

Abstract
The cytokines interleukin (IL)-2, IL-4, IL-6, IL-7, and IL-15 have all previously been shown to inhibit resting T cell death in vitro. We have found a difference in the response of T cells to IL-6, depending on the activation status of the cells. IL-6 inhibited the death of naive T cells, but had no effect on the death of either superantigen-activated T cells, or T cells bearing memory markers. This was true even when the resting and activated T cells were isolated from the same animal; thus, the determining factor for IL-6 insensitivity was the activation status or activation history of the cell, and not the milieu in the animal from which the cells were isolated. Activated T cells expressed lower levels of IL-6 receptors on their surfaces, yet there were sufficient levels of receptors for signaling, as we observed similar levels of signal transducer and activator of transcription (Stat)3 phosphorylation in resting and activated T cells treated with IL-6. However, there was profound inhibition of IL-6–induced Stat1 phosphorylation in activated T cells compared with resting T cells. These data suggest that there is activation-induced inhibition of IL-6 receptor signaling in T cells. This inhibition appears to be specific for some but not all of the IL-6–mediated signaling cascades in these cells.
Keywords: cytokine, survival, memory, death, signal transducer and activator of transcription 3
Introduction
Mouse T cells survive in vitro for only a few days, whereas the life span of resting mouse T cells in vivo is in the order of weeks to months 1 2 3 4 5. Therefore, T cells appear to have a default death mechanism, and sustained signaling from survival factors in vivo may promote the prolonged life span observed in animals. The death rate of activated T cells is very different from that of resting T cells 6 7 8 9 10. After activation, T cells die rapidly in vivo or in vitro by a process referred to as activation-induced cell death. Many researchers have focused on the mechanisms of the enhanced death rates of activated T cells, and several processes that lead to their death have now been described 11 12 13 14 15.
In our own experiments on this subject, we have used a system in which the death in culture of either resting or in vivo–activated T cells is used as a tool to screen cytokines and growth factors for their ability to provide survival signals to T cells 16 17 18 19. We used bacterial superantigens to activate easily trackable populations of T cells bearing particular TCR Vβs in normal mice or to activate the entire T cell population in TCR transgenic mice. We then followed the survival of these T cells in tissue culture in the presence or absence of various soluble or plate-bound factors. Although both resting and superantigen-activated T cells died in vitro, there was a substantial increase in the rate of death of activated T cells in vitro compared with the rate of death of resting T cells.
Experiments on the processes that led to the accelerated death of these activated T cells showed that, unexpectedly, the cells were not dying in vivo or in vitro because of engagement of Fas or TNF receptors and caspase activation. Instead, the cells were dying because they contained increased levels of reactive oxygen species which, directly or indirectly, led to the destruction of the cells 20. In unpublished work, similar results have been obtained for T cells stimulated with conventional antigens, so this caspase-independent death is not restricted to T cells activated with superantigens.
The search for the factors that regulate the survival of resting and activated T cells has implicated several mechanisms. Engagement of their TCRs by MHC has been reported to be required for the prolonged survival of resting T cells in vivo 21 22. Engagements of CD28, other cell surface molecules, or the cytokines that bind the common γ chain cytokine receptor family appear to be capable of blocking activation-induced T cell death in several systems 23 24 25 26.
Using superantigen-activated T cells, we identified several cytokines that inhibited the death of T cells. These included members of the IL-2 family of cytokines, IL-2, IL-4, IL-7, and IL-15 17 19. IL-4 and IL-7 were more efficient inhibitors of resting T cell death than IL-2 and IL-15, probably because resting T cells do not bear high affinity receptors for these latter cytokines. Of these cytokines, IL-2 is known to make T cells in vivo more susceptible to activation-induced cell death 27. The fact that this cytokine prevents the death of activated T cells in vitro suggests that activated T cells may be dying for different reasons in animals than in tissue culture dishes. However, we do not believe this is so because we do not think that IL-2 is the effector of either death or survival of the activated T cells in animals. IL-2 is not being made in large quantities at the time the activated cells are about to die, and the activated T cells do not contain the high levels of Bcl2 characteristic of IL-2 family exposure 28.
We previously showed that the proinflammatory cytokine IL-6 prevented the death of resting T cells 16; however, we did not then study the effect of IL-6 on activated or memory T cells. Since we previously showed that an LPS-induced proinflammatory environment could provide rescue of activated T cells from in vivo deletion, it was of interest to us to determine if IL-6 could rescue activated T cells in vitro. IL-6 was singled out as a good candidate because none of the other proinflammatory cytokines we tested had been capable of rescuing the resting T cells.
In this paper, we report that IL-6 had no effect on the survival of T cells, either activated in vivo or bearing memory markers. Bystander resting T cells from mice containing activated T cells could be rescued from death by IL-6; therefore, T cell activation appeared to be the determining factor for IL-6 insensitivity. We found only a moderate loss of IL-6 receptors after in vivo T cell activation with superantigen. We show that the loss of IL-6 sensitivity in the activated cells was not due to a complete lack of IL-6 receptor signaling, as IL-6 induced phosphorylation of signal transducer and activator of transcription (Stat) 3 in the activated cells. There was a loss of Stat1 phosphorylation in response to IL-6 in the activated T cells; thus, T cell activation appears to elicit an inhibitor specific for Stat1 signaling.
In sum, these results show that activation has several effects on T cells. Activation reduces T cells' ability to respond to a survival signal, IL-6, supplied by activation of innate immunity while increasing their ability to respond to signals (provided in part by activated T cells themselves) that include some members of the IL-2 family of cytokines.
Materials and Methods
Reagents and Mice.
Recombinant mouse IL-2, IL-4, IL-6, and IL-7 were purchased from R&D Systems. FITC- and PE-coupled antibodies against TCR Vβ4, Vβ5, Vβ6, Vβ8, Vβ11, and Vβ14, biotinylated antibodies against IL-6 receptor α chain, and biotinylated isotype control antibodies (rat IgG2bκ) were purchased from PharMingen. B220-specific FITC- and Cychrome®-coupled antibodies were purchased from PharMingen. The secondary staining reagent streptavidin-Cychrome® was also purchased from PharMingen. The anti-TCR Vβ3 mAb KJ25a, the anti-TCR mAb H57-597 (H597), and the anti–I-Ab mAb Y3P were purified from hybridoma supernatants and FITC coupled in our laboratory. The phospho-Stat3 (Tyr705), phospho-Stat3 (Ser727), and phospho-Stat1 (Tyr701) rabbit polyclonal anti–mouse antibodies were from New England Biolabs. The rabbit polyclonal anti–mouse Stat3 and Stat1 antibodies were from Santa Cruz Biotechnology. The peroxidase-labeled goat anti–rabbit IgG antibody was from Kirkegaard & Perry Laboratories. FITC–annexin V was purchased from R&D Systems. Propidium iodide (PI) was from Sigma Chemical Co. The protease and phosphatase inhibitors PMSF, Na3VO4, NaF, aprotinin, α1 antitrypsin, and leupeptin were from Sigma Chemical Co. C57BL/10SnJ (C57BL/10), BALB/cByJ (BALB/c), and B10.BR/SgSnJ (B10.BR) female mice were purchased from The Jackson Laboratory. B10.BR mice transgenic for the Vβ3+ TCR, AD10, were bred from mice provided by Dr. S. Hedrick (University of California, San Diego, CA). FBS was purchased from Intergen or from Summit Biotechnology. MEM culture medium was purchased from GIBCO BRL. Staphylococcal enterotoxin A (SEA) and SEB were purchased from either Sigma Chemical Co. or ToxinTech.
T Cell Activation.
Cells were activated in vivo with bacterial superantigens. We have found that mouse strain and superantigen (SEB versus SEA) are irrelevant in our in vitro system of T cell death and survival. B10.BR female mice were injected either intraperitoneally or intravenously with the amount of SEA indicated in the figure legends. C57BL/10 female mice were injected intravenously or intraperitoneally with 150 μg SEB. T cells were isolated after the times indicated in the figure legends.
T Cell Isolation and Culture.
LNs from B10.BR or C57BL/10 mice were removed and pressed through cell strainers (Falcon; Becton Dickinson) to produce single cell suspensions. The cell suspensions were enriched for T cells by passage through nylon wool columns 29. Cells were suspended in MEM supplemented with 10% FBS and nonessential amino acids (CTM) and plated in 96-well plates (Falcon; Becton Dickinson) at the densities indicated in the figure legends. Cells were cultured in the presence or absence of the indicated cytokines and/or inhibitors at a final volume of 200 μl per well.
Isolation and Culture of Memory versus Naive T Cells.
Spleens were harvested from 6-mo-old BALB/c female mice. We used BALB/c mice in these experiments because we needed mice that contained a reasonable number of memory cells and BALB/c mice of this age had such cells and were available. The spleens were passed through cell strainers, and cells were enriched for T cells by passage through nylon wool columns. The cells were then stained with anti-B220–FITC, anti–MAC-1–FITC, anti-CD44–Cychrome, and either anti-CD45RB–PE or anti-CD62L–PE. The cells were sorted using a MoFlo™ cell sorter from Cytomation. The cells were gated on live cells and all FITC+ cells were dumped. The remaining cells were sorted into naive (CD44lowCD45RBhigh or CD44lowCD62Lhigh) and memory (CD44highCD45RBlow or CD44highCD62Llow) populations. The cells were cultured at 37°C for 30 h at 105 cells per well in CTM containing heat-inactivated FBS and the cytokines indicated in the figures.
Cell Death Analysis.
Immediately after isolation from the mouse or after in vitro culture under various conditions, cells were analyzed for death by PI exclusion or FITC–annexin V binding. In the PI exclusion assays, the cells were first stained on ice with FITC-coupled anti-TCR antibodies, washed, and then incubated on ice with 0.5 μg/ml PI for at least 15 min before analysis. In the FITC–annexin V assays, cells were stained with FITC–annexin V in annexin V binding buffer (BSS supplemented with 2.5 mM CaCl2) at room temperature for 15 min. The cells were then washed with cold binding buffer and immediately put on ice until analyzed. Flow cytometry analysis was performed with a Becton Dickinson FACScan™ instrument (Becton Dickinson). Data were analyzed using either PC Lysis II™ software (Becton Dickinson) or CELLQuest™ software (Becton Dickinson).
IL-6 Receptor Staining.
T cells were isolated from either untreated mice or mice injected with superantigen for the lengths of time indicated in the figure legends. Immediately after isolation from LNs, the T cells were incubated on ice with either biotin-coupled anti–IL-6 receptor α chain mAb or a biotinylated isotype-matched rat IgG2bκ control antibody. The cells were washed twice and then incubated on ice with streptavidin-Cychrome.
Western Blot Analysis of Stat Phosphorylation.
T cells were isolated as described above from LNs of C57BL/10 female mice treated with 150 μg SEB for 2 d (phospho-Stat3 [Ser727] blot, phospho-Stat1 [Tyr701] blot) or 3 d (phospho-Stat3 [Tyr705] blot). The cells were sorted into resting and activated T cell populations using a MoFlo™ cell sorter. Only live gated cells were sorted and the cells were stained with anti-B220–Cy-chrome to dump residual B cells. In the phospho-Stat3 (Ser727) assay, the cells were stained with anti-Vβ5.x–PE to label resting T cells and anti-Vβ8.x–FITC to label activated T cells. In the phospho-Stat3 (Tyr705) assay, the cells were stained with a combination of FITC-coupled antibodies against Vβ4, Vβ6, Vβ11, and Vβ14 to label resting T cells and anti-Vβ8.1,2–PE to label activated T cells. In the phospho-Stat1 (Tyr701) assays, the LN cells from SEB-treated C57BL/10 mice were stained with FITC-coupled anti–I-Ab mAb Y3P instead of B220 to gate out APCs and costained with anti-TCR Vβ8.x mAb F23.1 to sort out activated T cells. Resting T cells were sorted from untreated C57BL/10 LNs using FITC-coupled Y3P and anti-TCR mAb H597.
After sorting, the cells were pelleted once and resuspended into serum-free medium. The cells were incubated at 37°C for 20 min in the presence of medium alone or medium plus IL-6. Cells were pelleted and lysed on ice for 15 min in lysis buffer containing 1% NP-40, 10 mM Tris, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, 2 mM Na3VO4, 10 mM NaF, 1 μg/ml aprotinin, 1 μg/ml α1 antitrypsin, and 1 μg/ml leupeptin. The lysates were then centrifuged at 4°C for 15 min, and supernatants were removed and immediately added to 4× SDS sample buffer containing β-ME. The lysates were boiled for 5 min and stored at −80°C.
Lysates were run in 8% polyacrylamide gels. The gels were transferred onto Immobilon-P™ polyvinylidene difluoride membranes (Millipore) using a Trans-Blot SD Semi-Dry electrophoretic transfer cell from Bio-Rad. Membranes were blocked at room temperature for 1–2 h with BLOTTO (5% milk and 1% FBS in Tris-buffered saline [TBS] plus 0.05% Tween-20). Membranes were washed with TBS-T (TBS plus 0.05% Tween-20) and incubated at 4°C overnight with TBS-T containing 5% BSA and a 1:1,000 dilution of the phospho-Stat antibodies. The blots were washed with TBS-T and incubated for 45 min at room temperature with BLOTTO containing a 1:10,000 dilution of the peroxidase-labeled goat anti–rabbit IgG antibody. Membranes were washed with TBS-T and developed for 1 min with ECL™ reagent (Amersham Pharmacia Biotech). Blots were visualized using Hyperfilm™-ECL™ (Amersham Pharmacia Biotech) and a Fuji RGII x-ray film processor.
The phospho-Stat blots were then stripped for 30 min at 60°C using a stripping buffer that contained 100 mM β-ME, 2% SDS, and 62.5 mM Tris-HCl, pH 6.7. The blots were washed with TBS-T and blocked with BLOTTO. The membranes were then probed for total Stat1 or Stat3 using the protocol described above.
Results
Activated T Cells Die More Rapidly In Vitro Than Resting T Cells.
To assess the effects of cytokines on the survival of resting or activated T cells, we used a system in which T cells were isolated from either normal mice or mice that had been treated 2–3 d previously with a superantigen (either SEA, which targets T cells bearing Vβ3, or SEB, which targets T cells bearing Vβ8s). These cells were cultured under various conditions and then stained with anti-Vβ antibodies. PI was used as a costain to measure cell permeability and death. As shown in Fig. 1Vβ3+ T cells isolated from an untreated B10.BR mouse were 10.5% dead at the start of the assay compared with 36.8% after 26 h in culture. The Vβ3+ T cells isolated from a SEA-treated B10.BR mouse were 25.4% dead at the start of the assay and showed accelerated death compared with the resting cells, with 94.4% dead after 26 h in culture. The increase in dead cells seen at the start of culture in the SEA-activated population correlated with the in vivo deletion of target Vβ3+ cells that has been previously shown to occur after day 2 of SEA activation 7. Fig. 1 illustrates both the in vivo expansion of the Vβ3+ cells by SEA treatment (3.8% in the untreated vs. 13.0% in the SEA-treated) and the enhanced death that occurred in the SEA-activated T cells. Similar results have also been obtained using PI-saponin (DNA degradation) and FITC–annexin V methods to analyze death in this system and with Vβ8+ T cells activated with SEB.
Figure 1.

Death of resting and activated T cells in tissue culture. LN T cells isolated from an untreated B10.BR mouse (A and B) or a B10.BR mouse treated with SEA (C and D) were analyzed for death at the start of culture (A and C) and after 26 h in tissue culture (B and D). Cells were stained with FITC–anti-TCR Vβ3 and PI as described in Materials and Methods.
IL-6 Inhibits In Vitro Death of Resting but Not Activated T Cells.
To analyze the effect of IL-6 on superantigen-activated or resting T cells, we incubated T cells from either superantigen-treated or untreated B10.BR or C57BL/10 mice in the presence or absence of IL-6 (Table ). We found that IL-6 rescued resting T cells from death but did not inhibit the death of activated T cells. Table shows six independent experiments that were completed to compare the effect of IL-6 on resting cells from untreated mice with its effect on resting and activated T cells from superantigen-treated mice. In all the experiments, the resting cell death was inhibited by IL-6. Inhibition of death averaged 39.5% for resting cells isolated from untreated mice, and 30.4% for resting cells isolated from superantigen-treated mice. The activated T cells, isolated from superantigen-treated mice, bearing the target TCR Vβ (Vβ3 for SEA, Vβ8 for SEB) were unaffected by IL-6 in these assays. Since the nontarget (resting) T cells from the superantigen-treated mice were rescued by IL-6, the insensitivity to IL-6 in the activated T cells could not have been solely mediated by cytokines expressed during activation. Therefore, these data suggest that signaling through the TCR rather than the cytokine milieu in the mouse was the determining factor for loss of IL-6 sensitivity in these survival assays.
Table 1.
IL-6 Inhibits the Death of Resting but Not Activated T Cells
| Superantigenmouse strain | Time in culture | IL-6 treatment | Resting T cells from untreated mice | Resting T cells from superantigen-treated mice | Activated T cells from superantigen-treated mice | |
|---|---|---|---|---|---|---|
| h | % Dead | % Dead | % Dead | |||
| Exp. 1 | SEA (3 d) | 24 | − | 40.3 | 51.7 | 94.3 |
| B10.BR | + | 19.3 | 31.6 | 95.3 | ||
| Exp. 2 | SEA (2 d) | 24 | − | 46.7 | 53.7 | 91.2 |
| B10.BR | + | 31.5 | 39.7 | 94.9 | ||
| Exp. 3 | SEA (2 d) | 34 | − | 53.3 | 53.5 | 87.4 |
| B10.BR | + | 32.7 | 39.8 | 87.8 | ||
| Exp. 4 | SEA (3 d) | 26 | − | 36.8 | 41.9 | 94.7 |
| B10.BR | + | 21.5 | 26.6 | 95.8 | ||
| Exp. 5 | SEA (2 d) | 29 | − | 67.2 | 50.9 | 87.6 |
| B10.BR | + | 45.6 | 33.5 | 93.8 | ||
| Exp. 6 | SEB (2 d) | 27 | − | 51.6 | ND | 89.4 |
| C57BL/10 | + | 30.9 | ND | 85.6 |
T cells were isolated from mouse LNs and cultured in vitro for the indicated times in the presence or absence of IL-6. Cell death was analyzed by PI exclusion in experiments (Exp.) 1, 3, 4, and 6. Cell death was analyzed by annexin V staining in experiments 2 and 5. The dose of IL-6 was either 10 or 100 ng/ml in each experiment.
Loss of IL-6 sensitivity was not unique to T cells activated in vivo by superantigens. Insensitivity to IL-6 could also be induced in vitro. For example, T cells activated in vitro with PMA and ionomycin were completely insensitive to IL-6 rescue, whereas their death was prevented by IL-2, IL-4, or IL-7 (data not shown). Thus, induction of insensitivity to IL-6 did not depend on a nonlymphoid phenomenon present in animals but not present in tissue culture.
Sensitivity to Various Cytokines for Survival Depends on Activation of the T Cells.
Although the doses of IL-6 used in Table were sufficient for death inhibition of resting T cells 16, it is possible that a greater concentration of IL-6 was required for survival of activated T cells. This question is addressed in Fig. 2, in which we assessed the dose–responses of resting and activated T cells to IL-6, IL-2, IL-4, and IL-7. As previously reported, IL-2 was effective at mediating survival of resting T cells only at higher concentrations (50 ng/ml or greater), whereas IL-4 and IL-7 effectively inhibited death of resting T cells at amounts as low as 100–500 pg/ml. Low concentrations of IL-6 also inhibited resting T cell death; however, the percentage of T cells rescued was lower than that rescued by IL-4 and IL-7, suggesting that there was a subpopulation of resting T cells that was IL-6 insensitive before activation.
Figure 2.
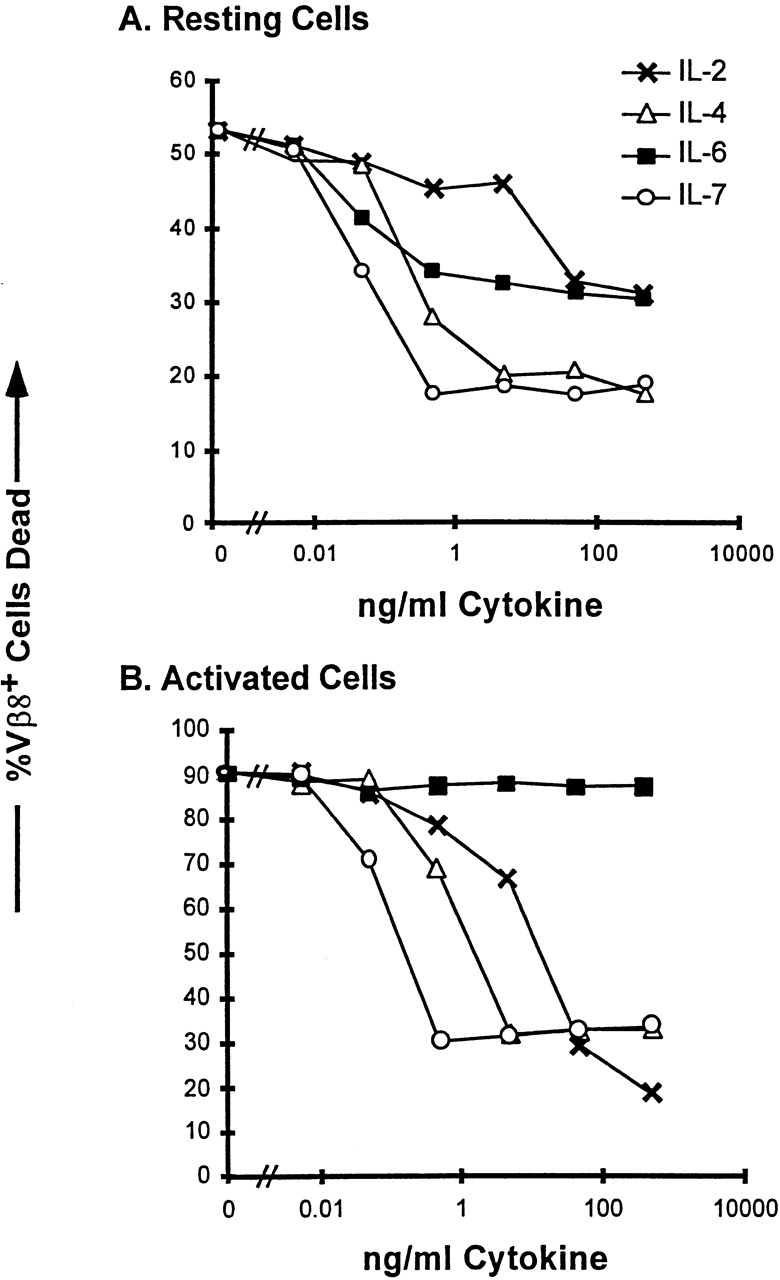
Reciprocal effects of IL-6– and IL-2–related cytokines on resting and activated T cells. LN T cells were isolated from untreated C57BL/10 mice (A) or from C57BL/10 mice 2 d after intravenous injection with 150 mg SEB (B). T cells were plated with the indicated amounts of cytokines at a density of 2 × 105 cells per well in a 96-well plate. After 36 h in culture, the cells were stained for Vβ8.1,2 and analyzed by PI exclusion for cell death as described in Materials and Methods. Cells were plated in duplicate wells and the mean of each duplicate is indicated. This figure shows one of three experiments with similar results.
IL-6 had no rescuing effect on activated T cells even at the highest concentrations added (0.5 μg/ml; Fig. 2 B). On the other hand the cytokines IL-2, IL-4, and IL-7 were very effective at inhibiting the death of the activated T cells. These data suggest that the IL-6 insensitivity shown in Table was not due simply to a requirement for a higher concentration of IL-6 by the activated T cells. These data also illustrate the different regulation of effects of the various cytokines depending on the activation state of the target cell. A higher dose of IL-2 was required to rescue resting cells than to rescue activated T cells. This is presumably due to the upregulation of high affinity IL-2 receptors on activated T cells.
Resting T Cells Bearing Memory Markers Are Also Insensitive to IL-6 Rescue.
Since there was a subpopulation of resting T cells that was unresponsive to IL-6 (shown in Fig. 2), we were interested in determining if this subpopulation consisted of previously activated T cells. To address this possibility, we sorted naive and memory phenotype T cells from untreated, 6-mo-old BALB/c mice. The markers CD44, CD62L, and CD45RB have all been reported to be useful in discriminating memory from naive T cells 30 31 32. Memory T cells express higher levels of CD44 and lower levels of both CD62L and CD45RB compared with resting T cells. In Fig. 3, we cultured memory and naive T cells in the presence or absence of 10 ng/ml IL-6 for 30 h. Cell survival was assessed in these assays by FITC–annexin V staining as described in Materials and Methods. Fig. 3 A shows that CD44lowCD45RBhigh (naive) T cells were rescued from apoptosis by IL-4, IL-2, and IL-6. In Fig. 3 B the CD44highCD45RBlow (memory) T cells were not rescued by IL-6, but were rescued by IL-2 and IL-4. When CD44lowCD62Lhigh (naive) T cells were compared with CD44highCD62Llow (memory) T cells (Fig. 3C and Fig. D), again only the naive T cells were rescued from death by IL-6, whereas IL-2 and IL-4 were capable of rescuing both naive and memory T cells. These results suggest that it is not just the activation state of a T cell, but perhaps the activation history of a T cell, that determines its sensitivity to IL-6 survival signaling. Although we identified a subpopulation of resting T cells (memory cells) that was unresponsive to IL-6, there was still a significant population of IL-6–unresponsive T cells in the naive population. Thus, there may be additional factors that can shut off sensitivity to IL-6. Alternatively, perhaps CD44 and CD45RB or CD62 levels do not define all memory T cells.
Figure 3.

IL-6 rescues naive but not memory T cells from death in vitro. Splenic T cells were isolated from untreated BALB/c mice. The cells were sorted into naive and memory populations as described in Materials and Methods. The cells were plated at a density of 105 cells per well in a 96-well plate in the presence of medium alone, 20 ng/ml IL-4, 20 ng/ml IL-6, or 200 ng/ml IL-2. Cell death was measured by FITC–annexin V staining after 30 h in culture. (A) Naive (CD44lowCD45RBhigh) T cells. (B) Memory (CD44highCD45RBlow) T cells. (C) Naive (CD44lowCD62Lhigh) T cells. (D) Memory (CD44highCD62Llow) T cells. The error bars represent the SD of triplicate wells. This figure represents one of two experiments with similar results.
We were interested in determining the mechanism by which activated T cells lost sensitivity to IL-6 survival signaling. There were three possibilities. The first possibility was that protein(s) induced after T cell activation interfered with the IL-6 signaling cascades and thus blocked the ability of IL-6 to rescue cells from death. A family of intracellular suppressors of cytokine signaling (SOCS) has recently been described 33 34 35 36 37 38. Of particular relevance to this paper were the reports demonstrating that SOCS-1 can suppress IL-6 signaling in M1 monocytic leukemia cells 34 35 37 38. The second possibility was that the activated T cells lost expression of IL-6 receptors and were not receiving IL-6 signals at all. Loss of IL-6 receptor expression on T cells activated in vitro, or after long-term cell culture, has been described previously 39 40. The third possibility was that IL-6 signaling was intact in the activated T cells and simply could not interfere with the mechanism that led to death of the activated cells. Such a hypothesis implies that activated and resting T cells die by different routes.
We addressed these three hypotheses by measuring SOCS-1 mRNA levels, IL-6 receptor expression, and IL-6 receptor signaling (Stat phosphorylation) in resting versus activated T cells.
Analysis of SOCS Expression in Activated versus Resting T Cells.
We attempted to find out whether SOCS-1 played a role in the suppression of IL-6 signaling in activated T cells. Fig. 4 shows a reverse transcription (RT)-PCR analysis of SOCS-1 mRNA expression in resting and SEA-activated T cells. In this experiment, AD10 TCR transgenic mice were used as a source of T cells. T cells from these mice all express TCR Vβ3 and can therefore be activated with SEA. Like the B10.BR and C57BL/10 mice, only resting and not activated T cells from these mice could be rescued from in vitro death by IL-6 (data not shown). The AD10 mice were injected with 4 μg SEA; 47 h after injection total RNA was isolated from spleen and LN T cells, and cDNA was synthesized as described in Materials and Methods. We performed RT-PCR for the indicated cycles to compare the levels of SOCS-1 and control hypoxanthine phosphoribosyltransferase (HPRT) mRNA expression in T cells from the activated and untreated AD10 mice. Fig. 4 shows that there was not an upregulation of SOCS-1 mRNA expression in the activated T cells. SOCS-1 was expressed in both the resting and activated T cells. The amount of SOCS-1 mRNA by comparison with that of the housekeeping gene, HPRT, was actually slightly higher in the resting than in the activated T cells. We also found SOCS-2 and SOCS-3 to be expressed in both populations (data not shown).
Figure 4.

Activated and resting T cells contain approximately the same amount of SOCS-1 mRNA. AD10 TCR transgenic mice were injected intraperitoneally with 4 μg SEA, and LN and spleen T cells were isolated and pooled from either untreated mice or 47-h SEA–treated mice. Total RNA and cDNA were obtained from 5 × 107 cells as described in Materials and Methods. RT-PCR was performed with oligos specific for control HPRT or SOCS-1 for the indicated number of cycles. PCR products were separated in a 1.5% agarose gel and analyzed for ethidium bromide intensity using a Nucleovision Imaging Workstation from Nucleotech.
The SOCS data did not disprove the idea that there was active suppression of cytokine signaling, as other SOCS have recently been described and other mechanisms of cytokine receptor signaling downregulation such as STAT masking 41 42 and induction of the protein inhibitor of activated Stat (PIAS)3 protein 43 have been reported. Instead of pursuing other potential mechanisms of inhibition of receptor signaling, we directly addressed the questions of whether the IL-6 receptor was expressed on activated T cells and, if so, whether it was capable of signaling at all.
Downregulation of IL-6 Receptor α Chain after T Cell Activation.
We used flow cytometry to find out whether loss of sensitivity to IL-6 survival signals correlated with a loss of IL-6 receptor. To do this, T cells were isolated from normal B10.BR mice, or B10.BR mice 2 d after injection of SEA. The cells were stained with anti-Vβ3 or anti-Vβ4 antibody and antibody to the IL-6 receptor α chain. As shown in Fig. 5, the levels of IL-6 receptor α on some of the Vβ3+ T cells were reduced by activation but remained normal on bystander resting Vβ4+ T cells. Since resting T cells have a higher expression of both SOCS-1 and IL-6 receptor than do the activated T cells we believe the ratio of SOCS-1 to IL-6 receptor in the two populations to be fairly similar. It seems unlikely to us that the partial downregulation of IL-6 receptor was fully responsible for the complete insensitivity of the activated T cells to IL-6. Therefore, we next addressed the question of whether this level of receptor was capable of signaling in the activated T cells.
Figure 5.

Activated T cells bear ∼50% fewer IL-6 receptors than resting T cells. LN T cells were isolated from an untreated (A and C) or SEA-treated (B and D) B10.BR mouse. The mouse was injected with 1 μg of SEA intraperitoneally 3 d before harvest of LNs. T cells were stained with anti-Vβ4–FITC, anti-Vβ3–FITC, and either anti–IL-6 receptor α–biotin or control IgG2bκ-biotin mAb followed by streptavidin-CyChrome as described in Materials and Methods. The IL-6 receptor α histograms are shown in white and the control mAb histograms are shown in black. The values above each histogram represent the median fluorescence intensities. A and B show the receptor stains of cells gated on Vβ3+ T cells; C and D show the receptor stains of the Vβ4+ T cells. This is one of nine separate experiments that showed downregulation of IL-6 receptor α after T cell activation.
IL-6 Induces Phosphorylation of Stat3 in Both Activated and Resting T Cells.
One of the ways that IL-6 signals is via phosphorylation and activation of the kinases Janus kinase (Jak)2 and Tyk2. These kinases in turn phosphorylate Stat3 at Tyr705 44 45. To address whether IL-6 signaled in the activated T cells, we performed phospho-Stat3 Western blots of lysates from resting and activated T cells. LN T cells from SEB-treated mice were sorted into resting and activated populations as described in Materials and Methods. Like unsorted cultures of cells, these cultures showed IL-6 rescue only in the resting and not the activated populations, whereas IL-4 rescued both populations from death (data not shown). Fig. 6 shows that both activated and resting T cells phosphorylated Stat3 Tyr705 after a 20-min incubation with 10 ng/ml IL-6. Similar levels of total Stat3 were observed in all the lanes after stripping and reprobing of the same blot (Fig. 6, bottom).
Figure 6.

IL-6 induced phosphorylation of Stat3 at Tyr705 in both activated and resting T cells. T cells from C57BL/10 LNs were sorted into resting (Vβ4,6,11,14) and activated (Vβ8) as described in Materials and Methods. Cells were treated with 10 ng/ml IL-6 or medium alone for 20 min and lysed. Each lane contains 2.5 × 106 cell equivalents. The top panels show the anti–phospho-Stat3 (Tyr705) blot (P-Tyr Stat3). The bottom panels show the total Stat3 in each lane after stripping and reprobing. This is one of three experiments with similar results.
Other signaling cascades, besides the Jaks, can be turned on after IL-6 receptor binding. Independent of Stat3, Tyr705 phosphorylation by Jak2 and Tyk2 and subsequent translocation of Stat3 to the nucleus Stat3 can also be phosphorylated on Ser727. This latter phosphorylation event is believed to be mediated by a mitogen-activated protein kinase–independent serine kinase 46. We show in Fig. 7 that both resting and activated T cells phosphorylated Stat3 on Ser727 after a 20-min exposure to 100 ng/ml IL-6, and thus this signaling cascade, like that involved in the tyrosine phosphorylation of Stat3, was apparently intact in the activated T cells. Fig. 7 (bottom) shows that similar levels of total Stat3 were present in all the lanes of the blot.
Figure 7.

IL-6 induced phosphorylation of Stat3 at Ser727 in both activated and resting T cells. T cells from C57BL/10 LNs were sorted into resting (Vβ5) and activated (Vβ8) as described in Materials and Methods. Cells were treated with 100 ng/ml IL-6 or medium alone for 20 min and lysed. Each lane contains 6 × 105 cell equivalents. The top show the anti–phospho-Stat3 (Ser727) blot (P-Ser Stat3). The bottom panels show the total Stat3 in each lane after stripping and reprobing. This is one of three experiments with similar results.
Inhibition of IL-6–induced Stat1 Phosphorylation in Activated T Cells.
Another DNA-binding protein known to be phosphorylated in response to IL-6 is Stat1 43 47 48 49 50. We addressed whether Stat1 signaling was also intact in the activated T cells by performing anti–phospho Stat1 Western blots. LN T cells from untreated or SEB-treated mice were sorted into resting and activated populations, as described in Materials and Methods. Fig. 8 shows the levels of Stat1 phosphorylation and total Stat1 for the activated and resting T cells after a 20-min exposure to 10 ng/ml IL-6 or medium alone. There was no phosphorylation seen in either population in the absence of IL-6 treatment, but very significant phosphorylation seen in the resting T cells treated with IL-6. Interestingly, the IL-6–induced Stat1 phosphorylation in the activated T cells was reduced to a barely detectable level. This result suggests that there is active inhibition of IL-6–mediated Stat1 signaling after T cell activation. Preliminary experiments show that this reduction in Stat1 phosphorylation after IL-6 exposure is also true for memory T cells (data not shown).
Figure 8.

IL-6–induced phosphorylation of Stat1 at Tyr701 was inhibited in the activated T cells by comparison with resting T cells. T cells from C57BL/10 LNs were sorted into resting and activated populations as described in Materials and Methods. Cells were treated with 10 ng/ml IL-6 or medium alone for 20 min and lysed. Each lane contains 2 × 106 cell equivalents. The top panels show the anti–phospho-Stat1(Tyr701) blot (P-Tyr Stat1). The bottom panels show the total Stat1 in each lane after stripping and reprobing. The arrows indicate the larger and smaller forms of Stat1 that are recognized by these antibodies. This is one of three experiments with similar results.
Since the activated T cells bear lower levels of IL-6 receptor, it was possible that Stat1 phosphorylation was simply delayed in response to IL-6 in activated T cells compared with their resting counterparts. To test this, we examined Stat1 and Stat3 phosphorylation in resting and activated T cells 2 h after addition of IL-6 (Fig. 9). As before, Stat1 was much less phosphorylated in activated than resting T cells 20 min after addition of IL-6. In fact, no phosphorylated Stat1 could be detected in either type of cell 2 h after addition of IL-6. In contrast, Stat3 was well phosphorylated in both types of cell 20 min after IL-6 addition.
Figure 9.

Activation of T cells does not lead to delayed Stat1 (Tyr701) phosphorylation. Resting (Rest) and activated (Act) T cells were prepared and analyzed as described in the legends to Fig. 7 and Fig. 8 except that the cells were harvested after 20 min or 2 h incubation in IL-6.
Discussion
The fact that T cells die in vitro at a much faster rate than in vivo suggests that there are survival factors in vivo that are absent in standard tissue culture medium. Several extracellular and intracellular factors have been identified that are capable of blocking or delaying apoptosis of T cells in vitro 16 17 19 23 25 51 52 53. Engagement of CD28 or any one of multiple cytokines can prevent or delay the death of T cells after activation. Several cytokines have also been reported to prolong survival of resting T cells in vitro. Although sufficient for survival in vitro, it is still unclear whether any of these factors are necessary for the survival of activated or resting T cells in vivo. It is likely that in vivo a T cell integrates multiple survival and death signals in its decision to proliferate, differentiate, survive, or die.
We previously showed that T cells activated in the presence of a proinflammatory environment induced by LPS were protected from deletion 18. Although the LPS effect on the activated T cells was indirectly mediated by the proinflammatory cytokines IL-1α and TNF-α it was unclear whether the direct mediators of T cell survival were soluble or cell bound in this system. We previously reported that IL-6 produced by endothelial cells was a survival factor for resting T cells 16. Since IL-6 is a proinflammatory cytokine, we were interested in its effects on activated T cells. We show in this paper that IL-6 could rescue only naive resting T cells and was ineffective at promoting survival of activated T cells or T cells bearing memory markers. Thus, the IL-6 effect is unlike the effect of the IL-2 family of cytokines (IL-2, IL-4, IL-7, and IL-15), which does rescue activated T cells 19 51 and memory T cells (Fig. 3).
It is possible that these effects of IL-6 were not due to direct action of IL-6 on resting T cells, but rather due to induction by IL-6 of another molecule that itself was responsible for the differential survival of resting versus activated T cells. However, we do not think this is the case for several reasons. First, the IL-6 helped purified resting T cells survive. Therefore, if the effects of IL-6 were mediated by a second molecule, this would have had to be induced by IL-6 on resting T cells. Second, as shown earlier and discussed below, survival of resting versus activated T cells was accompanied by differential phosphorylation of Stat1, a signal transduction protein known to be affected by the IL-6 receptor. Third, one of the best-characterized factors induced by IL-6 is IL-4 39. Although IL-4 is a survival factor for resting T cells, it also, unlike IL-6, acts as such for activated T cells. Moreover, we have shown that IL-6 is a survival factor for resting T cells from IL-4 knockout mice. Therefore, we do not believe the effects of IL-6 reported here are secondary to induction by IL-6 of some other factor.
We addressed whether the loss of sensitivity to IL-6 was restricted to the T cells that engaged antigen. We found that bystander resting T cells, isolated from the same mouse as the activated T cells, were rescued by IL-6. These data strongly suggested that engagement of the TCR was required for the mechanism driving the IL-6 insensitivity, and that cytokines or upregulated adhesion molecules after T cell activation in the mouse were insufficient to promote the IL-6 insensitivity.
A lot is known about signaling via the IL-6 receptor. The binding of the IL-6 receptor α chain to IL-6 initiates association with gp130 (the IL-6 receptor signaling subunit) to form an IL-6–specific signaling complex on the cell surface. After homodimerization and phosphorylation of gp130, there is recruitment and activation of several kinases that in turn phosphorylate and activate DNA binding proteins such as the Stats 47 54. IL-6 signaling is known to be mediated by the tyrosine kinases Jak1, Jak2, and Tyk2, as well as serine kinases 45 47 55. The activation of these kinases leads to phosphorylation of Stat1 on Tyr701 47 48 49 50 56 and phosphorylation of Stat3 on both Tyr705 and Ser727 46 55 56 57 58. Immediately after phosphorylation, the Stat proteins form either homodimers or heterodimers and translocate to the nucleus where they bind DNA and drive transcription 47. Inhibition of IL-6 receptor signaling at any step before phosphorylation of either Stat1 or Stat3 could result in the inhibition of all or some of the IL-6 effects on a cell.
With this information in mind, we tested whether IL-6 fails to prevent the death of activated T cells because of a defect in its signaling pathway in these cells. Activated T cells bore about half the number of IL-6 receptors per cell than resting T cells. However, we do not think this was responsible for the failure of IL-6 to rescue activated cells because IL-6 binding induced the phosphorylation of Stat3 on both Tyr 705 and Ser727 in both activated and resting T cells. Thus, the receptor on activated cells could deliver signals effectively.
Notably, engagement of IL-6 failed to induce phosphorylation of Stat1 tyrosines in activated T cells, whereas this phosphorylation occurred efficiently in resting T cells. This failure was not due to loss of Stat1 protein, and was thus caused by a real difference in signaling via the IL-6 receptor in activated T cells. The loss of Stat1 phosphorylation would prevent the transcription of genes that require Stat1 homodimer or Stat1/Stat3 heterodimer binding, but leave genes induced by Stat3 alone or in combination with other binding proteins intact. Thus, Stat1-induced genes are probably required for IL-6–mediated T cell survival. It is interesting to consider this result in light of our recent finding that type 1 IFNs slow the rates of death of activated but not resting T cells 59. As type 1 IFNs also induce Stat1 phosphorylation, perhaps T cell death can be slowed by some combination of signaling by Stat1 and another, unknown factor(s) which is induced in resting T cells by IL-6 and in activated T cells by type 1 IFNs.
Mechanisms of IL-6 receptor signaling downregulation such as Stat masking 41 42, induction of the PIAS1 60 and PIAS3 proteins 46, and the SOCS family of Jak binders 33 34 35 36 37 38 have been recently described. The Stat masking and PIAS mechanisms of inhibition do not prevent the phosphorylation of Stats, whereas the SOCS inhibitors do. SOCS-1 has been reported to be an effective inhibitor of IL-6–induced Stat phosphorylation in a myeloid leukemia cell line and was shown to be upregulated by several cytokines 35 37 38. We are interested in determining whether SOCS-1 induction after T cell activation was responsible for the lack of IL-6–mediated survival or Stat1 phosphorylation in the activated T cells.
We did not find an induction of SOCS-1 mRNA after T cell activation. SOCS-1 was expressed in the T cells even before activation and could not be ruled out as a potential mediator of the insensitivity to IL-6 signaling found in many of the resting T cells. It is also possible that the functional regulation of these SOCS proteins is posttranscriptional or posttranslational. Currently, we are investigating the possibility that a SOCS-like inhibitor is recruited to one of the IL-6 receptor–associated Jaks after T cell activation. The selective inhibition of Stat1 signaling in the activated cells may be due to a lack of activation or recruitment of some but not all of the Jaks normally associated with gp130. This type of inhibition would allow recruitment and activation of some Stat proteins but not others.
In sum, these results suggest that some aspects of the IL-6 receptor are unaffected by T cell activation, whereas at least one signaling pathway is lost. This suggests that T cell activation leaves the T cells able to go through some, but not all, of the differentiative processes associated with IL-6. This is consistent with previous reports showing that IL-6 can induce differentiation of activated CD8+ T cells into cytotoxic T cells 61 62 63 64, can promote activated CD4+ cell differentiation to the Th2 phenotype 39, and with the data shown here, cannot promote the survival of activated T cells.
A probable teleological reason for the lack of IL-6 rescue of activated cells is to provide selective protection of nonspecific T cells during antigen-driven cell death in vivo. Many activated T cells die during immune responses to antigen, perhaps to avoid accumulation of once useful but now functionless cells, or to prevent shock during a second exposure to the same antigen. Insensitivity to IL-6 may contribute to the loss of activated cells, as their death would not be prevented by this cytokine. Naive T cells may depend on IL-6 for maintenance of survival in specific environments in vivo such as spleen, LNs, or during travel through the circulatory system. Many cell types, such as endothelial cells and fibroblasts, constitutively secrete low levels of IL-6, thus providing resting T cells with plenty of opportunities to engage IL-6 16 65 66 67 68.
Acknowledgments
We thank Ella Kushnir and Chris Brown for typing the AD10 mice. We thank Anne Mette Buhl for assisting with the Western blots, and Dr. John Cambier for advice on cytokine signaling.
This work was supported by US Public Health Service grants AI17134, AI18785, and AI22295.
Footnotes
Abbreviations used in this paper: HPRT, hypoxanthine ribosyltransferase; Jak, Janus kinase; PI, propidium iodide; PIAS, protein inhibitor of activated Stat; SEA and SEB, staphylococcal enterotoxin A and B; SOCS, suppressor of cytokine signaling; Stat, signal transducer and activator of transcription.
References
- McDonagh M., Bell E.B. The survival and turnover of mature and immature CD8 T cells. Immunology. 1995;84:514–520. [PMC free article] [PubMed] [Google Scholar]
- Tough D.F., Sprent J. Life span of naive and memory T cells. Stem Cells. 1995;13:242–249. doi: 10.1002/stem.5530130305. [DOI] [PubMed] [Google Scholar]
- Tough D.F., Sprent J. Turnover of naive- and memory-phenotype T cells. J. Exp. Med. 1994;179:1127–1135. doi: 10.1084/jem.179.4.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprent J. T and B memory cells. Cell. 1994;76:315–322. doi: 10.1016/0092-8674(94)90338-7. [DOI] [PubMed] [Google Scholar]
- von Boehmer H., Hafen K. The life span of naive α/β T cells in secondary lymphoid organs. J. Exp. Med. 1993;177:891–896. doi: 10.1084/jem.177.4.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabe Y., Ochi A. Programmed cell death and extrathymic reduction of Vβ8+ CD4+ T cells in mice tolerant to Staphylococcus enterotoxin B. Nature. 1991;349:245–248. doi: 10.1038/349245a0. [DOI] [PubMed] [Google Scholar]
- McCormack J.E., Callahan J.E., Kappler J., Marrack P. Profound deletion of mature T cells in vivo by chronic exposure to exogenous superantigen. J. Immunol. 1993;150:3785–3792. [PubMed] [Google Scholar]
- Radvanyi L.G., Mills G.B., Miller R.G. Religation of the T cell receptor after primary activation of mature T cells inhibits proliferation and induces apoptotic cell death. J. Immunol. 1993;150:5704–5715. [PubMed] [Google Scholar]
- Kabelitz D., Pohl T., Pechhold K. Activation-induced cell death (apoptosis) of mature peripheral T lymphocytes. Immunol. Today. 1993;14:338–339. doi: 10.1016/0167-5699(93)90231-9. [DOI] [PubMed] [Google Scholar]
- Ucker D.S., Ashwell J.D., Nickas G. Activation-driven T cell death. I. Requirements for de novo transcription and translation and association with genome fragmentation. J. Immunol. 1989;143:3461–3469. [PubMed] [Google Scholar]
- Singer G.G., Abbas A.K. The Fas antigen is involved in peripheral but not thymic deletion of T lymphocytes in T cell receptor transgenic mice. Immunity. 1994;1:365–371. doi: 10.1016/1074-7613(94)90067-1. [DOI] [PubMed] [Google Scholar]
- Zheng L., Fisher G., Miller R.E., Peschon J., Lynch D.H., Leonardo M.J. Induction of apoptosis in mature T cells by tumor necrosis factor. Nature. 1995;377:348–351. doi: 10.1038/377348a0. [DOI] [PubMed] [Google Scholar]
- Duke R.C., Cohen J.J. IL-2 addictionwithdrawal of growth factor activates a suicide program in dependent T cells. Lymphokine Res. 1986;5:289–299. [PubMed] [Google Scholar]
- Dhein J., Walczak H., Baumler C., Debatin K., Krammer P.H. Autocrine T-cell suicide mediated by APO-1/(Fas/CD95) Nature. 1995;373:438–441. doi: 10.1038/373438a0. [DOI] [PubMed] [Google Scholar]
- Colotta F., Polentarutti N., Sironi M., Mantovani A. Expression and involvement of c-fos and c-jun protooncogenes in programmed cell death induced by growth factor deprivation in lymphoid cell lines. J. Biol. Chem. 1992;267:18278–18283. [PubMed] [Google Scholar]
- Teague T.K., Marrack P., Kappler J., Vella A.T. IL-6 rescues resting mouse T cells from apoptosis. J. Immunol. 1997;158:5791–5796. [PubMed] [Google Scholar]
- Vella T., Teague T.K., Ihle J., Kappler J., Marrack P. IL-4 or IL-7 prevents the death of resting T cellsStat6 is probably not required for the effect of IL-4. J. Exp. Med. 1997;186:325–330. doi: 10.1084/jem.186.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vella A.T., McCormack J.E., Linsley P.S., Kappler J.W., Marrack P. Lipopolysaccharide interferes with the induction of peripheral T cell death. Immunity. 1995;2:261–270. doi: 10.1016/1074-7613(95)90050-0. [DOI] [PubMed] [Google Scholar]
- Vella A.T., Dow S., Potter T., Kappler J., Marrack P. Cytokine-induced survival of activated T cells in vitro and in vivo. Proc. Natl. Acad. Sci. USA. 1998;95:3810–3815. doi: 10.1073/pnas.95.7.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildeman D.A., Mitchell T., Teague T.K., Henson P., Day B.J., Kappler J., Marrack P.C. Reactive oxygen species regulate activation-induced T cell apoptosis. Immunity. 1999;10:735–744. doi: 10.1016/s1074-7613(00)80072-2. [DOI] [PubMed] [Google Scholar]
- Takeda S., Rodewald H.-R., Arakawa H., Bluethmann H., Shimizu T. MHC class II molecules are not required for survival of newly generated CD4+ T cells, but affect their long-term life span. Immunity. 1996;5:217–228. doi: 10.1016/s1074-7613(00)80317-9. [DOI] [PubMed] [Google Scholar]
- Kirberg J., Berns A., von Boehmer H. Peripheral T cell survival requires continual ligation of the T cell receptor to major histocompatibility complex–encoded molecules. J. Exp. Med. 1997;186:1269–1275. doi: 10.1084/jem.186.8.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agea E., Bistoni O., Bini P., Migliorati G., Nicoletti I., Bassotti G., Riccardi C., Bertotto A., Spinozzi F. Costimulation of CD3/TcR complex with either integrin or nonintegrin ligands protects CD4+ allergen-specific T-cell clones from programmed cell death. Allergy. 1995;50:677–682. doi: 10.1111/j.1398-9995.1995.tb02585.x. [DOI] [PubMed] [Google Scholar]
- Boise L.H., Minn A.J., June C.H., Lindsten T., Thompson C.B. Growth factors can enhance lymphocyte survival without committing the cell to undergo cell division. Proc. Natl. Acad. Sci. USA. 1995;92:5491–5495. doi: 10.1073/pnas.92.12.5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boise L.H., Minn A.J., Noel P.J., June C.H., Accavitti M.A., Lindsten T., Thompson C.B. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity. 1995;3:87–98. doi: 10.1016/1074-7613(95)90161-2. [DOI] [PubMed] [Google Scholar]
- Cohen D.J., Tian Y., Ooi B.S., Henkart P.A. Differential effects of costimulator signals and interleukin-2 on T cell receptor-mediated cell death of resting and activated CD4+ murine splenic T cells. Transplant. 1996;61:486–491. doi: 10.1097/00007890-199602150-00029. [DOI] [PubMed] [Google Scholar]
- Lenardo M.J. Interleukin-2 programs mouse αβ T lymphocytes for apoptosis. Nature. 1991;353:858–861. doi: 10.1038/353858a0. [DOI] [PubMed] [Google Scholar]
- Mitchell T., Kappler J., Marrack P. Bystander virus infection prolongs activated T cell survival. J. Immunol. 1999;162:4527–4535. [PubMed] [Google Scholar]
- Julius M.H., Simpson E., Herzenberg L. A rapid method for the isolation of functional thymus-derived murine lymphocytes. Eur. J. Immunol. 1973;3:645–649. doi: 10.1002/eji.1830031011. [DOI] [PubMed] [Google Scholar]
- Ernst D.N., Weigle W.O., Noonan D.J., McQuitty D.N., Hobbs M.V. The age-associated increase in IFN-gamma synthesis by mouse CD8+ T cells correlates with shifts in the frequencies of cell subsets defined by membrane CD44, CD45RB, 3G11, and MEL-14 expression. J. Immunol. 1993;151:575–587. [PubMed] [Google Scholar]
- Jung T.M., Gallatin W.M., Weissman I.L., Dailey M.O. Down-regulation of homing receptors after T cell activation. J. Immunol. 1988;141:4110–4117. [PubMed] [Google Scholar]
- Kelly K., Shortman K., Scollay R. The surface phenotype of activated T lymphocytes. Immunol. Cell Biol. 1988;66:297–306. doi: 10.1038/icb.1988.39. [DOI] [PubMed] [Google Scholar]
- Hilton D.J., Richardson R.T., Alexander W.S., Viney E.M., Willson T.A., Sprigg N.S., Starr R., Nicholson S.E., Metcalf D., Nicola N.A. Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc. Natl. Acad. Sci. USA. 1998;95:114–119. doi: 10.1073/pnas.95.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohya K.i., Kajigaya S., Yamashita Y., Miyazato A., Hatake K., Miura Y., Ikeda U., Shimada K., Ozawa K., Mano H. SOCS-1/JAB/SSI-1 can bind to and suppress Tec protein-tyrosine kinase. J. Biol. Chem. 1997;272:27178–27182. doi: 10.1074/jbc.272.43.27178. [DOI] [PubMed] [Google Scholar]
- Starr R., Willson T.A., Viney E.M., Murray L.J., Rayner J.R., Jenkins B.J., Gonda T.J., Alexander W.S., Metcalf D., Nicola N.A., Hilton D.J. A family of cytokine-inducible inhibitors of signalling. Nature. 1997;387:917–921. doi: 10.1038/43206. [DOI] [PubMed] [Google Scholar]
- Minamoto S., Ikegame K., Ueno K., Narazaki M., Naka T., Yamamoto H., Matsumoto T., Saito H., Hosoe S., Kishimoto T. Cloning and functional analysis of new members of STAT induced STAT inhibitor (SSI) familySSI-2 and SSI-3. Biochem. Biophys. Res. Commun. 1997;237:79–83. doi: 10.1006/bbrc.1997.7080. [DOI] [PubMed] [Google Scholar]
- Naka T., Narazaki M., Hirata M., Matsumoto T., Minamoto S., Aono A., Nishimoto N., Kajita T., Taga T., Yoshizaki K. Structure and function of a new STAT-induced STAT inhibitor. Nature. 1997;387:924–929. doi: 10.1038/43219. [DOI] [PubMed] [Google Scholar]
- Endo T.A., Masuhara M., Yokouchi M., Suzuki R., Sakamoto H., Mitsui K., Matsumoto A., Tanimura S., Ohtsubo M., Misawa H. A new protein containing an SH2 domain that inhibits JAK kinases. Nature. 1997;387:921–924. doi: 10.1038/43213. [DOI] [PubMed] [Google Scholar]
- Rincon M., Anguita J., Nakamura T., Fikrig E., Flavell R.A. Interleukin (IL)-6 directs the differentiation of IL-4–producing CD4+ T cells. J. Exp. Med. 1997;185:461–469. doi: 10.1084/jem.185.3.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulie P.G., Stevens M., Van Snick J. High and low affinity receptors for murine interleukin 6. Eur. J. Immunol. 1989;19:2107–2114. doi: 10.1002/eji.1830191121. [DOI] [PubMed] [Google Scholar]
- Rayanade R.J., Ndubuisi M.I., Etlinger J.D., Sehgal P.B. Regulation of IL-6 signaling by p53STAT3- and STAT5-masking in p53-Val135-containing human hepatoma Hep3B cell lines. J. Immunol. 1998;161:325–334. [PubMed] [Google Scholar]
- Rayanade R.J., Patel K., Ndubuisi M., Sharma S., Omura S., Etlinger J.D., Pine R., Sehgal P.B. Proteasome- and p53-dependent masking of signal transducer and activator of transcription (STAT) factors. J. Biol. Chem. 1997;272:4659–4662. doi: 10.1074/jbc.272.8.4659. [DOI] [PubMed] [Google Scholar]
- Chung C.D., Liao J., Liu B., Rao X., Jay P., Berta P., Shuai K. Specific inhibition of Stat3 signal transduction by PIAS3. Science. 1997;278:1803–1805. doi: 10.1126/science.278.5344.1803. [DOI] [PubMed] [Google Scholar]
- Zhong Z., Wen Z., Darnell J.E., Jr. Stat3a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 1994;264:95–98. doi: 10.1126/science.8140422. [DOI] [PubMed] [Google Scholar]
- Ihle J.N. The Janus protein tyrosine kinase family and its role in cytokine signaling. Adv. Immunol. 1995;60:1–35. doi: 10.1016/s0065-2776(08)60582-9. [DOI] [PubMed] [Google Scholar]
- Chung J., Uchida E., Grammer T.C., Blenis J. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol. Cell. Biol. 1997;17:6508–6516. doi: 10.1128/mcb.17.11.6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich P.C., Behrmann I., Muller-Newen G., Schaper F., Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 1998;334:297–314. doi: 10.1042/bj3340297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujitani Y., Nakajima K., Kojima H., Nakae K., Takeda T., Hirano T. Transcriptional activation of the IL-6 response element in the junB promoter is mediated by multiple Stat family proteins. Biochem. Biophys. Res. Commun. 1994;202:1181–1187. doi: 10.1006/bbrc.1994.2053. [DOI] [PubMed] [Google Scholar]
- Lutticken C., Wegenka U.M., Yuan J., Buschmann J., Schindler C., Ziemiecki A., Harpur A.G., Wilks A.F., Yasukawa K., Taga T. Association of transcription factor APRF and protein kinase Jak1 with the interleukin-6 signal transducer gp130. Science. 1994;263:89–92. doi: 10.1126/science.8272872. [DOI] [PubMed] [Google Scholar]
- Harroch S., Revel M., Chebath J. Interleukin-6 signaling via four transcription factors binding palindromic enhancers of different genes. J. Biol. Chem. 1994;269:26191–26195. [PubMed] [Google Scholar]
- Akbar A.N., Borthwick N.J., Wickremasinghe R.G., Panayoitidis P., Pilling D., Bofill M., Krajewski S., Reed J.C., Salmon M. Interleukin-2 receptor common gamma-chain signaling cytokines regulate activated T cell apoptosis in response to growth factor withdrawalselective induction of anti-apoptotic (bcl-2, bcl-xL) but not pro-apoptotic (bax, bcl-xS) gene expression. Eur. J. Immunol. 1996;26:294–299. doi: 10.1002/eji.1830260204. [DOI] [PubMed] [Google Scholar]
- Radvanyi L.G., Shi Y., Vaziri H., Sharma A., Dhala R., Mills G.B., Miller R.G. CD28 costimulation inhibits TCR-induced apoptosis during primary T cell response. J. Immunol. 1996;156:1788–1798. [PubMed] [Google Scholar]
- Cory S. Regulation of lymphocyte survival by the bcl-2 gene family. Annu. Rev. Immunol. 1995;13:513–543. doi: 10.1146/annurev.iy.13.040195.002501. [DOI] [PubMed] [Google Scholar]
- Hirano T., Matsuda T., Nakajima K. Signal transduction through gp130 that is shared among the receptors for the interleukin 6 related cytokine subfamily. Stem Cells. 1994;12:262–277. doi: 10.1002/stem.5530120303. [DOI] [PubMed] [Google Scholar]
- Boulton T.G., Zhong Z., Wen Z., Darnell J.E., Jr., Stahl N., Yancopoulos G.D. STAT3 activation by cytokines utilizing gp130 and related transducers involves a secondary modification requiring an H7-sensitive kinase. Proc. Natl. Acad. Sci. USA. 1995;92:6915–6919. doi: 10.1073/pnas.92.15.6915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Z., Zhong Z., Darnell J.J. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82:241–250. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- Wen Z., Darnell J.E., Jr. Mapping of Stat3 serine phosphorylation to a single residue (727) and evidence that serine phosphorylation has no influence on DNA binding of Stat1 and Stat3. Nucleic Acids Res. 1997;25:2062–2067. doi: 10.1093/nar/25.11.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Blenis J., Li H.C., Schindler C., Chen-Kiang S. Requirement of serine phosphorylation for formation of STAT-promoter complexes. Science. 1995;267:1990–1994. doi: 10.1126/science.7701321. [DOI] [PubMed] [Google Scholar]
- Marrack P., Kappler J., Mitchell T. Type I interferons keep activated T cells alive. J. Exp. Med. 1999;189:521–530. doi: 10.1084/jem.189.3.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B., Liao J., Rao X., Kushner S.A., Chung C.D., Chang D.D., Shuai K. Inhibition of Stat1-mediated gene activation by PIAS1. Proc. Natl. Acad. Sci. USA. 1998;95:10626–10631. doi: 10.1073/pnas.95.18.10626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renauld J.C., Vink A., Van Snick J. Accessory signals in murine cytolytic T cell responses. Dual requirement for IL-1 and IL-6. J. Immunol. 1989;143:1894–1898. [PubMed] [Google Scholar]
- Quentmeier H., Klaucke J., Muhlradt P.F., Drexler H.G. Role of IL-6, IL-2, and IL-4 in the in vitro induction of cytotoxic T cells. J. Immunol. 1992;149:3316–3320. [PubMed] [Google Scholar]
- Ming J.E., Steinman R.M., Granelli-Piperno A. IL-6 enhances the generation of cytolytic T lymphocytes in the allogeneic mixed leucocyte reaction. Clin. Exp. Immunol. 1992;89:148–153. doi: 10.1111/j.1365-2249.1992.tb06894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski T.F., Renauld J.C., Van Pel A., Boon T. Costimulation with B7-1, IL-6, and IL-12 is sufficient for primary generation of murine antitumor cytolytic T lymphocytes in vitro. J. Immunol. 1995;154:5637–5648. [PubMed] [Google Scholar]
- Fabry Z., Fitzsimmons K.M., Herlein J.A., Moninger T.O., Dobbs M.B., Hart M.N. Production of the cytokines interleukin 1 and 6 by murine brain microvessel endothelium and smooth muscle pericytes. J. Neuroimmunol. 1993;47:23–34. doi: 10.1016/0165-5728(93)90281-3. [DOI] [PubMed] [Google Scholar]
- Kishimoto T. The biology of interleukin-6. Blood. 1989;74:17–26. [PubMed] [Google Scholar]
- Kruger-Krasagakes S., Moller A., Kolde G., Lippert U., Weber M., Henz B.M. Production of interleukin-6 by human mast cells and basophilic cells. J. Invest. Dermatol. 1996;106:75–79. doi: 10.1111/1523-1747.ep12327815. [DOI] [PubMed] [Google Scholar]
- Tosato G., Seamon K.B., Goldman N.D., Sehgal P.B., May L.T., Washington G.C., Jones K.D., Pike S.E. Monocyte-derived human B-cell growth factor identified as interferon-beta 2 (BSF-2, IL-6) Science. 1988;239:502–504. doi: 10.1126/science.2829354. [DOI] [PubMed] [Google Scholar]