

Abstract
Conserved differences between the transmembrane and cytoplasmic domains of membrane immunoglobulin (Ig)M and IgG may alter the function of antigen receptors on naive versus memory B cells. Here, we compare the ability of these domains to signal B cell allelic exclusion and maturation in transgenic mice. A lysozyme-binding antibody was expressed in parallel sets of mice as IgM, IgG1, or a chimeric receptor with IgM extracellular domains and transmembrane/cytoplasmic domains of IgG1. Like IgM, the IgG1 or chimeric IgM/G receptors triggered heavy chain allelic exclusion and supported development of mature CD21+ B cells. Many of the IgG or IgM/G B cells became CD21high and downregulated their IgG and IgM/G receptors spontaneously, resembling memory B cells and B cells with mutations that exaggerate B cell antigen receptor signaling. Unlike IgM-transgenic mice, “edited” B cells that carry non–hen egg lysozyme binding receptors preferentially accumulated in IgG and IgM/G mice. This was most extreme in lines with the highest transgene copy number and diminished in variant offspring with fewer copies. The sensitivity of B cell maturation to transgene copy number conferred by the IgG transmembrane and cytoplasmic domains may explain the diverse phenotypes found in other IgG-transgenic mouse strains and may reflect exaggerated signaling.
Keywords: B lymphocyte, development, isotype switch, allelic exclusion, transgene
Introduction
B cell antigen receptors (BCRs) undergo stereotypic changes in their constant regions during development of each B lymphocyte. B cells of the preimmune repertoire express first IgM and then IgM and IgD receptors of the same antigen specificity 1 2 3 4, whereas memory B cells either express IgM or switch to downstream isotypes such as IgG, IgA, or IgE 5 6 7. The constant regions of these isotypes differ markedly not only in the CH1, hinge, and Fc domains but also in the membrane-proximal spacer, transmembrane, and cytoplasmic segments required for cell surface expression. Interestingly, the latter domains are highly conserved within isotypes and between species 8 9 10 11 12 13 14 15. For example, the different IgG subtypes have an almost identical amino acid sequence in their extracellular spacer, transmembrane, and cytoplasmic regions in mice and humans, whereas they are more divergent in the hinge and Fc domains 16 17 18. In addition, the cytoplasmic tails of IgM and IgD receptors consist of only three amino acids (KVK), whereas receptors expressed uniquely on memory cells have more extended cytoplasmic tails of either 28 (IgG, IgE) or 14 (IgA) evolutionarily conserved amino acids 17 18 19. The highly conserved and isotype-specific nature of the membrane proximal regions suggests stage-specific signaling or intracellular trafficking functions attributed to the different isotypes 17 18 19 20. In support of this notion, IgG1 or IgE memory B cell production and antibody responses in mice are diminished by mutations that truncate the cytoplasmic tail of IgG1 or IgE, respectively 21 22.
Comparing the function of naive and memory BCR isotypes in vivo is normally complicated by differences in BCR affinity, specificity, B cell location, and activation state that are also brought about by immunization. Any difference between the behavior of naive IgM-bearing B cells and primed IgG-bearing cells would be difficult to ascribe to the BCR isotype or to priming. These variables can be separated, in principle, by constructing transgenic mice in which IgG or another downstream isotype is constitutively expressed as the sole BCR on B cells independent of any need for immunization. A number of research groups have constructed IgG-transgenic mice with different VH/VL specificities, but these have yielded variable results and conclusions 23 24 25 26 27 28 29 30. In some studies, the γ heavy chain (Hc) signaled allelic exclusion, at least to the extent that endogenous IgM expression was inhibited in the bone marrow 27 28 30 31, whereas in other studies allelic exclusion was not apparent 23 24. Of the mice that failed allelic exclusion, the presence of membrane IgG was either not directly assessed 23 or very low 24, suggesting that the failure of allelic exclusion in these mice may be a reflection of little or no surface IgG expression. The polyadenylation sites of the M2 exon for Cγ1, Cγ2b, and Cγ3 have not yet been precisely located by DNA sequencing, and failure to achieve surface IgG expression in some experiments may have been due to lack of the pA sites in the DNA constructs 24 or lack of inclusion of membrane exons due to their deletion during the integration process 23.
Another variable finding in IgG-transgenic mice has concerned the capacity of IgG to substitute for IgM in supporting B cell maturation from immature B cells in the bone marrow through to mature recirculating cells in the spleen and lymph nodes. The most detailed studies to date are those of Roth and coworkers 27 29 in multiple lines of transgenic mice carrying the IgG2b Hc for an anti-Pseudomonas antibody. With the exception of one unusual variant line, the transgenic IgG receptor did not support maturation, and the majority of spleen and lymph node B cells that developed expressed low levels of IgG and bore endogenous IgM and IgD receptors 27 28 29. Similarly, very few IgG only–bearing cells were found in the periphery of mice constructed by Yamamura et al., Tsao et al., Offen et al., and Battegay et al. 23 24 25 30. By contrast, mature B cells expressing exclusively IgG were found in large numbers in one transgenic line carrying an anti-Pseudomonas IgG2b Hc 28 29, and modest numbers were found in transgenic mice carrying an antibacterial phosphorylcholine IgG2b transgene (Tg; reference 28). The reason for these differences is unclear, leaving unresolved the extent to which IgG may differ from IgM or IgD in its capacity to signal B cell maturation in the preimmune repertoire.
To compare the in vivo function of IgG1 and IgM as antigen receptors on B cells directly, independent of any differences in VH/VL specificity, microenvironment, state of priming, or antigenic experience, we have generated transgenic mice carrying Hc and Lc (light chain) genes encoding a well characterized lysozyme-binding antibody 32 33 of IgG1 isotype. These mice could then be directly compared with previously established IgM-transgenic mice carrying the same antilysozyme V regions. To examine the role of the conserved IgG transmembrane/cytoplasmic tail region in isolation, an additional set of transgenic mice was made expressing an IgM/G chimeric receptor comprising the IgM CH1 and Fc regions and the IgG1 extracellular spacer, transmembrane, and cytoplasmic domains. We find that the IgG1 and IgM/G receptors can substitute for IgM in supporting generation of large numbers of recirculating B cells in spleen and lymph nodes. Unlike IgM- or IgD-transgenic mice, the numbers of mature B cells expressing transgenic BCRs in IgG1 and IgM/G mice is very sensitive to Tg copy number. Few are present in blood, spleen, or lymph node in higher copy number lines, where they are replaced by B cells with different BCRs. The characteristics of B cell development in these animals are most consistent with enhanced signaling by IgG BCRs conferred in part by the unique membrane/tail domains.
Materials and Methods
Gene Constructs.
IgM-transgenic mice were produced previously by coinjecting Hc (VH10–μ) and Lc (Vk10–Ck) Ig gene constructs into the germline of C57BL/6 (Hc b-allotype, IgHb) mice 34 35. These constructs together encode IgM (Hc a-allotype, IgHa) carrying the antigen binding site of the high-affinity (1.5 × 109 M−1) anti–hen egg lysozyme (HEL) mAb HyHEL10 32 33. The IgG1 gene construct was produced from a plasmid, pTB6, which contained a genomic clone of the productive H locus from hybridoma HyHEL10, carrying the promoter, L–VDJ exons, the μ/γ1 switch recombination region, the γ1 constant domains, and the first γ1 membrane exon (from Drs. T. Lavoie and S. Smith-Gill, National Institutes of Health, Bethesda, MD). The insert from pTB6 was cloned into plasmid pSVG-γM2 containing the γ1 M2 exon plus ∼2.5 kb of downstream sequence, derived from λ phage clone G1.2 36. The Vκ10–Cκ gene construct was described previously 34.
The chimeric IgM/G Hc gene construct was generated by “sticky feet–directed mutagenesis” 37. In brief, oligonucleotide primers were synthesized in which the 5′ 30 nucleotides corresponded to the IgM nucleotides flanking the IgG1 insertion site (uppercase letters), and the 3′ 15 nucleotides corresponded with the DNA flanking the IgG1 insertion sequences (lowercase letters). The primers used were as follows: forward, 5′-GACCCTCCCTCTCTGTGTCCCTTCATAGAGgggctgcaactggacgag-3′; reverse: 5′-GTCTCTGCTGTCCTTCCATGCTGAGAGctagggcgcttgcccaatc-3′. After mutagenesis, a KpnI–SpeI fragment containing the modified membrane exons was inserted into a pSVG plasmid containing the IgM constant domain exons and HyHEL10 V region 35.
Transgenic Lines.
For microinjection, IgG1 and IgM/G gene constructs were separated from vector sequences by digestion with ClaI and NotI restriction enzymes and purified as described 34. IgG1 (VH10–γ + Vk10–Ck) and IgM/G (VH10–μγ + Vk10–Ck) founder Ig-transgenic mice were generated after microinjection of embryos from (C57BL/6 × CBA/J)F1 females with an equimolar mixture of Hc and Lc constructs as described 34 38. Screening for founder Ig-transgenic mice and initial progeny was performed by Southern blot analysis of tail DNA 34 38. Routine screening for transgenic Hc was done using PCR (reference 39; for details see http://jcsmr.anu.edu.au/group_pages/mgc/). Transgenic lines were maintained by backcrossing to C57BL/6J. In addition, two Ig-transgenic lines, IgM/G (μγ2) and IgG1 (γγ4), were crossed with recombinase activating gene (RAG-2)−/− mice originally provided by Dr. Fred Alt (Children's Hospital, Boston, MA). Generation of IgM-transgenic line was described previously 35.
For Hc copy number estimates, Southern blots of XbaI-digested tail DNA were probed with a 912 bp HindIII–XbaI fragment from the germline JH4 region. The JH probe identified a 3.6-kb fragment derived from unrearranged endogenous Hc genes in all mice and a single 3.0-kb fragment from the rearranged Hc Tg. For Lc copy number estimates, the same XbaI-digested Southern blots were reprobed with a 650-bp AvaI–XbaI fragment from the germline Jκ4 region, revealing a 3.8-kb endogenous κ fragment and a 2.6-kb Tg κ band. Autoradiographs in the linear range of the film were scanned on a Molecular Dynamics scanning densitometer, areas under the curves were measured, and copy numbers were expressed by assigning the endogenous signals a copy number of two. For visualizing loss of array copies, Southern blots of SacI-digested tail DNA were probed with a 912-bp HindIII–XbaI fragment from the germline JH4 region. Because there is no Sac1 site 5′ to the JH probe within the microinjected Hc construct, this results in an 11-kb fragment generated by head-to-tail junction fragments with upstream Hc copies in tandem arrays and a unique fragment at the 5′ end of the array.
FACS® Analysis.
The following antibodies were used (specificities in parentheses): RS3.1 (IgMa; reference 40); AMS-9.1 (IgDa; reference 41); AFS122.2 (IgDb); RA3-6B2 (B220; reference 42); HyHEL9 (HEL; reference 43); S7 (CD43); M1/69 (HSA); and 7G6 (CR1/2; reference 44). Spleen and bone marrow cells were prepared and stained for FACS® analysis as previously indicated 34. HEL binding was detected with 200 ng/ml of HEL followed by HyHEL9–tricolor conjugate. Analysis was performed on a FACScan™ flow cytometer (Becton Dickinson) with FACS® desk software (Beckman Center Shared FACS® Facility).
Results
Constructs and Transgenic Mice.
To allow direct comparison between the function of IgM and IgG1 antigen receptors, we modified a rearranged Hc gene construct that was used previously to produce IgM Hc- and Lc-transgenic mice 35. The original μ construct and the two modified Hc constructs used in this study, γ1 and μ/γ1, employed identical VDJH variable region elements derived from the original lysozyme-binding hybridoma, HyHEL10 32. The chimeric μ/γ1 construct was generated by selectively replacing the μ membrane coding elements with γ1 membrane coding sequences using sticky feet–directed mutagenesis to preserve the μM1 splice acceptor and the μM2 3′ untranslated regions and poly A signals. As illustrated in Fig. 1, the extracellular CH1–CH4 portion of the μγ1 Hc was identical to the IgM extracellular region, thereby enabling use of identical reagents for analysis and allowing any phenotypic differences to be directly attributed to the γ1 membrane-coding segment. The IgG1 Hc construct was derived from genomic DNA of the original lysozyme-binding HyHEL10 IgG1+ hybridoma, which had retained the μ/γ switch regions (Fig. 1 C). Inclusion of the switch region has been reported to be essential for high-level expression of human IgG1 Tgs 26. An additional genomic fragment encompassing ∼2.5 kb 3′ of the γM2 exon was also included to ensure presence of the γM poly A signals. As done previously with the IgM construct 35, the modified Hc constructs were microinjected with the κ Lc gene construct derived from the HyHEL10 hybridoma. Multiple founders were obtained with each construct, and breeding lines were established from each (Table ). Different numbers of copies of the Hc and Lc Tgs were present in each line, and these Hc and Lc genes cosegregated in all progeny, indicating cointegration in each of the founders.
Figure 1.

Schematic representation of Hc constructs used to generate antilysozyme IgM- (A), IgG1- (B), and IgM/G-transgenic (C) mice. Wide boxes indicate protein coding regions, medium boxes indicate untranslated regions or switch recombination sequences, and filled oval represents the μ intron enhancer. Unfilled boxes indicate μ-specific sequences, black boxes indicate γ1 sequences, and gray boxes are shared sequences. E, EcoRI; S, SpeI, K, KpnI. (D) Schematic representation of membrane receptors generated from constructs diagrammed in A, B, and C after coinjecting the different Hc genes with the same antilysozyme κ Lc gene.
Table 1.
Splenic B Cells with HEL-binding Receptors in Lines of IgM-, IgM/G-, and IgG1-transgenic Mice
| Encoded receptor (line) | Tg copy nos.Hc/Lc | No. of mice | Mean age | No. of B cells/spleen | HEL-binding cells/spleen | B cells/spleen | HEL-binding cells/spleen | B cells that bind HEL |
|---|---|---|---|---|---|---|---|---|
| wk | ×10−7‡ | % | % | % | ||||
| Nontransgenic | 0/0 | 11 | 7 ± 2 | 6.1 ± 2.8 | 0.2 ± 0.1 | 34.8 ± 8.0 | 0.2 ± 0.1 | 0.3 ± 0.1 |
| IgM | 2/2 | 7 | 12 ± 4 | 1.4 ± 1.8 | 1.2 ± 0.3 | 13.4 ± 3.6 | 11.2 ± 4.0 | 83 ± 12 |
| IgM/G (μγ6) | 2/2 | 5 | 10 ± 2 | 2.4 ± 1.1 | 1.2 ± 0.9 | 20.9 ± 5.4 | 9.9 ± 5.2 | 48 ± 27 |
| IgM/G (μγ2) | 3/1 | 13 | 12 ± 2 | 1.1 ± 0.5 | 0.5 ± 0.3 | 15.3 ± 3.7 | 7.3 ± 2.9 | 47 ± 10 |
| IgM/G (μγ1) | 8/4 | 6 | 9 ± 4 | 2.0 ± 1.3 | 0.2 ± 0.2 | 25.7 ± 7.8 | 2.0 ± 0.7 | 11 ± 8 |
| IgM/G (μγ8A) | 15/12 | 5 | 13 ± 3 | 1.6 ± 0.7 | 0.2 ± 0.1 | 25.1 ± 9.9 | 1.6 ± 0.7 | 9 ± 5 |
| IgG1 (γγ2) | 2/2 | 2 | 15 ± 1 | 6.0 ± 2.2 | 2.5 ± 0.6 | 32.8 ± 12.3 | 13.7 ± 3.5 | 42 ± 5 |
| IgG1 (γγ4) | 2/3 | 4 | 16 ± 4 | 2.7 ± 0.9 | 0.7 ± 0.3 | 26.5 ± 2.6 | 7.2 ± 0.9 | 27 ± 5 |
| IgG1 (γγ3) | 2/3 | 4 | 17 ± 7 | 1.8 ± 0.3 | 0.04 ± 0.03 | 22.3 ± 4.6 | 0.5 ± 0.3 | 2 ± 2 |
| IgG1 (γγ1) | 6/5 | 3 | 25 ± 9 | 3.2 ± 1.6 | 0.08 ± 0.05 | 19.1 ± 4.2 | 0.9 ± 0.7 | 5 ± 4 |
Expression of Transgenic Receptors on Peripheral B Cells in Transgenic Mice.
Transgenic offspring from each of the founders were analyzed by flow cytometry for the presence of splenic B cells that expressed high-affinity lysozyme-binding receptors encoded by pairing of the transgenic Hcs and Lcs. B cells that stained intensely with 10 nM HEL, which selectively identifies high-affinity cells carrying the transgenic Hcs and Lcs 45, were present in most of the lines, although the level of HEL-binding receptors per cell was four- to fivefold lower on mature IgG1 cells compared with IgM-transgenic mice (Fig. 2). As is illustrated in Fig. 2 and Table , a much lower proportion of B cells bound HEL in the preimmune splenic repertoire of IgG1- and IgM/G-transgenic mice compared with the representative IgM-transgenic mouse line, MM4. Whereas 80% of peripheral B cells in IgM-transgenic mice stained intensely with lysozyme (Fig. 2A and Fig. E), between 2 and 50% of splenic B cells in IgG1 and IgM/G mice showed appreciable HEL binding (Fig. 2B, Fig. C, Fig. F, and Fig. G). The absolute number of HEL-binding B cells in the spleen was nevertheless comparable in IgM and the lower copy number IgM/G and IgG1 lines, but it was also markedly lower in high copy number IgM/G and IgG1 lines (Table ).
Figure 2.

Representative flow cytometric analysis of peripheral B cells in IgM-, IgM/G-, and IgG1-transgenic mice. (A–H) Spleen cells were stained for high-affinity HEL binding, which depends on surface expression of receptors comprised of the transgenic Hc and Lc (y-axis), and for B220 (x-axis). Numbers next to boxes indicate the percentages of splenocytes that were HEL-binding B lymphocytes (upper boxes) or non–HEL-binding B lymphocytes (lower boxes). (I–L) B220+ splenocytes from IgM (I and K), IgM/G μγ2 (J), or IgG1 γγ4 (L) were stained for HEL binding (y-axis) versus Tg-encoded Hc expression (IgMa, x-axis for upper profiles; IgG1, x-axis for lower profiles). Note that in IgG1-transgenic mice, most B cells that do not bind lysozyme lack cell surface expression of the transgenic IgG1 Hc, whereas in the IgM/G mice the non–HEL-binding cells still bear the transgenic IgMa Hc.
Three-color staining confirmed that HEL-binding B cells in each case expressed Tg-encoded Hcs (Fig. 2I–L). Thus, in the IgM/G mice, the HEL binding B cells all carried IgM of the Tg-encoded a-allotype (IgMa; Fig. 2 J), and the staining for IgMa on these cells was tightly correlated with HEL binding amongst individual spleen cells, falling on a diagonal line. Similarly, HEL-binding B cells in the IgG-transgenic lines stained proportionally for IgG1 Hcs (Fig. 2 L). By contrast, the B cells that lacked HEL-binding receptors differed between the IgG1- and IgM/G-transgenic lines in their expression of transgenic Hc. In the IgG1-transgenic lines, lack of HEL binding was largely due to absence of Tg-encoded IgG1 Hcs on the cell surface (Fig. 2 L). By contrast, most B cells that lacked HEL-binding receptors in the IgM/G lines still expressed the Tg-encoded a-allotype Hc, as demonstrated by off-diagonal B cells that stained for IgMa but not HEL (Fig. 2 J).
Endogenous Hcs were present on most non–HEL-binding B cells in IgG1 mice but not in IgM/G mice, but in both cases, cells bearing HEL-binding receptors in these mice appeared allelically excluded and lacked detectable endogenous Hc expression. Fig. 3 shows histograms for staining of IgD on HEL-binding B cells (bold line) and non–HEL-binding B cells (thin line) from IgM-, IgG1-, and nontransgenic mice (upper panels) and IgM-, IgM/G-, and nontransgenic mice (lower panels). IgD is a useful measure of allelic exclusion because it can only be expressed if rearrangement of endogenous Hc genes occurs. Thus, HEL-binding B cells from all three types of transgenic mice, IgM, IgG1, and IgM/G, were negative for IgD expression (Fig. 3D, Fig. E, Fig. J, and Fig. K), indicating allelic exclusion of endogenous receptor expression.
Figure 3.
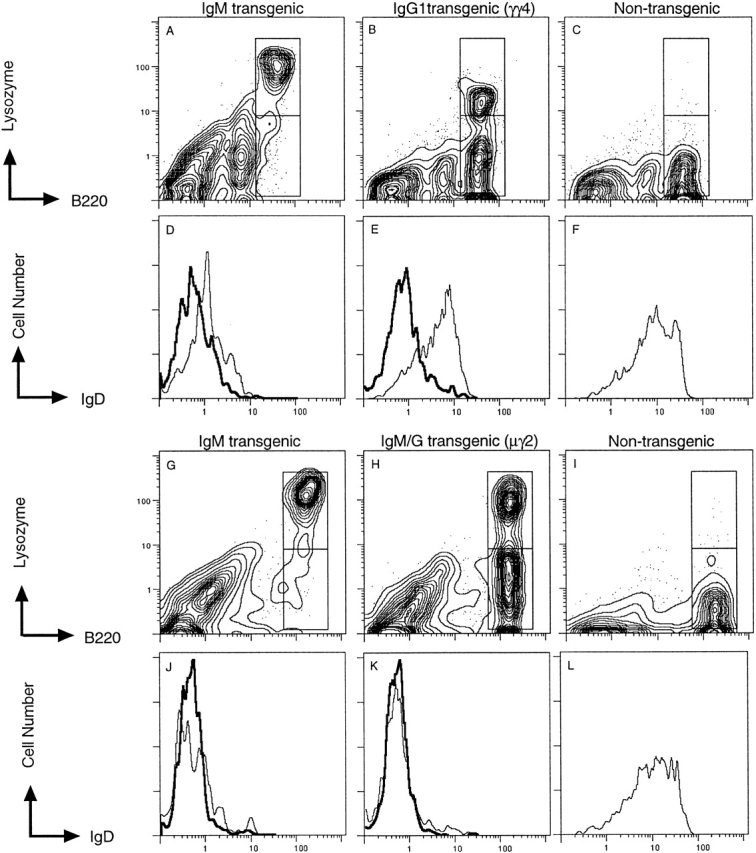
Flow cytometric analysis of endogenous Hc expression by HEL-binding and non–HEL-binding B cells. Splenocytes were stained for HEL binding (lysozyme), B220, and IgD (a combination of IgDa and IgDb). For each spleen, HEL-binding (upper boxed region) and non–HEL-binding (lower boxed region) B220+ cells were analyzed for IgD expression, which is displayed as a histogram below each profile. Histograms for HEL-binding B cells are shown by the bold line and for non–HEL-binding B cells by the thin line.
Influence of Hc Tg Copy Number on Peripheral B Cell Composition.
Analysis of multiple animals from each of the IgG1- and IgM/G-transgenic lines revealed consistent line-to-line differences in the proportion and number of HEL-binding B cells in the spleen (Table ). There was a clear tendency for these to be lowest in mice carrying more copies of the IgM/G or IgG1 Hc Tgs. For example, in the IgM/G (μγ6) line with 2 Hc copies, 48% of spleen B cells (1.2 × 107 cells) were HEL binding, whereas in the IgM/G (μγ8) line with 15 Hc copies, 9% of spleen B cells (0.2 × 107 cells) were HEL binding. No such effect was previously observed in the set of 10 Hc- and Lc-transgenic mice established with the IgM construct despite a similar range of copy numbers (reference 35 and Goodnow, C., unpublished observations). The inverse correlation between copy number and number of HEL-binding cells was not perfect, however, because one IgG1 line, γγ3, contained very few HEL-binding cells despite containing only two copies of the Hc and three copies of the Lc Tgs (Table ). It is conceivable that the Tgs in this mouse had integrated in a site or way that was nonpermissive to transcription.
Loss-of-copy variant offspring from high copy lines allowed the effect of copy number to be examined independently from integration site. Flow cytometry and Southern blot analysis revealed two separate low copy variant mice from 32 transgenic offspring of the μγ8A IgM/G line. The parent line contained 15 copies of the Hc Tg and 12 Lc copies (Table ), and only 6% of B cells in the blood had HEL-binding receptors (Fig. 4A and Fig. E). One variant offspring, μγ8C, carried only five copies of the Hc Tg (Fig. 4 D), and the frequency of HEL-binding B cells was increased to 52% (Fig. 4 C). Another variant, 8B, carried seven Hc copies and three Lc copies, and the proportion of HEL-binding B cells was increased to 18%. The 8B variant mouse was successfully bred as a subline, and the offspring consistently produced threefold higher proportions of peripheral HEL-binding B cells than the parental line (Fig. 4 E).
Figure 4.

Increased numbers of HEL-binding B cells in spontaneous copy loss variants of a high copy IgM/G line. Two spontaneous copy loss variants were noted during Southern blot typing of offspring from the IgM/G (μγ8A) strain. (A–C) B220+ cells in tail blood from the parental IgM/G (μγ8A) mouse line (A) and from the two variant offspring (B and C) were stained for HEL binding and Hc Tg (IgMa) expression. The percentage of B220+ cells falling in the windows is shown. (D) Southern blot analysis of SacI-digested tail DNA from the same mice, probed with JH fragment. The endogenous Hc germline band of 3 kb (two copies per cell) provides an internal standard for loading. The Hc Tg array yields many more copies of an 11-kb fragment resulting from head–tail junctions between copies. Smaller fragments of variable abundance presumably result from internal deletions and junctions with flanking DNA. (E) Percent of B220+ cells with HEL-binding BCRs in peripheral blood from eight offspring of the parental IgM/G line, μγ8A, and from eight offspring of the variant mouse, μγ8B. Data point marked with a single asterisk denotes variant mouse μγ8C. The mouse denoted by double asterisk was not analyzed by Southern blot.
A similar loss-of-copy variant was observed in one of four transgenic offspring analyzed from the high copy γγ1 IgG1 line, which contained six copies of Hc Tg and five copies of Lc Tg. In three Tg+ γγ1 offspring, <3.6% of spleen B cells bound lysozyme, whereas in the fourth Tg+ mouse, >14% of spleen B cells were HEL binding (not shown). Analysis of genomic DNA from these mice by Southern analysis revealed that the mouse with more HEL-binding B cells had lost two copies of the Hc Tg, resulting in four copies of Hc Tg and five copies of Lc Tg (not shown).
HEL-binding IgG1 or IgM/G Receptors Are Expressed on Most Immature B Cells, and the Levels on Early Pro-B Cells Correlate with Tg Copy Number.
In contrast to the variable proportion of mature B cells expressing the transgenic Hcs and Lcs in IgG1- or IgM/G-transgenic mice, the majority of immature B cells in the bone marrow nevertheless expressed HEL-binding surface receptors (Fig. 5). Moreover, the number of surface receptors on pro-BI B cells correlated with Tg copy number. Fig. 5 shows representative staining for HEL-binding receptors on the surfaces of pro-BI cells (Hardy's fraction A; HSAlowB220lowS7+; reference 46) in the bone marrow from IgM- and IgM/G-transgenic mice. Whereas pro-BI cells from IgM-transgenic mice did not express detectable HEL-binding receptors (Fig. 5 B), pro-BI cells from IgM/G-transgenic mice with 2, 8, and 15 copies exhibited increasing levels of Tg-encoded receptors (Fig. 5C–E, and Table ). A similar correlation of Tg copy number with surface expression on pro-BI cells was observed in IgG1 lines, where IgG1 with six copies of Hc Tg (γγ1) exhibited sixfold higher levels of HEL binding than the IgG1 line with two Tg copies (Table ).
Figure 5.

Expression of lysozyme-binding surface receptors on pro-BI cells is correlated with Hc Tg copy number. Bone marrow cells were analyzed by four-color analysis for S7, HSA, B220, and lysozyme binding. In profiles A–E, S7+B220+ cells were gated to display HSA and lysozyme binding. The most immature fraction A/pro-BI cells (HSAloS7+B220+) are in the boxed regions. Note that little lysozyme binding can be detected on these cells from IgM or low copy number IgM/G mice, but this increases 10-fold in the high copy strain. No consistent differences in the frequencies of fraction A cells in the bone marrow were noted between the different copy number lines, as the mean frequencies were: MM4, 0.91%; μγ2, 1.28%; μγ1, 1.20%; and μγ8, 0.97%.
Table 2.
Mean Fluorescence of HEL-binding Receptor Levels in IgM/G Pro-B Cells and IgG1 Pro-B Cells
| Tg line | Tg copy no. | Analysis 1 | Analysis 2 | Analysis 3 | Analysis 4 | Analysis 5 | Analysis 6 | Mean ± SD |
|---|---|---|---|---|---|---|---|---|
| IgM | 2 | 0.15 | 0.15 | 0.15 | 0.15 | – | 2.0 | 0.5 ± 0.8 |
| IgM/G μγ2 | 3 | 1.8 | 2.0 | 3.0 | 1.5 | 4.0 | 3.5 | 2.6 ± 1.0 |
| IgM/G μγ1 | 8 | 8.0 | 6.0 | 8.0 | 7.0 | 15.0 | 15.0 | 10.4 ± 4.3 |
| IgM/G μγ8 | 15 | – | 15.0 | – | 25.0 | 25.0 | 30.0 | 23.8 ± 6.3 |
| IgM | 2 | 0.15 | 0.15 | – | – | – | – | 0.15 ± 0.8 |
| IgG1 γγ4 | 2 | 0.5 | 0.6, 0.5, 0.5 | – | – | – | – | 0.5 ± 0.1 |
| IgG1 γγ1 | 6 | 4.0 | 3.5, 3.0, 1.5 | – | – | – | – | 3.0 ± 1.1 |
Bone marrow cells were stained as in Fig. 5. Mean fluorescence levels of HEL-binding receptors on pro-B1 (B220+S7+HSAlow) cells from six (IgM/G) and two (IgG1) separate FACS® analyses were tabulated.
Rescue of B Cell Maturation in RAG-2− /− Mice.
The data above indicated that large numbers of mature B cells bearing HEL-binding IgM/G or IgG receptors could develop in low copy transgenic lines, and that the majority of these HEL-binding cells did not express detectable endogenous Hcs. To confirm that the IgG1 and IgM/G Hcs in these low copy lines were indeed sufficient to provide the signals necessary for B cell maturation in the preimmune repertoire, representative low copy lines were crossed to RAG-2 gene–deficient mice 47. B cells from RAG-2−/− mice are blocked in development in the bone marrow at the c-kit+IL-2R−S7hiCD45R(B220)low large pro-B cell stage of development (Fig. 6B and Fig. F) due to lack of a signal normally transmitted by the pre-B cell receptor 47 48. As previously found with IgM/IgD-transgenic mice 49 50, inheritance of the anti-HEL IgM, IgG1, or IgM/G antigen receptor Tgs was sufficient to rescue pro-B cell development in RAG-2−/− mice. Thus, the appearance of S7lowB220low small B cells was restored in IgG1 RAG-2−/− bone marrow (Fig. 6C and Fig. G, boxed areas and histograms) and in IgM and IgM/G RAG-2−/− mice (not shown).
Figure 6.
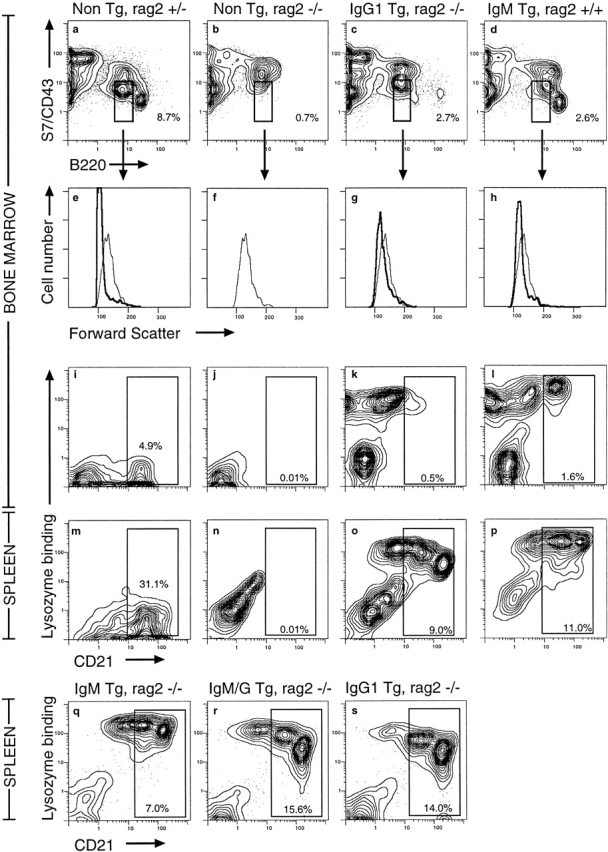
Tg-encoded IgG1 antigen receptors rescue bone marrow B cell development and B maturation in RAG-2−/− mice. (a–h) Bone marrow cells from nontransgenic, RAG-2+/− (a and e), nontransgenic, RAG-2−/− (b and f), IgG1-transgenic, RAG-2−/− (c and g), or IgM-transgenic, RAG-2+/+ mice (d and h) were stained with anti-CD43 (S7) and anti-CD45 (B220) antibody and analyzed by flow cytometry. Boxed cells in a–d were gated to yield forward scatter histograms in e–h as a measure of cell size (bold lines). For reference, the thin line in each display is the histogram of the S7lowB220low nontransgenic RAG-2−/− pro-B cell population. Numbers represent percentage of bone marrow cells falling within boxed region. (i–p) Bone marrow (i–l) and spleen cells (m–p) of the same mice were stained for B220, HEL binding, and CD21. Profiles show gated B220+ cells, and the percentage of bone marrow or spleen cells falling within the CD21+ mature B cell window is indicated. (q–s) Comparison of CD21 expression and HEL-binding receptors on B220+ cells in the spleens of RAG-2−/− mice carrying the IgM, IgM/G, or IgG BCR Tgs.
To determine whether IgG1 antilysozyme antigen receptors could also provide the signals necessary for immature B cells to complete their development into mature circulating B cells 51 52, spleen and bone marrow HEL-binding B cells were stained for expression of the mature B cell marker CD21. Immature B cells are marked by low or negative levels of CD21, whereas mature B cells express medium to high levels, the latter being primarily marginal zone B cells 53 54. B lineage cells from nontransgenic RAG-2−/− mice are blocked at the CD21−stage in the bone marrow (Fig. 6 J). By contrast, B cells from IgM RAG-2−/−, IgG1 RAG-2−/−, and IgM/G RAG-2−/− mice mature to the CD21low stage in the bone marrow without any discernible difference (Fig. 6 K and data not shown) and proceed to the CD21med and CD21high stages in the spleen in comparable numbers (Fig. 6Q). These results establish that IgG1 antigen receptors can provide the necessary signals for B cell maturation.
Spontaneous downregulation of surface IgG1 antigen receptors compared with IgM receptors accompanied B cell maturation in RAG-2−/− IgG1-transgenic mice, as noted above in the RAG-2+/+ background (Fig. 2). Thus, IgM and IgG1 B cells initially carried similar surface receptor levels at the CD21− immature B cell stage in the bone marrow (Fig. 6K and Fig. L). As the cells matured and increased CD21 expression, IgM receptor levels increased twofold (Fig. 6L, , and Q), whereas IgG1 receptor levels decreased threefold (Fig. 6K, Fig. O), and IgM/G receptor levels decreased twofold (Fig. 6 R).
Discussion
In this study, we have compared the function of a memory isotype BCR with its naive IgM counterpart during B cell development in vivo, independent of confounding factors such as prior antigenic experience, specificity differences, and microenvironment. A well characterized lysozyme-specific antibody combining site 32 33 was used to generate transgenic mice expressing IgG1 BCRs or chimeric IgM/G receptors composed of IgM extracellular regions and IgG1 membrane and cytoplasmic regions. The data show that IgG1 antigen receptors can substitute for IgM in supporting B cell allelic exclusion, development, and maturation in the preimmune repertoire. Thus, large numbers of IgG1 or IgM/G Tg receptor–expressing B cells were present in the periphery, and these cells did not coexpress endogenous IgM or IgD receptors. Several features of B cell development nevertheless differed markedly in IgG1- and IgM/G-transgenic mice compared with IgM-transgenic animals, indicating that functional differences between these isotypes do exist and are due at least in part to the unique IgG transmembrane region and cytoplasmic tail.
IgG1 Antigen Receptor Transmits Signals Required for B Cell Development and Allelic Exclusion.
The absolute requirement for membrane μ to signal the transition from the pro-B to pre-B cell stage and to cease further Hc gene rearrangement was demonstrated in recent years using mutant mice generated by targeted disruption of genes involved in expression or signaling of μ Hc 47 55 56 57 58 59. In all of these mice, development was blocked at the pro-B cell/large pre-B cell stage, with few small pre-B cells or immature B cells detected in the bone marrow. Given the structural differences in the IgM and IgG membrane-proximal regions, it has been suggested that the IgG Hc may not be able to provide the necessary signals for pro-B to B cell transition 27 29 and allelic exclusion 23 24 26. The findings here demonstrate that IgG1 can transmit the signals for allelic exclusion and B cell maturation, but this is complicated in mice with supraphysiological gene dosage. Thus, expression of IgG1 antigen receptor in low copy transgenic mice rescued B cell maturation in RAG-2−/− mice, as demonstrated by appearance of S7lowB220low small B cells in bone marrow and production of mature CD21+ B cells in the periphery. Secondly, analysis of surface receptor isotypes expressed on mature B cells of IgG1 mice did not reveal a significant population of cells coexpressing endogenous receptors with IgG1 receptors, demonstrating that IgG1 can mediate allelic exclusion.
Sensitivity of IgG B Cell Maturation to Excessive Tg Copy Number.
Unlike IgM anti-HEL transgenic mice, where typically more than 80% of peripheral B cells bound HEL and carried the transgenic receptor, less than half of peripheral B cells from IgG1 or IgM/G low copy number mice bound HEL. This phenotype was further exaggerated in mice with higher Hc Tg copy numbers, such that in IgM/G mice containing 15 Hc gene copies, <9% of B cells bound HEL. In IgG-transgenic mice that contained six copies of Hc, only 3% of splenic B cells bound HEL. The lower frequency of Tg-expressing B cells was specific to mice expressing the γ1 transmembrane and cytoplasmic tail, as it has not been observed in other transgenic mice carrying HEL-binding IgM, IgD, or chimeric IgM/D receptors containing the IgD transmembrane and cytoplasmic regions (reference 35 and Pogue, S.L., and C.C. Goodnow, unpublished observations). FACS® analysis of the non–HEL-binding B cells in the IgG1- or IgM/G-transgenic mice revealed that they carried antigen receptors containing endogenous Hc or transgenic Hc presumably paired with endogenous Lc.
The appearance of large numbers of B cells bearing endogenous receptors does not reflect failure of the IgG or IgM/G receptors to trigger allelic exclusion in either the low or high copy lines. Thus, those B cells that carried HEL-binding receptors did not carry endogenous Hcs (Fig. 4), and B cells with endogenous receptors lacked transgenic HEL-binding IgG (Fig. 3 B). Moreover, B cells that bore exclusively transgenic receptors were fully mature even in high copy lines, indicating that the receptor was in each case able to support the preimmune phase of maturation to varying degrees. Instead, the IgG1 and IgM/G Tgs appear to favor accumulation of “edited” B cells that have lost transgenic Hc or Lc expression and gained expression of endogenous Hc or Lc, especially in higher copy lines. This loss must occur at or before the developmental stage in which RAG expression occurs, thereby allowing rearrangement of endogenous Hc. It is not known whether loss of Tg expression reflects deletion of the Tg DNA, suppression of transcription, or competitive protein assembly and transport to the cell surface.
The sensitivity to gene dose observed here may explain the variability among IgG-expressing transgenic mouse strains reported previously 23 24 25 26 27 28 29 30. One of the most extensive analyses of IgG-expressing transgenic mice was conducted in the laboratory of Roth and coworkers 27 29 and by Kenny et al. 28. In transgenic mice from two of these lines, 343-1 and 348A, carrying 8–16 or 22–60 stably integrated copies of the Hc Tg, respectively, the number of splenic B cells was 10–30% of normal. The mature B cells that were present in these mice expressed high levels of IgM receptors derived from endogenous gene rearrangements and carried only low levels of the transgenic IgG2b on their cell surfaces 27 28 29 60. When the 343-1 line was crossed with mice homozygous for a targeted disruption of the IgM membrane exons, few mature B cells developed, indicating that the γ2b Hc did not allow maturation or accumulation of preimmune cells in the absence of membrane IgM. By contrast, almost normal numbers of splenic B cells developed bearing exclusively the transgenic IgG2b receptors in a third line with the same IgG2b Hc gene, 348C 28 29. Unlike the 343-1 and 348A mice, the 348C line carried only three copies of the Tg and was derived from an unstable line, 348-4-8 (348B), carrying 90 Tg copies 60. The conclusion favored at the time was that IgG is intrinsically unable to signal for B cell maturation normally, but this defect was somehow complemented by an integration site effect in the 348C variant line.
The evidence that IgG cannot signal for B cell maturation is supported by data similar to that from the 343-1 line, obtained in mice carrying an undetermined number of copies of an IgG2a/κ Tg encoding an anti-CD8.2 antibody 30. In these mice, surface IgM expression was inhibited in the bone marrow, but the number of mature splenic B cells was 15% of normal, and these expressed high levels of endogenous IgM and IgD and lower levels of IgG2a.
By contrast, IgG did support maturation of small numbers of B cells in the absence of endogenous IgM in an antiphosphorylcholine IgG2b/κ transgenic line carrying an unspecified number of Tg copies 28. The recovery of B cell maturation in several independent copy loss variants from high copy number lines observed here appears similar to the history of the copy loss variant 348C line described by Roth et al. 29. Loss of Tg copies is likely to result from intrachromosomal recombination within the Tg array before or during meiosis, as postulated for copy reduction variants arising among somatic cells 61 62.
Possible Signaling Differences Conferred by the IgG Membrane and Cytoplasmic Domains.
The varied numbers and frequency of HEL-binding mature B cells in IgG1- or IgM/G-transgenic mice could reflect either too little of a BCR signal needed for cell maturation or too much of a BCR signal that inhibits maturation. BCR signaling by IgM (or IgD) is required by preimmune B cells, first to trigger allelic exclusion and B cell maturation in the bone marrow and then to promote survival and accumulation in the spleen, lymph nodes, and blood 48 51 63. The nature of these intracellular positive signals is unclear, although the tyrosine kinase Syk is required for maturation in the bone marrow 58 59, and the tyrosine kinase Btk and the src-activating tyrosine phosphatase CD45 are required for survival and accumulation of mature cells in the spleen 51 64. If these positive signals were not sufficiently activated by BCRs with the IgG transmembrane and cytoplasmic regions, that could in principle favor maturation of B cells that had lost the Tg and replaced it with an endogenous IgM/IgD receptor or non–HEL-binding IgM/G receptor, as observed in the IgG and IgM/G mice, respectively. Other data makes this interpretation less likely: (a) a comparable preferential accumulation of variant B cells is not observed in anti-HEL IgM/IgD-transgenic mice when positive selection signals are crippled by mutations in Btk or CD45; and (b) spontaneous downregulation of IgM BCRs on mature B cells, as occurs during maturation of IgG and IgM/G cells, does not occur but is in fact suppressed when positive selection signals are crippled (51, 64, and Goodnow, C., unpublished data).
We believe the alternative interpretation, namely exaggerated signaling by IgG and IgM/G BCRs, represents a better explanation for the data. Maturation and accumulation of IgM- or IgD-bearing B cells is inhibited by chronic activation of inhibitory signals triggered either by self-antigen binding 53 65 66 67 or by exaggeration of basal or antigen-induced BCR signaling in the absence of the inhibitory tyrosine phosphatase, Src homology 2 domain–containing protein tyrosine phosphatase (SHP)-1 54. This promotes the formation of edited B cells that have lost or reduced expression of the transgenic Hc or Lc genes and replaced them by an endogenously encoded Hc or Lc 62 68 69 70 71. Interestingly, Lc gene editing predominates in anti-DNA IgM-transgenic mice with weakly DNA-reactive BCRs, whereas Hc editing predominated in anti-DNA transgenic mice with V region substitutions that conferred stronger DNA reactivity 62 69 70. It is interesting that the variant B cells accumulating in the spleens of IgG-transgenic mice have primarily lost the transgenic Hc, whereas the variant cells accumulating in the spleens of IgM/G mice retain the transgenic Hc and lose HEL binding, presumably due to loss of the transgenic Lc (Fig. 3). Based on the above, we hypothesize that IgG has an enhanced ability to trigger signals that inhibit immature B cell maturation when compared with IgM, and that the IgM/G chimera has the same characteristic but to a lesser degree. In the lines of IgG- and IgM/G-transgenic mice with supraphysiological gene dosage, higher amounts of BCR protein are made, as judged by surface staining of pro-B cells (Fig. 5 and Table ). The fact that fewer B cells mature with the transgenic receptor and more develop with the edited phenotype in these high copy lines is consistent with a further exaggeration of signals that inhibit maturation as a consequence of overexpressed IgG and IgM/G receptors.
The notion that IgG and IgM/G have exaggerated signaling is also consistent with the spontaneous downregulation of these receptors on mature B cells (Fig. 6). Similar decreases in surface display of IgMHEL receptors occurs during progression from immature to mature B cells in response to chronic BCR signaling. This can occur as a result of continuous binding of soluble HEL autoantigen 34, in which case chronic signaling and downregulation is suppressed in the absence of the stimulatory tyrosine phosphatase CD45 51 67. IgM receptors also downregulate spontaneously when BCR signaling is exaggerated by lack of the inhibitory phosphatase SHP-1 54, absence of CD22, which recruits SHP-1 to the BCR signaling complex 72 73 74 75, or overexpression of CD19, which augments BCR signaling 76. It is interesting that spontaneous downregulation of BCRs on mature B cells is more extreme for IgG than for IgM/G, in line with the more extreme editing that occurs in the IgG mice (above).
The decreased surface expression of IgG BCRs observed here on mature B cells matches a characteristic of isotype-switched memory B cells in vivo 77. Hayakawa et al. found that isotype-switched memory B cells in the spleens of immunized mice had four- to fivefold fewer κ chains on the cell surface compared with naive IgM+IgD+ B cells in the same animals. It is interesting that, without immunization, the IgG and IgM/G B cells also tend to accumulate in the CD21high marginal zone subset in the spleen (Fig. 6), as this subset contains many of the memory B cells in immunized animals 78 79 80. The low copy number transgenic mouse lines described here make possible future studies to explore the function of switched BCR isotypes in later phases of B cell development, particularly during B cell activation and differentiation into plasma cells and memory cells.
Acknowledgments
We wish to thank Karen Canaan and Denise Leong for expert technical assistance in producing transgenic mice and Jeffrey Grein for breeding and genotyping mice. We thank Dr. T. Lavoie and Dr. S. Smith-Gill for the original pTB6 plasmid, Dr. G. Adams for the gift of plasmid G1.2, Dr. F. Alt for the gift of RAG-2−/− mice, and Drs. Richard Glynne, Bennett Weintraub, Jason Cyster, and Michael Tomlinson for informative discussion and critical review of this manuscript.
This project was supported by National Institutes of Health (NIH) grant AI19512. S. Pogue was supported by a predoctoral fellowship, NIH training grant 5 T32 AI07328. C.C. Goodnow was an investigator of the Howard Hughes Medical Institute.
Footnotes
Abbreviations used in this paper: BCR, B cell antigen receptor; Hc, heavy chain; HEL, hen egg lysozyme; Lc, light chain; RAG, recombinase activating gene; SHP, Src homology 2 domain–containing protein tyrosine phosphatase; Tg, transgene.
References
- Goding J.W., Layton J.E. Antigen-induced co-capping of IgM and IgD-like receptors on murine B cells. J. Exp. Med. 1976;144:852–857. doi: 10.1084/jem.144.3.852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffman R.L., Cohn M. The class of surface immunoglobulin on virgin and memory B lymphocytes. J. Immunol. 1977;118:1806–1815. [PubMed] [Google Scholar]
- Kearney J.F., Cooper M.D., Klein J., Abney E., Parkhouse R.M.E., Lawton A.R. Ontogeny of Ia and IgD on IgM-bearing B lymphocytes in mice. J. Exp. Med. 1977;146:297–301. doi: 10.1084/jem.146.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitetta E.S., Uhr J.W. IgD and B cell differentiation. Immunol. Rev. 1977;37:50–88. doi: 10.1111/j.1600-065x.1977.tb00245.x. [DOI] [PubMed] [Google Scholar]
- Kraal G., Weissman I.L., Butcher E.C. Germinal centre B cellsantigen specificity and changes in heavy chain class expression. Nature. 1982;298:377–379. doi: 10.1038/298377a0. [DOI] [PubMed] [Google Scholar]
- Apel M., Berek C. Somatic mutations in antibodies expressed by germinal centre B cells early after primary immunization. Int. Immunol. 1990;2:813–819. doi: 10.1093/intimm/2.9.813. [DOI] [PubMed] [Google Scholar]
- Korthauer U., Graf D., Mages H., Briere F., Padayachee M., Malcolm S., Ugazio A., Notarangelo L., Levinsky R., Kroczek R. Defective expression of T-cell CD40 ligand causes X-linked immunodeficiency with hyper-IgM. Nature. 1993;361:539–541. doi: 10.1038/361539a0. [DOI] [PubMed] [Google Scholar]
- Harding F., Amemiya C., Litman R., Cohen N., Litman G. Two distinct immunoglobulin heavy chain isotypes in a primitive, cartilaginous fish, Raja erinacea . Nucleic Acids Res. 1990;18:6369–6376. doi: 10.1093/nar/18.21.6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson M., Marcuz A., van Ginkel F., Miller N., Clem L., Middleton D., Warr G. The immunoglobulin M heavy chain constant region gene of the channel catfish, Ictalurus punctatusan unusual form of the molecule. Nucleic Acids Res. 1990;18:5227–5233. doi: 10.1093/nar/18.17.5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokubu F., Hinds K., Litman R., Shamblott M., Litman G. Complete structure and organization of immunoglobulin heavy chain constant region genes in a phylogenetically primitive vertebrate. EMBO (Eur. Mol. Biol. Organ.) J. 1988;7:1979–1988. doi: 10.1002/j.1460-2075.1988.tb03036.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K., Saxon A., Max E. Two unusual forms of human immunoglobulin E encoded by alternate RNA splicing of epsilon heavy chain membrane exons. J. Exp. Med. 1992;176:233–243. doi: 10.1084/jem.176.1.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita K., Shimizu A., Honjo T. The membrane exons of the pseudo-gamma-chain gene of the human immunoglobulin are apparently functional and highly homologous to those of the gamma 1 gene. Immunol. Lett. 1991;27:151–155. doi: 10.1016/0165-2478(91)90143-x. [DOI] [PubMed] [Google Scholar]
- Hellman L. Characterization of four novel epsilon chain mRNA and a comparative analysis of genes for immunoglobulin E in rodents and man. Eur. J. Immunol. 1993;23:159–167. doi: 10.1002/eji.1830230126. [DOI] [PubMed] [Google Scholar]
- Bensmana M., Lefranc M. Gene segments encoding membrane domains for the human immunoglobulin gamma 3 and alpha chains. Immunogenetics. 1990;32:321–330. doi: 10.1007/BF00211646. [DOI] [PubMed] [Google Scholar]
- Akahori Y., Kurosawa Y. Nucleotide sequence of all the gamma gene loci of murine immunoglobulin heavy chains. Genomics. 1997;41:100–104. doi: 10.1006/geno.1997.4606. [DOI] [PubMed] [Google Scholar]
- Yamawaki-Kataoka Y., Kataoka T., Takahashi N., Obata M., Honjo T. Complete nucleotide sequence of immunoglobulin γ2b chain gene cloned from newborn mouse DNA. Nature. 1980;283:786–789. doi: 10.1038/283786a0. [DOI] [PubMed] [Google Scholar]
- Yamawaki-Kataoka Y., Nakai S., Miyata T., Honjo T. Nucleotide sequences of gene segments encoding membrane domains of immunoglobulin gamma chains. Proc. Natl. Acad. Sci. USA. 1982;79:2623–2627. doi: 10.1073/pnas.79.8.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler B.M., Cowman A.F., Gerondakis S.D., Adams J.M., Bernard O. mRNA for surface immunoglobulin gamma chains encodes a highly conserved transmembrane sequence and a 23-residue intracellular domain. Proc. Natl. Acad. Sci. USA. 1982;79:2008–2012. doi: 10.1073/pnas.79.6.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Word C.J., Mushinski J.F., Tucker P.W. The murine immunoglobulin alpha gene expresses multiple transcripts from a unique membrane exon. EMBO (Eur. Mol. Biol. Organ.) J. 1983;2:887–896. doi: 10.1002/j.1460-2075.1983.tb01518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiser P., Muller R., Braun U., Reth M. Endosomal targeting by the cytoplasmic tail of membrane immunoglobulin. Science. 1997;276:407–409. doi: 10.1126/science.276.5311.407. [DOI] [PubMed] [Google Scholar]
- Kaisho T., Schwenk F., Rajewsky K. The roles of gamma1 heavy chain membrane expression and cytoplasmic tail in IgG1 responses. Science. 1997;276:412–415. doi: 10.1126/science.276.5311.412. [DOI] [PubMed] [Google Scholar]
- Achatz G., Nitschke L., Lamers M. Effect of transmembrane and cytoplasmic domains of IgE on the IgE response. Science. 1997;276:409–411. doi: 10.1126/science.276.5311.409. [DOI] [PubMed] [Google Scholar]
- Yamamura K.-I., Kudo A., Ebihara T., Kamino K., Araki K., Kumahara Y., Watanabe T. Cell-type-specific and regulated expression of a human γ1 heavy-chain immunoglobulin gene in transgenic mice. Proc. Natl. Acad. Sci. USA. 1986;83:2152–2156. doi: 10.1073/pnas.83.7.2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao B.P., Ohnishi K., Cheroutre H., Mitchell B., Teitell M., Mixter P., Kronenberg M., Hahn B.H. Failed self-tolerance and autoimmunity in IgG anti-DNA transgenic mice. J. Immunol. 1992;149:350–358. [PubMed] [Google Scholar]
- Offen D., Spatz L., Escowitz H., Factor S., Diamond B. Induction of tolerance to an IgG autoantibody. Proc. Natl. Acad. Sci. USA. 1992;89:8332–8336. doi: 10.1073/pnas.89.17.8332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gram H., Zenke G., Geisse S., Kleuser B., Burki K. High-level expression of a human immunoglobulin γ1 transgene depends on switch region sequences. Eur. J. Immunol. 1992;22:1185–1191. doi: 10.1002/eji.1830220512. [DOI] [PubMed] [Google Scholar]
- Roth P.E., Doglio L., Manz J.T., Kim J.Y., Lo D., Storb U. Immunoglobulin γ2b transgenes inhibit heavy chain gene rearrangement, but cannot promote B cell development. J. Exp. Med. 1993;178:2007–2021. doi: 10.1084/jem.178.6.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny J., Stall A., Fischer R., Derby E., Yang M., Tucker P., Longo D. Igγ2b transgenes promote B cell development but alternate developmental pathways appear to function in different transgenic lines. J. Immunol. 1995;154:5694–5705. [PubMed] [Google Scholar]
- Roth P., Kurtz B., Lo D., Storb U. λ5, but not μ, is required for B cell maturation in a unique γ2b transgenic mouse line. J. Exp. Med. 1995;181:1059–1070. doi: 10.1084/jem.181.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battegay M., Fiedler P., Kalinke U., Brombacher F., Zinkernagel R., Peter H., Korher G., Eibel H. Non-tolerant B cells cause autoimmunity in anti-CD8 IgG2a-transgenic mice. Eur. J. Immunol. 1996;26:250–258. doi: 10.1002/eji.1830260139. [DOI] [PubMed] [Google Scholar]
- Iliev A., Spatz L., Ray S., Diamond B. Lack of allelic exclusion permits autoreactive B cells to escape deletion. J. Immunol. 1994;153:3551–3556. [PubMed] [Google Scholar]
- Smith-Gill S.J., Lavoie T.B., Mainhart C.R. Antigenic regions defined by monoclonal antibodies correspond to structural domains of avian lysozyme. J. Immunol. 1984;133:384–393. [PubMed] [Google Scholar]
- Padlan E.A., Silverton E.W., Sheriff S., Cohen G.H., Smith-Gill S.J., Davies D.R. Structure of an antibody-antigen complexcrystal structure of the HyHEL-10 Fab-lysozyme complex. Proc. Natl. Acad. Sci. USA. 1989;86:5938–5942. doi: 10.1073/pnas.86.15.5938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodnow C.C., Crosbie J., Adelstein S., Lavoie T.B., Smith-Gill S.J., Brink R.A., Pritchard-Briscoe H., Wotherspoon J.S., Loblay R.H., Raphael K. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature. 1988;334:676–682. doi: 10.1038/334676a0. [DOI] [PubMed] [Google Scholar]
- Brink R., Goodnow C.C., Crosbie J., Adams E., Eris J., Mason D.Y., Hartley S.B., Basten A. Immunoglobulin M and D antigen receptors are both capable of mediating B lymphocyte activation, deletion, or anergy after interaction with specific antigen. J. Exp. Med. 1992;176:991–1005. doi: 10.1084/jem.176.4.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams J.M., Webb E., Gerondakis S., Cory S. Cloned embryonic DNA sequences flanking the mouse immunoglobulin Cγ3 and Cγ1 genes. Nucleic Acids Res. 1980;8:6019–6031. doi: 10.1093/nar/8.24.6019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clackson T., Winter G. ‘Sticky feet’-directed mutagenesis and its application to swapping antibody domains. Nucleic Acids Res. 1989;17:10163–10170. doi: 10.1093/nar/17.24.10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan B., Costantini F., Lacy E. Manipulating the Mouse Embryo. A Laboratory Manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1986. [Google Scholar]
- Chen S., Evans G.A. A simple screening method for transgenic mice using the polymerase chain reaction. Biotechniques. 1990;8:32–33. [PubMed] [Google Scholar]
- Schuppel R., Wilke J., Weiler E. Monoclonal anti-allotype antibody towards BALB/c IgM. Analysis of specificity and site of a V-C crossover in recombinant strain BALB-Igh-Cb. Eur. J. Immunol. 1987;17:739–741. doi: 10.1002/eji.1830170527. [DOI] [PubMed] [Google Scholar]
- Stall A., Loken M. Allotypic specificities of murine IgD and IgM recognized by monoclonal antibodies. J. Immunol. 1984;132:787–795. [PubMed] [Google Scholar]
- Coffman R.L. Surface antigen expression and immunoglobulin gene rearrangement during mouse pre-B cell development. Immunol. Rev. 1982;69:5–23. doi: 10.1111/j.1600-065x.1983.tb00446.x. [DOI] [PubMed] [Google Scholar]
- Smith-Gill S.J., Wilson A.C., Potter M., Feldmann R.J., Mainhart C.R. Mapping the antigenic epitope for a monoclonal antibody against avian lysozyme. J. Immunol. 1982;128:314–322. [PubMed] [Google Scholar]
- Kinoshita T., Thyphronitis G., Tsokos G.C., Finkelman F.D., Hong K., Sakai H., Inoue K. Characterization of murine complement receptor type 2 and its immunological cross-reactivity with type 1 receptor. Int. Immunol. 1990;2:651–659. doi: 10.1093/intimm/2.7.651. [DOI] [PubMed] [Google Scholar]
- Hartley S.B., Goodnow C.C. Censoring of self-reactive B cells with a range of receptor affinities in transgenic mice expressing heavy chains for a lysozyme-specific antibody. Int. Immunol. 1994;6:1417–1425. doi: 10.1093/intimm/6.9.1417. [DOI] [PubMed] [Google Scholar]
- Hardy R.R., Carmack C.E., Shinton S.A., Kemp J.D., Hayakawa K. Resolution and characterization of pro-B and pre-pro-B cell stages in normal mouse bone marrow. J. Exp. Med. 1991;173:1213–1225. doi: 10.1084/jem.173.5.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkai Y., Rathbun G., Lam K.-P., Oltz E.M., Stewart V., Mendelsohn M., Charron J., Datta M., Young F., Stall A.M. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell. 1992;68:855–867. doi: 10.1016/0092-8674(92)90029-c. [DOI] [PubMed] [Google Scholar]
- Rajewsky K. Clonal selection and learning in the antibody system. Nature. 1996;381:751–758. doi: 10.1038/381751a0. [DOI] [PubMed] [Google Scholar]
- Young F., Ardman B., Shinkai Y., Lansford R., Blackwell T.K., Mendelsohn M., Rolink A., Melchers F., Alt F.W. Influence of immunoglobulin heavy- and light-chain expression on B-cell differentiation. Genes. Dev. 1994;8:1043–1057. doi: 10.1101/gad.8.9.1043. [DOI] [PubMed] [Google Scholar]
- Spanopoulou E., Roman C.A., Corcoran L.M., Schissel M.S., Silver D.P., Nemazee D., Nussenzweig M.C., Shinton S.A., Hardy R.R., Baltimore D. Functional immunoglobulin transgenes guide ordered B-cell differentiation in Rag-1-deficient mice. Genes. Dev. 1994;8:1030–1042. doi: 10.1101/gad.8.9.1030. [DOI] [PubMed] [Google Scholar]
- Cyster J.G., Healy J.I., Kishihara K., Mak T.W., Thomas M.L., Goodnow C.C. CD45 sets thresholds for negative and positive selection of B lymphocytes. Nature. 1996;381:325–328. doi: 10.1038/381325a0. [DOI] [PubMed] [Google Scholar]
- Torres R.M., Flaswinkel H., Reth M., Rajewsky K. Aberrant B cell development and immune response in mice with a compromised BCR complex. Science. 1996;272:1804–1808. doi: 10.1126/science.272.5269.1804. [DOI] [PubMed] [Google Scholar]
- Hartley S.B., Cooke M.P., Fulcher D.A., Harris A.W., Cory S., Basten A., Goodnow C.C. Elimination of self-reactive B lymphocytes proceeds in two stagesarrested development and cell death. Cell. 1993;72:325–335. doi: 10.1016/0092-8674(93)90111-3. [DOI] [PubMed] [Google Scholar]
- Cyster J.G., Goodnow C.C. Protein tyrosine phosphatase 1C negatively regulates antigen receptor signaling in B lymphocytes and determines thresholds for negative selection. Immunity. 1995;2:13–24. doi: 10.1016/1074-7613(95)90075-6. [DOI] [PubMed] [Google Scholar]
- Mombaerts P., Iacomini J., Johnson R.S., Herrup K., Tonegawa S., Papaioannou V.E. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- Kitamura D., Roes J., Kühn R., Rajewsky K. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin μ chain gene. Nature. 1991;350:423–426. doi: 10.1038/350423a0. [DOI] [PubMed] [Google Scholar]
- Kitamura D., Kudo A., Schaal S., Müller W., Melchers F., Rajewsky K. A critical role of λ5 protein in B cell development. Cell. 1992;69:823–831. doi: 10.1016/0092-8674(92)90293-l. [DOI] [PubMed] [Google Scholar]
- Cheng A., Rowley B., Pao W., Hayday A., Bolen J., Pawson T. Syk tyrosine kinase required for mouse viability and B-cell development. Nature. 1995;378:303–306. doi: 10.1038/378303a0. [DOI] [PubMed] [Google Scholar]
- Turner M., Mee P., Costello P., Williams O., Price A., Duddy L., Furlong M., Geahlen R., Tybulewicz V. Perinatal lethality and blocked B-cell development in mice lacking the tyrosine kinase Syk. Nature. 1995;378:298–302. doi: 10.1038/378298a0. [DOI] [PubMed] [Google Scholar]
- Tsang H., Pinkert C., Hagman J., Lostrum M., Brinster R.L., Storb U. Cloning of a γ2b gene encoding anti-Pseudomonas aeruginosa H chains and its introduction into the germ line of mice. J. Immunol. 1988;141:308–314. [PubMed] [Google Scholar]
- Sangren E., Palmiter R., Heckel J., Daugherty C., Brinster R., Degen J. Complete hepatic regeneration after somatic deletion of an albumin-plasminogen activator gene. Cell. 1991;66:245–256. doi: 10.1016/0092-8674(91)90615-6. [DOI] [PubMed] [Google Scholar]
- Chen C., Radic M.Z., Erikson J., Camper S.A., Litwin S., Hardy R.R., Weigert M. Deletion and editing of B cells that express antibodies to DNA. J. Immunol. 1994;152:1970–1980. [PubMed] [Google Scholar]
- Wienands J., Larbolette O., Reth M. Evidence for a preformed transducer complex by the B cell antigen receptor. Proc. Natl. Acad. Sci. USA. 1996;93:7865–7870. doi: 10.1073/pnas.93.15.7865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan W., Sideras P., Rosen F., Alt F. The role of Bruton's tyrosine kinase in B cell development and function in mice and man. Ann. NY Acad. Sci. 1995;764:27–38. doi: 10.1111/j.1749-6632.1995.tb55802.x. [DOI] [PubMed] [Google Scholar]
- Cyster J.G., Hartley S.B., Goodnow C.C. Competition for follicular niches excludes self-reactive cells from the recirculating B-cell repertoire. Nature. 1994;371:389–395. doi: 10.1038/371389a0. [DOI] [PubMed] [Google Scholar]
- Fulcher D.A., Basten A. Reduced life span of anergic self-reactive B cells in a double-transgenic model. J. Exp. Med. 1994;179:125–134. doi: 10.1084/jem.179.1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy J., Dolmetsch R., Timmerman L., Cyster J., Thomas M., Crabtree G., Lewis R., Goodnow C. Different nuclear signals are activated by the B cell receptor during positive versus negative signaling. Immunity. 1997;6:419–428. doi: 10.1016/s1074-7613(00)80285-x. [DOI] [PubMed] [Google Scholar]
- Tiegs S., Russell D., Nemazee D. Receptor editing in self-reactive bone marrow B cells. J. Exp. Med. 1993;177:1009–1020. doi: 10.1084/jem.177.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay D., Saunders T., Camper S., Weigert M. Receptor editingan approach by autoreactive B cells to escape tolerance. J. Exp. Med. 1993;177:999–1008. doi: 10.1084/jem.177.4.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C., Nagy Z., Luning-Prak E., Weigert M. Immunoglobulin heavy chain gene replacementa mechanism of receptor editing. Immunity. 1995;3:747–755. doi: 10.1016/1074-7613(95)90064-0. [DOI] [PubMed] [Google Scholar]
- Hertz M., Nemazee D. BCR ligation induces receptor editing in IgM+IgD− bone marrow B cells in vitro. Immunity. 1997;6:429–436. doi: 10.1016/s1074-7613(00)80286-1. [DOI] [PubMed] [Google Scholar]
- O'Keefe T., Williams G., Davies S., Neuberger M. Hyperresponsive B cells in CD22-deficient mice. Science. 1996;274:798–801. doi: 10.1126/science.274.5288.798. [DOI] [PubMed] [Google Scholar]
- Otipoby K.L., Andersson K.B., Draves K.E., Klaus S.J., Farr A.G., Kerner J.D., Perlmutter R.M., Law C.L., Clark E.A. CD22 regulates thymus-independent responses and the lifespan of B cells. Nature. 1996;384:634–637. doi: 10.1038/384634a0. [DOI] [PubMed] [Google Scholar]
- Sato S., Miller A., Inaoki M., Bock C., Jansen P., Tang M., Tedder T. CD22 is both a positive and negative regulator of B lymphocyte antigen receptor signal transductionaltered signaling in CD22-deficient mice. Immunity. 1996;5:551–562. doi: 10.1016/s1074-7613(00)80270-8. [DOI] [PubMed] [Google Scholar]
- Nitschke L., Carsetti R., Ocker B., Kohler G., Lamers M.C. CD22 is a negative regulator of B-cell receptor signalling. Curr. Biol. 1997;7:133–143. doi: 10.1016/s0960-9822(06)00057-1. [DOI] [PubMed] [Google Scholar]
- Sato S., Steeber D.A., Jansen P.J., Tedder T.F. CD19 expression levels regulate B lymphocyte developmenthuman CD19 restores normal function in mice lacking endogenous CD19. J. Immunol. 1997;158:4662–4669. [PubMed] [Google Scholar]
- Hayakawa K., Ishii R., Yamasaki K., Kishimoto T., Hardy R. Isolation of high-affinity memory B cellsphycoerythrin as a probe for antigen-binding cells. Proc. Natl. Acad. Sci. USA. 1987;84:1379–1383. doi: 10.1073/pnas.84.5.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.-J., Oldfield S., MacLennan I.C.M. Memory B cells in T cell-dependent antibody responses colonize the splenic marginal zones. Eur. J. Immunol. 1988;18:355–362. doi: 10.1002/eji.1830180306. [DOI] [PubMed] [Google Scholar]
- Liu Y., Barthelemy C., Bouteiller O., Arpin C., Durand I., Banchereau J. Memory B cells from human tonsils colonize mucosal epithelium and directly present antigen to T cells by rapid up-regulation of B7-1 and B7-2. Immunity. 1995;2:239–248. doi: 10.1016/1074-7613(95)90048-9. [DOI] [PubMed] [Google Scholar]
- Spencer J., Finn T., Pulford K.A., Mason D.Y., Isaacson P.G. The human gut contains a novel population of B lymphocytes which resemble marginal zone cells. Clin. Exp. Immunol. 1985;62:607–612. [PMC free article] [PubMed] [Google Scholar]