

Abstract
Activation-induced cell death (AICD) is a mechanism of peripheral T cell tolerance that depends upon an interaction between Fas and Fas ligand (FasL). Although c-Jun NH2-terminal kinase (JNK) and p38 mitogen-activated protein kinase (MAPK) may be involved in apoptosis in various cell types, the mode of regulation of FasL expression during AICD in T cells by these two MAPKs is incompletely understood. To investigate the regulatory roles of these two MAPKs, we analyzed the kinetics of TCR-induced p38 MAPK and JNK activity and their regulation of FasL expression and AICD. We report that both JNK and p38 MAPK regulate AICD in T cells. Our data suggest a novel model of T cell AICD in which p38 MAPK acts early to initiate FasL expression and the Fas-mediated activation of caspases. Subsequently, caspases stimulate JNK to further upregulate FasL expression. Thus, p38 MAPK and downstream JNK converge to regulate FasL expression at different times after T cell receptor stimulation to elicit maximum AICD.
Keywords: Fas ligand, apoptosis, p38 MAPK, JNK, T cells
Introduction
Lymphocyte development is regulated not only by proliferation and differentiation but also by cell death or apoptosis 1. Lymphocyte apoptosis is believed to maintain homeostasis and self-tolerance in the immune system. For example, thymocytes that fail to rearrange their TCR gene will die by neglect, and those that recognize self-antigens will be eliminated by apoptosis, a process called negative selection. In peripheral T cells, a form of apoptosis induced by repeated TCR stimulation, known as activation-induced cell death (AICD), may be responsible for the peripheral deletion of autoreactive T cells 2 3. AICD results from the interaction between Fas and Fas ligand (FasL), and activated T cells expressing both Fas and FasL are killed either by themselves or by interacting with each other 4 5 6. Thus, a defect in AICD of effector T cells may result in the development of an autoimmune disease 7 8. In support of this notion, mutations in Fas or FasL result in a systemic lupus–like autoimmune disease in mice 9 and humans 10 11, illustrating a critical role for AICD in the maintenance of self-tolerance.
The signaling mechanisms of Fas-mediated cell death are not fully understood, but caspases play a crucial role in apoptotic processes 12 13 14. At least two distinct caspase activities are required for completion of the apoptotic pathway 15 16 17 18 19 20 21. One preferentially cleaves a YVAD tetrapeptide substrate, whereas the other preferentially cleaves a DEVD tetrapeptide substrate 22. In the simplest scheme, binding of FasL to Fas induces trimerization of the Fas receptor, and Fas-associated death domain (FADD) binds to the trimerized Fas cytoplasmic region through the interaction of the respective death domains. Procaspase-8 is then recruited to FADD through binding of the death effector domains. This is a death-inducing signaling complex in which the aggregated procaspase-8 transactivates 23. The active caspase-8 then acts to cleave and activate the downstream caspases that culminate in apoptosis 1 24 25. However, the mechanism by which the apoptotic process is executed is unclear.
Signal transduction by mitogen-activated protein kinases (MAPKs) plays a key role in a variety of cellular responses, including proliferation, differentiation, and cell death 26 27. Recently, two subfamily members of the MAPK superfamily, c-Jun NH2-terminal kinase (JNK) and p38 MAPK, have been implicated in several types of apoptosis 28 29 30 31 32 33. The role of JNK in apoptosis has been debated for several reasons 34 35 36 37 38. First, thymocytes from stress-activated protein kinase/extracellular signal–regulated kinase (an upstream activator of JNK) deficient mice are more susceptible to anti-CD3–induced apoptosis 37. Second, JNK and activator protein (AP)-1 do not mediate Fas-induced apoptosis in human Jurkat T cells 34. Third, activation of JNK by TNF occurs independent of TNF-induced apoptosis 38. Thus, the activation of JNK that ensues upon anti-Fas stimulation may occur subsequent to the activation of the caspase cascade, and a JNK cascade may be redundant or arise secondary to cellular damage associated with apoptosis 1 35 39 40 41.
Recent evidence indicates that JNK may commit T cells to stress-induced apoptosis through FasL expression 42 43. Most importantly, AICD in T cells is significantly impaired in JNK-1– and JNK-2–deficient mice 44 45, suggesting that JNK is actively involved in the regulation of AICD in primary T cells. However, the mechanism by which JNK regulates T cell AICD remains to be elucidated. Studies using Jurkat T cells suggest that Fas-dependent activation of p38 MAPK requires caspase-1–like family members in Jurkat T cells 46 and that MAP kinase kinase (MKK)3 or MKK6b but not p38 MAPK is involved in anti-Fas–induced apoptosis 46 47. Note that because Jurkat T cells constitutively express a high level of Fas, the mechanism of cell death induced by anti-Fas stimulation of these T cells may differ from that induced by FasL ligation. Notwithstanding, we found in pilot studies that other T cells that express low levels of Fas and FasL, including DO11.10 T hybridoma cells and primary naive splenic T cells, are relatively resistant to anti-Fas–induced T cell death. Moreover, we also showed that Jurkat T cells and primary T cells differ in their requirements for MAPK activation, and that p38 MAPK is a critical molecule involved in T cell activation in primary T cells 27. Inasmuch as the upregulation of FasL expression in primary T cells requires activation signals through the TCR 48 49, it is possible that p38 MAPK plays an important role in the regulation of FasL expression. Thus, the regulation of FasL expression during AICD by p38 MAPK and JNK is not well understood and merits further investigation.
In this study, we investigated the role of p38 MAPK and JNK in the control of FasL expression during TCR-induced AICD. We demonstrate that p38 MAPK plays a key role in the initiation of FasL expression and the Fas-mediated activation of caspases and JNK. Caspase inhibitors do not block the activation of p38 MAPK during AICD, and JNK activation requires caspase activity. Moreover, inhibition of p38 MAPK activity by SB203580 blocks the activation of caspases. Our observations suggest that (a) p38 MAPK is an upstream regulator of caspases and (b) JNK functions downstream of the caspases. Interestingly, FasL expression is downregulated by caspase inhibitors and a dominant negative (DN) JNK. These data favor a model of T cell AICD in which p38 MAPK and downstream JNK converge to regulate FasL expression at different times after TCR stimulation to elicit maximum T cell AICD.
Materials and Methods
Mice.
C57BL/6J (B6) mice were obtained from Taconic Farms Inc., and B6Smn.C3H-FasLgld (gld) mice were purchased from The Jackson Laboratory. The mice were bred in the Animal Care Facility at the University of Western Ontario (London, ON, Canada). All mice used in these experiments were 6–8 wk old.
Reagents and Cell Culture.
The following reagents were purchased from Santa Cruz Biotechnology: rabbit polyclonal antibodies against mouse extracellular signal–regulated kinase (ERK)1, p38 MAPK, caspase-1 (M20), glutathione–agarose, glutathione-S-transferase (GST)–c-Jun (1-79), and GST–ATF-2 (activating transcription factor 2; 1-505). YVAD–chloromethyl ketone, DEVD–fluoromethyl ketone (FMK), and zVAD–FMK were purchased from Cedarlane Labs. The 145-2C11 anti-CD3 and 37.51 anti-CD28 mAbs were purified by protein G affinity chromatography (Santa Cruz Biotechnology) of the supernatants of the B cell hybridomas supplied by Dr. J. Bluestone (University of Chicago, Chicago, IL) and Dr. J. Allison (University of California, Berkeley, CA), respectively. An anti-CPP32 (caspase-3) antiserum was a gift from Dr. R.-P. Sékaly (University of Montreal, Montreal, PQ, Canada). The p38 MAPK inhibitor 203580 and its inactive form, SKF106978, were provided by Dr. P. Young (SmithKline Beecham Pharmaceuticals, King of Prussia, PA). Recombinant human IL-2 (rIL-2) and myelin basic protein (MBP) were purchased from Sigma Chemical Co. Ac-YVAD–AMC (7-amino-4-methylcoumarin) and Ac-DEVD–AFC (7-amino-4-trifluoromethylcoumarin) were purchased from PharMingen. A Fas-Fc chimeric protein was obtained from Dr. D. Green (La Jolla Institute for Allergy and Immunology, San Diego, CA). The DO11.10 T cell hybridoma was obtained from Dr. B. Singh (University of Western Ontario, London, ON, Canada) and grown in RPMI 1640 medium supplemented with 10% heat-inactivated FCS, 10 mM Hepes, 0.1 mg/ml streptomycin, 100 U/ml penicillin, 0.05 mM 2-ME, and 2 mM glutamine (all purchased from GIBCO BRL). rIL-2 was purchased from Sigma Chemical Co.
Plasmids.
The DN p38 mutant (p38 [M], K57 to M) in pcDNA3 was supplied by Dr. J. Han (Scripps Research Institute, La Jolla, CA). The wild-type (WT) stress-activated protein kinase (SAPK)α and DN SAPKα constructs were gifts from Dr. E. Nishida (Kyoto University, Kyoto, Japan). The DN JNK (SAPKα–VPF) was constructed by replacement of the phosphorylation sites threonine and tyrosine with valine and phenylalanine, respectively. The WT or DN SAPKα coding region was inserted into the pSRα-HA1 vector as described 50. Hereafter, we will refer to the WT SAPKα or DN SAPKα constructs as WT–JNK-2 and DN–JNK-2, respectively. The human FasL promoter construct, which contains 511 bp 5′ upstream of the FasL gene cloned into the luciferase reporter construct pGL3, was a gift from Dr. J. Ashwell (National Cancer Institute, National Institutes of Health, Bethesda, MD). This 511-bp sequence contains the major regulatory element(s) for the FasL gene 51.
T Cell Isolation and Activation.
Splenic T cells from naive B6 mice were isolated (purity ≥98% as determined by FACS® analysis of CD3+ cell surface expression) on T cell enrichment columns (R & D Systems, Inc.). For in vitro activation, B6 T cells (2 × 106 per milliliter) were stimulated with plate-bound anti-CD3 (10 μg/ml) and anti-CD28 (5 μg/ml) mAbs in 24-well plates and then cultured for 48 h in complete RPMI 1640. Viable cells were recovered for restimulation.
Induction and Analysis of AICD.
DO11.10 T hybridoma cells (2 × 106 per milliliter) were cultured in triplicate in 24-well plates precoated with 10 μg/ml anti-CD3 for either 16 h or the times indicated (see Fig. 1 Fig. 2 Fig. 3 Fig. 4 Fig. 5). Alternatively, in vitro–activated B6 splenic T cells were either left unstimulated or were restimulated with plate-bound anti-CD3 for 16 h in the presence of rIL-2 (20 U/ml). Viability was assessed by addition of 10 μg/ml propidium iodide (PI) and immediate analysis using a FACScan™ (Becton Dickinson). Apoptotic cells were identified as those cells that stained positive for PI. Chromosomal DNA was isolated using a DNA purification kit (QIAGEN Inc.) according to the manufacturer's instructions, and T cell AICD was monitored by DNA fragmentation visualized after analysis of the samples on a 2% agarose gel in Tris-borate–EDTA.
Figure 1.
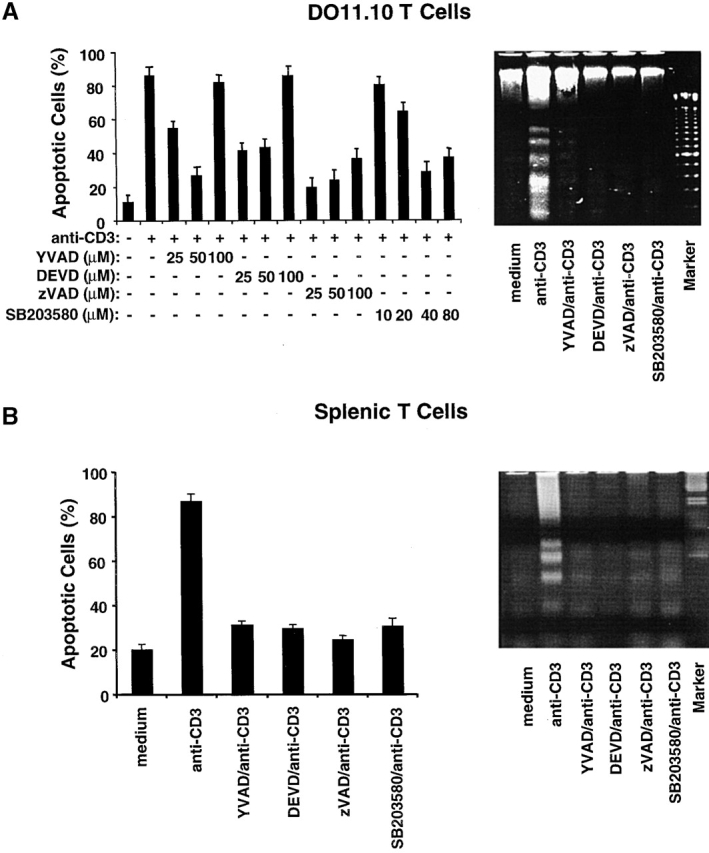
p38 MAPK is required for T cell AICD. (A) DO11.10 T hybridoma cells were pretreated with different concentrations of SB203580 or caspase inhibitors YVAD, DEVD, and zVAD and then stimulated with plate-bound anti-CD3 mAb (10 μg/ml) for 16 h. The number of dead cells was quantitated by PI staining, and the results are expressed as percent apoptotic cells (left panel). Data are shown as mean values ± SEM and are from one of two independent experiments. DO11.10 T hybridoma cells were pretreated and then stimulated as in A. Chromosomal DNA was extracted and analyzed on a 2% agarose gel to detect DNA fragmentation (right panel). (B) Purified splenic T cells from B6 mice were activated with plate-bound anti-CD3 and anti-CD28 mAbs for 48 h. Activated T cells were then restimulated in wells coated with anti-CD3 for 16 h in the presence of either the caspase inhibitors YVAD, DEVD, or zVAD or the p38 MAPK inhibitor SB203580. rIL-2 (20 U/ml) was added to unstimulated and restimulated cells. The percent apoptotic cells was determined by PI staining (left panel). Data are shown as mean values ± SEM and are from one of three independent and reproducible experiments. DNA fragmentation (right panel) was detected as in A. Data shown are from one of three independent and reproducible experiments.
Figure 2.
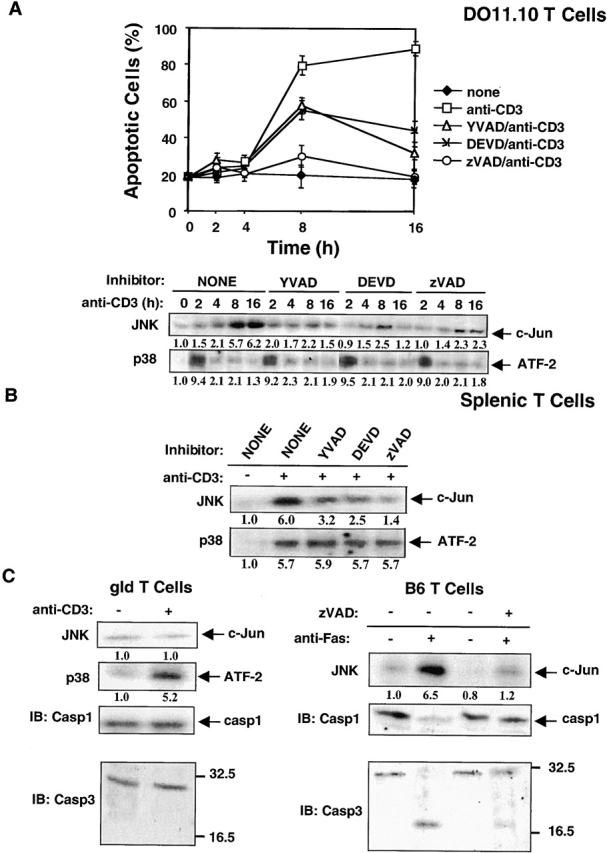
p38 MAPK and JNK are activated during AICD in DO11.10 T hybridoma cells and primary splenic T cells. (A) DO11.10 T cells were pretreated with the caspase inhibitors YVAD, DEVD, or zVAD and were then cultured in anti-CD3 (10 μg/ml)-coated plates for varying times. The percent of apoptotic cells was determined by PI staining as in Fig. 1. p38 MAPK was immunoprecipitated with an anti-p38 MAPK, and p38 MAPK activity in immunoprecipitates was measured using GST–ATF-2 as substrate. JNK activity was measured using a solid-phase JNK assay. Cell lysates were reacted for 4 h at 4°C with GST–c-Jun precoupled to glutathione–agarose beads, followed by an in vitro kinase reaction. Data are shown as mean values ± SEM and are from one of two independent and reproducible experiments. The relative activities of the p38 MAPK and JNK were quantitated by densitometric scanning and are shown below the respective gel lanes. (B) Splenic T cells from B6 mice were activated with plate-bound anti-CD3 and anti-CD28 mAbs for 48 h. Activated T cells were then restimulated in wells coated with anti-CD3 for 16 h in the presence of the caspase inhibitors YVAD, DEVD, or zVAD. For in vitro kinase assays of p38 MAPK and JNK, activated T cells were restimulated with plated-bound anti-CD3 for 2 h and 8 h, respectively, and the relative activities of p38 MAPK and JNK were assayed and quantitated as in A. (C) Splenic T cells from gld mice (left panel) were activated with plate-bound anti-CD3 and anti-CD28 mAbs for 48 h. Activated T cells were then restimulated in wells coated with anti-CD3 for 2 and 8 h. JNK and p38 MAPK activities were assayed as in A. Cell lysates were immunoblotted with anti–caspase-1 (Casp1) or anti-CPP32 (Casp3), respectively (left panel). Alternatively, B6 splenic T cells (right panel) were activated as in B, and activated T cells were then treated with plate-bound anti-Fas (5 μg/ml) for 8 h in the presence or absence of zVAD (50 μ/M). Activities of JNK and p38 MAPK and cleavage of caspase-1 and -3 were assayed as above. A and B, p29, line 4–5: in the presence of the caspase inhibitors YVAD, DEVD, and zVAD.
Figure 3.
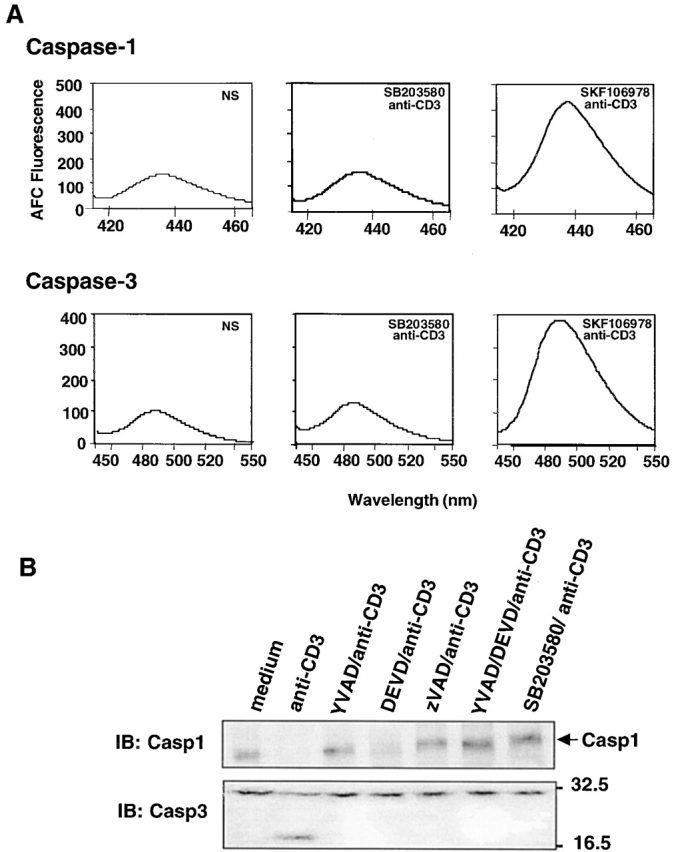
p38 MAPK regulates caspase activities. (A) DO11.10 T cells were pretreated for 1 h with 40 μM SB203580 or SKF106978 and then stimulated for 8 h with plate-bound anti-CD3. The activities of caspase-1 and caspase-3 were assayed using Ac-YVAD–AMC and Ac-DEVD–AFC as substrates. (B) DO11.10 T cells were pretreated with YVAD, DEVD, zVAD, and SB203580 for 1 h and then stimulated with plate-bound anti-CD3 for 8 h. The cells were lysed, and the cleavage of procaspase-1 and procaspase-3 were detected by immunoblotting with anti–caspase-1 (Casp1) and anti-CPP32 (Casp3) antibodies. Data shown are from one of three independent and reproducible experiments.
Figure 4.
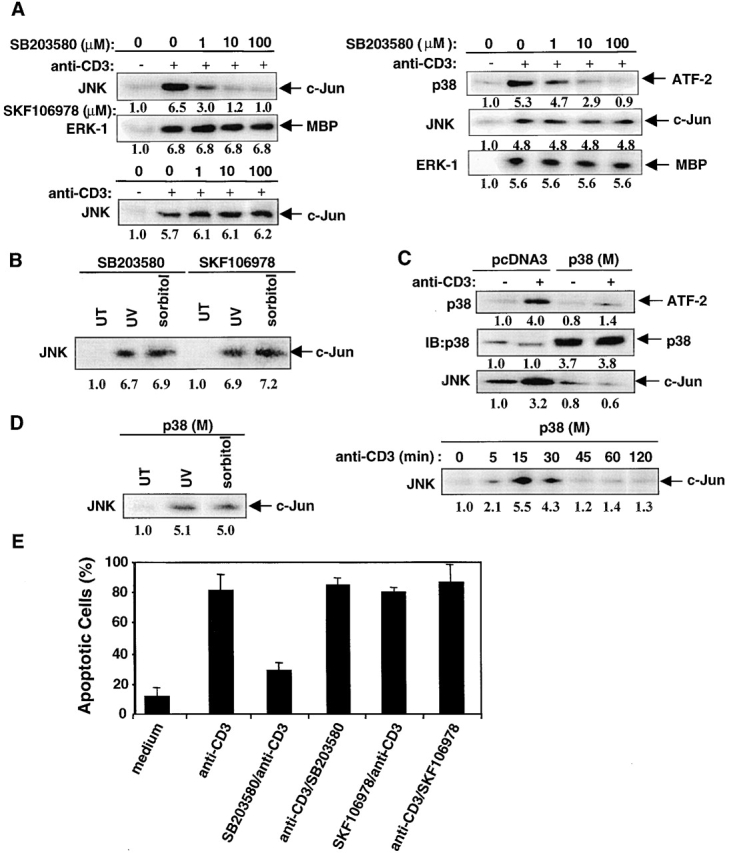
p38 MAPK is required for JNK activation during AICD. (A) Left panel, DO11.10 T cells were pretreated with SB203580 (0–100 μM) and then stimulated with plate-bound anti-CD3 for 8 h. JNK activity was assayed as in Fig. 2 A, and ERK1 activity was measured using MBP as substrate. Alternatively, JNK activity was assayed in DO11.10 T cells pretreated with SKF106978 (0–100 μM) and then stimulated with plate-bound anti-CD3 for 8 h. The relative activities of JNK and ERK1 were quantitated as in Fig. 2 A. Right panel, the activities of p38 MAPK, JNK, and ERK1 were assayed in DO11.10 T cells pretreated with SB203580 (0–100 μM) and then stimulated with anti-CD3 for 15 min. (B) DO11.10 T cells were pretreated with 40 μM of SB203580 or SKF106798 for 15 min and then treated with 0.4 mM sorbitol for 30 min. The cells were either left untreated (UT) or exposed to UV irradiation at 40 J/m2 and were then maintained for a further 30 min at 37°C. JNK activity in cell lysates was assayed and quantitated as in Fig. 2 A. (C) DO11.10 T cells were stably transfected with a control plasmid pcDNA3 or with pcDNA3 encoding p38 (M). DO11.10 transfectants were stimulated with plate-bound anti-CD3 for either 2 or 8 h for the p38 MAPK and JNK assays, respectively, which were performed and quantitated as in Fig. 2 A. The amounts of p38 MAPK proteins loaded were determined by anti-p38 MAPK immunoblotting. (D) p38 (M) DO11.10 transfectants were treated with UV irradiation or sorbitol as in B (left panel) or were stimulated with anti-CD3 for 0–120 min (right panel) and lysed. JNK activity was assayed and quantitated as in Fig. 2 A. (E) SB203580 or SKF106978, each at a concentration of 40 μM, was either added at 1 h before anti-CD3 stimulation or 4 h after anti-CD3 stimulation of DO11.10 T cells. The percent of apoptotic cells was scored by FACS® analysis of PI-stained cells. Data are shown as mean values ± SEM and are from one of three independent and reproducible experiments.
Figure 5.
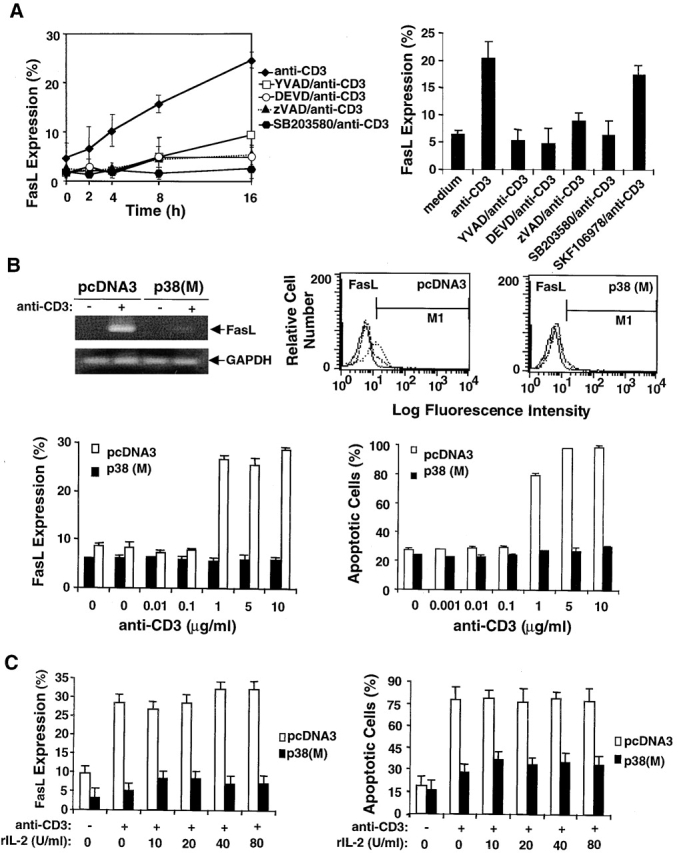
p38 MAPK regulates FasL expression. (A) Left panel, DO11.10 T cells were pretreated with SB203580 or caspase inhibitors YVAD, DEVD, and zVAD for 1 h at 37°C and then incubated with plate-bound anti-CD3 for varying times. At each time point, cells were harvested, stained with PE–anti-FasL mAb, and analyzed by flow cytometry. Data are shown as mean values ± SEM and are from one of three independent and reproducible experiments. Right panel, B6 splenic T cells were preactivated with plate-bound anti-CD3 and anti-CD28 for 48 h. Activated T cells were pretreated with either SB203580 or SKF106978 or caspase inhibitors YVAD, DEVD, and zVAD and then cultured in wells coated with anti-CD3. After 16 h, the cells were assayed for surface FasL expression as in A. Data are shown as mean values ± SEM and are from one of three independent and reproducible experiments. (B) DO11.10 T cells stably transfected with p38 (M) or control plasmid pcDNA3 were either left unstimulated or were stimulated with plate-bound anti-CD3 for 16 h. Cells were harvested, and RNA was prepared from all samples, reverse transcribed, amplified using primers specific for FasL and GAPDH, and electrophoresed in 1.5% agarose gels. Alternatively, cells were stained with PE–anti-FasL mAb or PI and analyzed by flow cytometry (center and right panels, respectively). The FasL expression data (fluorescence intensity) are shown as overlays (solid line, negative control; dashed line, unstimulated cells; and dotted line, stimulated cells) and are from one of three independent experiments. Alternatively, DO11.10 T cells stably transfected with p38 (M) or control plasmid pcDNA3 were either left unstimulated or stimulated with different concentrations of plate-bound anti-CD3 (0.001–10 μg/ml), and FasL expression and apoptosis were analyzed by flow cytometry. The data shown are from one of three independent and reproducible experiments. (C) p38 (M)- or pcDNA3-transfected DO11.10 T cells were cultured in wells coated with anti-CD3 in the presence of different concentrations of rIL-2 (10–80 U/ml) for 16 h. Percent FasL expression and apoptotic cells was quantitated as in B. Data shown are from one of three independent and reproducible experiments.
In Vitro Kinase Assay.
After stimulation, cells were lysed in ice-cold lysis buffer containing 1% Triton X-100, 10 mM Tris, pH 7.5, 150 mM NaCl, 2 mM EGTA, 50 mM β-glycerophosphate, 2 mM Na3VO4, 10 mM NaF, 1 mM dithiothreitol (DTT), 1 mM PMSF, 10 μg/ml leupeptin, and 10 μg/ml aprotinin. Lysates were clarified by centrifugation at 12,000 rpm for 10 min at 4°C, and their protein content was determined by the Bradford assay using BSA as a standard. Kinase assays for ERK1 and p38 MAPK and solid-phase JNK assays were performed essentially as described 27. Equal loading of precipitated proteins was confirmed by probing the blots with specific antibodies, and phosphorylation of the fusion protein or MBP bands was quantitated using a Molecular Imager System and Molecular Analyst imaging software (Bio-Rad Labs.).
Western Blotting.
Cell lysates were resolved on SDS-PAGE and transferred to nitrocellulose membranes (Hy-bond C Super; Amersham). Blots were blocked for 1 h at 23°C in PBS containing 2% BSA and 0.05% Tween-20. Membranes were incubated overnight with rabbit antisera to human CPP32 (caspase-3), caspase-1, or other rabbit-specific antibodies and were then washed three times in PBS containing 0.05% Tween-20. Caspase-1 and caspase-3 were detected using horseradish peroxidase–conjugated goat anti–rabbit IgG (Santa Cruz Biotechnology). After three washes in PBS containing 0.05% Tween-20, signals were revealed by ECL Western blotting (Amersham) and visualized by autoradiography.
Cell Transfection.
DO11.10 T cells (5 × 105 per milliliter) were transfected with LipofectAMINE PLUS (GIBCO BRL) as recommended by the manufacturer. Exponentially growing cells were washed twice with PBS and resuspended in Opti-MEM (GIBCO BRL). The plasmids (5 μg/ml) were preincubated with PLUS reagent for 15 min at 23°C, and the precomplexed plasmids were then incubated for 5 h at 37°C with a mixture of 1 μg/ml pCMV vector expressing green fluorescence protein (GFP) and 6 μl of LipofectAMINE in 1.5 ml of Opti-MEM. Cells were washed twice with complete RPMI 1640, incubated for 36 h, and then selected in 600 μg/ml G418 for 3 wk. Transfection efficiencies were assessed by FACScan™ analyses of GFP expression and in vitro kinase assays of enzyme activity.
Luciferase Assay.
p38 (M) or pcDNA3 DO11.10 stable transfectants (5 × 105 cells per milliliter) were transfected with 1 μg of a FasL promoter–reporter construct. Cells were grown in complete RPMI 1640 medium for 48 h at 37°C. The cells were either left unstimulated or were stimulated with plate-bound anti-CD3 for 16 h, washed, and then lysed in luciferase buffer. Luciferase activity was monitored using a luciferase reporter gene assay kit (Roche Labs.), and luminescence was detected in a scintillation counter (Beckman Instruments). Transfection efficiency was monitored by cotransfection of a β-galactosidase–encoding plasmid (pUC19) and measurement of the relative β-galactosidase activity.
Assay of Caspase Activity.
DO11.10 T cells (2 × 106 per milliliter) were treated with plate-bound anti-CD3 for 8 h, washed three times with PBS, and lysed in a buffer containing 10 mM Tris, pH 7.5, 130 mM NaCl, 1% Triton X-100, 10 mM NaPi, and 10 mM NaPPi. Cell lysates were centrifuged at 13,000 g, and the cleared supernatants were collected for protease assays. The lysates (50 μg) and 10 μg Ac-YVAD–AMC or Ac-DEVD–AFC were added to 1 ml protease assay buffer containing 20 mM Hepes, pH 7.5, 10% glycerol, and 10 mM DTT for the caspase-1 assay or 20 mM Pipes, pH 7.2, 100 mM NaCl, 10 mM DTT, 1 mM EDTA, 0.1% (wt/vol) Chaps, and 10% sucrose for the caspase-3 assay. The mixtures were incubated for 1 or 2 h at 37°C for the caspase-1 and caspase-3 assays, respectively. Caspase (protease) activities were determined by monitoring either the release of AMC at an excitation wavelength of 380 nm and an emission wavelength of 430–460 nm or the release of AFC at an excitation of 400 nm and an emission wavelength of 480–520 nm, using a spectrofluorometer (RF-M2004; Photon Technology International).
FasL Expression.
DO11.10 T cells or preactivated B6 splenic T cells were restimulated as indicated (see Fig. 2 and Fig. 5), washed twice with PBS containing 1% BSA and 0.05% sodium azide, and then incubated with anti-mFasL–PE antibodies (PharMingen) for 45 min at 4°C. After rinsing twice with PBS, the cells were fixed in PBS/1% paraformaldehyde and analyzed for FasL staining with a FACScan™. An isotype-matched control antibody was used to evaluate background staining.
FasL mRNA expression was detected using a semiquantitative PCR assay. Total RNA was purified from tissues by a guanidinium isothyocyanate/silica gel–based membrane RNeasy method (QIAGEN Inc.) according to the manufacturer's instructions. The resulting RNA (1 μg) was reversed transcribed with Superscript II (GIBCO BRL) and oligo (dT) primers (GIBCO BRL) as per the manufacturer's recommendations. PCR reaction conditions were as follows. Equivalent amounts of cDNA were added in addition to the following components: 20 mM Tris-Cl, pH 8.0, 50 mM KCl, 0.75 mM MgCl2, 0.2 mM of each dNTP, 0.5 μM of each primer (sense primer, 5′-CAGCTCTTCCACCTGCAGAAGG-3′; antisense primer, 5′-AGATTCCTCAAAATTGATCAGAGAGAG-3′), and 2.5 U of Taq DNA polymerase (GIBCO BRL). Templates were denatured at 95°C for 3 min, primer annealing was carried out at 54°C for 1 min, and extension was performed at 72°C for 1 min. The cycle was repeated 34 times, followed by a final extension at 72°C for 7 min. The number of amplification cycles chosen for each reaction were determined to be within the linear range of the assay. To verify that equivalent amounts of RNA were added to each FasL PCR reaction, amplification of GAPDH housekeeping gene was performed. Standardization of cDNA was determined by PCR amplification of GAPDH (sense primer, 5′-CCATGGAGAAGGCTGGGG-3′; antisense primer, 5′-CAAAGTTGTCATGGATGACC-3′) in each sample. Amplification conditions were as follows: 95°C for 2 min, 94°C for 40 s, 62°C for 40 s, and 72°C for 1 min, repeated for 25 cycles. PCR amplification products were separated on 1.5% agarose Tris-borate–EDTA gels and stained with ethidium bromide, and product band intensities were quantified by Gel Doc (Bio-Rad Labs.).
Results
p38 MAPK Is Required for AICD in T Cells.
To determine whether p38 MAPK mediates TCR stimulation–induced AICD in T cells, we analyzed the effect of the p38 MAPK–specific inhibitor SB203580, a pyridinyl imidazole compound 52, on AICD in murine DO11.10 T hybridoma cells. Several caspase inhibitors, i.e., YVAD for caspase-1, DEVD for caspase-3, and zVAD for pan-caspases, were used as positive controls. T cell AICD as assayed by PI staining was optimally inhibited by YVAD and DEVD at a concentration of 50 μM (Fig. 1 A, left panel). A higher dose (100 μM) of YVAD and DEVD proved toxic to the T cells, as this concentration of inhibitors did not block AICD. The pan-caspase inhibitor zVAD inhibited AICD more effectively than either YVAD or DEVD, suggesting that many caspases are involved in AICD. Interestingly, although 10 μM SB203580 did not inhibit AICD, increasing the concentration of SB203580 to 40 μM did inhibit AICD efficiently. Consistent with this observation, DNA fragmentation induced by TCR ligation in DO11.10 T cells was blocked not only by caspase inhibitors but also by SB203580 (Fig. 1 A, right panel). To test whether this is also the case in primary T cells, B6 splenic T cells were preactivated with plate-bound anti-CD3 and anti-CD28 mAbs for 48 h and then restimulated with plate-bound anti-CD3 for 16 h in the presence of SB203580 or caspase inhibitors. About 20% of B6 T cells displayed spontaneous apoptosis in the absence of any stimulation or inhibitors, and TCR-induced AICD in B6 T cells was significantly inhibited by both SB203580 and the caspase inhibitors (Fig. 1 B). These data imply a role for p38 MAPK in T cell AICD.
To further investigate how p38 MAPK mediates T cell AICD as well as the relationship between p38 MAPK, caspases, and JNK in this process, the kinetics of induction of AICD and activation of p38 MAPK and JNK induced by TCR stimulation were analyzed in the presence or absence of caspase inhibitors. Whereas only ∼20–30% apoptosis was evident at 2 h after stimulation, this level increased to 70–80% apoptosis by 8 h and to 90% apoptosis by 16 h (Fig. 2 A, top panel). All three caspase inhibitors suppressed apoptosis, but again zVAD was the most effective inhibitor. Most interestingly, JNK activation was detectable by 2 h (1.5-fold increase) but was maximal at 8 and 16 h (about a sixfold increase) after stimulation (Fig. 2 A, bottom panel). The kinetics of JNK activation correlated closely with the kinetics of anti-CD3–induced apoptosis of DO11.10 T cells. Activation of JNK during T cell AICD was inhibited by the three caspase inhibitors, indicating that the activities of many caspases are required to activate JNK. In contrast, the peak of p38 MAPK activation (seven- to eightfold increase) occurred at 2 h after stimulation, i.e., ∼6 h before maximal JNK activation. The activity of p38 MAPK returned to a basal level by 8–16 h (Fig. 2 A, bottom panel). Note that p38 MAPK activation was not inhibited by any of the caspase inhibitors used. Interestingly, although the caspase inhibitors did not reduce p38 MAPK activity in the stimulated B6 T cells, JNK activity was diminished appreciably (two- to fourfold) after treatment with each of these inhibitors (Fig. 2 B).
Next, we further analyzed the role of caspases in JNK activation during AICD using two approaches. First, we reasoned that if JNK activity is dependent on the activation of caspases initiated by Fas–FasL interaction, JNK activity may be very weak or absent in FasL-deficient gld T cells stimulated with anti-CD3. Splenic T cells from gld mice were preactivated for 48 h with plate-bound anti-CD3 and anti-CD28 and restimulated for 2 or 8 h with plate-bound anti-CD3, and their p38 MAPK and JNK activities were determined by in vitro kinase assays. Indeed, JNK activity was not induced, whereas p38 MAPK was activated significantly (fivefold) by CD3 ligation in gld T cells (Fig. 2 C, left panel). Thus, JNK activation induced by Fas–FasL interaction is impaired in gld T cells, whereas TCR-mediated p38 MAPK activation remains intact. Second, we examined whether the cross-linking of Fas activates caspase-1, caspase-3, and JNK in preactivated B6 splenic T cells. Activation of procaspase-1 and procaspase-3 was determined by their ability to be cleaved into their respective mature caspases after anti-Fas stimulation. Cross-linking Fas with plate-bound anti-Fas antibody resulted in the activation of caspase-1, caspase-3, and JNK (six- to sevenfold) in B6 T cells (Fig. 2 C, right panel). Pretreatment of B6 T cells with zVAD completely inhibited JNK activation. In contrast, neither procaspase-1 nor procaspase-3 was cleaved in anti-CD3–restimulated gld T cells (Fig. 2 C, left panel). Consistent with this observation, no AICD was seen in anti-CD3–restimulated gld T cells (data not shown). These data clearly demonstrate that p38 MAPK is active upstream of caspases and JNK and that JNK activity lies downstream of caspases in primary T cells.
p38 MAPK Regulates the Activation of Caspases and JNK.
The data presented above raise the possibility that p38 MAPK is an upstream regulator of caspases and JNK. To test this possibility, DO11.10 T cells were pretreated with SB203580 for 1 h and then stimulated with plate-bound anti-CD3 mAb for 8 h. Caspase-1 and caspase-3 activities were assayed using specific fluorescent substrates. SB203580 pretreatment greatly reduced caspase-1 and caspase-3 activities, as evidenced by decreased emission peaks of their fluorescent substrates. (Fig. 3 A). Moreover, procaspase-1 was cleaved after CD3 ligation, and YVAD, zVAD, and SB203580 but not DEVD inhibited this cleavage (Fig. 3 B, top panel). Similarly, CD3 ligation of DO11.10 T cells induced the cleavage of procaspase-3, as revealed by the detection of a 17-kD band (Fig. 3 B, bottom panel). This cleavage of procaspase-3 was undetectable when the T cells were stimulated in the presence of either the caspase inhibitors or the p38MAPK inhibitor SB203580. The inhibition of procaspase-3 cleavage by YVAD but lack of inhibition of procaspase-1 cleavage by DEVD suggests that caspase-1 or a caspase-1–like member(s) lies upstream of caspase-3. Taken together, these data further indicate that p38 MAPK is an upstream regulator of caspases.
If p38 MAPK indeed regulates the activity of caspases as well as JNK, we would expect that the inhibition of p38 MAPK activity by SB203580 should also abrogate JNK activation during T cell AICD. To address this issue, in vitro kinase activities of JNK and ERK1 were assayed in lysates of DO11.10 T cells previously treated with SB203580 for 1 h and then stimulated with plate-bound anti-CD3 for 8 h at 37°C. SB203580 pretreatment inhibited JNK activity in a dose-dependent manner (Fig. 4 A, left panel). This inhibition was specific, as SB203580 did not block ERK1 activation, and SKF106978, an inactive inhibitor of p38 MAPK, did not interfere with JNK activation during AICD. Moreover, pretreatment of DO11.10 T cells with either SB203580 or SKF106978 did not affect JNK activation induced by either CD3 ligation for 15 min (Fig. 4 A, right panel) or in response to stress stimuli such as UV and sorbitol (Fig. 4 B). In additional control experiments, SB203580 inhibited p38 MAPK but not ERK1 activity in anti-CD3–stimulated DO11.10 T cells (Fig. 4 A, right panel).
To further exclude the possibility that JNK is inhibited nonspecifically by SB203580, DO11.10 T cells were stably transfected with the DN p38 MAPK (p38 [M]) mutant, and the activities of p38 MAPK and JNK were assayed during AICD. JNK activity was markedly suppressed by p38 (M) but not the control pcDNA3 vector (Fig. 4 C). Note that this suppression of JNK activity was not due to different amounts of p38 MAPK proteins loaded, as about a fourfold increase in the amounts of p38 MAPK was seen in p38 (M)-transfected cells compared with that in pcDNA3-transfected cells (Fig. 4 C, center panel). On the other hand, JNK was activated normally in response to stress stimuli (Fig. 4 D, left panel) or after a short stimulation (15–30 min) with anti-CD3 (Fig. 4 D, right panel). As the peak of p38 MAPK activation during T cell AICD occurs at 2 h after CD3 ligation, one would predict that the addition of SB203580 at ≥2 h after T cell stimulation should not affect the amount of T cell apoptosis observed. To test this possibility, SB203580 was added to DO11.10 T cells that were previously stimulated with anti-CD3 for 4 h. As expected, addition of SB203580 after TCR stimulation did not inhibit AICD, in contrast to what was observed when SB203580 was added to T cells before TCR stimulation. In control cultures, addition of SKF106978 either before or after TCR stimulation had no effect on AICD (Fig. 4 E). These data suggest that p38 MAPK may be responsible for initiating the activation of caspases and JNK and that p38 MAPK may be an important regulator of TCR-induced AICD in T cells.
p38 MAPK Regulates FasL Expression.
AICD is mediated by Fas–FasL interaction, and JNK has been proposed to regulate FasL expression in response to stress stimulation 42 43. Since we observed that p38 MAPK may regulate the activation of caspases and JNK, it was of interest to examine the effect of p38 MAPK on FasL expression during AICD in T cells. We found that the expression of FasL was upregulated in a time-dependent manner during TCR-stimulated AICD in DO11.10 T cells (Fig. 5 A). Notably, the kinetics of stimulated FasL expression closely paralleled that of AICD shown in Fig. 2 A. Moreover, a Fas-Fc chimeric protein that can block Fas–FasL interaction 42 significantly inhibited AICD in DO11.10 T cells (data not shown). These results confirm that TCR-induced T cell AICD is directly dependent on Fas–FasL interaction. Pretreatment of DO11.10 T cells with either caspase inhibitors or SB203580 reduced FasL expression about three- to fivefold and 10-fold, respectively (Fig. 5 A, left panel). Similar results were obtained with primary B6 splenic T cells (Fig. 5 A, right panel). Therefore, inhibition of p38 MAPK activity may suppress TCR-induced AICD by the inhibition of FasL expression and consequent disruption of Fas–FasL interaction.
This possibility was addressed by analyzing FasL expression and AICD induced by CD3 ligation in p38 (M) stably transfected DO11.10 T cells. In support of a role for p38 MAPK in the control of FasL expression and AICD in T cells, we found that the capacity of these p38 (M) DO11.10 transfectants to express FasL both at the level of mRNA level and at the cell surface was significantly impaired (Fig. 5 B, top). To substantiate these observations, we analyzed FasL expression and the amount of apoptosis in the pcDNA3 and p38 (M) transfectants in response to different doses of anti-CD3 (0.001–10 μg/ml). Maximum FasL expression and apoptosis occurred at 1–10 μg/ml of anti-CD3 stimulation in pcDNA3-transfected cells, and this response was completely abrogated in p38 (M)-transfected cells (Fig. 5 B, bottom).
It has been shown that IL-2 can upregulate FasL expression during AICD 53. To investigate whether p38 (M) blocks FasL expression by downregulating IL-2 production, rIL-2 was added to p38 (M)-transfected DO11.10 T cells before CD3 ligation in an attempt to restore FasL expression. Addition of rIL-2 over a wide concentration range had only a marginal effect on the stimulation of FasL expression and AICD in either the p38 (M) or pcDNA3 control vector transfectants (Fig. 5 C). The failure of rIL-2 to further increase FasL expression and AICD in the latter control transfectants is likely due to the high amount of IL-2 secreted by these cells (our unpublished observations). Thus, the p38 (M)-mediated suppression of FasL expression and AICD can not be ascribed simply to a lack of available IL-2, but rather may result from a direct effect of p38 MAPK on the regulation of FasL gene transcription. Evidence in support of this idea was obtained by demonstrating that FasL promoter transcriptional activity was significantly reduced after transfection of a FasL promoter–luciferase reporter gene construct into p38 (M) DO11.10 T cell stable transfectants (Fig. 6). In contrast, FasL promoter activity was enhanced upon TCR stimulation in DO11.10 cells transfected with the control pcDNA3 vector.
Figure 6.

p38 MAPK regulates FasL promoter activity. p38 (M) or pcDNA3 DO11.10 stable transfectants were transiently transfected with a FasL-511 luciferase construct, maintained in medium for 36 h at 37°C, and were then either left unstimulated or were stimulated with plate-bound anti-CD3 for 16 h. Luciferase activity in whole cell lysates was assayed and is shown as mean value ± SEM. Transfection efficiency was monitored by cotransfection of a pUC19 plasmid encoding β-galactosidase. Similar results were obtained in three independent and reproducible experiments.
JNK Activity Is Required for FasL Expression.
Earlier in this report, we showed that inhibition of caspase activation by YVAD, DEVD, or zVAD not only blocks T cell AICD (Fig. 1) but also downregulates FasL expression in T cells (Fig. 5 A). Since JNK functions downstream of caspases, we next determined whether a block in JNK activity also suppresses FasL expression and AICD. FasL expression and AICD were assayed in DO11.10 T cells stably transfected with DN–JNK-2 or WT–JNK-2. The activity of JNK but not p38 MAPK was selectively inhibited in the DN–JNK-2 DO11.10 transfectants (Fig. 7 A). Both TCR-induced FasL expression and AICD were inhibited virtually completely in DN–JNK-2 but not control WT–JNK-2 DO11.10 transfectants (Fig. 7 B). Thus, JNK also regulates FasL expression during AICD in T cells.
Figure 7.

JNK regulates FasL expression and AICD. (A) DO11.10 T cells transfected with WT–JNK-2 or DN–JNK-2 were cultured for 8 h in the absence or presence of plate-bound anti-CD3. Cell lysates were reacted with 10 μg of either GST–c-Jun (left panel) precoupled to glutathione–agarose beads for 4 h at 4°C or immunoprecipitated with anti-p38 MAPK (right panel) and were then assayed for JNK or p38 MAPK activity, respectively. (B) DO11.10 T cells stably transfected with WT–JNK-2 or DN–JNK-2 were either left unstimulated or were stimulated with different concentrations of plate-bound anti-CD3 (0.001–10 μg/ml) for 16 h. The cells were harvested, stained with PE–anti-FasL or PI, and analyzed for percent surface Fas expression and apoptotic cells by flow cytometry (solid line, negative control; dashed line, unstimulated cells; and dotted line, stimulated cells). Data are shown as mean values ± SEM and are from one of three independent and reproducible experiments.
Discussion
We analyzed the mechanism of regulation of FasL expression and its role in the FasL–Fas-dependent signaling cascade by which TCR stimulation potentiates AICD in T cells. Our results demonstrate that p38 MAPK plays a pivotal role in T cell AICD by initiating FasL expression, which then leads to the FasL–Fas-dependent activation of caspases and JNK. JNK functions downstream of caspases and together with p38 MAPK upregulates FasL expression and elicits caspase-mediated AICD in T cells.
JNK activity has been shown to mediate AICD in primary T cells 44 45, but the mechanism involved is unknown. In addition, stress-induced FasL expression in T cells is mediated by a MEKK1-regulated response element in the FasL promoter 42, and AP-1 transcriptional activation site binding proteins, including c-Jun and ATF-2, regulate transcription of this promoter 43. We investigated the regulation of FasL expression by p38 MAPK in T cells and found that both the transcription and surface expression of FasL in DO11.10 T cells are significantly inhibited by the DN p38 MAPK (p38 [M]) mutant. Since IL-2 can enhance FasL expression during AICD 53, we considered the possibility that inhibition of FasL expression by p38 (M) results from a low level of IL-2 in DO11.10 T cells. However, the addition of rIL-2 at varying concentrations to p38 (M) DO11.10 transfectants failed to restore FasL expression, suggesting that blockade of FasL expression by p38 (M) was not due to a lack of sufficient IL-2. Rather, our FasL reporter gene assays demonstrated that p38 MAPK directly regulates FasL promoter activity. Recently, the cyclosporin A–sensitive and Ca2+-dependent transcription factors NFAT (nuclear factor and activator of transcription) and Egr3 were reported to regulate FasL transcription by binding positive regulatory elements upstream of the FasL coding sequence 51 54 55. Since we 27 and others 56 have shown that optimal p38 MAPK activation in T cells requires costimulation by Ca2+, p38 MAPK may phosphorylate ATF-2 and Ca2+-dependent transcription factors or otherwise induce the expression of c-Jun 27. ATF-2 and c-Jun bind to the FasL promoter and activate FasL gene transcription. Thus, p38 MAPK elicits caspase activation via the regulation of FasL expression. This notion is supported by our observation that the activities of both caspase-1 and caspase-3 during AICD were significantly inhibited by SB203580.
Both caspases and stress-related MAPKs are involved in apoptosis in various cell types, but the relationship between them has not been fully elucidated. JNK- as well as p38 MAPK–dependent pathways may be activated during Fas-induced cell death in Jurkat T cells 40 46 47. Whereas activation of p38 MAPK is dependent upon caspase-1–like members, p38 MAPK activity is not required for Fas-mediated apoptosis 46 47. However, the upstream activator of p38 MAPK, MKK3 or MKK6b, is involved in Fas-induced cell death 46 47, indicating that p38 MAPK may mediate some cellular responses that precede Fas-mediated apoptosis 40 47. It is noteworthy that Jurkat T cells and primary T cells differ in terms of their requirements for activation of MAPKs 27. Moreover, JNK has been implicated in an important role in the regulation of AICD in primary T cells 44 45, but it is not required for anti-Fas–induced Jurkat T cell death 35. These findings indicate that the signaling mechanism(s) of TCR-induced AICD in primary T cells may be different from that of anti-Fas–induced cell death in Jurkat T cells. Both primary naive T cells and DO11.10 T cells express low levels of Fas and FasL, and the expression of Fas and FasL is upregulated upon TCR ligation in primary T cells and DO11.10 T cells. Therefore, the mechanism of TCR-induced AICD in DO11.10 T cells may be similar to that in primary T cells.
We examined the relationship between the activation of p38 MAPK, caspase, and JNK during AICD. Analyses of T cells from FasL-deficient gld mice, anti-Fas–induced Fas cross-linking in B6 splenic T cells, and DO11.10 T cells demonstrated that p38 MAPK is active upstream of caspases and JNK and that JNK activity lies downstream of caspases. Furthermore, JNK activity was shown to require caspase activity, as pretreatment of T cells with YVAD, DEVD, and zVAD before TCR stimulation blocked the activation of JNK but not p38 MAPK. Interestingly, a block in caspase activation resulted in the downregulation of FasL expression. Caspases are upstream regulators of JNK 35 40 41, and JNK may participate in stress-induced cell death by the regulation of FasL expression 42 43. Moreover, p21-activated kinase (PAK) is required for Fas-induced JNK activation in Jurkat T cells 41. Since JNK acts downstream of PAK, the latter finding suggests that JNK functions downstream of caspase activity. Thus, it is possible that caspases regulate FasL expression via JNK during AICD. Indeed, we found that DN–JNK significantly inhibits both FasL expression and AICD. Consistent with this model, T cells from JNK-1– and JNK-2–deficient mice exhibit decreased AICD, suggesting a role for JNK in the regulation of T cell apoptosis in primary T cells 44 45.
Our observations are consistent with a novel model in which p38 MAPK and JNK both regulate FasL expression during TCR-induced T cell AICD (Fig. 8). In this model, p38 MAPK acts early during the first 2 h after TCR stimulation in the apoptotic cascade to activate FasL expression. Subsequently, 2–8 h after TCR stimulation, FasL–Fas interaction and the Fas-mediated activation of caspases stimulate JNK to further upregulate FasL expression, possibly via the phosphorylation of c-Jun and/or ATF-2, which each bind to the FasL promoter. Thus, p38 MAPK and downstream JNK appear to converge to regulate FasL expression at different times after TCR stimulation to elicit maximal AICD observed at ≥16 h.
Figure 8.

Model of regulation of FasL expression during TCR-induced T cell AICD. In this model, TCR ligation leads to the activation of PTKs, which results in the activation of p38 MAPK. p38 MAPK then phosphorylates several nuclear transcription factors (e.g., ATF-2), which may bind to the FasL promoter and activate FasL gene transcription. Transcribed FasL protein translocates from the cytoplasm to the plasma membrane, interacts with Fas which then recruits FADD to bind to its death domain. This FasL–Fas interaction initiates a caspase cascade that subsequently activates JNK. Activated JNK promotes AICD by regulating FasL expression. Thus, p38 MAPK and JNK may preferentially regulate FasL expression at early and later times after activation, respectively, perhaps by the phosphorylation and activation of distinct transcription factors for the FasL promoter. Consistent with this model, we observed that inhibition (⊗) of p38 MAPK, JNK, and caspase activity blocks T cell AICD.
The model proposed supports our previous finding that p38 MAPK activity is essential for the TCR-stimulated induction of c-Jun expression during the activation of primary T cells, perhaps due to its ability to phosphorylate various transcription factors (e.g., ATF-2) that control c-Jun gene activation 27. This may explain why both c-Jun and ATF-2 are involved in the regulation of FasL promoter activity. An increase in FasL promoter activity potentiated by p38 MAPK via ATF-2, c-Jun, or other transcription factors would enable FasL to be synthesized initially at a low level and then translocate to the plasma membrane, where it can bind to Fas. Membrane-associated FasL–Fas interactions will recruit FADD, activate caspases, and lead to enhanced JNK activity and the phosphorylation of c-Jun and ATF-2. This may allow c-Jun and ATF-2 to function synergistically in a positive feedback loop to further upregulate FasL promoter activity and enable the synthesis of sufficient FasL to trigger the caspase cascade that results in T cell apoptosis. Consistent with this model, it is interesting that p38 MAPK was recently shown to regulate FasL expression during AICD of murine T cell hybridomas 57. Analyses of the role of ATF-2 and c-Jun, as well other transcription factors, in transcriptional complexes that bind to the FasL promoter region are required to formally prove this model.
In summary, we demonstrate that the biological relevance of our studies is the novel observation that the integration of p38 MAPK– and caspase-dependent JNK activities results in optimal FasL expression and consequent T cell AICD. p38 MAPK and JNK may each preferentially phosphorylate and activate different transcription factors, e.g., ATF-2 and c-Jun, respectively, which may interact in a transcription complex to regulate FasL expression. These findings are particularly interesting in view of our previous report that p38 MAPK mediates signal integration of TCR/CD28 costimulation in primary murine T cells, and that this integration includes the activation of JNK for optimum T cell responsiveness 27. Thus, it will prove important to further elucidate how p38 MAPK and JNK activities govern whether T cells follow a pathway of (a) deletion via AICD leading to immune tolerance or (b) activation leading to immune responsiveness.
Acknowledgments
We thank Drs. J.P. Allison, J.D. Ashwell, B. Singh, J. Bluestone, D.R. Green, J. Han, E. Nishida, R.-P. Sékaly, and P. Young for their kind gifts of various reagents that made this work possible, Dr. Jeffrey Dixon for caspase assays, Ms. A. Leaist for her valuable assistance in the preparation of this manuscript, and all members of our laboratory for their advice and encouragement.
This work was supported by the Vern Bruder grant from the Canadian Diabetes Association, a grant from the Juvenile Diabetes Foundation International, and a Diabetes Interdisciplinary Research Program grant from the Medical Research Council of Canada and Juvenile Diabetes Foundation International. Jian Zhang and Konstantin Salojin were recipients of a postdoctoral fellowship from the Juvenile Diabetes Foundation International.
Footnotes
J. Zhang's present address is Dept. of Orthopedic Surgery, Rush-Presbyterian-St.-Luke's Medical Center, 1653 W. Congress Parkway, Chicago, IL 60612.
Abbreviations used in this study: AICD, activation-induced cell death; ATF-2, activating transcription factor 2; DN, dominant negative; ERK, extracellular signal–regulated kinase; FADD, Fas-associated death domain; FasL, Fas ligand; GST, glutathione-S-transferase; JNK, c-Jun NH2-terminal kinase; MAPK, mitogen-activated protein kinase; MBP, myelin basic protein; PI, propidium iodide; SAPK, stress-activated protein kinase; WT, wild-type.
References
- Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- Green D.R., Scott D.W. Activation-induced apoptosis in lymphocytes. Curr. Opin. Immunol. 1994;6:476–487. doi: 10.1016/0952-7915(94)90130-9. [DOI] [PubMed] [Google Scholar]
- Russell J.H. Activation-induced death of mature T cells in the regulation of immune responses. Curr. Opin. Immunol. 1995;7:382–388. doi: 10.1016/0952-7915(95)80114-6. [DOI] [PubMed] [Google Scholar]
- Brunner T., Mogil R.J., LaFrace D., Yoo N.J., Mahboubl A., Echeverri F., Martin S.J., Force W.R., Lynch D.H., Ware C.F. Cell-autonomous Fas (CD95)/Fas-ligand interaction mediates activation-induced apoptosis in T-cell hybridomas. Nature. 1995;373:441–444. doi: 10.1038/373441a0. [DOI] [PubMed] [Google Scholar]
- Dhein J., Walczak H., Baumler C., Debatin K.-M., Krammer P.H. Autocrine T-cell suicide mediated by APO-1/(Fas/CD95) Nature. 1995;373:438–441. doi: 10.1038/373438a0. [DOI] [PubMed] [Google Scholar]
- Ju S.-T., Panka D.J., Cui H., Ettinger R., El-Katib M., Sherr D.H., Stanger B.Z., Marshak-Rothstein A. Fas (CD95)/FasL interactions required for programmed cell death after T-cell activation. Nature. 1995;373:444–448. doi: 10.1038/373444a0. [DOI] [PubMed] [Google Scholar]
- Delovitch T.L., Singh B. The nonobese diabetic mouse as a model of autoimmune diabetesimmune dysregulation gets the NOD. Immunity. 1997;7:727–738. doi: 10.1016/s1074-7613(00)80392-1. [DOI] [PubMed] [Google Scholar]
- Salojin K.V., Zhang J., Madrenas J., Delovitch T.L. T-cell anergy and altered T-cell receptor signalingeffects on autoimmune disease. Immunol. Today. 1998;19:468–473. doi: 10.1016/s0167-5699(98)01326-7. [DOI] [PubMed] [Google Scholar]
- Nagata S., Suda T. Fas and Fas ligandlpr and gld mutations. Immunol. Today. 1995;16:39–43. doi: 10.1016/0167-5699(95)80069-7. [DOI] [PubMed] [Google Scholar]
- Fisher G.H., Rosenberg F.J., Strauss S.E., Dale J.K., Middleton L.A., Lin A.Y., Strober W., Lenardo M.J., Puck J.M. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune syndrome. Cell. 1995;81:935–946. doi: 10.1016/0092-8674(95)90013-6. [DOI] [PubMed] [Google Scholar]
- Rieux-Laucat F., LeDeist F., Hivroz C., Roberts I.A., Debatin K.M., Fisher A., de Villartay J.P. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science. 1995;268:1347–1349. doi: 10.1126/science.7539157. [DOI] [PubMed] [Google Scholar]
- Henkart P.A. ICE family proteasesmediators of all apoptotic cell death? Immunity. 1996;4:195–201. doi: 10.1016/s1074-7613(00)80428-8. [DOI] [PubMed] [Google Scholar]
- Miller D.K. The role of the caspase family of cysteine proteases in apoptosis. Semin. Immunol. 1997;9:35–49. doi: 10.1006/smim.1996.0058. [DOI] [PubMed] [Google Scholar]
- Salvesen G.S., Dixit V.M. Caspasesintracellular signaling by proteolysis. Cell. 1997;91:443–446. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- Chow S.C., Weis M., Kass G.E., Holmstrom T.H., Ericksson J.E., Orrenius S. Involvement of multiple proteases during Fas-mediated apoptosis in T lymphocytes. FEBS Lett. 1995;364:134–138. doi: 10.1016/0014-5793(95)00370-o. [DOI] [PubMed] [Google Scholar]
- Los M., Van de Craen M., Penning L.C., Schenk H., Westendorp M., Baeuerle P.A., Droge W., Krammer P.H., Fiers W., Schulze-Osthoff K. Requirement of an ICE/CED-3 protease for Fas/Apo-1-mediated apoptosis. Nature. 1995;375:81–83. doi: 10.1038/375081a0. [DOI] [PubMed] [Google Scholar]
- Nicholson D.W., Ali A., Thornberry N.A., Vaillancourt J.P., Ding C.K., Gallant N., Gareau Y., Griffin P.R., Labelle M., Lazebnik Y.A. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- Tewari N., Quan L.T., O'Rourke K., Desnoyers S., Zeng Z., Beidler D.R., Poirier G.G., Salvesen G.S., Dixit V.M. Yama/CPP32β, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- Enari M., Hug H., Nagata S. Involvement of an ICE-like protease in Fas-mediated apoptosis. Nature. 1996;375:78–81. doi: 10.1038/375078a0. [DOI] [PubMed] [Google Scholar]
- Hasegawa J., Kamada S., Kamiike W., Shimizu S., Imazu T., Matsuda H., Tsujimoto Y. Involvement of CPP32/Yama (-like) proteases in Fas-mediated apoptosis. Cancer Res. 1996;56:1713–1718. [PubMed] [Google Scholar]
- Schlegel J., Peters I., Orrenius S., Miller D.K., Thornberry N.A., Yamin T.-T., Nicholson D.W. CPP32/apopain is a key interleukin 1β converting enzyme-like protease involved in Fas- mediated apoptosis. J. Biol. Chem. 1996;271:1841–1844. doi: 10.1074/jbc.271.4.1841. [DOI] [PubMed] [Google Scholar]
- Enari M., Talanian R.V., Wong W.W., Nagata S. Sequential activation of ICE-like and CPP32-like proteases during Fas-mediated apoptosis. Nature. 1996;380:723–726. doi: 10.1038/380723a0. [DOI] [PubMed] [Google Scholar]
- Muzio M., Chinnaiyan A.M., Kischkel F.C., O'Rourke K., Shevchenko A., Ni J., Scaffidi C., Bretz J.D., Zhang M., Gentz R. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is regulated to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- Depraetere V., Golstein P. Fas and other death signaling pathways. Semin. Immunol. 1997;9:93–107. doi: 10.1006/smim.1997.0062. [DOI] [PubMed] [Google Scholar]
- Green D.R. Apoptotic pathwaysthe road to ruin. Cell. 1998;94:695–698. doi: 10.1016/s0092-8674(00)81728-6. [DOI] [PubMed] [Google Scholar]
- Whitmarsh A.J., Davis R.J. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J. Mol. Med. 1996;74:589–607. doi: 10.1007/s001090050063. [DOI] [PubMed] [Google Scholar]
- Zhang J., Salojin K.V., Gao J.-X., Cameron M.J., Bergerot I., Delovitch T.L. p38 mitogen-activated protein kinase mediates signal integration of TCR/CD28 costimulation in primary murine T cells. J. Immunol. 1999;162:3819–3829. [PubMed] [Google Scholar]
- Xia Z., Dickens M., Raingeaud J., Davis R.J., Greenberg M.E. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- Chen Y.-R., Meyer C.F., Tan T.-H. Persistent activation of c-Jun N-terminal kinase 1 (JNK1) in gamma radiation-induced apoptosis. J. Biol. Chem. 1996;271:631–634. doi: 10.1074/jbc.271.2.631. [DOI] [PubMed] [Google Scholar]
- Verheij M., Bose R., Lin X.H., Yao B., Jarvis W.D., Grant S., Birrer M.J., Szabo E., Zon L.I., Kyriakis J.M. Requirement for ceramide-initiated SAPK/JNK signaling in stress-induced apoptosis. Nature. 1996;380:75–79. doi: 10.1038/380075a0. [DOI] [PubMed] [Google Scholar]
- Frasch S.C., Nick J.A., Fadok V.A., Bratton D.L., Worthen G.S., Henson P.M. p38 mitogen-activated protein kinase-dependent and independent intracellular signal transduction pathways leading to apoptosis in human neutrophils. J. Biol. Chem. 1998;273:8389–8397. doi: 10.1074/jbc.273.14.8389. [DOI] [PubMed] [Google Scholar]
- Lei W., Yu R., Mandlekar S., Kong A.-N.T. Induction of apoptosis and activation of interleukin 1β-converting enzyme/Ced-3 protease (caspase-3) and c-Jun NH2-terminal kinase 1 by benzo(a)pyrene. Cancer Res. 1998;58:2102–2106. [PubMed] [Google Scholar]
- Aoshiba K., Yasui S., Hayashi M., Tamaoki J., Nagai A. Role of p38-mitogen-activated protein kinase in spontaneous apoptosis of human neutrophils. J. Biol. Chem. 1999;162:1692–1700. [PubMed] [Google Scholar]
- Khwaja A., Downward J. Lack of correlation between activation of Jun-NH2-terminal kinase and induction of apoptosis after detachment of epithelial cells. J. Cell Biol. 1997;17:1017–1023. doi: 10.1083/jcb.139.4.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenczowski J.M., Dominguez L., Eder A.M., King L.B., Zacharchuk C.M., Ashwell J.D. Lack of a role for Jun kinase and AP-1 in Fas-induced apoptosis. Mol. Cell. Biol. 1997;17:170–181. doi: 10.1128/mcb.17.1.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki I., Tani E., Ikemoto H., Kitagawa H., Fujikawa H. Activation of stress-activated protein kinase/c-Jun-NH2-terminal kinase and p38 kinase in calphostin C-induced apoptosis requires caspase-3-like proteases but is indispensable for cell death. J. Biol. Chem. 1998;274:5310–5317. doi: 10.1074/jbc.274.9.5310. [DOI] [PubMed] [Google Scholar]
- Nishina N., Fischer K.D., Radvanyl L., Shahinlan A., Hakem R., Rubie E.A., Bernstein A., Mak T.W., Woodgett J.R., Penninger J.M. Stress-signaling kinase Sek1 protects thymocytes from apoptosis mediated by CD95 and CD3. Nature. 1997;385:350–353. doi: 10.1038/385350a0. [DOI] [PubMed] [Google Scholar]
- Liu Z., Hsu H., Goeddel D.V., Karin M. Dissection of TNF receptor 1 effector functionsJNK activation is not linked to apoptosis while NF-κB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- Cahill M.A., Peter M.E., Kischkel F.C., Chinnaiyan A.M., Dixit V.M., Krammer P.H., Nordheim A. CD95 (APO-1/Fas) induces activation of SAP kinase downstream of ICE-like proteases. Oncogene. 1996;13:2087–2096. [PubMed] [Google Scholar]
- Toyoshima F., Moriguchi T., Nishida E. Fas induces cytoplasmic apoptotic responses and activation of the MKK7-JNK/SAPK and MKK6-p38 pathways independent of CPP32-like proteases. J. Cell Biol. 1997;139:1005–1015. doi: 10.1083/jcb.139.4.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudel T., Zenke F.T., Chuang T.H., Bokoch G.M. p21-activated kinase (PAK) is required for Fas-induced JNK activation in Jurkat cells. J. Immunol. 1998;160:7–11. [PubMed] [Google Scholar]
- Faris M., Latinis K.M., Kempiak S.J., Koretzky G.A., Nel A. Stress-induced Fas ligation expression in T cells is mediated through a MEK kinase 1-regulated response element in the Fas ligand reporter. Mol. Cell. Biol. 1998;18:5414–5424. doi: 10.1128/mcb.18.9.5414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasibhatla S., Brunner T., Genestier L., Echeverri F., Mahboubi A., Green D.R. DNA damaging agents induce expression of Fas ligand and subsequent apoptosis in T lymphocytes via the activation of NF-kappa B and AP-1. Mol. Cell. 1998;1:543–551. doi: 10.1016/s1097-2765(00)80054-4. [DOI] [PubMed] [Google Scholar]
- Dong C., Yang D.D., Wysk M., Whitmarsh A.J., Davis R.J., Flavell R.A. Defective T cell differentiation in the absence of Jnk1. Science. 1998;282:2092–2095. doi: 10.1126/science.282.5396.2092. [DOI] [PubMed] [Google Scholar]
- Sabapathy K., Hu Y., Kallunki T., Schreiber M., David J.P., Jochum W., Wagner E.F., Karin M. JNK2 is required for efficient T-cell activation and apoptosis but not for normal lymphocyte development. Curr. Biol. 1999;11:116–125. doi: 10.1016/s0960-9822(99)80065-7. [DOI] [PubMed] [Google Scholar]
- Huang S., Jiang Y., Li Z., Nishida E., Mathias P., Lin S., Ulevitch R.J., Nemerow G.R., Han J. Apoptosis signaling pathway in T cells is composed of ICE/Ced-3 family proteases and MAP kinase kinase 6b. Immunity. 1997;6:739–749. doi: 10.1016/s1074-7613(00)80449-5. [DOI] [PubMed] [Google Scholar]
- Juo P., Kuo C.J., Reynolds S.E., Konz R.F., Raingeaud J., Davis R.J., Biemann H.-P., Blenis J. Fas activation of the p38 mitogen-activated protein kinase signaling pathway requires ICE/CED-3 family proteases. Mol. Cell. Biol. 1997;17:24–35. doi: 10.1128/mcb.17.1.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong B., Choi Y. Pathways leading to cell death in T cells. Curr. Opin. Immunol. 1997;9:358–364. doi: 10.1016/s0952-7915(97)80082-9. [DOI] [PubMed] [Google Scholar]
- Miyawaki T., Uehara T., Nibu R., Tsuji T., Yachie A., Yonehara S., Taniguchi N. Differential expression of apoptosis-related Fas antigen on lymphocyte subpopulations in human peripheral blood. J. Immunol. 1992;149:3753–3758. [PubMed] [Google Scholar]
- Moriguchi T., Kuroyanagi N., Yamaguchi K., Gotoh Y., Irie K., Kano T., Shirakabe K., Muro Y., Shibuya H., Matsumoto K. A novel kinase cascade mediated by mitogen-activated protein kinase kinase 6 and MKK3. J. Biol. Chem. 1996;271:13675–13679. doi: 10.1074/jbc.271.23.13675. [DOI] [PubMed] [Google Scholar]
- Mittelstadt P.R., Ashwell J.D. Cyclosporin A-sensitive transcription factor Egr-3 regulates Fas ligand expression. Mol. Cell. Biol. 1998;18:3744–3751. doi: 10.1128/mcb.18.7.3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.C., Laydon J.T., McDonnell P.C., Gallagher T.F., Kumar S., Green D., McNulty D., Blumenthal M.J., Heys J.R., Landvatter S.W. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–743. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- Refaeli Y., Parijs L.V., London C.A., Tschopp J., Abbas A.K. Biochemical mechanisms of IL-2-regulated Fas-mediated T cell apoptosis. Immunity. 1998;8:615–623. doi: 10.1016/s1074-7613(00)80566-x. [DOI] [PubMed] [Google Scholar]
- Latinis K.M., Carr L.L., Peterson E.J., Norian L.A., Eliason S.L., Koretzky G.A. Regulation of CD95 (Fas) ligand expression by TCR-mediated signaling events. J. Immunol. 1997;158:4602–4611. [PubMed] [Google Scholar]
- Latinis K.M., Norian L.A., Eliason S.L., Koretzky G.A. Two NFAT transcription factor binding sites participate in the regulation of CD95 (Fas) ligand expression in activated human T cells. J. Biol. Chem. 1997;272:31427–31434. doi: 10.1074/jbc.272.50.31427. [DOI] [PubMed] [Google Scholar]
- Matsuda S., Moriguchi T., Shigeo S., Nishida E. T lymphocyte activation signals for interleukin-2 production involve activation of MKK6-p38 and MKK7-SAPK/JNK signaling pathways sensitive to cyclosporin A. J. Biol. Chem. 1998;273:12378–12382. doi: 10.1074/jbc.273.20.12378. [DOI] [PubMed] [Google Scholar]
- Hsu S.-C., Gravrilin M.A., Tsai M.-H., Han J., Lai M.-Z. p38 mitogen-activated protein kinase is involved in Fas ligand expression. J. Biol. Chem. 1999;274:25769–25776. doi: 10.1074/jbc.274.36.25769. [DOI] [PubMed] [Google Scholar]