

Abstract
In lymphocytes, the Rel transcription factor is essential in establishing a pattern of gene expression that promotes cell proliferation, survival, and differentiation. Here we show that mitogen-induced expression of interferon (IFN) regulatory factor 4 (IRF-4), a lymphoid-specific member of the IFN family of transcription factors, is Rel dependent. Consistent with IRF-4 functioning as a repressor of IFN-induced gene expression, the absence of IRF-4 expression in c-rel −/− B cells coincided with a greater sensitivity of these cells to the antiproliferative activity of IFNs. In turn, enforced expression of an IRF-4 transgene restored IFN modulated c-rel −/− B cell proliferation to that of wild-type cells. This cross-regulation between two different signaling pathways represents a novel mechanism that Rel/nuclear factor κB can repress the transcription of IFN-regulated genes in a cell type–specific manner.
Keywords: Rel/NF-κB, lymphocytes, IRF-4, interferon, transcription
Introduction
During antigen and mitogen stimulation of lymphocytes, specific genetic programs are established by the coordinated activation of various signal transduction pathways, which in turn, lead to morphologic changes, cell division, differentiation, and the acquisition of immunological function. The temporal patterns of gene expression that underlie these processes in lymphocytes collectively span a time frame that extends from minutes to days, with the induction of immediate early and early response genes coinciding with that period of mitogenic stimulation required to commit a cell to a program of activation 1.
One group of transcription factors essential for lymphocyte activation and immune function is the Rel/nuclear factor (NF)-κB proteins 2, homodimers and heterodimers composed of related subunits that are encoded by a small multigene family. Rel/NF-κB proteins regulate gene expression by binding to specific decameric sequences (κB elements) located within the transcriptional regulatory regions of cellular genes, particularly those encoding proteins involved in immune, acute phase, and inflammatory responses 3 4. The five mammalian Rel/NF-κB subunits share a conserved NH2-terminal domain (Rel homology domain [RHD]) that encompass sequences essential for DNA binding, dimerization, and nuclear localization 3. The 50- and 52-kD forms of NF-κB1 and NF-κB2, respectively, comprise the RHD and lack intrinsic transcriptional transactivating properties, whereas Rel, RelA, and RelB all possess transactivation domains within their divergent COOH termini 3. In most cell types, the majority of Rel/NF-κB is sequestered as an inactive cytoplasmic complex with inhibitor or IκB proteins 5 6. Diverse stimuli promote the nuclear translocation of cytoplasmic Rel/NF-κB by activating an IκB kinase complex (for a review, see reference 2) that phosphorylates conserved serine residues on the IκB proteins, targeting them for ubiquitin-dependent proteosome-mediated degradation 5 6.
Rel is dispensable for normal embryonic development and hemopoiesis, but is essential for the division, survival, differentiation, and immune function of mature lymphocytes and macrophages 7 8 9 10 11. To date, most of the genes known to be direct transcriptional targets of Rel encode cytokines, cytokine receptors, and immune-regulatory molecules such as IL-2, IL-3, GM-CSF, IL-6, TNF-α, IL-2Rα, and inducible nitric oxide synthase (iNOS [2, 3]). However, impaired expression of these proteins only partly explains the immune defects exhibited by c-rel −/− mice. For example, the B cell proliferative defect resulting from the absence of Rel is due to a cell cycle block and enhanced apoptosis 10. This impaired mitogenic response is a result of the combined failure to induce the expression of prosurvival genes such as A1 11, and Rel-regulated genes necessary for G1 progression, the latter of which remain to be identified.
As part of a strategy aimed at identifying genes regulated by Rel, the expression of genes normally induced early in lymphocyte activation were examined in c-rel −/− B and T cells. Among those surveyed, IFN regulatory factor 4 (IRF-4), a lymphoid-restricted member of the IFN family of transcription factors, emerged as a promising candidate. IRF-4 expression is rapidly induced in resting lymphocytes by mitogens 12 with kinetics that closely follow the nuclear induction of Rel 13. Like Rel, it also appears to be critical in lymphocyte proliferation, as IRF-4 −/− B and T cells respond poorly to mitogens 14 and deregulated overexpression of IRF-4 due to the translocation of the gene into the Ig CH locus occurs in a subset of human multiple myelomas 15. Proteins comprising the IRF family, of which there are at least 10 members, function either as transcriptional activators or repressors 16. All share a conserved NH2-terminal domain required for the recognition and binding of a specific DNA consensus sequence, the IFN stimulation response element (ISRE) found in the promoters of IFN-regulated genes 17 18. Although originally identified as mediating the diverse biological activities of type I and type II IFNs 19 20, subsequent studies have revealed roles for IRFs that are independent of IFN signaling. These include controlling susceptibility to oncogenic transformation, promoting cell cycle progression, and the induction of growth arrest and programmed cell death 18. Here we show that IRF-4, a repressor of ISRE-mediated transcription, while normally rapidly upregulated in mitogen stimulated B and T cells is not induced in activated c-rel −/− lymphocytes. This impaired expression of IRF-4, which coincides with a hyperresponsiveness of c-rel −/− lymphocytes to type I and type II IFNs and enhanced expression of certain IFN-induced genes, establishes a novel mechanism that Rel/NF-κB can regulate IFN-induced gene expression in a cell type–specific fashion.
Materials and Methods
Mice.
The generation of the c-rel −/− 7 and nfkbl1 −/− 21 mice has been described previously. All mutant mouse strains have been backcrossed for greater than nine generations with C57BL/6 mice.
Genomic Clones, Plasmid Constructs, and Retroviral Expression Vector.
Phage genomic clones encompassing the murine IRF-4 gene 12 were a gift of Prof. Tak Mak (Ontario Cancer Institute, Toronto, Canada). Plasmids IRF4 Pst1–chloramphenicol acetyltransferase (CAT), IRF4 H3-CAT, and IRF4 Sph1-CAT consisted of the 2644-bp Pst1–Avr1, 1195-bp Hind3–Avr1, and 498-bp Sph1–Avr1 genomic fragments, respectively, from the murine IRF-4 5′ flanking sequence inserted upstream of CAT in the promoterless reporter plasmid pBCAT3 22. IRF4 κB1m-CAT and IRF4 κB2m-CAT are derivatives of IRF4 Pst1-CAT, in which NF-κB binding sites κB1 (5′-GGGGATCCAC-3′ at −1733 to −1724) and κB2 (5′-GGGATCCCCC-3′ at −686 to −657) were altered by in vitro mutagenesis 23 to 5′-GGTCATAAAC-3′ and 5′-GTCATCAACC-3′, respectively, or in the case of IRF4 κB1m/κB2m-CAT, where both sites have been mutated. The reporter plasmid pA3luc-ISRE comprising four copies of the ISRE consensus sequence 5′-GTACCAGTTTCGGTTTCCCTTG-3′ inserted upstream of the minimal mouse H2Dk promoter in the reporter plasmid pGem-luc (Promega) was provided by H. Thomas (Walter and Eliza Hall Institute for Medical Research). The expression plasmid pCMDIRF-4 was generated by inserting a 2-kb Xba1–Hind3 murine IRF-4 cDNA encompassing the entire IRF-4 coding region into pCMD-8. A cDNA encompassing the murine IRF-4 coding region fused in frame with an NH2-terminal FLAG tag was cloned as a BglII–EcoR1 insert into the retroviral expression vector pMSCV-IRES–green fluorescent protein (GFP) (a gift of Dr. L. Van Parijs, California Institute of Technology, Pasadena, CA).
Purification of Primary B and T Lymphocytes.
Small resting B and T lymphocytes were purified from the spleens of 6–8-wk-old wild-type, nfkb1 −/−, and c-rel −/− mice by negative sorting 10 on a FACS® or FACStarPLUS™ cell sorter (Becton Dickinson). The purity of all sorted B and T lymphocytes was verified by staining a portion of the cells with PE-labeled anti-B220 or anti-Thy1 antibodies, respectively (Caltag Labs) and ranged between 95 and 99%.
Cell Culture and Lymphocyte Activation in Tissue Culture.
The Jurkat T cell line was maintained in RPMI 1640 supplemented with 8% FCS, and 293T fibroblasts were grown in DMEM/8% FCS. The B cell lines 404.1 and B1.1 have been described previously 11 and were cultured in DMEM/10% FCS/50 μM BME. Primary B and T lymphocytes were cultured in the high glucose version of DMEM supplemented with 13 mM folic acid, 250 mM l-asparagine, 50 μM 2-ME, and 10% FCS. Spleen cells were cultured at an initial concentration of 106 cells/ml, and FACS®-purified B or T lymphocytes were cultured at an input concentration of 3 × 105 cells/ml. All primary B or T cells were stimulated in vitro with LPS (Difco), affinity-purified goat anti–mouse IgM (Fab′) fragment (Jackson ImmunoResearch Laboratories), rat anti–mouse radioprotective (RP) mAbs 24, Con A (Amersham Pharmacia Biotech), PMA and ionomycin (Sigma Chemical Co.), or plate-coated mouse anti-CD3 and anti-CD28 mAbs as described previously 11. In proliferation assays for which the effect of IFNs were assessed, either recombinant IFN-α or IFN-γ (gifts of Drs. P. Hertzog, Monash University, Melbourne, Australia, and T. Kay, The Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia) were added at increasing 10-fold concentrations (1–103 U/ml) with the mitogen combinations anti-RP/LPS or Con A/IL-2 (for B and T cells, respectively) for 72 h. Cellular proliferation was measured by adding 0.5 μCi of [3H]thymidine for 6 h. Afterwards, cells were harvested onto glass fiber filters, and incorporated radioactivity was quantitated by scintillation counting.
Transfections, CAT Assays, and Luciferase Assays.
Jurkat T cells and 293T fibroblasts were transiently transfected using Superfect (QIAGEN) as described previously 11. Equimolar amounts (1–2 μg) of IRF4-CAT plasmids or the pluc-ISRE reporter plasmid (1 μg) were transfected alone or with a threefold molar excess of pDAMP56, PDAMP56c-rel 25, pCTax 26, pCDMIκB-αm 27, pCDMIRF-1 19, pCDMIRF-2 20, or pCDMIRF-4. After 48 h, Jurkat and 293T cells were harvested and CAT or luciferase assays were performed on cell extracts that had been standardized for protein content. Transfections were performed five times, with a maximum variance of ∼15% observed between replicate experiments.
Retroviral Production and Infection.
Retroviral stocks were obtained by Superfect (QIAGEN)-mediated transfection of the pMSCV-IRES-GFP and pMSCV-IRES-GFP:IRF-4 vectors into BOS23 cells as described 28. B cell lines 404.1 and B1.1 were infected by incubating each of 106 cells for 2 h with 0.25 ml (∼106 virus/ml) of viral stock in 1 ml of media containing 5 μg/ml of polybrene. Cells were washed once, resuspended in 3 ml of complete medium, and incubated for 48 h. Infected cells were identified as GFP+ and then cloned in 96-well plates using the cell deposition unit of the FACStarPLUS™.
Immunoblotting.
Total cell extracts from ∼106 cells boiled in 0.5% SDS were electrophoresed on 10% SDS-polyacrylamide gels and transferred to nitrocellulose membranes as described 13. Filters were incubated with affinity-purified mouse anti-FLAG M2 specific monoclonal Ig (Sigma Chemical Co.) for detection of FLAG-tagged IRF-4. Bound antibody was then revealed by horseradish peroxidase–conjugated goat anti–mouse Ig (Silenus) using enhanced chemiluminescence (Amersham Pharmacia Biotech).
Northern Blot Analysis.
Northern blot hybridization on total RNA isolated from cells using RNAgents (Promega) was performed essentially as described 11. Filters were washed in 0.2× SSC, 0.1% SDS at 65°C and exposed to autoradiography at −70°C. For successive hybridizations, filters were boiled in 10 mM EDTA, 0.1% SDS to remove the bound probe before the rehybridization. The probes were 2-kb Xbal–Hind3 murine IRF-4 cDNA 29, and 1.1-kb Pst1 rat glyceraldehyde 3-phosphate dehydrogenase (GAPDH) cDNA 30 inserts radiolabeled with [α-32P]dATP by primer extension.
Semiquantitative Reverse Transcription PCR.
Total RNA was isolated from mouse primary splenic B cells (106) and embryonic fibroblasts (3 × 105) that were nonactivated or stimulated for 8 h with 103 IU/ml of recombinant IFN-α in the absence or presence of 50 μg/ml of LPS using RNAgents (Promega). cDNA synthesis on equivalent amounts of total RNA was performed essentially as described 9. For semiquantitative PCR, cDNA was added to a cocktail comprising 50 mM KCl, 2 mM MgCl2, 10 mM Tris-HCl, pH 8.3, 0.01% (wt/vol) gelatin, 0.5 mM dNTPs, 1 U of Taq polymerase (Ampli Taq: Perkin-Elmer Cetus) and 1 μM of each oligonucleotide in a final volume of 50 μl. After an initial 5-min denaturation at 94°C, the cDNA was amplified for 25 cycles with each cycle programmed for denaturation at 94°C for 45 s, annealing at 58°C for 60 s, followed by elongation at 72°C for 90 s. Samples were then fractionated on a 1% agarose gel. The sequence of the oligonucleotides used for the amplification of murine 2′–5′ oligoadenylate synthetase (OAS) and β-actin mRNA were: 2′-5′OAS, 5′ oligo CGCTGGACAAGTTCATAGAGGATT (nucleotides 70–93, according to the sequence of Ichii et al. 31), 3′ oligo TTTCCTTGATGAACTCTCCCCGTC (nucleotides 319–342, according to the sequence of Ichii et al. [31]); and β-actin, 5′ oligo CTGAAGTACCCATTGAACATGGC (nucleotides 278–303, according to the sequence of Tokunaga et al. 32), 3′ oligo CAGAGCAGTAATCTCCTTCTGCAT (nucleotides 1016–1040 [32]); the 2′-5′OAS and β-actin PCR products are 272 and 762 nucleotides in length, respectively.
Electrophoretic Mobility Shift Assays.
IRF4 κB1 and IRF4 κB2 probes were prepared by end-labeling the double stranded oligonucleotides 5′-TCACTTCTGGGGGATCCACACAACGAG-3′ and 5′-TGAGTACTCAGGGATCCCCCATCTCTT-3′, respectively, and electrophoretic mobility shift assays reactions were performed with 1–2 μg of nuclear extract as described 13. For competition analysis, a 50-fold excess of unlabeled IRF4 κB1, IRF4 κB2, IRF4 κB1m (5′-TCACTTCTGGGTCATAAACACAACGAG-3′), or IRF4 κB2m (5′-TGAGTACTCAGTCATCAACCATCTCTT-3′) competitor DNA was added to the reaction at room temperature 15 min before the addition of radiolabled probe. For supershift analysis, antibodies that specifically recognize NF-κB1, RelA, Rel 13, or NF-κB2 (sc 298; Santa Cruz Biotechnology) were incubated on ice for 30 min before the addition of radiolabeled probe. All electrophoretic mobility shift assay reactions were incubated for 20 min at room temperature, 2 μl of Ficoll dye was added, and the reactions were fractionated on 5% nondenaturing polyacrylamide gels. Gels were then dried and exposed to autoradiography at −70°C.
Results
Rel Induction of Murine IRF-4 Transcription Is Mediated by κB Elements in the Promoter.
In an attempt to identify novel Rel-regulated genes, RNA hybridization studies were performed on c-rel −/− lymphocytes to examine the expression of genes normally induced during mitogen stimulation with kinetics that parallel Rel translocation into the nucleus. One gene, IRF-4 (also referred to as Pip 29, ICSAT [IFN consensus binding protein in activated T cells; 33], or LSIRF [lymphoid IFN stimulatory response factor; 12]), encodes a lymphoid-specific member of the IFN family of transcription factors that is strongly upregulated by various mitogens in normal and nfkb1 −/− B and T cells within 2 h, but is barely induced in stimulated c-rel −/− lymphocytes (Fig. 1).
Figure 1.
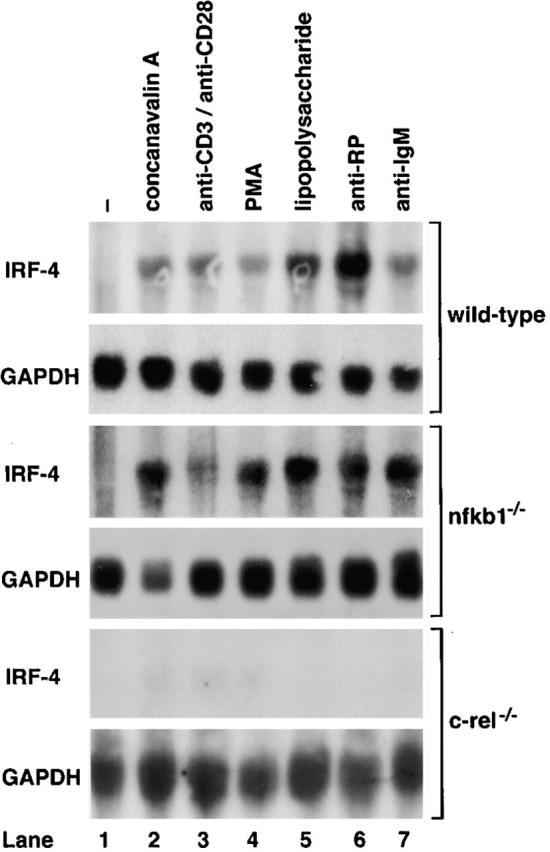
Mitogen-induced expression of IRF-4 mRNA is markedly reduced in c-rel −/− lymphocytes. 10-μg samples of total RNA isolated from untreated or mitogen-stimulated wild-type, nfkb1 −/−, or c-rel −/− splenocytes activated in culture for 2 h were analyzed by Northern blot hybridization. Filters were sequentially hybridized with murine IRF-4 and rat GAPDH cDNA probes and exposed to autoradiography for 24–48 h. Untreated cells (lane 1) and mitogen-activated cells (lanes 2–7). The stimuli were Con A (lane 2); anti-CD3 plus anti-CD28 antibodies (lane 3); phorbol ester (lane 4); LPS (lane 5); anti-RP antibodies (lane 6); and anti-IgM antibodies (lane 7).
To determine if Rel directly regulates IRF-4 transcription, a nested set of murine IRF-4 promoter truncations extending upstream of the 5′ untranslated region (+177), inserted 5′ of CAT in a promoterless reporter plasmid (Fig. 2 A; 22) were transiently transfected into Jurkat T cells with or without a c-rel expression vector. Basal activity of the full-length promoter, IRF4 Pst1-CAT (−2465 to +177) was equivalent to that of the truncated clones, IRF4 H3-CAT (−1010 to +177) and IRF4 Sph1-CAT (−320 to +177) (Fig. 2 B, lanes 3, 5, and 7), indicating that the sequences necessary for basal transcription appear to reside within 320 nucleotides upstream of the transcription start site. In contrast, whereas IRF4 Pst1-CAT was induced 3.5-fold by Rel, the Rel-dependent expression of IRF4 H3-CAT and IRF4 Sph1-CAT was significantly reduced or completely ablated (Fig. 2 B, compare lanes 4, 6, and 8). These findings indicated that at least two sequences residing between −2465 and −320 were mediating Rel-dependent transcription. As this region of the murine IRF-4 promoter encompassed two putative Rel/NF-κB binding sites at −1733 (κB1: 5′-GGGGATCCAC-3′) and −686 (κB2: 5′-GGGATCCCC-3′) 12, the requirement of these elements for Rel-mediated transcription was tested directly by mutating either or both of these sequences. Promoter constructs, IRF4 κB1m-CAT, IRF4 κB2m-CAT, and IRF4 κB1m/κB2m-CAT, that contain either or both mutant κB sites within the context of the full-length promoter retained normal basal promoter activity (Fig. 2 B, lanes 3, 9, 11, and 13). However, mutations in κB1 (lane 10) or κB2 (lane 12) diminished the Rel-dependent induction of IRF4 Pst1-CAT by twofold, while the disruption of both sites (lane 14) eliminated Rel-induced transcription. These results demonstrate that these two κB elements are both necessary and sufficient for Rel-dependent IRF-4 transcription.
Figure 2.
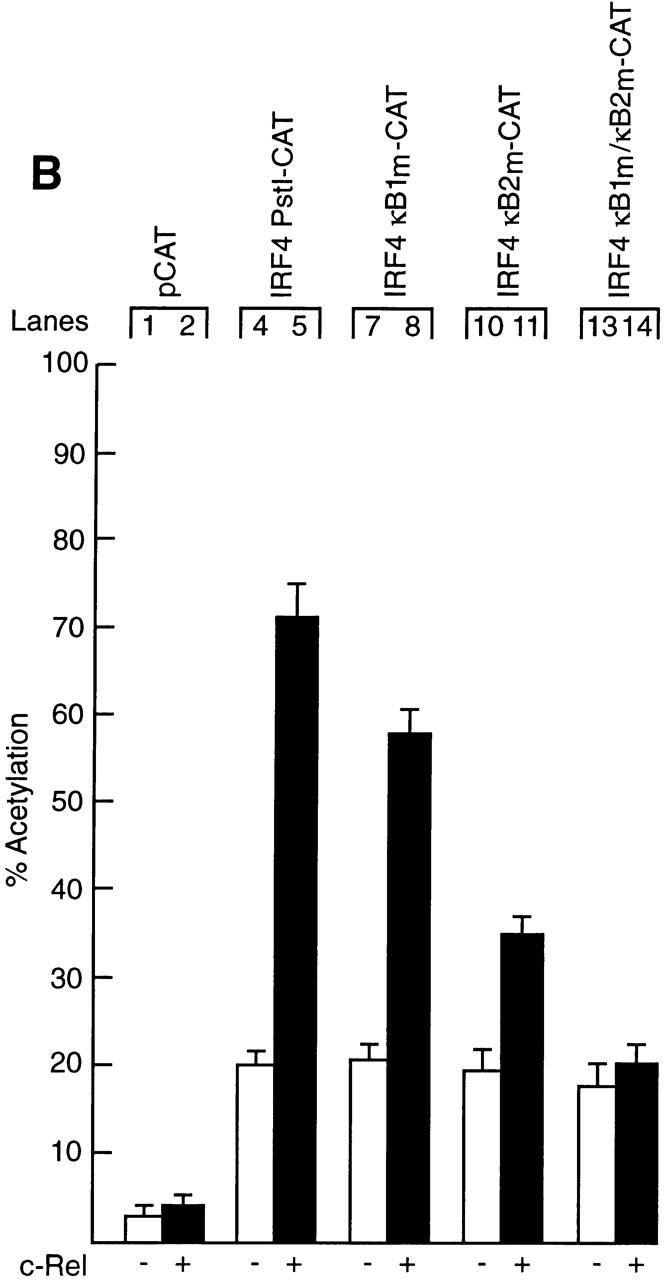

Functional analysis of the murine IRF-4 promoter. (A) Schematic representation of the IRF-4 5′ flanking region and the CAT reporter plasmids. The numbers in parenthesis indicate the position of restriction enzyme sites in the murine IRF-4 5′ flanking sequence according to the sequence of Matsuyama et al. (reference 12). The filled boxes represent the two putative NF-κB binding sites κB1 (5′-GGGGATCCAC-3′: −1733 to −1724) and κB2 (5′-GGGATCCCCC-3′: −686 to −677), whereas the corresponding symbol with a cross represents the mutated motifs κB1m (5′-GGTCATAAAC-3′) and κB2m (5′-GTCATCAACC-3′). The arrow denotes the transcription initiation site and the CAT gene is depicted as an open box. Plasmid nomenclature is indicated to the right of each construct. (B) Both IRF-4 NF-κB motifs contribute to Rel-dependent transcription in T cells. Jurkat cells were transiently transfected with 2 μg of the reporter plasmids pCAT (lanes 1 and 2), IRF4 Pst1-CAT (lanes 3 and 4), IRF4 H3-CAT (lanes 5 and 6), IRF4 Sph1-CAT (lanes 7 and 8), IRF4 κB1m-CAT (lanes 9 and 10), IRF4 κB2m-CAT (lanes 11 and 12), or IRF4 κB1m/κB2m-CAT (lanes 13 and 14) plus 10 μg of the expression plasmid DAMP56 containing no insert (lanes 1, 3, 5, 7, 9, 11, and 13) or c-rel (lanes 2, 4, 6, 8, 10, 12, and 14). Chloramphenicol acetylation for transfections with pDAMP56 or pDAMP56c-rel is indicated by white and black bars, respectively. Results represent the mean percentage of chloramphenicol acetylation ± SD obtained from six separate sets of transient transfections.
Mitogen-induced Nuclear Rel Complexes Bind to Both κB Elements in the IRF-4 Promoter.
The ability of the two IRF-4 κB sites to bind nuclear Rel complexes was examined by electrophoretic mobility shift assays (Fig. 3). Single major complexes expressed in quiescent wild-type and c-rel −/− T cells (Fig. 3 A, lanes 1 and 3) bound κB1 (denoted C2) and κB2 (denoted C4) probes. Within 2 h of anti-CD3/anti-CD28 stimulation, novel complexes (C1 and C3 for κB1 and κB2 probes) were detected in wild-type (Fig. 3 A, lane 2) but not c-rel −/− T cells (lane 4). The binding specificity of all complexes was demonstrated in normal (Fig. 3 A, lanes 5, 6, 9, and 10) and c-rel −/− (lanes 7, 8, 11, and 12) T cells by competition with excess unlabeled IRF-4 κB1, IRF-4 κB2 (lanes 9–12), or the corresponding mutant probes (lanes 5–8). Equivalent results were obtained for normal and c-rel −/− B cells activated with anti-IgM antibodies (results not shown).
Figure 3.


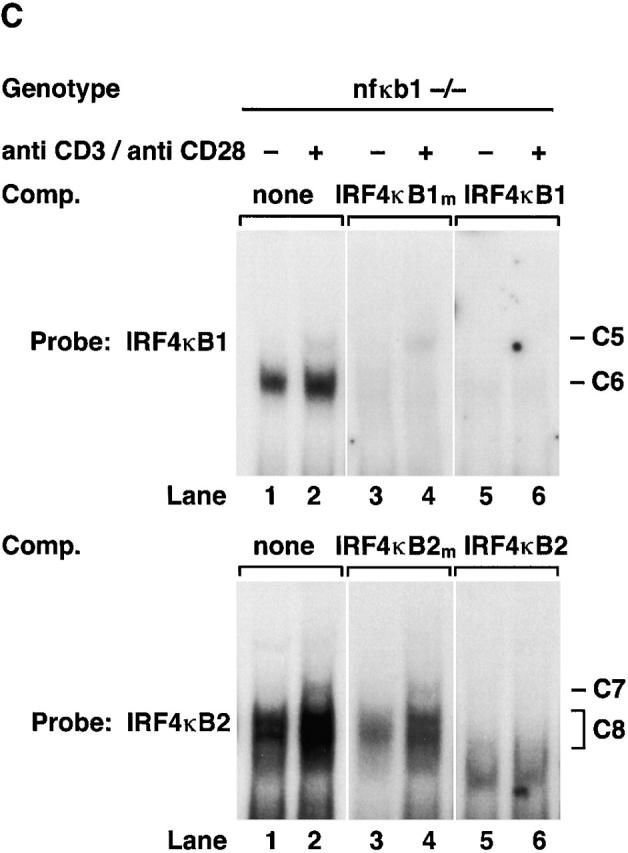
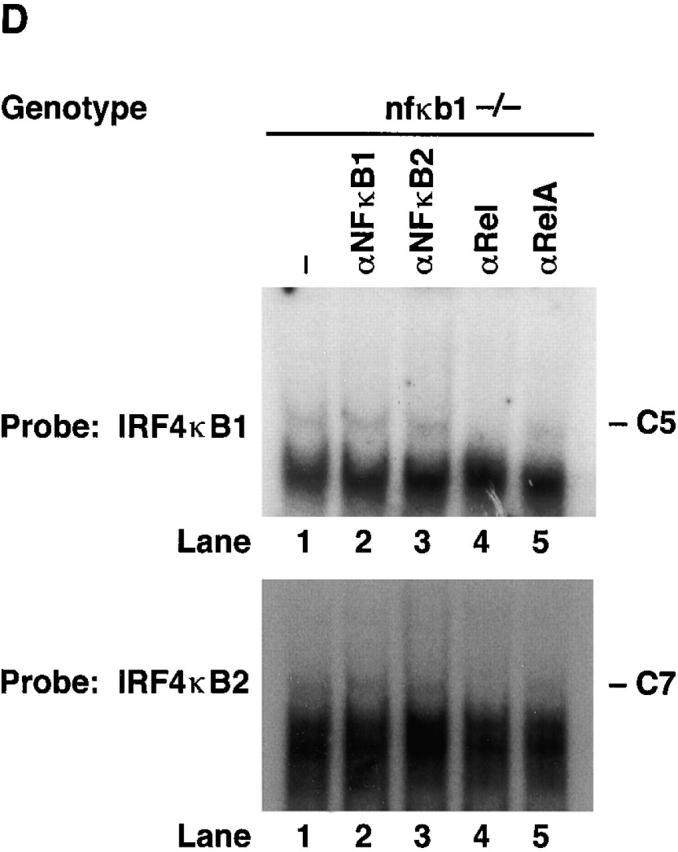
Rel/NF-κB complexes in resting and mitogen-stimulated T cells. Nuclear extracts (1–2 μg) isolated from purified normal, c-rel −/−, and nfkb1 −/− splenic T cells stimulated with anti-CD3 plus anti-CD28 antibodies for 2 h, then incubated with 32P-radiolabeled IRF4 κB1 or IRF4 κB2 probes were resolved on 5% nondenaturing polyacrylamine gels and exposed to autoradiography for 8–24 h at −70°C. (A) Nuclear complexes rapidly induced by mitogen that bind IRF4 κB1 and IRF4 κB2 are absent in c-rel −/− T cells. Nuclear extracts from resting (lanes 1, 3, 5, 7, 9, and 11) and anti-CD3/anti-CD28–stimulated (lanes 2, 4, 6, 8, 10, and 12) normal (lanes 1, 2, 5, 6, 9, and 10) and c-rel −/− (lanes 3, 4, 7, 8, 11, and 12) T cells were preincubated in the absence (lanes 1–4) or presence of a 50-fold molar excess of unlabeled mutant (κB1m, κB2m: lanes 5–8) or wild-type (κB1, κB2: lanes 9–12) probe before adding radiolabeled IRF4 κB1 or IRF4 κB2. The inducible slow mobility complexes binding to κB1 and κB2 are designated C1 and C3, respectively, while the constitutive fast mobility complexes binding κB1 and κB2 are designated C2 and C4, respectively. (B) Inducible complexes C1 and C3 are Rel/NF-κB1 heterodimers. Nuclear extracts from resting (lanes 1–4) and anti-CD3/anti–CD28 stimulated (lanes 5–8) wild-type splenic T cells were incubated with preimmune (lanes 1 and 5) or NF-κB1 (lanes 2 and 6), Rel (lanes 3 and 7) or RelA (lanes 4 and 8)–specific sera before adding radiolabeled IRF4 κB1 or IRF4 κB2 probes. (C) Mitogen-induced κB binding complexes in nfkbl −/− T cells. Nuclear extracts from resting (lanes 1, 3, and 5) and anti-CD3/anti–CD28 stimulated (lanes 2, 4, and 6) nfkbl −/− T cells were preincubated in the absence (lanes 1 and 2) or presence of a 50-fold molar excess of unlabeled mutant (κB1m, κB2m: lanes 3 and 4) or wild-type (κB1 or κB2: lanes 5 and 6) binding sites before adding radiolabeled IRF4 κB1 or IRF4 κB2 probes. The inducible slow mobility complexes binding to κB1 and κB2 are designated C5 and C7, respectively, while the constitutive fast mobility complexes binding κB1 and κB2 are designated C6 and C8, respectively. (D) Inducible κB binding complexes C5 and C7 in nfkbl −/− T cells are Rel homodimers. Nuclear extracts from anti-CD3/anti-CD28–stimulated nfkbl −/− T cells were incubated with preimmune (lane 1) or NF-κB1 (lane 2), NF-κB2 (lane 3), Rel (lane 4), or RelA (lane 5)–specific sera before adding radiobalelled IRF4 κB1 or IRF4 κB2 probes.
The composition of complexes binding IRF-4 κB1 and κB2 in normal resting and mitogen-stimulated T lymphocytes was examined by supershift analysis using antibodies specific for the different Rel/NF-κB subunits (Fig. 3 B). Mitogen-induced complexes C1 and C3 were supershifted with antibodies specific for NF-κB1 (lane 6) and Rel (lane 7), but not RelA (lane 8), demonstrating that both complexes predominantly comprise NF-κB1/Rel heterodimers. Although C2 and C4 have similar mobilities to NF-κB1 homodimers, neither complex was supershifted with various NF-κB1–specific antibodies or antibodies directed to other Rel/NF-κB proteins. The identity of protein(s) in C2 and C4 remains to be determined.
Although mitogen-induced IRF-4 expression coincided with the binding of NF-κB1/Rel to IRF-4 κB1 and κB2, the normal induction of IRF-4 mRNA in mitogen-stimulated nfkb1 −/− lymphocytes prompted us to examine the basis of NF-κB1–independent IRF-4 transcription. DNA binding assays performed with nuclear extracts from resting or anti-CD3/anti-CD28 antibody–stimulated nfkb1 −/− T lymphocytes are shown in Fig. 3 C. IRF-4 κB1 and κB2 bound complexes in resting cells (lane 1, denoted C6 and C8) and novel complexes of slower mobility (lane 2, denoted C5 and C7) induced in mitogen activated cells. Supershift studies (Fig. 3 D) indicated that the induced complexes C5 and C7 appear to be Rel homodimers, whereas C6 and C8 were not recognized by Rel/NF-κB–specific antisera (lanes 2, 3, 4, and 5). While the roles of C6 and C8 in IRF-4 transcription are unknown, ablation of C6 binding by competition with unlabeled IRF-4 κB1 mutant probe (Fig. 3 C, lane 4) indicates that this protein(s) does not bind the IRF-4 κB1 probe via the consensus κB site. Collectively, these data establish that mitogen induced IRF-4 expression coincides with the binding of Rel complexes to both κB elements in the IRF-4 promoter.
Rel-deficient B Cells Exhibit Increased Responsiveness to Type I and Type II IFNs.
IFN-induced gene expression is controlled by the coordinated interplay of transcriptional activators and repressors 18. IFN-regulated transactivators IRF-1 and ISGF3 induce transcription by binding to ISREs found in the promoters and enhancers of IFN-regulated genes 18, while a negative regulator such as IRF-2 is thought to repress transcription by competitive binding to these motifs 17. Since IRF-4 functions as a repressor of IFN-induced gene expression 33 34, one consequence of the inability to upregulate IRF-4 expression during the mitogenic activation of c-rel −/− lymphocytes may be increased responsiveness of these cells to the action of IFNs. Inhibition of normal lymphocyte proliferation by high concentrations of type I and type II IFNs 35 36 prompted us to compare the proliferative response of normal and c-rel −/− B cells over a wide range of IFN-α and IFN-γ concentrations (Fig. 4). The stimulus anti-RP plus LPS was chosen for these experiments because in contrast to individual mitogens which fail to induce c-rel −/− B cell proliferation 7, certain mitogen or mitogen plus cytokine combinations function synergistically to promote the proliferation of Rel-deficient cells 7 10. Inhibition of B cell proliferation over a wide range of IFN-α and IFN-γ concentrations (Fig. 4A and Fig. B) was significantly greater in c-rel −/− cultures. This heightened sensitivity of c-rel −/− B cells to the antiproliferative activity of both cytokines was concentration-dependent. While inhibition of c-rel −/− B cell proliferation was more pronounced with increasing concentrations of IFN-γ (Fig. 4 C), the difference in the inhibition of normal and c-rel −/− B cells by IFN-α was maximal at 102 IU/ml (Fig. 4 B). Consistent with the specificity of IRF-4 induction by Rel in lymphocytes and its proposed role in modulating IFN responsiveness, proliferating c-rel −/− T cells but not c-rel −/− fibroblasts display a heightened sensitivity to the antiproliferative activity of IFNs (Grumont, R., unpublished results).
Figure 4.
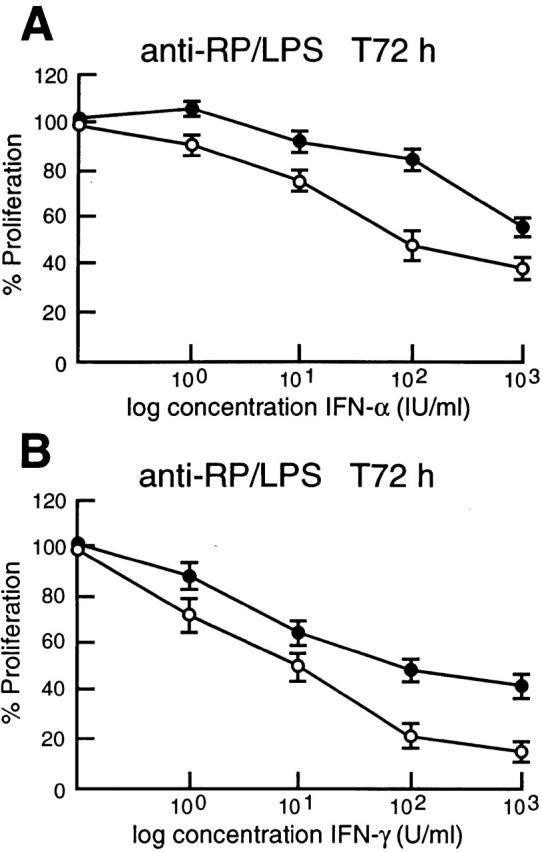
c-rel −/− B cells display greater sensitivity to the antiproliferative activity of type I and type II IFNs. Resting splenic lymphocytes from normal (filled circles) and c-rel −/− (open circles) mice were stimulated for 72 h with optimal concentrations of anti-RP antibodies plus LPS in the absence or presence of 1, 10, 100, or 1,000 U/ml of IFN-α or IFN-γ. Cellular proliferation was measured at 24-h intervals over 72 h by [3H]thymidine incorporation. Proliferation of normal and c-rel−/− B cells in the presence of a particular concentration of IFN at the 72-h time point is represented in this figure as a percentage of [3H]thymidine incorporated by normal or c-rel −/− cells, respectively, in the absence of IFN. All results represent the mean ± SD from five experiments.
Enforced IRF-4 Expression in c-rel−/− B Cells Restores Normal IFN Responsiveness.
We have previously shown that surface IgM+ c-rel −/− B cell lines, like primary c-rel −/− B cells undergo apoptosis upon B cell receptor engagment due in part to an absence of Rel-induced A1 expression 11. To ascertain if these cell lines could also be useful models in determining whether the Rel-dependent induction of IRF-4 in B cells has a direct role in modulating the responsiveness of proliferating B cells to IFNs, we first examined the expression of IRF-4 in control and c-rel −/− B cell lines. In contrast to the constitutive expression of IRF-4 in the control B cell line W404.1, IRF-4 mRNA was not detected in the c-rel −/− B cell line B1.1 (Fig. 5 A). To determine if an absence of IRF-4 expression in B1.1 was associated with an increased sensitivity to the antiproliferative action of IFNs, both W404.1 and B1.1 were treated with a range of IFN-α and IFN-γ concentrations for 72 h and proliferation assessed by tritiated thymidine incorporation (Fig. 5 B). Consistent with the trends observed for primary B cells, after 72 h, proliferation of the c-rel −/− B cell line B1.1 was significantly less over a range of IFN-α and IFN-γ concentrations compared with that of W404.1. The role of IRF-4 in reducing the antiproliferative action of IFNs was examined by infecting the W404.1 and B1.1 cell lines with a retrovirus expressing NH4-terminally Flag-tagged IRF-4. Clones of each cell line expressing comparable levels of exogeneous IRF-4 (Fig. 5 C) were then treated with IFN-α or IFN-γ for 72 h and proliferation assessed (Fig. 5 D). These experiments, which showed that enforced expression of IRF-4 restores c-rel −/− B cell proliferation, established that IRF-4 can modulate IFN-induced inhibition of lymphocyte proliferation to that of c-rel +/+ B cells in the presence of IFN.
Figure 5.
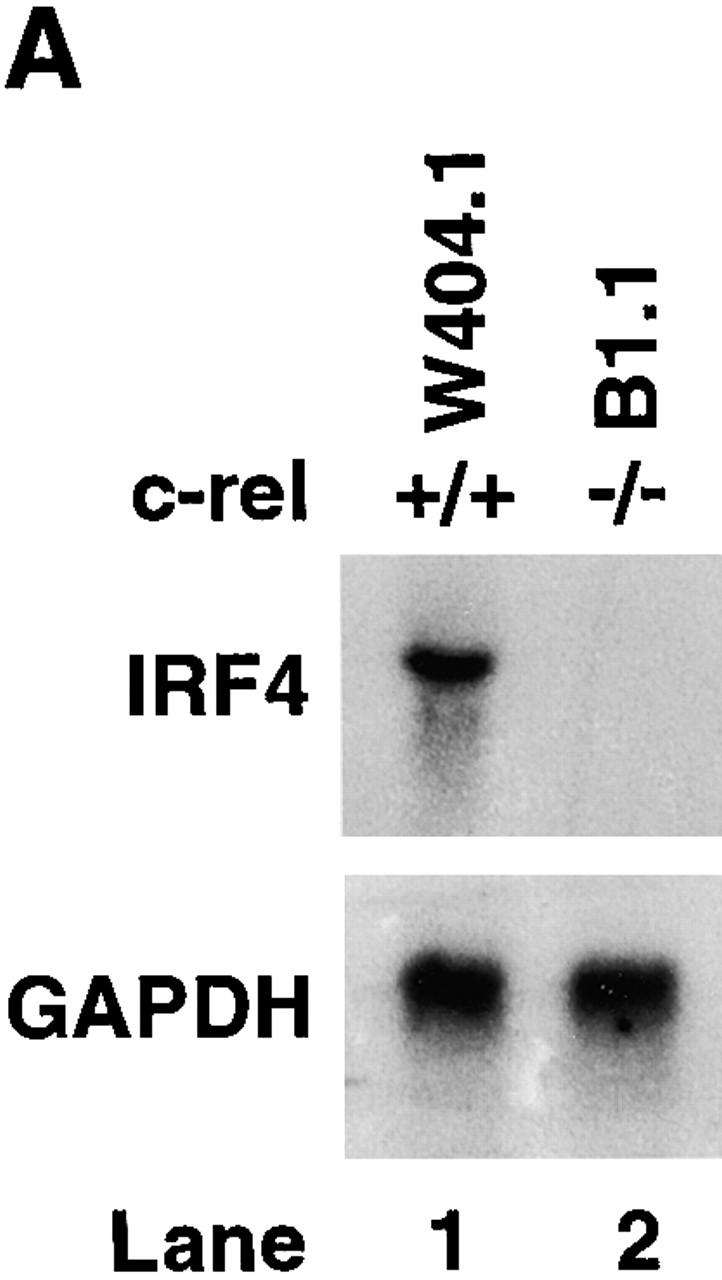
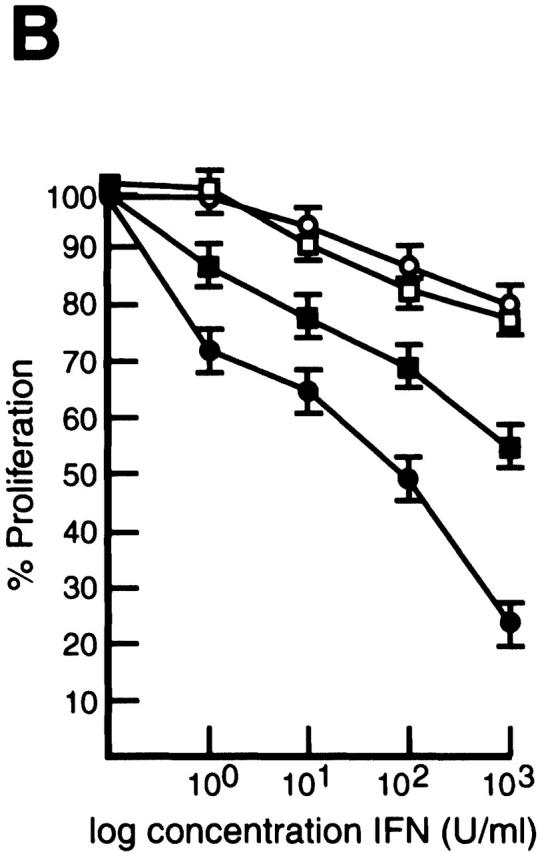


Constitutive IRF-4 expression promotes c-rel −/− B cell proliferation in the presence of IFNs. (A) IRF-4 is not expressed in c-rel −/− B cell lines. 5 μg samples of total RNA isolated from the immortalized B cell lines W404.1 (c-rel +/+; lane 1) and B1.1 (c-rel −/−; lane 2) were analyzed by Northern blot hybridization. Filters were sequentially hybridized with murine IRF-4 and rat GAPDH cDNA probes and exposed for autoradiography for 24 h. The results obtained with these two cell lines were representative of other independent c-rel +/+ and c-rel −/− cell lines. (B) c-rel −/− B cell lines are more sensitive to the antiproliferative activity of IFNs. The W404.1 (c-rel +/+; open symbols) and B1.1 (c-rel −/−; filled symbols) B cell lines were either untreated or stimulated for 72 h with 1, 10, 100, or 1,000 U/ml of IFN-α (circles) or IFN-γ (squares). Cellular proliferation was measured at 24-h intervals over 72 h by [3H]thymidine incorporation. Proliferation of all cell lines in the presence of a particular concentration of IFN at the 72-h time point is represented in this figure as a percentage of [3H]thymidine incorporated by normal or c-rel −/− cells, respectively, in the absence of IFN. All results represent the mean ± SD from four experiments. (C) Expression of exogeneous IRF-4. Whole cell extracts from W404.1 (c-rel +/+; lanes 1 and 2) and B1.1 (c-rel −/−; lanes 3 and 4) cells that had been infected with a control retrovirus (lanes 1 and 3) or a retrovirus expressing NH2-terminally FLAG-tagged IRF-4 (lanes 2 and 4) were resolved by SDS-PAGE and subjected to Western blot analysis using mAbs directed to the FLAG epitope. (D) Enforced IRF-4 expression overcomes the heightened anti-proliferative action of IFNs that result from the loss of Rel. The cell lines W404.1 (circles) and B1.1 (triangles) that were infected with a control retrovirus (open symbols) or a virus expressing FLAG-tagged IRF-4 (filled symbols) were either untreated or stimulated for 72 h with 1, 10, 100, or 1,000 U/ml of IFN-α or IFN-γ. The data shown represents the 72-h time point and are expressed as a percentage of [3H]thymidine incorporated by normal or c-rel −/− cells, respectively, in the absence of IFN. All results represent the mean ± SD from four experiments.
Rel Induction of IRF-4 Coincides with a Repression of IFN-regulated Gene Expression.
The heightened responsiveness of c-rel −/− lymphocytes to the action of IFN led us to determine if Rel could be shown to modulate ISRE-dependent transcription. This was determined by comparing ISRE-dependent transcription in Jurkat T cells or 293T embryonic kidney epithelial cells transiently transfected with an ISRE-regulated reporter plasmid and various combinations of expression vectors encoding IRF-1, IRF-2, IRF-4, and Rel (Fig. 6 A). IRF-1–induced ISRE-dependent transcription was reduced significantly in both cell types by cotransfection with an equimolar concentration of IRF-2 or IRF-4 plasmids (compare lanes 2 and 7 with lanes 3, 4, 8, and 9). However, Rel was only able to repress IRF-1–induced gene expression (sixfold, lane 5) in Jurkat T cells. Consistent with Rel repressing IRF-1–dependent transcription in a lymphoid-specific manner, Northern blot analysis of transiently transfected Jurkat and 293T cells (Fig. 6 B) revealed that IRF-4 mRNA levels were only upregulated in Jurkat T cells transfected with c-rel (lane 4). Collectively, these findings are consistent with Rel repressing ISRE-dependent transcription in a lymphoid-restricted manner by upregulating IRF-4.
Figure 6.
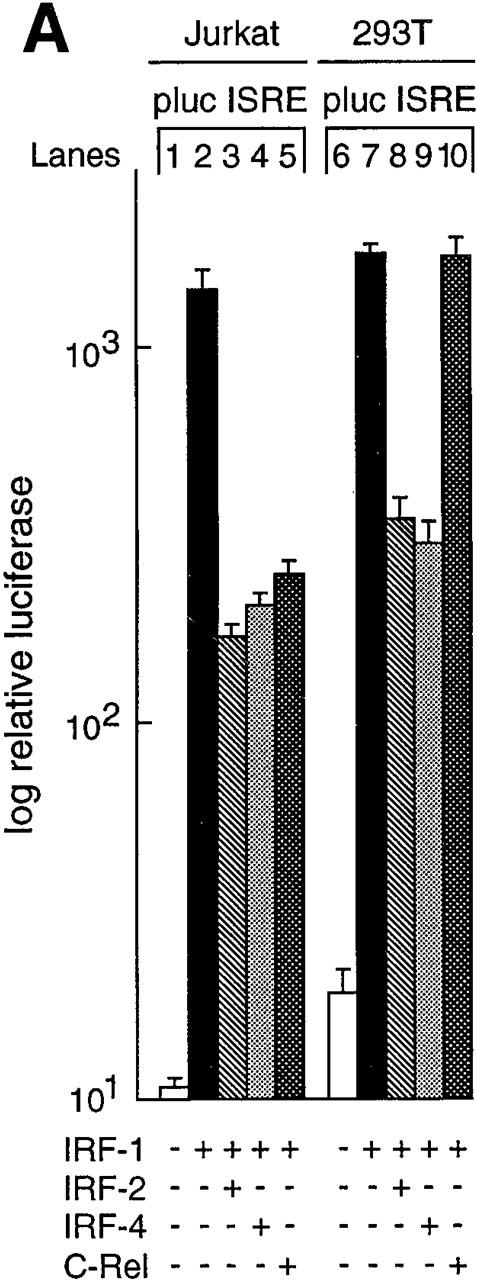

Rel regulation of IRF-1–dependent transcription. (A) Rel repression of IRF-1–dependent transcription is lymphoid specific. Jurkat T cells (lanes 1–5) and 293T fibroblasts (lanes 6–10) were transiently transfected with 2 μg of the ISRE-regulated reporter plasmid plucISRE in the absence (lanes 1 and 6) or presence of an equimolar concentration of expression vectors encoding IRF-1 (lanes 2 and 7) IRF-1 plus IRF-2 (lanes 3 and 8), IRF-1 plus IRF-4 (lanes 4 and 9), or IRF-1 plus c-rel (lanes 5 and 10). Luciferase activity for transfections with vector control (white bars), IRF-1 (black bars), IRF-1 plus IRF-2 (hatched bars), IRF-1 plus IRF-4 (stippled bars), or IRF-1 plus c-rel (cross-hatched bars) represent the mean activity ± SD obtained from four separate sets of transient transfections. (B) Rel repression of IRF-1–regulated transcription in T cells coincides with IRF-4 induction. 10-μg samples of total RNA isolated from Jurkat and 293T cells transiently transfected with 15 μg of pDAMP56 (vector control) or pDAMP56 c-rel after 48 h were analyzed by Northern blot hybridization with murine IRF-4 and rat GAPDH cDNA probes. Filters were exposed to autoradiography for 24 h.
IFN-regulated endogeneous gene expression was also examined in c-rel −/− cells. We focused on the gene encoding the 1.7-kb transcript for 2′-5′OAS because the molecular basis of its IFN- or IRF-1–induced transcription has been well studied 30 36 37 38 and transcription of the human gene is repressed by IRF-4 33. The IFN induction of 2′-5′OAS mRNA was examined in normal and c-rel −/− fibroblasts and B cells stimulated with IFN-α or IFN-α plus LPS. A representative example of these experiments is shown in Fig. 7. Consistent with previous findings for various cell types 37, 2′-5′OAS expression is absent or low in unstimulated normal and mutant fibroblasts or lymphocytes (lanes 1, 2, 7, and 8). While the induction of 2′-5′OAS mRNA by IFN-α or LPS plus IFN-α is equivalent in wild-type and c-rel −/− fibroblasts (lanes 3-6), mRNA levels are between four- and fivefold higher than normal in c-rel −/− B cells activated with IFN-α and LPS (lanes 11 and 12). This is in contrast to stimulation with IFN-α only, where 2′-5′OAS mRNA levels are equivalent in wild-type and mutant B cells (lanes 9 and 10). Since IRF-4 is upregulated by mitogens but not IFNs 12, reduced expression of 2′-5′OAS in normal but not c-rel −/− B cells treated with LPS and IFN-α is consistent with repression of 2′-5′OAS transcription arising from the Rel-dependent induction of IRF-4.
Figure 7.

Rel modulates 2′-5′OAS expression in lymphocytes. Total mRNA isolated from wild-type (lanes 1, 3, 5, 7, 9, and 11) or c-rel −/− (lanes 2, 4, 6, 8, 10, and 12) mouse embryonic fibroblasts (lanes 1–6) and B cells (lanes 7–12) that were non-activated (lanes 1, 2, 7, and 8) or stimulated with IFN-α (lanes 3, 4, 9, and 10) or LPS plus IFN-α (lanes 5, 6, 11, and 12) for 8 h was subjected to semiquantitative reverse transcription PCR using 2′-5′OAS– and β-actin–specific primers. PCR products were fractionated on a 1% agarose gel.
Induction of IRF-4 by Tax Is Rel/NF-κB Dependent.
Since IRF gene expression is often induced in cells in response to viral infection 20 38, it was noteworthy that IRF-4 is expressed constitutively in HTLV-1–induced T cell leukemias, but not in other T cell tumors of varying etiology 33. Consistent with this finding, Tax, a potent HTLV-1–encoded transcriptional activator essential for viral mediated oncogenesis upregulates IRF-4 expression in Jurkat cells 33. As Tax induces the expression of various cellular genes by activating or enhancing the activity of cellular transcription factors including Rel/NF-κB proteins 39 40, we examined whether Tax-induced IRF-4 transcription was mediated by Rel/NF-κB (Fig. 8). While Tax upregulated the IRF4 Pst1-CAT reporter plasmid fourfold in Jurkat cells (compare lanes 4 and 5), an IκB-α super repressor reduced the Tax-dependent induction of the IRF-4 promoter to near basal levels (lane 6). Cotransfections of tax and the IRF-4 κB promoter mutants established that both IRF-4 κB1 and κB2 are essential for Tax-dependent IRF-4 transcription. Whereas Tax-mediated transactivation of IRF4 κB1m-CAT (lane 8) and IRF4 κB2m-CAT (lane 11) was 70 and 50%, respectively, of wild-type promoter activity, the loss of both κB sites completely abolished transactivation (lane 14). As was the case with IRF4 Pst1-CAT, the diminished Tax-induced activation of IRF-4 reporter plasmids containing κB1 or κB2 mutations was further reduced to basal levels by IκB-αm (lanes 9 and 12). These findings establish that the Tax induction of IRF-4 transcription in T cells is mediated by Rel/NF-κB.
Figure 8.
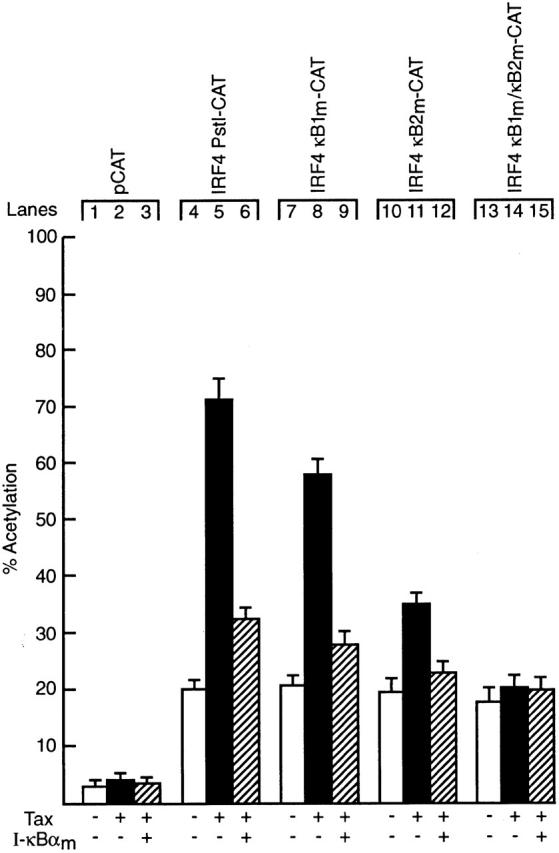
Tax-induced IRF-4 transcription is Rel/NF-κB-dependent. Jurkat cells were transiently transfected with 2 μg of the reporter plasmids pCAT (lanes 1–3), IRF4 Pst1-CAT (lanes 4–6), IRF4 κB1m-CAT (lanes 7–9), IRF4 κB2m-CAT (lanes 10–12) or IRF4 κB1m/κB2m-CAT (lanes 13–15) in the absence (lanes 1, 4, 7, 10, and 13) or presence of a threefold molar excess of an expression plasmid for HTLV-1 Tax (lanes 2, 5, 8, 11, and 14) or expression plasmids for Tax plus IκB-αm (lanes 3, 6, 9, 12, and 15). Chloramphenicol acetylation for transfections with pCDM-8 (vector control), pCTax or pCDM IκB-αm are indicated by white, black, and hatched bars, respectively. Results represent the mean percentage of chloramphenicol acetylation ± SD obtained from five separate sets of transient transfections.
Discussion
Previous studies have shown that in lymphocytes, Rel is crucial in promoting cellular activation and the acquisition of immunological function 2. Consistent with the immunological component of Rel function, cytokine genes such as IL-2, IL-3, and GM-CSF are transcriptional targets of Rel 7 8. Here we show that expression of IRF-4, a lymphoid-specific IFN regulatory factor is directly induced by Rel in activated lymphocytes and that an absence of Rel coincides with a heightened responsiveness of lymphocytes to type I and type II IFNs. The implications of this finding for lymphocyte proliferation, viral replication, and the cross-regulation of IFN signaling by Rel/NF-κB are discussed.
Rel-dependent Transcription.
Here we show mitogen-induced IRF-4 transcription is Rel-dependent and is regulated by two κB elements located in the IRF-4 promoter that bind Rel/NF-κB complexes. Although the major nuclear complex induced in lymphocytes that binds to both IRF-4 κB elements is NF-κB1/Rel, normal upregulation of IRF-4 in nfkb1 −/− lymphocytes that results from Rel homodimers binding to these sites indicates that NF-κB1 is a nonessential heterodimeric partner of Rel in controlling the transcription of this gene. A similar finding has been made for mitogen-induced regulation of A1 expression, which is also under the transcriptional control of Rel/NF-κB 11. Together, this establishes that under normal physiological conditions, Rel homodimers can function as transcriptional activators.
Rel/NF-κB binding sites 5′-GGGG/ARRNNA/CC-3′ comprise a conserved 5′ half site, GGGA/GR, and a divergent 3′ half site. Both dimer partners contribute to DNA binding, with different subunits binding to the half sites with varying affinity 41 42. Since NF-κB1 preferentially interacts with the conserved 5′ half site 41, NF-κB1 homodimers bind with strongest affinity to symmetrical motifs 41. By contrast, DNA sequences randomly selected in vitro to bind recombinant Rel homodimers with high affinity (5′-NGGRNA/TTTCC-3′) exhibit considerable sequence variation 43. The IRF-4 and A1 κB sites, all of which bind NF-κB1 homodimers, Rel homodimers, and NF-κB1/Rel heterodimers, comprise the consensus sequence 5′-G/AGGGATCCA/C A/C-3′. As each of these sites shares the invariant core motif 5′-GGGATCC-3′, we suggest that this may represent a signature sequence for functional Rel/NF-κB sites that bind Rel homodimers. Interestingly, the A1 and IRF4 κB sites do not conform to the artificial high-affinity Rel consensus sequences, but instead are more closely related to NF-κB1 homodimer binding sites 43. While the reason for this difference is unclear, one plausible explanation may be that physiological κB sites need to bind Rel homodimers with lower affinity than the artificial sites which were selected in vitro to favor high-affinity protein–DNA interactions.
IRF-4, a Transcriptional Target of Rel: Implications for the Immune Response and Cell Division.
Our findings establish that Rel/NF-κB transcription factors are dichotomous regulators of IFN signaling. While NF-κB has been shown to be important in viral-induced human IFN-β transcription 44, we show here that Rel can modulate IFN-regulated gene expression in lymphocytes by inducing IRF-4, a repressor of ISRE-regulated transcription. While higher than normal 2′5′OAS expression in mitogen plus IFN-activated c-rel −/− lymphocytes is consistent with a role for IRF-4 in the transcriptional repression of this gene, the expression of certain other IFN-induced genes is not altered in c-rel −/− B and T cells (Grumont, R., unpublished results). The reason for the selective modulation of IFN-regulated gene expression in c-rel −/− lymphocytes remains to be determined. For example, it may reflect the mechanism(s) by which IRF-4 blocks IFN-induced transcription, which is thought to occur either by direct competition with activators for binding to ISREs 33 34 or through protein–protein interactions with activators such as IRF-1 or ISGF3, as has been proposed for IFN consensus sequence binding protein (ICSBP)-mediated repression 45.
In contrast to other IRF genes, the expression of IRF-4 is not regulated by type I or type II IFNs, but instead is induced by Rel/NF-κB in response to mitogenic stimulation. As IRF-4 is required for lymphocyte proliferation 14 and inhibits IFN-induced transcription 33, it had been proposed that this novel mode of IRF regulation represents a mechanism that permits lymphocyte activation under conditions normally refractory to cellular proliferations 29 34. Particularly as the growth inhibitory activity of IFNs 35 would be detrimental to lymphocyte function during an immune response, attenuating the effects of IFN by upregulating IRF-4 could be envisaged as a means of maximizing the antiviral immune response of B and T cells in a microenvironment with high local concentrations of these cytokines. Certainly, such a dual role for IRF-4 in regulating lymphocyte proliferation and IFN gene expression is compatible with the finding that IRF-4 is a cellular gene induced by HTLV-1 Tax. As retroviral replication is dependent on the division of target cells 46, Tax induction of IRF-4 would contribute in maintaining an activated cellular state, while aiding HTLV-1–infected cells to evade the anti-viral response by repressing IFN-regulated gene expression.
Although IFR-4 is a transcriptional target of Rel that regulates lymphocyte responsiveness to IFNs during cellular activation, the finding that mature B and T cells from young IRF-4 −/− mice, like c-rel −/− lymphocytes, proliferate poorly when treated with diverse mitogens 14 indicates that IRF-4 is also important in controlling IFN-independent lymphocyte proliferation. Although these observations support a model in which c-rel −/− B cell proliferative defects are in part due to an inability to upregulate IRF-4, this appears to be at odds with the finding that lymphoproliferative disorders develop in older IRF-4 −/− mice 14, but not c-rel −/− animals 7. The apparent paradox that an absence of IRF-4 can both reduce and increase lymphocyte proliferation supports an emerging idea that normal cellular proliferation requires the balanced expression of IRF transcriptional activators and repressors, and that altering this balance perturbs cell growth 18. Such a model would also account for the observation that dysregulated overexpression of IRF-4, resulting from a translocation of the gene into the Ig CH locus contributes to the development of multiple myeloma in humans 15. The finding that IRF-4 is a transcriptional target of Rel may also have important implications for v-Rel–mediated transformation. Because v-Rel lacks part of the Rel transactivation domain, the transcriptional activity of the viral oncoprotein differs from that of Rel 47 48. Consequently, a v-Rel–induced change in the normal pattern of IRF-4 expression could perturb cell growth and may help to explain the basis of lymphoid-specific transformation by v-Rel.
Although it remains to be determined which IRF-4–regulated genes are involved in the control of lymphocyte proliferation, 2′5′OAS represents an interesting candidate. 2′5′OAS functions to convert ATP into 2′-5′-oligoadenylate, which in turn activates the latent endonuclease RNase L 49. In addition to the 2′5′OAS/RNase L pathway being a key mechanism for inhibiting viral replication 50, activation of this enzyme is also likely to be important in degrading cellular RNA and as a consequence modulating normal cellular functions. Since overexpression of 2′5′OAS can inhibit cellular proliferation 51, the enhanced expression of 2′5′OAS in mitogen-activated c-rel −/− lymphocytes treated with IFN-α may account in part for the increased sensitivity of these cells to the antiproliferative action of this cytokine. Ultimately, the identification of direct targets of Rel and genes further along the transcriptional cascade will offer important insights into understanding how Rel regulates cellular processes in response to a variety of extracellular signals.
Acknowledgments
We thank Prof. David Baltimore for making the nfkb1l −/− mice available, Prof. Tak Mak for murine IRF-4 genomic clones, Dr. Harinder Singh for the mouse IRF-4 cDNA, Dr. Kensuke Miyake for the kind gift of anti-RP antibodies, Drs. Tom Kay, Alain Israel, and Francis Shannon for various plasmids, Drs. Tom Kay and Paul Hertzog for IFN-γ and IFN-α, Dr. Frank Battye and colleagues for assistance with cell sorting and Julie Merryfull for animal husbandry. We also wish to acknowledge Dr. Andreas Strasser for crucial discussions at the beginning of this study and for his comments together with those of Drs. David Huang and Jane Visvader on the manuscript.
This work was supported by the National Health and Medical Research Council (Australia), the Anti-Cancer Council of Victoria, Commonwealth AIDS Research Grant 971274, and the International Association for Cancer Research (Saint Andrews, UK).
Footnotes
Abbreviations used in this paper: CAT, chloramphenicol acetyltransferase; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; GFP, green fluorescent protein; IRF, IFN regulatory factor; ISRE, IFN stimulation response element; NF, nuclear factor; OAS, oligoadenylate synthetase; RHD, Rel homology domain; RP, radioprotective.
References
- Crabtree G.R. Contingent genetic regulatory events in T lymphocyte activation. Science. 1989;243:355–361. doi: 10.1126/science.2783497. [DOI] [PubMed] [Google Scholar]
- Gerondakis S., Grumont R., Rourke I., Grossmann M. The regulation and roles of Rel/NF-κB transcription factors during lymphocyte activation. Curr. Opin. Immunol. 1998;10:353–359. doi: 10.1016/s0952-7915(98)80175-1. [DOI] [PubMed] [Google Scholar]
- Baeuerle P.A., Henkel T. Function and activation of NF-kappa B in the immune system. Annu. Rev. Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- Baldwin A.S., Jr. The NF-kappa B and I kappa B proteinsnew discoveries and insights. Annu. Rev. Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- Finco T.S., Baldwin A.S. Mechanistic aspects of NF-kappa B regulationthe emerging role of phosphorylation and proteolysis. Immunity. 1995;3:263–272. doi: 10.1016/1074-7613(95)90112-4. [DOI] [PubMed] [Google Scholar]
- Verma I.M., Stevenson J.K., Schwarz E.M., Van Antwerp D., Miyamoto S. Rel/NF-kappa B/I kappa B familyintimate tales of association and dissociation. Genes Dev. 1995;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- Kontgen F., Grumont R.J., Strasser A., Metcalf D., Li R., Tarlinton D., Gerondakis S. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. 1995;9:1965–1977. doi: 10.1101/gad.9.16.1965. [DOI] [PubMed] [Google Scholar]
- Gerondakis S., Strasser A., Metcalf D., Grigoriadis G., Scheerlinck J.Y., Grumont R.J. Rel-deficient T cells exhibit defects in production of interleukin 3 and granulocyte-macrophage colony-stimulating factor. Proc. Natl. Acad. Sci. USA. 1996;93:3405–3409. doi: 10.1073/pnas.93.8.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoriadis G., Zhan Y., Grumont R.J., Metcalf D., Handman E., Cheers C., Gerondakis S. The Rel subunit of NF-kappaB-like transcription factors is a positive and negative regulator of macrophage gene expressiondistinct roles for Rel in different macrophage populations. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:7099–7107. [PMC free article] [PubMed] [Google Scholar]
- Grumont R.J., Rourke I.J., O'Reilly L.A., Strasser A., Miyake K., Sha W., Gerondakis S. B lymphocytes differentially use the Rel and nuclear factor kappaB1 (NF-kappaB1) transcription factors to regulate cell cycle progression and apoptosis in quiescent and mitogen-activated cells. J. Exp. Med. 1998;187:663–674. doi: 10.1084/jem.187.5.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumont R.J., Rourke I.J., Gerondakis S. Rel-dependent induction of A1 transcription is required to protect B cells from antigen receptor ligation-induced apoptosis. Genes Dev. 1999;13:400–411. doi: 10.1101/gad.13.4.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama T., Grossman A., Mittrucker H.W., Siderovski D.P., Kiefer F., Kawakami T., Richardson C.D., Taniguchi T., Yoshinaga S.K., Mak T.W. Molecular cloning of LSIRF, a lymphoid-specific member of the interferon regulatory factor family that binds the interferon-stimulated response element (ISRE) Nucleic Acids Res. 1995;23:2127–2136. doi: 10.1093/nar/23.12.2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumont R.J., Gerondakis S. The subunit composition of NF-kappa B complexes changes during B-cell development. Cell Growth Differ. 1994;5:1321–1331. [PubMed] [Google Scholar]
- Mittrucker H.W., Matsuyama T., Grossman A., Kundig T.M., Potter J., Shahinian A., Wakeham A., Patterson B., Ohashi P.S., Mak T.W. Requirement for the transcription factor LSIRF/IRF4 for mature B and T lymphocyte function. Science. 1997;275:540–543. doi: 10.1126/science.275.5299.540. [DOI] [PubMed] [Google Scholar]
- Iida S., Rao P.H., Butler M., Corradini P., Boccadoro M., Klein B., Chaganti R.S., Dalla-Favera R. Deregulation of MUM1/IRF4 by chromosomal translocation in multiple myeloma. Nat. Genet. 1997;17:226–230. doi: 10.1038/ng1097-226. [DOI] [PubMed] [Google Scholar]
- Nguyen H., Hiscott J., Pitha P.M. The growing family of interferon regulatory factors. Cytokine Growth Factor Rev. 1997;8:293–312. doi: 10.1016/s1359-6101(97)00019-1. [DOI] [PubMed] [Google Scholar]
- Tanaka N., Kawakami T., Taniguchi T. Recognition DNA sequences of interferon regulatory factor 1 (IRF-1) and IRF-2, regulators of cell growth and the interferon system. Mol. Cell. Biol. 1993;13:4531–4538. doi: 10.1128/mcb.13.8.4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi T., Lamphier M.S., Tanaka N. IRF-1the transcription factor linking the interferon response and oncogenesis. Biochim. Biophys. Acta. 1997;1333:M9–M17. doi: 10.1016/s0304-419x(97)00014-0. [DOI] [PubMed] [Google Scholar]
- Fujita T., Kimura Y., Miyamoto M., Barsoumian E.L., Taniguchi T. Induction of endogenous IFN-alpha and IFN-beta genes by a regulatory transcription factor, IRF-1. Nature. 1989;337:270–272. doi: 10.1038/337270a0. [DOI] [PubMed] [Google Scholar]
- Harada H., Fujita T., Miyamoto M., Kimura Y., Maruyama M., Furia A., Miyata T., Taniguchi T. Structurally similar but functionally distinct factors, IRF-1 and IRF- 2, bind to the same regulatory elements of IFN and IFN-inducible genes. Cell. 1989;58:729–739. doi: 10.1016/0092-8674(89)90107-4. [DOI] [PubMed] [Google Scholar]
- Sha W.C., Liou H.C., Tuomanen E.I., Baltimore D. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell. 1995;80:321–330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- Luckow B., Schutz G. CAT constructions with multiple unique restriction sites for the functional analysis of eukaryotic promoters and regulatory elements. Nucleic Acids Res. 1987;15:5490. doi: 10.1093/nar/15.13.5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho S.N., Hunt H.D., Horton R.M., Pullen J.K., Pease L.R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- Miyake K., Yamashita Y., Hitoshi Y., Takatsu K., Kimoto M. Murine B cell proliferation and protection from apoptosis with an antibody against a 105-kD moleculeunresponsiveness of X-linked immunodeficient B cells. J. Exp. Med. 1994;180:1217–1224. doi: 10.1084/jem.180.4.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumont R.J., Richardson I.B., Gaff C., Gerondakis S. rel/NF-kappa B nuclear complexes that bind κB sites in the murine c-rel promoter are required for constitutive c-rel transcription in B-cells. Cell Growth Differ. 1993;4:731–743. [PubMed] [Google Scholar]
- Bohnlein E., Siekevitz M., Ballard D.W., Lowenthal J.W., Rimsky L., Bogerd H., Hoffman J., Wano Y., Franza B.R., Greene W.C. Stimulation of the human immunodeficiency virus type 1 enhancer by the human T-cell leukemia virus type I tax gene product involves the action of inducible cellular proteins. J. Virol. 1989;63:1578–1586. doi: 10.1128/jvi.63.4.1578-1586.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteside S.T., Ernst M.K., LeBail O., Laurent-Winter C., Rice N., Israel A. N- and C-terminal sequences control degradation of MAD3/I kappa B alpha in response to inducers of NF-kappa B activity. Mol. Cell. Biol. 1995;15:5339–5345. doi: 10.1128/mcb.15.10.5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pear W.S., Nolan G.P., Scott M.L., Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. USA. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenbeis C.F., Singh H., Storb U. Pip, a novel IRF family member, is a lymphoid-specific, PU.1-dependent transcriptional activator. Genes Dev. 1995;9:1377–1387. doi: 10.1101/gad.9.11.1377. [DOI] [PubMed] [Google Scholar]
- Piechaczyk M., Blanchard J.M., Marty L., Dani C., Panabieres F., El Sabouty S., Fort P., Jeanteur P. Post-transcriptional regulation of glyceraldehyde-3-phosphate-dehydrogenase gene expression in rat tissues. Nucleic Acids Res. 1984;12:6951–6963. doi: 10.1093/nar/12.18.6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichii Y., Fukunaga R., Shiojiri S., Sokawa Y. Mouse 2-5A synthetase cDNAnucleotide sequence and comparison to human 2-5A synthetase. Nucleic Acids Res. 1986;14:10117. doi: 10.1093/nar/14.24.10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga K., Taniguchi H., Yoda K., Shimizu M., Sakiyama S. Nucleotide sequence of a full-length cDNA for mouse cytoskeletal beta-actin mRNA. Nucleic Acids Res. 1986;14:2829. doi: 10.1093/nar/14.6.2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata T., Nishida J., Tanaka S., Sakai R., Mitani K., Yoshida M., Taniguchi T., Yazaki Y., Hirai H. A novel interferon regulatory factor family transcription factor, ICSAT/Pip/LSIRF, that negatively regulates the activity of interferon- regulated genes. Mol. Cell. Biol. 1996;16:1283–1294. doi: 10.1128/mcb.16.4.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brass A.L., Kehrli E., Eisenbeis C.F., Storb U., Singh H. Pip, a lymphoid-restricted IRF, contains a regulatory domain that is important for autoinhibition and ternary complex formation with the Ets factor PU.1. Genes Dev. 1996;10:2335–2347. doi: 10.1101/gad.10.18.2335. [DOI] [PubMed] [Google Scholar]
- Lindahl-Magnusson P., Leary P., Gresser I. Interferon inhibits DNA synthesis induced in mouse lymphocyte suspensions by phytohaemagglutinin or by allogeneic cells. Nature New Biol. 1972;237:120–121. doi: 10.1038/newbio237120a0. [DOI] [PubMed] [Google Scholar]
- Belardelli F., Gresser I. The neglected role of type I interferon in the T-cell responseimplications for its clinical use. Immunol. Today. 1996;17:369–372. doi: 10.1016/0167-5699(96)10027-X. [DOI] [PubMed] [Google Scholar]
- Williams B.R. Transcriptional regulation of interferon-stimulated genes. Eur. J. Biochem. 1991;200:1–11. doi: 10.1111/j.1432-1033.1991.tb21041.x. [DOI] [PubMed] [Google Scholar]
- Miyamoto M., Fujita T., Kimura Y., Maruyama M., Harada H., Sudo Y., Miyata T., Taniguchi T. Regulated expression of a gene encoding a nuclear factor, IRF-1, that specifically binds to IFN-beta gene regulatory elements. Cell. 1988;54:903–913. doi: 10.1016/s0092-8674(88)91307-4. [DOI] [PubMed] [Google Scholar]
- Smith M.R., Greene W.C. Molecular biology of the type I human T-cell leukemia virus (HTLV-I) and adult T-cell leukemia. J. Clin. Invest. 1991;87:761–766. doi: 10.1172/JCI115078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M., Suzuki T., Fujisawa J., Hirai H. HTLV-1 oncoprotein tax and cellular transcription factors. Curr. Top. Microbiol. Immunol. 1995;193:79–89. doi: 10.1007/978-3-642-78929-8_4. [DOI] [PubMed] [Google Scholar]
- Urban M.B., Schreck R., Baeuerle P.A. NF-kappa B contacts DNA by a heterodimer of the p50 and p65 subunit. EMBO (Eur. Mol. Biol. Organ.) J. 1991;10:1817–1825. doi: 10.1002/j.1460-2075.1991.tb07707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen S.K., Nerlov C., Zabel U., Verde P., Johnsen M., Baeuerle P.A., Blasi F. A novel complex between the p65 subunit of NF-kappa B and c-Rel binds to a DNA element involved in the phorbol ester induction of the human urokinase gene. EMBO (Eur. Mol. Biol. Organ.) J. 1992;11:205–213. doi: 10.1002/j.1460-2075.1992.tb05043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunsch C., Ruben S.M., Rosen C.A. Selection of optimal kappa B/Rel DNA-binding motifsinteraction of both subunits of NF-kappa B with DNA is required for transcriptional activation. Mol. Cell. Biol. 1992;12:4412–4421. doi: 10.1128/mcb.12.10.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenardo M.J., Fan C.M., Maniatis T., Baltimore D. The involvement of NF-kappa B in beta-interferon gene regulation reveals its role as widely inducible mediator of signal transduction. Cell. 1989;57:287–294. doi: 10.1016/0092-8674(89)90966-5. [DOI] [PubMed] [Google Scholar]
- Bovolenta C., Driggers P.H., Marks M.S., Medin J.A., Politis A.D., Vogel S.N., Levy D.E., Sakaguchi K., Appella E., Coligan J.E. Molecular interactions between interferon consensus sequence binding protein and members of the interferon regulatory factor family. Proc. Natl. Acad. Sci. USA. 1994;91:5046–5050. doi: 10.1073/pnas.91.11.5046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varmus H., Swanstrom R. Replication of retroviruses. In: Weiss R., Teich N., Varmus H., Coffin J., editors. RNA Tumor Viruses. 2nd ed. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1982. pp. 369–512. [Google Scholar]
- Inoue J., Kerr L.D., Ransone L.J., Bengal E., Hunter T., Verma I.M. c-rel activates but v-rel suppresses transcription from kappa B sites. Proc. Natl. Acad. Sci. USA. 1991;88:3715–3719. doi: 10.1073/pnas.88.9.3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore T.D. Role of rel family genes in normal and malignant lymphoid cell growth. Cancer Surv. 1992;15:69–87. [PubMed] [Google Scholar]
- Lengyel P. Biochemistry of interferons and their actions. Annu. Rev. Biochem. 1982;51:251–282. doi: 10.1146/annurev.bi.51.070182.001343. [DOI] [PubMed] [Google Scholar]
- Chebath J., Benech P., Hovanessian A., Galabru J., Revel M. Four different forms of interferon-induced 2′,5′-oligo(A) synthetase identified by immunoblotting in human cells. J. Biol. Chem. 1987;262:3852–3857. [PubMed] [Google Scholar]
- Rysiecki G., Gewert D.R., Williams B.R. Constitutive expression of a 2′,5′-oligoadenylate synthetase cDNA results in increased antiviral activity and growth suppression. J. Interferon Res. 1989;9:649–657. doi: 10.1089/jir.1989.9.649. [DOI] [PubMed] [Google Scholar]