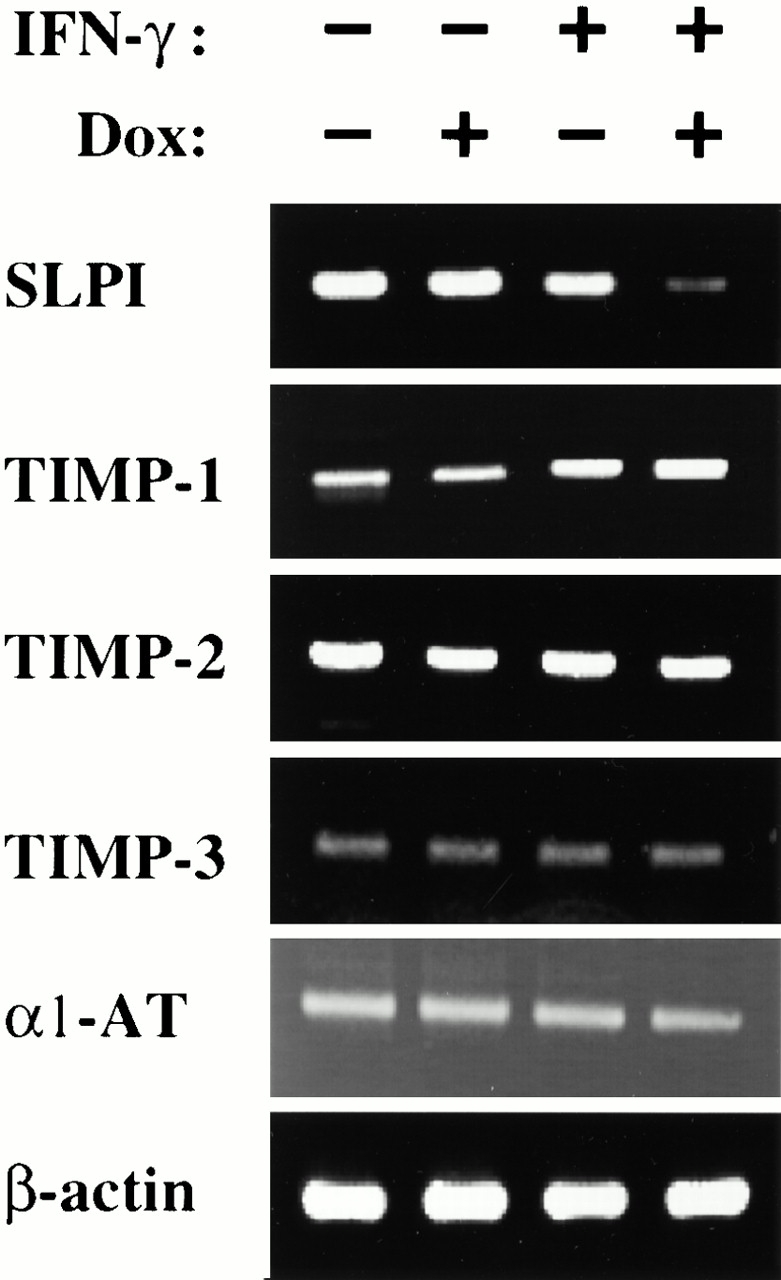
Effect of IFN-γ on respiratory antiproteases. RT-PCR was used to compare the levels of mRNA encoding respiratory-relevant antiproteases in 2-mo-old transgene-negative and transgene-positive CC10-rtTA-IFN-γ mice that received either normal water (−) or dox water (+) from 1–2 mo of age. The lack of effect of IFN-γ on TIMP-1, -2, and -3, and α1-AT, and the ability of IFN-γ to inhibit SLPI can be appreciated.
Discussion
Chronic inflammation and structural remodeling coexist and are believed to be linked in a cause and effect fashion in COPD tissues 7 12 13 14. Surprisingly, virtually nothing is known about the mechanism(s) by which COPD inflammation causes these structural alterations. We hypothesized that selected mediators of COPD tissue inflammation could directly induce pulmonary emphysema. To test this hypothesis, we used inducible overexpression transgenic modeling to target COPD-relevant inflammatory mediators to the adult murine lung. IFN-γ was chosen for these investigations. This choice was based on prior studies demonstrating that CD8+ lymphocyte infiltration is a prominent feature of the pathology of COPD, that lymphocytic number correlates with tissue destruction and airflow limitation in this disorder, and that CD8+ cells are potent producers of IFN-γ, and the belief that these cells elaborate type I cytokines 6 7 8 11 14 15. These studies demonstrate that the overexpression of IFN-γ causes a phenotype that mirrors human COPD with prominent emphysema, enlarged lungs, enhanced pulmonary compliance, and a mixed tissue and BAL inflammatory response containing increased numbers of macrophages, lymphocytes, and neutrophils. They also demonstrate that IFN-γ shifts pulmonary protease/antiprotease balance in a proteolytic direction via inducing MMP-12 and a variety of cathepsins while inhibiting SLPI. These are the first studies to demonstrate that IFN-γ, or any type I cytokine, can cause COPD-relevant pulmonary structural remodeling and tissue inflammation. They are also the first studies to define the in vivo, protease/antiprotease alterations induced by IFN-γ in the lung. When viewed in combination, these studies demonstrate that IFN-γ is an important regulator of pulmonary protease/antiprotease balance and suggest that IFN-γ, and/or the pathways it activates, may contribute to the pathogenesis of pulmonary emphysema.
The type of immune response that develops in response to a foreign antigen can be attributed, at least in part, to the heterogeneity of responding T cell populations 33. Type I and type II polarized inflammatory responses represent contrasting and, at times, mutually antagonistic, responses of an organism to immunogenic insult. The specialized cytokine profiles of type 1 and type 2 lymphocytes that mediate these responses (for a review, see reference 34) are major determinants of the outcomes of many immune responses including autoimmune, allergic, and infectious diseases. This is nicely illustrated in our current concept of asthma pathogenesis, which suggests that CD4+ Th2-dominated inflammation is the cornerstone of this disorder 34. In contrast to asthma, CD8+ cells are the major lymphocytes in tissues from patients with COPD with airflow obstruction 8 11. Until recently, CD8+ cells were considered a uniform class whose major function was MHC class I–restricted (cytotoxic) lysis of infected or altered host cells, at least in part, via the production of IFN-γ 11. It is now known that excessive or inappropriate activation of CD8+ cells can also induce pathologic changes in lung tissues 35 and that CD8+ cells have a broader cytokine profile than suspected previously 36. Our studies demonstrate that IFN-γ causes emphysema while inducing and activating a variety of pulmonary proteolytic pathways. This observation suggests that emphysema may be a Tc1/IFN-γ–mediated disorder. However, this speculation must be viewed with caution because sputum and tissue eosinophilia are also seen in some patients with this disorder 10 37. This raises the possibility that type 2 cytokine mediators may also contribute to disease pathogenesis in some individuals. IL-13 is an attractive candidate mediator of this response because it induces tissue eosinophilia 25 and causes emphysema when targeted to the adult murine lung 37a. Interestingly, IL-13 can be produced by Tc1 as well as Tc2 cells 38. Thus, COPD, in these individuals, could be a mixed Tc1/Tc2 disorder or a Tc1-dominant disease in which IL-13 is produced and tissue eosinophilia is generated by Tc1 cells. These observations and our studies allow us, for the first time, to begin to view the spectrum of clinical presentations of COPD from the perspective of the Tc1/Tc2 cytokine paradigm. However, additional experimentation will be required to define the applicability of this paradigm to this complex disorder(s).
The recruitment and activation of neutrophils in the lung has been considered for years to be a major contributor to the pathogenesis of emphysema 16 39. This is based on studies demonstrating neutrophil accumulation in BAL fluids and, to a lesser extent, tissues from patients with COPD 1 6 7 16, correlations between neutrophil number and airflow obstruction in severe COPD 7, and the potent tissue-destructive and mucus-enhancing effects of neutrophil-derived serine proteases 13 40 41. Macrophage-derived leukotriene (LT)B4 42, epithelial-derived IL-8 16 43, and cigarette-derived nicotine 44 have been postulated to contribute to the generation of neutrophilia in COPD. Repetitive endotoxin exposure has also been shown to produce pulmonary neutrophilia and emphysema in some animal modeling systems 45. Our studies demonstrate that the targeted pulmonary expression of IFN-γ increases BAL neutrophils to levels comparable to those seen in human COPD. Several laboratories have demonstrated that tissue neutrophilia can be induced in the lung via the passive transfer and activation of polarized type I cells 46 and that IFN-γ augments tissue responses to endotoxin 47. These findings suggest that CD8+ cell IFN-γ is responsible, at least in part, for the recruitment and activation of neutrophils in the lungs of cigarette smokers and that the structural events that ensue are mediated, in part, by the consequences of these cells in alveolar structures. They also raise the possibility that IFN-γ–induced exaggerated responses to ambient levels of environmental endotoxin may contribute to this response. These hypotheses link, for the first time, the lymphocyte and neutrophil abnormalities in COPD.
To gain insight into the mechanisms of IFN-γ–induced pulmonary proteolysis, we determined if IFN-γ altered the levels of expression of lung-relevant MMPs. These studies demonstrate, for the first time, that IFN-γ is a potent and selective stimulator of MMP-12 in vivo. This observation compliments prior studies from our laboratories that demonstrated that MMP-12 is essential for cigarette smoke–induced inflammation and emphysema in the murine lung 27 and is expressed in an exaggerated fashion in alveolar macrophages from smokers and patients with emphysema 48. Our studies also demonstrate, for the first time, that IFN-γ stimulates BAL MMP-9 protein accumulation and that these protein alterations are not associated with simultaneous increases in MMP-9 mRNA. The dissociation between the effects of IFN-γ on mRNA and protein suggests that IFN-γ increases MMP-9 elaboration by acting on cells with pools of preformed enzyme such as the neutrophil 49. In accord with these observations, exaggerated levels of MMP-9 have been found in BAL fluids from patients with COPD and the neutrophil has been shown to be the major source of this enzyme in this setting 50. When viewed in combination, these observations suggest that cigarette smoke may induce MMP-12 and stimulate MMP-9 elaboration via the induction of IFN-γ and that the ability of these MMPs to degrade elastin, basement membrane components (fibronectin, laminin, entactin, vitronectin, and type IV collagen), and nonbasement membrane components (converting plasminogen to angiostatin and activating latent TNF [17, 51]) contributes to the pathogenesis of IFN-γ–mediated lesions. However, it is important to point out that induction and exaggerated expression of the MMP-2–MMP-14–TIMP-2 complex and mucus metaplasia are also seen in human COPD tissues 1 52 but not in our IFN-γ–treated murine lungs. These observations suggest that IFN-γ–independent mechanisms also exist and contribute to the pathogenesis of COPD.
Although cysteine proteases play a major role in lysosomal protein degradation, they also participate in extracellular biologic responses 53. Several investigators have suggested that cysteine proteinases play important roles in the pathogenesis of emphysema 12 53 54 55. This includes studies that demonstrate increased levels of cathepsin L protein, mRNA, and bioactivity in BAL cells and fluids from smokers versus nonsmokers 54 55. However, the pathogenetic importance of individual cathepsin moieties in animal models of COPD have not been established, their roles in human COPD have not been defined, and the mechanisms of cysteine proteinase induction in COPD have not been elucidated. Our studies demonstrate, for the first time, that IFN-γ is a potent in vivo stimulator of the production and activation of a variety of pulmonary cathepsins including cathepsins D, H, S, B, and L. The elastolytic, collagenolytic, and gelatin degrading properties of these enzymes make them tempting candidates for the proteolysis that is believed to underlie pulmonary emphysema. They also raise the possibility that IFN-γ, or the proteolytic pathways it activates, plays important roles in other pulmonary proteolytic responses. This could be particularly important in diseases characterized by type 1 cytokine–dominated tissue inflammation and tissue destruction or remodeling, including pulmonary infections such as tuberculosis and pulmonary inflammatory disorders such as advanced sarcoidosis.
To fully understand the protease/antiprotease alterations that lead to emphysema, we also characterized the levels of expression of a variety of important antiproteases in the lungs of IFN-γ–overexpressing animals. Significant alterations in TIMP-1, -2, and -3 and α1-AT were not noted. Interestingly, significant inhibition of SLPI was readily appreciated. SLPI is a 12-kD protein produced at epithelial surfaces. It is a potent inhibitor of elastase and serine proteinases, modulates tissue inflammation, and functions as a defensin-like antibiotic 56. It is thus reasonable to speculate that IFN-γ inhibition of pulmonary SLPI can contribute to the proteolysis, chronic inflammation, and recurrent infections that are seen in patients with COPD.
Our studies demonstrate that the transgenic overexpression of IFN-γ in the adult murine lung causes a phenotype compatible with human emphysema. However, it is important to point out that exaggerated IFN-γ production and/or CD 8+ cell accumulation are also noted in a variety of other pulmonary disorders, including sarcoidosis, tuberculosis, obliterative bronchiolitis, allograft rejection, pulmonary fibrosis, and HIV and other viral infections 57 58 59 60 61 62 63 64 65. In accord with our hypothesis that chronic IFN-γ production activates pulmonary proteolytic pathways, emphysema is seen in patients with HIV infection 64 and pulmonary cavitation is seen in patients with advanced tuberculosis and sarcoidosis. The lack of emphysema or overt pulmonary destruction in the other disorders may be due to differences in the site(s), timing, and/or magnitude of IFN-γ production in the different disorders. Alternatively, it is well known that cytokine effector functions are context dependent with the same cytokine having different effects in different organs and different effects in the same organ, depending on the other cells and or mediators that are present in the local microenvironment. In addition, it is increasingly appreciated that host genetic background and polymorphisms in crucial disease-modifying genes also regulate the phenotypic features that are induced by inflammation-regulating mediators. Thus, as yet unidentified features of the responses in these disorders may also serve to exaggerate or inhibit IFN-γ–induced pulmonary proteolysis. Additional investigation will be required to define the conditions under which IFN-γ induces emphysema and pulmonary proteolysis and the responses that augment and/or ameliorate these responses.
In summary, these studies demonstrate that the targeted expression of IFN-γ in the adult lung causes emphysema and a macrophage-, lymphocyte-, and neutrophil-rich response that mirrors, in many ways, the lesions seen in human COPD. They also demonstrate that this emphysematous response is associated with the induction and activation of MMP-12 and cathepsins B, H, S, D, and L, the enhanced elaboration of MMP-9, and the inhibition of SLPI. They suggest that IFN-γ may play an important role in the pathogenesis of pulmonary emphysema and provide the rationale for investigations of the role(s) of IFN-γ in the pathogenesis of human COPD.
Acknowledgments
The authors thank Kathleen Bertier for excellent secretarial and administrative assistance.
This work was supported by National Institutes of Health grants HL56389, HL61904, and HL64242 (J.A. Elias), National Institutes of Health grant K08-AI01555 and a research grant from the American Lung Association (R. Riese), and National Institutes of Health grants HL50472 (S. Shapiro) and HL48261 (H. Chapman).
Footnotes
Abbreviations used in this paper: α1-AT, α1 antitrypsin; BAL, bronchoalveolar lavage; CC10, clara cell 10-kD protein; COPD, chronic obstructive pulmonary disease; dox, doxycycline; hGH, human growth hormone; IP-10, IFN-γ–inducible protein 10; MMP, matrix metalloproteinase; Mig, monokine induced by IFN-γ; RT, reverse transcription; rtTA, reverse tetracycline transactivator; SLPI, secretory leukocyte proteinase inhibitor; Tc1, type 1 T cytotoxic; tet-O, tetracycline operator; TIMP, tissue inhibitor of metalloproteinase.
References
- Senior R.M., Shapiro S.D. Chronic obstructive pulmonary diseaseepidemiology, pathophysiology, and pathogenesis. In: Fishman A.P., Elias J.A., Fishman J.A., Grippi M.A., Kaiser L.R., Senior R.M., editors. Fishman's Pulmonary Diseases and Disorders. Vol. 1. McGraw-Hill Inc; New York: 1998. pp. 659–681. [Google Scholar]
- American Thoracic Society Definitions, epidemiology, pathophysiology, diagnosis, and staging. Am. J. Respir. Crit. Care Med. 1995;152:S78–S83. [Google Scholar]
- Jeffery P.K. Inflammation in chronic obstructive lung disease. Am. J. Respir. Crit. Care Med. 1999;160:53–54. [PubMed] [Google Scholar]
- Peto R., Chen Z.M., Boreham J. Tobaccothe growing epidemic. Nat. Med. 1999;5:15–17. doi: 10.1038/4691. [DOI] [PubMed] [Google Scholar]
- Gross C.P., Anderson G.F., Powe N.R. The relation between funding by the National Institutes of Health and the burden of disease. N. Engl. J. Med. 1999;340:1881–1887. doi: 10.1056/NEJM199906173402406. [DOI] [PubMed] [Google Scholar]
- Cosio M.G., Guerassimov A. Chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1999;160:S21–S25. doi: 10.1164/ajrccm.160.supplement_1.7. [DOI] [PubMed] [Google Scholar]
- Saetta M. Airway inflammation in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1999;160:517–520. doi: 10.1164/ajrccm.160.supplement_1.6. [DOI] [PubMed] [Google Scholar]
- Saetta M., Baraldo S., Corbino L., Turato G., Braccioni F., Rea F., Cavallesco G., Tropeano G., Mapp C.E., Maestrelli P. CD8+ve cells in the lungs of smokers with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1999;160:711–717. doi: 10.1164/ajrccm.160.2.9812020. [DOI] [PubMed] [Google Scholar]
- Saetta M., Di Stefano A., Maestrelli P., Ferraresso A., Drigo R., Potena A., Ciaccia A., Fabbri L.M. Activated T-lymphocytes and macrophages in bronchial mucosa of subjects with chronic bronchitis. Am. Rev. Respir. Dis. 1993;147:301–306. doi: 10.1164/ajrccm/147.2.301. [DOI] [PubMed] [Google Scholar]
- Saetta M., Di Stefano A., Maestrelli P., Turato G., Ruggieri M.P., Roggeri A., Calcagni P., Mapp C.E., Ciaccia A., Fabbri L.M. Airway eosinophilia in chronic bronchitis during exacerbations. Am. J. Respir. Crit. Care Med. 1994;150:1646–1652. doi: 10.1164/ajrccm.150.6.7952628. [DOI] [PubMed] [Google Scholar]
- O'Shaughnessy T.C., Ansari T.W., Barnes N.C., Jeffrey P.K. Inflammation in bronchial biopsies of subjects with chronic bronchitisinverse relationship of CD8+ T lymphocytes with FEV1. Am. J. Respir. Crit. Care Med. 1997;155:852–857. doi: 10.1164/ajrccm.155.3.9117016. [DOI] [PubMed] [Google Scholar]
- Senior R.M. Mechanisms of COPDconference summary Chest 117Suppl. 12000. 320S 323S 10843968 [Google Scholar]
- Jeffrey P.K. Comparison of the structural and inflammatory features of COPD and asthma Chest. 117Suppl. 12000. 251S 260S [DOI] [PubMed] [Google Scholar]
- Finkelstein R., Fraser R.S., Ghezzo H., Cosio M.G. Alveolar inflammation and its relation to emphysema in smokers. Am. J. Respir. Crit. Care Med. 1995;152:1666–1672. doi: 10.1164/ajrccm.152.5.7582312. [DOI] [PubMed] [Google Scholar]
- Boushey H.A. Glucocorticoid therapy for chronic obstructive pulmonary disease. N. Engl. J. Med. 1999;340:1990–1991. doi: 10.1056/NEJM199906243402509. [DOI] [PubMed] [Google Scholar]
- Stockley R.A. Neutrophils and protease/antiprotease imbalance. Am. J. Respir. Crit. Care Med. 1999;160:S49–S52. doi: 10.1164/ajrccm.160.supplement_1.13. [DOI] [PubMed] [Google Scholar]
- Shapiro S.D. The macrophage in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1999;160:S29–S32. doi: 10.1164/ajrccm.160.supplement_1.9. [DOI] [PubMed] [Google Scholar]
- Wright J.L., Churg A. Smoke-induced emphysema in guinea pigs is associated with morphometric evidence of collagen breakdown and repair. Am. J. Physiol. 1995;268:L17–L20. doi: 10.1152/ajplung.1995.268.1.L17. [DOI] [PubMed] [Google Scholar]
- MacNee W., Rahman I. Oxidants and antioxidants as therapeutic targets in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1999;160:S58–S65. doi: 10.1164/ajrccm.160.supplement_1.15. [DOI] [PubMed] [Google Scholar]
- Burri P.H. Structural aspects of prenatal and postnatal development and growth of the lung. In: McDonald J.A., editor. Lung Growth and Development. Vol. 100. Marcel Dekker, Inc.; New York: 1997. pp. 1–35. [Google Scholar]
- Snider G.L., Lucey E.C., Stone P.J. Pitfalls in antiprotease therapy of emphysema. Am. J. Respir. Crit. Care Med. 1994;150:S131–S137. doi: 10.1164/ajrccm/150.6_Pt_2.S131. [DOI] [PubMed] [Google Scholar]
- Ray P., Tang W., Wang P., Homer R., Kuhn C.I., Flavell R.A., Elias J.A. Regulated overexpression of interleukin-11 in the lunguse to dissociate development-dependent and -independent phenotypes. J. Clin. Invest. 1997;100:2501–2511. doi: 10.1172/JCI119792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W., Geba G.P., Zheng T., Ray P., Homer R., Kuhn C., Favell R.A., Elias J.A. Targeted expression of IL-11 in the murine airway causes airways obstruction, bronchial remodeling and lymphocytic inflammation. J. Clin. Invest. 1996;98:2845–2853. doi: 10.1172/JCI119113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L., Dey C.R., Wert S.E., Whitsett J.A. Arrested lung morphogenesis in transgenic mice bearing an SP-C-TGF-β1 chimeric gene. Dev. Biol. 1996;175:227–238. doi: 10.1006/dbio.1996.0110. [DOI] [PubMed] [Google Scholar]
- Zhu Z., Homer R.J., Wang Z., Chen Q., Geba G.P., Wang J., Zhang Y., Elias J.A. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities and eotaxin production. J. Clin. Invest. 1999;103:779–788. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon H.J., Zhu Z., Gwaltney J.M., Jr., Elias J.A. Rhinovirus regulation of IL-1 receptor antagonist in vivo and in vitroa potential mechanism of disease resolution. J. Immunol. 1999;162:7461–7469. [PubMed] [Google Scholar]
- Hautamaki R.D., Kobayashi D.K., Senior R.M., Shapiro S.D. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277:2002–2004. doi: 10.1126/science.277.5334.2002. [DOI] [PubMed] [Google Scholar]
- Senior R.M., Griffin G.L., Fliszar C.J., Shapiro S.D., Goldberg G.I., Welgus H.G. Human 92- and 72-kilodalton type IV collagenases are elastases. J. Biol. Chem. 1991;266:7870–7875. [PubMed] [Google Scholar]
- Shi G.-P., Munger J.S., Meara J.P., Rich D.H., Chapman H.A. Molecular cloning and expression of human alveolar macrophage cathepsin S, an elastinolytic cysteine protease. J. Biol. Chem. 1992;267:7258–7562. [PubMed] [Google Scholar]
- Kuhn C.I., Homer R.J., Zhu Z., Ward N., Flavell R., Elias J.A. Airways hyperresponsiveness and airways obstruction in transgenic micemorphologic correlates in mice overexpressing IL-11 and IL-6. Am. J. Respir. Cell Mol. Biol. 2000;22:289–295. doi: 10.1165/ajrcmb.22.3.3690. [DOI] [PubMed] [Google Scholar]
- Lee J.J., McGarry M.P., Farmer S.C., Denzler K.L., Larson K.A., Carrigan P.E., Brenneise I.E., Horton M.A., Haczku A., Gelfand E.W. Interleukin-5 expression in the lung epithelium of transgenic mice leads to pulmonary changes pathognomonic of asthma. J. Exp. Med. 1997;185:2143–2156. doi: 10.1084/jem.185.12.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider G.L., Kleinerman J., Thurlbeck W.M., Bengali Z.H. The definition of emphysemareport of a National Heart, Lung, and Blood Institute, Division of Lung Diseases workshop. Am. Rev. Respir. Dis. 1985;132:182–185. doi: 10.1164/arrd.1985.132.1.182. [DOI] [PubMed] [Google Scholar]
- Hosken N.A., Shibuya K., Heath A.W., Murphy K.M., O'Garra A. The effect of antigen dose on CD4+ T helper cell phenotype development in a T cell receptor-αβ–transgenic model. J. Exp. Med. 1995;182:1579–1584. doi: 10.1084/jem.182.5.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A., Cohn L. Th2 cells and GATA-3 in asthmanew insights into the regulation of airway inflammation. J. Clin. Invest. 1999;104:1001–1006. doi: 10.1172/JCI8204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs D., Bangham C.R.M., McMichael A.J. Cell mediated cytotoxic responses to respiratory syncytial virus in infants with bronchiolitis. Lancet. 1987;2:769–771. doi: 10.1016/s0140-6736(87)92502-5. [DOI] [PubMed] [Google Scholar]
- Kemeny D.M., Vyas B., Vukmanovic-Stejic M., Thomas M.J., Noble A., Loh L.-C., O'Connor B.J. CD8+ T cell subsets and chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1999;160:S33–S37. doi: 10.1164/ajrccm.160.supplement_1.10. [DOI] [PubMed] [Google Scholar]
- Lams B.E., Sousa A.R., Rees P.J., Lee T.H. Immunopathology of the small-airway submucosa in smokers with and without chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1998;158:1518–1523. doi: 10.1164/ajrccm.158.5.9802121. [DOI] [PubMed] [Google Scholar]
- Zheng T., Zhu Z., Wong Z., Homer R.J., Ma B., Riese R.J., Jr., Chapman H.A., Jr., Shapiro S.D., Elias J.A. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J. Clin. Invest. 2000;106:1081–1093. doi: 10.1172/JCI10458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minty A., Asselin S., Bensussan A., Shire D., Vita N., Vyakarnam A., Wijdenes J., Ferrara P., Caput D. The related cytokines interleukin-13 and interleukin-4 are distinguished by differential production and differential effects on T lymphocytes. Eur. Cytokine Netw. 1997;8:203–213. [PubMed] [Google Scholar]
- Liou T.G., Campbell E.J. Quantum proteolysis resulting from release of single granules by human neutrophils. J. Immunol. 1996;157:2624–2631. [PubMed] [Google Scholar]
- Fischer B., Voynow J. Neutrophil elastase induces MUC5AC messenger RNA expression by an oxidant-dependent mechanism Chest. 117Suppl. 12000. 317S 320S [DOI] [PubMed] [Google Scholar]
- Stockley R.A. The role of proteinases in the pathogenesis of chronic bronchitis. Am. J. Respir. Crit. Care Med. 1994;150:S109–S113. doi: 10.1164/ajrccm/150.6_Pt_2.S109. [DOI] [PubMed] [Google Scholar]
- Hubbard R.C., Fells G., Gadek J., Pacholok S., Humes J., Crystal R.G. Neutrophil accumulation in the lung in alpha-1-antitrypsin deficiencyspontaneous release of leukotrine B4 by alveolar macrophages. J. Clin. Invest. 1991;88:891–897. doi: 10.1172/JCI115391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keatings V.M., Collins P.D., Scott D.M., Barnes P.J. Differences in interleukin-8 and tumor necrosis factor-α in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am. J. Respir. Crit. Care Med. 1996;153:530–534. doi: 10.1164/ajrccm.153.2.8564092. [DOI] [PubMed] [Google Scholar]
- Totti N., McCusker K.T., Campbell E.J., Griffin G.L., Senior R.M. Nicotine is chemotactic for neutrophils and enhances neutrophil responses to chemotactic peptides. Science. 1984;223:169–171. doi: 10.1126/science.6318317. [DOI] [PubMed] [Google Scholar]
- Stolk J, Rudolphus A., Davies P., Osinga D., Dijkman J.H., Agarwal L., Keenan K.P., Fletcher D., Kramps J.A. Induction of emphysema and bronchial mucus cell hyperplasia by intratracheal instillation of lipopolysaccharide in the hamster. J. Pathol. 1992;167:349–356. doi: 10.1002/path.1711670314. [DOI] [PubMed] [Google Scholar]
- Cohn L., Homer R.J., Marinov A., Rankin J., Bottomly K. Induction of airway mucus production by T helper 2 (Th2) cellsa critical role for interleukin 4 in cell recruitment but not mucus production. J. Exp. Med. 1997;186:1737–1747. doi: 10.1084/jem.186.10.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paludan S.R. Synergistic action of pro-inflammatory agentscellular and molecular aspects. J. Leukoc. Biol. 2000;67:18–25. doi: 10.1002/jlb.67.1.18. [DOI] [PubMed] [Google Scholar]
- Shapiro S.D. Animal models for chronic obstructive pulmonary diseaseage of klotho and marlboro mice. Am. J. Respir. Cell Mol. Biol. 2000;22:4–7. doi: 10.1165/ajrcmb.22.1.f173. [DOI] [PubMed] [Google Scholar]
- Graubert T., Johnston J., Berliner N. Cloning and expression of the cDNA encoding mouse neutrophil gelatinasedemonstration of coordinate secondary granule gene expression during terminal neutrophil maturation. Blood. 1993;82:3192–3197. [PubMed] [Google Scholar]
- Finlay G.A., Russell K.J., McMahon K.J., D'arcy E.M., Masterson J.M., FitzGerald M.X., O'Connor C.M. Elevated levels of matrix metalloproteinases in bronchoalveolar lavage fluid of emphysematous patients. Thorax. 1997;52:502–506. doi: 10.1136/thx.52.6.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler S.J., Cossins J., Lury J., Wells G. Matrix metalloelastase degrades matrix and myelin proteins and processes a tumor necrosis factor alpha fusion protein. Biochem. Biophys. Res. Commun. 1996;228:421–429. doi: 10.1006/bbrc.1996.1677. [DOI] [PubMed] [Google Scholar]
- Ohnishi K., Takagi M., Kurokawa Y., Satomi S., Konttinen Y.T. Matrix metalloproteinase-mediated extracellular matrix protein degradation in human pulmonary emphysema. Lab. Invest. 1998;78:1077–1087. [PubMed] [Google Scholar]
- Chapman H.A., Riese R.J., Shi G.-P. Emerging roles for cysteine proteases in human biology. Am. Rev. Physiol. 1997;59:63–88. doi: 10.1146/annurev.physiol.59.1.63. [DOI] [PubMed] [Google Scholar]
- Takeyabu K., Betsuyaku T., Nishimura M., Yoshioka A., Tanino M., Miyamoto K., Kawakami Y. Cysteine proteinases and cystatin C in bronchoalveolar lavage fluid from subjects with subclinical emphysema. Eur. Respir. J. 1998;12:1033–1039. doi: 10.1183/09031936.98.12051033. [DOI] [PubMed] [Google Scholar]
- Takahashi H., Ishidoh K., Muno D., Ohwada A., Nukiwa T., Kominami E., Kira S. Cathepsin L activity is increased in alveolar macrophages and bronchoalveolar lavage fluid of smokers. Am. Rev. Respir. Dis. 1993;147:1562–1568. doi: 10.1164/ajrccm/147.6_Pt_1.1562. [DOI] [PubMed] [Google Scholar]
- Zitnik R.J., Zhang J., Kashem M.A., Kohno T., Lyons D.E., Wright C.D., Rosen E., Goldberg I., Hayday A.C. The cloning and characterization of a murine secretory leukocyte protease inhibitor cDNA. Biochem. Biophys. Res. Commun. 1997;232:687–697. doi: 10.1006/bbrc.1997.6358. [DOI] [PubMed] [Google Scholar]
- Moller D.R., Forman J.D., Liu M.C., Noble P.W., Greenlee B.M., Vyas P., Holden D.A., Forrester J.M., Lazarus A., Wysocka M. Enhanced expression of IL-12 associated with Th1 cytokine profiles in active pulmonary sarcoidosis. J. Immunol. 1996;156:4952–4960. [PubMed] [Google Scholar]
- Neuringer I.P., Walsh S.P., Mannon R.B., Gabriel S., Aris R.M. Enhanced T cell cytokine gene expression in mouse airway obliterative bronchiolitis. Transplantation. 2000;69:399–405. doi: 10.1097/00007890-200002150-00016. [DOI] [PubMed] [Google Scholar]
- Davis G.S., Pfeiffer L.M., Hemenway D.R. Expansion of interferon-gamma-producing lung lymphocytes in mouse silicosis. Am. J. Respir. Cell Mol. Biol. 1999;20:813–824. doi: 10.1165/ajrcmb.20.4.3407. [DOI] [PubMed] [Google Scholar]
- Majumdar S., Li D., Ansari T., Pantelidis P., Black C.M., Gizycki M., duBois R.M., Jeffery P.K. Different cytokine profiles in cryptogenic fibrosing alveolitis and fibrosing alveolitis associated with systemic sclerosisa quantitative study of open lung biopsies. Eur. Respir. J. 1999;14:251–257. doi: 10.1034/j.1399-3003.1999.14b03.x. [DOI] [PubMed] [Google Scholar]
- Ross D.J., Moudgil A., Bagga A., Toyoda M., Marchevsky A.M., Kass R.M., Jordan S.C. Lung allograft dysfunction correlates with gamma-interferon gene expression in bronchoalveolar lavage. J. Heart Lung Transplant. 1999;18:627–638. doi: 10.1016/s1053-2498(99)00007-8. [DOI] [PubMed] [Google Scholar]
- Wang J., Wakeham J., Harkness R., Xing Z. Macrophages are a significant source of type 1 cytokines during mycobacterial infection. J. Clin. Invest. 1999;103:1023–1029. doi: 10.1172/JCI6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helene M., Lake-Bullock V., Zhu J., Hao H., Cohen D.A., Kaplan A.M. T cell independence of bleomycin-induced pulmonary fibrosis. J. Leukoc. Biol. 1999;65:187–195. doi: 10.1002/jlb.65.2.187. [DOI] [PubMed] [Google Scholar]
- Diaz P.T., King M.A., Pacht E.R., Wewers M.D., Gadek J.E., Nagaraja H.N., Drake J., Clanton T.L. Increased susceptibility to pulmonary emphysema among HIV-seropositive smokers. Ann. Intern. Med. 2000;132:369–372. doi: 10.7326/0003-4819-132-5-200003070-00006. [DOI] [PubMed] [Google Scholar]
- Podlech J., Holtappels R., Pahl-Seibert M.F., Steffens H.P., Reddehase M.J. Murine model of interstitial cytomegalovirus pneumonia in syngeneic bone marrow transplantationpersistence of protective pulmonary CD8-T-cell infiltrates after clearance of acute infection. J. Virol. 2000;74:7496–7507. doi: 10.1128/jvi.74.16.7496-7507.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]