

Abstract
Developing B cells undergo dramatic changes in their responses to chemoattractant cytokines (chemokines) and in expression of chemokine receptors. Bone marrow pre–pro-B cells (AA4.1+/natural killer 1.1− Fraction A cells) and cells capable of generating pro-B colonies in the presence of interleukin 7 and flt3 ligand migrate to thymus-expressed chemokine (TECK), a response lost in later stages of B cell development. B cell–attracting chemokine 1 (BCA-1) responses correlate with CXC chemokine receptor (CXCR)5 expression, are first displayed by a pro-B cell subset, are lost in pre-B cells, and then are regained just before and after egress from the marrow. All peripheral B cell subsets, including follicular and germinal center as well as marginal zone and peritoneal B1 B cells, respond to BCA-1, implying that responsiveness to this follicular chemokine is not sufficient to predict follicle localization. Responses to the CC chemokine receptor (CCR)7 ligands secondary lymphoid tissue chemoattractant (SLC) and macrophage inflammatory protein (MIP)-3β, implicated in homing to lymphoid tissues, are upregulated before B cell exit from the marrow, but increase further in the periphery and are shared by all peripheral B cells. In contrast, responsiveness to MIP-3α and expression of CCR6 are acquired only after emigration to the periphery and during maturation into the recirculating B cell pool. Chemotaxis to stromal cell–derived factor 1α is observed at all stages of B cell differentiation. Thus, unique patterns of chemokine responses may help define developing B cell populations and direct their maturation in the marrow and migration to the periphery.
Keywords: B lymphocyte, chemokines, chemotaxis, bone marrow, hematopoiesis
Introduction
B cells provide a two-pronged defense against invading pathogens. B cells internalize specific antigens via their surface Ig (sIg) receptors, process the antigen into peptides, and present the peptides to T cells in the context of the B cells' class II MHC. In this manner, they act as sentinels and survey the body for the presence of nonself-antigens. If nonself-antigens are detected, B cells undergo a complex developmental program to generate large quantities of high-affinity neutralizing antibodies that help in clearing the pathogen from the host. To encounter antigen and to obtain help in generating a potent humoral response, the fulfillment of these two complex tasks requires that B cells migrate into specific areas at specific times during their development. Although the physical site(s) of B cell precursor development and differentiation in the bone marrow (BM) remain poorly defined, mature peripheral B cell subsets occupy characteristic microenvironments or domains. The major B cell microenvironment in all secondary lymphoid tissues is the primary follicle. The follicle contains recirculating naive B cells that sample secondary lymphoid organs such as the spleen, LNs, and Peyer's patches (PPs) for the presence of nonself-antigens and contribute to T cell–dependent humoral responses 1. In the spleen, an additional B cell population occupies the marginal zone (MZ) that surrounds the white pulp and are positioned near the efficient antigen-trapping marginal sinus macrophages. MZ B cells are thought to sample blood-borne T cell–independent antigens that flow through the marginal sinus 2 3 4. They turn over more slowly than follicular B cells and recirculate poorly to other lymphoid organs 4 5, but are translocated rapidly into the splenic white pulp in response to endotoxin stimulation 6. During active immune responses, the follicle or follicular mantle surrounds another distinct B cell microenvironment, the germinal center (GC). GCs are sites of antigen-driven proliferation, affinity maturation, and memory B cell generation 7 8. Finally, serosal surface cavities are populated by B1 or CD5+ B cells, which may represent an alternative B cell development pathway. They are the primary source of natural antibodies and contribute to auto-antibody production in autoimmune diseases 9 10. The unique and distinctive microenvironmental location of each of these well-defined B cell populations requires that they display differential homing properties.
Chemotactic signals are thought to play important roles in leukocyte navigation by regulating migration from the blood into tissues, as well as subsequent microenvironmental localization within those tissues. The list of characterized leukocyte chemoattractants has grown rapidly with the identification of the chemoattractant cytokine (chemokine) superfamily 11. Chemokines send directional signals to leukocytes by binding to seven transmembrane receptors that are coupled to pertussis toxin–inhibitable Gαi G protein heterotrimers. Most leukocytes express multiple chemokine and other chemoattractant receptors in overlapping patterns as a function of their development and activation state. Cells expressing multiple receptors can navigate in a step-by-step fashion through spatial arrays of overlapping chemokine and chemoattractant gradients 12. Thus, the migration and microenvironmental targeting of leukocytes are thought to be determined as a function (in part) of the set of chemokines to which they can respond: their chemokine response profile or “fingerprint.”
Several chemokines attract primary B cells or B cell lines and have been hypothesized to play a role in the homing and microenvironmental localization of B cell subsets at different stages of antigen-independent and -dependent B cell differentiation 13 14 15 16 17 18 19 20. To explore the signals that guide B cells, we identified chemokines to which developing and mature B cells respond and assessed how those responses change as the B cells develop and populate various locales throughout the body. Our results reveal dramatic developmental switches in chemotactic response profiles during B cell development and suggest that altered chemotactic responses may play a major role in determining the migration patterns of developing B cells emigrating to the periphery after differentiation in the BM. Conversely, the major peripheral mature B cell subsets (follicular, MZ, GC, and B1 B cells) display surprisingly similar responses to the known secondary lymphoid and follicular chemokines, suggesting that microenvironmental localization in the periphery involves additional uncharacterized elements.
Materials and Methods
Mice.
Male and female C57BL/6 mice were housed and bred at the Veterans Affairs Palo Alto Health Care Systems mouse facility under specific pathogen-free conditions.
Flow Cytometric and Chemokine Reagents.
The following antibodies were used (specificities and modifications in parentheses), all from PharMingen: RA3-6B2 (allophycocyanin [APC]-conjugated rat anti–mouse CD45R/B220); 145-2C11 (biotin-conjugated Armenian hamster anti–mouse CD3ε); AF6-78 (PE-conjugated rat anti–mouse IgMb); 53-7.3 (PE-conjugated anti–mouse CD5); 1D3 (PE-conjugated rat anti–mouse CD19); B3B4 (PE-conjugated rat anti–mouse CD23); M1/69 (PE-conjugated rat anti–mouse CD24 [heat-stable antigen, HSA]); 11-26c.2a (FITC-conjugated rat anti–mouse IgD); II/41 (FITC-conjugated rat anti–mouse IgM); 145-2C11 (FITC-conjugated Armenian hamster anti–mouse CD3ε); 7G6 (FITC-conjugated rat anti–mouse CD21); M1/69 (FITC-conjugated rat anti–mouse CD24 [HSA]); S7 (FITC-conjugated rat anti–mouse CD43); and PK136 (FITC-conjugated rat anti–mouse NK1.1). Purified rabbit anti–mouse CXC chemokine receptor (CXCR)5 was obtained from Dr. Jason Cyster (University of California at San Francisco, San Francisco, CA), and biotin-conjugated goat anti–rabbit IgG was purchased from PharMingen. Rat anti–mouse CXCR3 (5B4) and CC chemokine receptor (CCR)6 (1C12) mAb supernatants were obtained from LeukoSite, and biotin-conjugated mouse anti–rat IgG H+L was purchased from Jackson Immunoresearch Laboratories. Peridinine chlorophyll protein (PerCP)-conjugated streptavidin (SAv; Becton Dickinson) was used to visualize biotinylated anti–mouse CD3ε, anti–mouse CXCR3, anti–mouse CXCR5, and anti–mouse CCR6. FITC-conjugated peanut agglutinin (PNA; EY Laboratories) was used to visualize GC cells. Technical grade rat IgG was purchased from Sigma Chemical Co.
The following chemokines were purchased from PeproTech: recombinant mouse eotaxin, human I-309, mouse JE (monocyte chemoattractant protein [MCP]-1), mouse KC (Gro-α), human macrophage inflammatory protein (MIP)-3α (liver and activation-regulated chemokine [LARC]), mouse regulated on activation, normal T cell expressed and secreted (RANTES), and human thymus and activation-regulated chemokine (TARC). The following chemokines were purchased from R&D Systems: recombinant human B cell–attracting chemokine 1 (BCA-1), recombinant mouse B lymphocyte chemoattractant (BLC), mouse monokine induced by IFN-γ (MIG), mouse MIP-1β, rat MIP-3α (LARC), mouse MIP-3β (EBV-induced molecule 1 ligand chemokine [ELC]), mouse secondary lymphoid tissue chemoattractant (SLC, 6Ckine), and mouse thymus-expressed chemokine (TECK). Human stromal cell–derived factor (SDF)-1α was either provided by Gryphon or purchased from PeproTech.
Lymphocyte Isolation.
6–10-wk-old mice of both sexes were killed by cervical dislocation. Peripheral LNs (subiliac, proper axillary, accessory axillary, and cervical), mesenteric LNs, PPs, and spleen were harvested, minced with scissors in RPMI/10% bovine calf serum (BCS), and pressed through a wire mesh screen with a rubber syringe plunger. Peritoneal cavity cells were harvested by injecting 10 ml of RPMI/10% BCS into the peritoneal cavity using a 27-gauge needle, agitating the cavity periodically for 5 min, and removing the lymphocyte-rich solution with a 19-gauge needle. BM cells were harvested by dissecting the femur and tibia, removing all muscle from the bone, cutting the ends off the bones, flushing with 3 ml of RPMI/10% BCS through both ends of the bone, and passing the disrupted marrow though a wire mesh screen. Splenocytes and BM cells were depleted of erythrocytes by lysis in ACK buffer (5 min at room temperature). The lymphocytes were allowed to incubate in RPMI/10% BCS for 1 h at 37°C in a CO2 incubator in T-75 flasks to remove tissue culture flask adherent cells and to allow time for resensitization of potentially desensitized chemokine responses.
Chemotaxis Assay and Quantitation.
Chemotaxis assays were performed in 5-μm pore transwell inserts as described by Campbell et al. 21, with the following modifications. Chemokines were used at the following concentrations: 500 nM BCA-1, 1 nM JE, 100 nM MCP-3, 100 nM MIG, 10 nM MIP-1α, 3 nM MIP-1β, 100 nM MIP-3α, 100 nM MIP-3β, 100 nM RANTES, 50 nM SDF-1α, 100 nM SLC, and 300 nM TECK. The chemotaxis assay proceeded for 2 h, a known number of counting beads were added to each chemotaxis well after Transwell insert removal, and the contents of the chemotaxis well were transferred to a polypropylene pointed bottom tube. The beads and cells were centrifuged at 200 g for 10 min, excess medium was removed, and the cells were resuspended in 25 μl of cold Staining Buffer (PBS/1 mM MgCl2/1 mM CaCl2/0.1% NaN3/2% Fraction V BSA) containing 30 μg of rat IgG (Sigma Chemical Co.). The cells were incubated on ice for 15 min and 25 μl of the appropriate antibody cocktail was added. The cells were further incubated on ice for 30 min, washed with 3 ml of Staining Buffer, centrifuged, and incubated with 50 μl of Staining Buffer containing 2.5 μl Sav-PerCP for 30 min. The cells were either washed, centrifuged, and analyzed by flow cytometry (non–PNA-stained cells), or incubated with 50 μl of PNA-FITC for 4–6 min, stopped by addition of 3 ml of PBS/2% formaldehyde or Staining Buffer, centrifuged, and analyzed by flow cytometry (for some PP B cell staining).
Chemokine Receptor Staining.
The indicated lymphoid organs were disrupted and lymphocytes were isolated as described. The cells were incubated for 1 h at 37°C to remove tissue culture plate–adherent cells. 5 × 105 lymphocytes were stained with either purified rabbit anti–mouse CXCR5 polyclonal antibody at 1:25 dilution or purified rabbit anti-R–ras (0.5 μg) as a negative control in 25 μl of Staining Buffer 22. Cells were washed and stained with biotin-conjugated goat anti–rabbit IgG. Cells were washed, blocked with 30 μg rat IgG, and incubated with the appropriate cocktail of conjugated antibodies (and SAv-PerCP) to visualize the relevant B cell populations.
106 lymphocytes were stained with supernatants from either rat anti–mouse CXCR3 mAb, rat anti–mouse CCR6 mAb, or irrelevant control supernatants (HECA-452, MK2.7). Cells were washed and stained with biotin-conjugated mouse anti–rat IgG H+L. Cells were washed, blocked with 30 μg rat IgG, and incubated with the appropriate cocktail of conjugated antibodies (and SAv-PerCP) to visualize the relevant B cell populations.
During chemokine receptor staining of the splenic Transition 1 population, anti–mouse CD3ε-FITC, was included with B220-APC, CD23-PE, and CD21-FITC to prevent B220+ T cells (which do not have CD23 or CD21) from contaminating the Transition 1 population (B220+CD23−CD21−/lo). During chemokine receptor staining of PP GC B cells, B220+ T cells were excluded from analysis by staining with B220-APC, CD19-PE, and PNA-FITC because B cells but not B220+ T cells express CD19.
Pro-B CFU Assay.
Unfractioned BM cells were isolated and allowed to migrate to chemokines as described. Responding cells from two chemotaxis wells (input 2E6 BM cells per Transwell insert) were harvested, pelleted by centrifugation, and resuspended in 75 μl of RPMI/10% BCS. 1.35 ml of Methocult M3630 containing 10 ng/ml recombinant human IL-7 (StemCell Technologies Inc.) was aliquoted into 12 × 75 mm polypropylene tubes, and 75 μl of recombinant murine flt3 ligand (Flt3-L; R&D Systems) was added to give a final concentration of 50 ng/ml Flt3-L (based on the methods of Hunte et al. 23 and Veiby et al. 24). Responding cells were added to the Methocult mixture, vortexed for 5 s, and aliquoted to 35 × 10 mm tissue culture polystyrene dishes using a 20-gauge needle. Multiple plates were placed in a 150 × 15 mm plate containing an uncovered 35 × 10 mm plate filled with water to prevent dehydration. The plates were incubated in a 37°C incubator for 12–17 d. Colonies were identified and counted using a dissecting microscope. Colonies were harvested by removing them with a pipette and stained with B220-APC, HSA-biotin/SAv-PerCP, CD19-PE, and Mac-1–FITC to confirm the pro-B colony identification. The TECK-responding pro-B CFUs only became apparent in the second week of the assay, and they were tightly compact colonies compared with a few very spread and disorganized B220− colonies that were found in all assays (including the Basal Medium control plates). Many pro-B CFU colonies had a multilobed shape or were a collection of three to six small colonies very close together. In all cases, these were counted as a single colony because the relatively few number of colonies per plate did not suggest that these colonies arose from multiple pro-B CFUs that happen to settle out close together.
Results
Developmental Shifts in Chemokine Response Profiles during B Cell Development in the BM.
B cells develop in the BM through sequential developmental stages, from stem cells to multipotent progenitors to pre–pro-B cells, to pro-B cells, to pre-B cells, and finally to immature B cells. After commitment to the B cell lineage, these stages have been associated with particular surface phenotypic markers according to Hardy et al. and Li et al. 25 26. To explore the developmental control of the chemotaxis response profiles during B cell development, we assessed the chemotactic responses of cells at each of these antigenically defined stages (Fig. 1 A). A panel of chemokines was selected that bound all of the currently described CC and CXC receptors (Table ). Naive splenic μδ+ follicular B cells (μ = sIgM heavy chain, and δ = sIgD heavy chain), the most abundant mature peripheral B cell population, were evaluated in parallel for comparison. Chemokines were titered from 0.1–100 nM (TECK was tested up to 1 μM and BCA-1 was tested up to 2.5 μM), and results shown here reflect responses to the concentrations that gave the optimal migration of BM and peripheral B cells (concentrations used are given in the legend of Table ). No significant difference was observed in the dose–response curves to any chemokine by any B cell subset analyzed in this paper (differences in the magnitude of response will be presented below).
Figure 1.

The chemotactic fingerprint of B cells undergoes a dramatic shift during development in the BM. (A) List of antigenically defined stages in B cell development, according to Hardy et al. and Li et al. (references 25 26). (B) AA4.1+NK1.1− Fraction A cells respond to RANTES, JE, MIG, and TECK. Unfractioned BM cells were prepared and added to inserts that were placed in wells containing the following chemokines: 100 nM RANTES, 1 nM JE, 100 nM MIG, or 300 nM TECK. Responding cells were harvested and stained with either B220-APC, unlabeled AA4.1/mouse anti–rat IgG-biotin/SAv-PerCP, HSA-PE, and NK1.1-FITC or AA4.1-APC, HSA-biotin/SAv-PerCP, B220-PE, NK1.1-FITC to identify the AA4.1+NK1.1− Fraction A population or with combinations of B220-APC, IgMb-PE, HSA-PE, and CD43-FITC to identify the later stages during B cell development. The data are the mean ± SEM from two to eight experiments. (C) Responses to peripheral lymphoid chemokines appear late during BM B cell development. Same procedure as above except the following chemokines were used: 100 nM MIP-3α, 100 nM SLC, 100 nM MIP-3β, 50 nM SDF-1α, and 500 nM BCA-1. The data are the mean ± SEM from 3–15 experiments. All symbols have error bars even though in some instances (i.e., Basal migration and pro-B/BCA-1 migration) the error bars are smaller than the symbol.
Table 1.
Chemokines Used and Their Known Receptors
| Chemokine | Receptor |
|---|---|
| CCR1 | MIP-1α, MCP-3 |
| CCR2 | JE (MCP-1), MCP-3 |
| CCR3 | Eotaxin |
| CCR4 | TARC |
| CCR5 | RANTES, MIP-1α, MIP-1β |
| CCR6 | MIP-3α (LARC) |
| CCR7 | MIP-3β (ELC), SLC (6Ckine) |
| CCR8 | I-309 |
| CCR9 | TECK |
| CXCR2 | KC |
| CXCR3 | MIG, SLC (6Ckine) |
| CXCR4 | SDF-1α |
| CXCR5 | BCA-1 (BLC) |
The chemokines were used at the following concentrations: 100 nM MIP-1α, 100 nM MCP-3, 1 nM JE, 100 nm Eotaxin, 100 nM TARC, 100 nM RANTES, 3 nM MIP-1β, 100 nM MIP-3α, 100 nM MIP-3β, 100 nM SLC, 100 nM I-309, 300 nM TECK, 100 nM KC, 100 nM MIG, 50 nM SDF-1α, or 500 nM BCA-1.
None of the BM B cell populations nor the splenic μδ+ B cells responded to eotaxin, TARC, I-309, or KC; migration to these chemokines at all concentrations tested was indistinguishable from background migration (data not shown). Non-B cell populations present in the BM and periphery responded to KC and TARC, confirming these chemokines' activity in our assay. MIP-3α, a well-characterized peripheral lymphocyte-active chemokine also failed to attract B lineage cells in the BM (but see next section). All populations responded fairly equally to SDF-1α, confirming their competence to migrate under our experimental conditions (Fig. 1 C).
Interestingly, cells of pre–pro-B cell phenotype (B220lo HSA−/loAA4.1+NK1.1−; a subpopulation within the original Fraction A population of Hardy et al. 25 that combines the A1 and A2 fractions 26) displayed substantial migration to RANTES, JE (mouse MCP-1), MIG, and TECK, chemokines associated with monocyte and/or activated T cell responses in the periphery. As illustrated in Fig. 1 B, these cells responded well, but the response to these chemokines was lost rapidly during progression to the pro-B cell stage. B cell migration to these chemokines was not observed in any more developed BM B cell or peripheral B cell population tested (follicular μδ+ B cells to the right of the dotted line are shown as an example of a peripheral B cell population). In preliminary experiments, MIP-1α, MIP-1β, and MCP-3 stimulated migration of Fraction A BM B cells (data not shown). As these chemokines are known to bind the same receptors as RANTES and JE (Table ), they were not used in subsequent experiments.
To confirm that the Fraction A1/A2 responses to these chemokines included B lineage cells, BM cells were allowed migrate to each of these chemokines and the responding cells were placed into subsequent assays that facilitated growth of B lineage progenitors into colonies of pro-B cells or pro-B CFUs by incubation with IL-7 and Flt3-L in methylcellulose 23 24. BM cells capable of generating pro-B CFUs responded to TECK (Fig. 2), whereas RANTES and JE were much weaker at attracting pro-B CFUs and no response over control was seen to MIG. The TECK-responding cells required both IL-7 and Flt3-L to generate colonies, as they yielded much fewer colonies in the CFU assay with only IL-7 (data not shown). Therefore, very early B lineage progenitors can respond to TECK and this response is lost as B cells enter into the pro-B compartment.
Figure 2.

Early B cells respond to TECK. Unfractioned BM cells were prepared and added to inserts that were placed in wells containing the following chemokines: 100 nM RANTES, 1 nM JE, 100 nM MIG, or 300 nM TECK. Responding cells were harvested and placed in methylcellulose containing IL-7 and Flt3-L. The number of colonies was scored on days 12–17 by isolating colonies and staining with B220-APC, HSA-biotin/SAv-PerCP, CD19-PE, and Mac-1–FITC to verify its pro-B CFU classification. The data are the mean ± SEM from three individual experiments.
The loss of response to TECK during B cell development occurred at the same stage as a gain of responsiveness to chemokines associated with homing in secondary lymphoid organs: SLC, MIP-3β, and BCA-1 16. Fig. 1 C demonstrates that Fraction A1/A2 cells responded relatively poorly to SLC and MIP-3β, chemokines implicated in lymphocyte homing into peripheral lymphoid organs. However, the responses increased dramatically in the further differentiated BM B cell populations and in peripheral μδ+ follicular B cells. Similar responses of pre-B and immature B cells to MIP-3β have been reported by Ngo et al. 17.
BCA-1 (BLC), a chemokine implicated in B cell entry into the follicles of PPs and spleen (but interestingly, not most PLNs 27) is of particular interest in the context of B cell behavior and microenvironmental homing. It is known to attract the majority of B cells in the periphery, as well as a small subset of CD4+ T cells. We found that there is a consistent but small response in the pro-B cell compartment (P < 0.02 by Mann-Whitney rank sum test) that is lost with progression to the pre-B cell stage. Regained BCA-1 responsiveness in the immature B cell compartment is consistently observed, but the magnitude was variable from experiment to experiment. Mature peripheral μδ+ B cells responded strongly to BCA-1.
The differential migration of gated B220+ BM subpopulations to chemokines is illustrated in Fig. 3. Responding B lineage cells (bold lines) are stained for various markers and compared with the B lineage cells in the initial input population (light line) after migration to SLC (Fig. 3, left) and BCA-1 (Fig. 3, right). BM B cells that respond to SLC and to BCA-1 are enriched for IgM expression, but they are not identical populations. SLC-responding BM B cells contain both IgM− and IgM+ cells, with an enrichment in IgM+ cells (Fig. 3 A). Conversely, only IgM+ cells are found after chemotaxis to BCA-1 (Fig. 3 B), consistent with later acquisition of BCA-1 responsiveness. As a control, we showed that incubation with various chemokines did not change the surface level expression of any of the markers used in this study (data not shown).
Figure 3.

Flow cytometry plots illustrate the selectivity of chemokine responses by antigenically defined B cell subsets. Unfractioned BM cells were prepared and added to inserts that were placed in wells containing either 100 nM SLC (left) or 500 nM BCA-1 (right). Initial input (light line) and responding (bold line) cells were harvested and stained with different combinations of B220-APC, IgMb-PE, and CD43-FITC. All panels were gated on B220+ cells. The presented histograms were obtained by overlaying individual histograms from three to four experiments, adding them together using CELLQuest™ software (Becton Dickinson) to obtain an average representative histogram, and normalizing them to the peak cell number. CTX, chemotaxis.
Receptor expression for one of the Fraction A1/A2 chemokines, MIG, was investigated in order to study the unexpected chemotactic responses by these cells in more detail. The only known receptor for MIG is CXCR3 11, and we used an anti–mouse CXCR3 mAb in combination with the staining cocktails described above to visualize CXCR3 expression at the different stages of B cell development in Fig. 4 A. Fraction A1/A2 cells (Fig. 4 A, first column) displayed very low to no staining with anti-CXCR3 mAb, whereas the AA4.1− Fraction A subpopulation displayed fairly uniform CXCR3 staining (data not shown). No B lineage cells at later stages of development in the BM or in the periphery (Fig. 4 A, last column) expressed CXCR3, consistent with their failure to migrate to MIG (Fig. 1 B).
Figure 4.

Chemokine receptor expression by B cells at different stages of development in the BM. Control staining (dashed line) and antichemokine receptor staining (bold line) for CXCR3 (A), CXCR5 (B), and CCR6 (C) are shown for the indicated BM populations identified by their surface expression of antigens listed in Fig. 1 A except for Fraction A1/A2, which were identified for chemokine receptor staining purposes using AA4.1-APC, B220-PE, and HSA-FITC. Fraction (Fxn) A1/A2 cells were identified as B220+HSA−/loAA4.1+.NK1.1+ AA4.1− immature NK precursors are excluded by these gates. The data are representative of two experiments.
As the biphasic response pattern of BM B cells to BCA-1 was unique among the peripherally important chemokines tested, we further explored this chemokine–chemokine receptor interaction by determining the expression pattern of the only known receptor for BCA-1, CXCR5 14 15. No detectable expression of CXCR5 above background was observed in the Fraction A1/A2 or pre-B cell population (Fig. 4 B, first and third column). CXCR5 expression was detectable on a subset of the pro-B cell population (Fig. 4 B, second column), although the magnitude of the shift was variable. The immature B cell population displayed CXCR5 (Fig. 4 B, fourth column) but the frequency of expression was again variable, consistent with their variable responses to BCA-1. In contrast, splenic μδ+ follicular B cells were uniformly and strongly CXCR5+ (Fig. 4 B, last column). No staining above controls was observed in any of the BM B cell populations (including pro-B cells) harvested from CXCR5 knockout mice (data not shown). Similar CXCR5 expression patterns for pre-B cell and immature B cell populations have been reported by Pevzner et al. 28 and Cyster et al. 22.
Taken together, these data demonstrate a shift in chemotactic responses during B cell maturation in the BM, characterized by the loss of responsiveness to TECK and an initial increase in responsiveness to the peripheral B cell chemokines SLC, MIP-3β, and BCA-1 in the later stages of BM B cell development (the immature BM B cell). However, neither the chemotactic responses nor the CXCR5 expression levels by immature B cells approached the high levels displayed by naive splenic μδ+ B cells, suggesting that further alterations in chemokine responses occur during developmental transition to mature peripheral B cells.
Major Shift to Peripheral Chemokine Responsiveness Associated with B Cell Emigration to Peripheral Lymphoid Tissues.
The large numbers of relatively short lived B cells that have recently emigrated from the adult BM, and which are transitioning into follicular B cells, can be found in the spleen 4 29 30. Transition 1 B cells are B220+, sIgMhi, sIgD−, CD21lo/−, and CD23−; Transition 2 B cells are B220+, sIgMhi, sIgDhi, CD21hi, CD23int; whereas, fully mature recirculating naive splenic B cells are B220hi, sIgMlo, sIgDhi, CD21int, CD23int, and HSAlo 1 4 29 30. We asked whether the chemotactic responses of splenic transitional B cells were different from those of either immature BM B cells (B220loCD43−sIgM+) or mature naive splenic μδ+ follicular B cells (B220hisIgMlosIgDhi).
Fig. 5 A illustrates that, like the sIgM+ but immature BM B cells that gave rise to them, Transition 1 recent BM emigrants (B220+CD21lo/−CD23−) to the spleen do not respond to MIP-3α. However, B cells that have progressed to Transition 2 B cells (B220+sIgMhisIgDhi), or that have become members of the naive, recirculating pool of follicular μδ+ B cells, show a greatly increased responsiveness to MIP-3α. Responses to SLC, MIP-3β, SDF-1α, and BCA-1 exhibited by immature BM B cells were increased in the Transition 1 population and increased further in both the Transition 2 and the naive, splenic μδ+ B cells (Fig. 5 B). Moreover, the responses of putative blood-borne peripheral B cells passing through the BM at the time of harvest (Fraction F in the classification scheme of Hardy et al. 25, B220hiCD43−sIgM+) were indistinguishable from those of splenic follicular μδ+ B cells (data not shown).
Figure 5.

Continued maturation of B cell chemotactic responses after exit from the BM. Unfractioned BM cells or splenic lymphocytes were prepared and added to inserts that were placed in wells containing 100 nM MIP-3α (A) or 100 nM SLC, 100 nM MIP-3β, 50 nM SDF-1α, or 500 nM BCA-1 (B). Responding cells were harvested and stained with combinations of B220-APC, CD3ε-biotin/SAv-PerCP, IgMb-PE, CD23-PE, IgD-FITC, and CD21-FITC to identify different cell populations. The data are the mean ± SEM from two to six experiments. (C) Control mAb staining (dashed line) and anti-CCR6 staining (bold line) are shown for the indicated BM and splenic populations. The data are representative of two experiments. (D) Control Ab staining (dashed line) and anti-CXCR5 staining (bold line) are shown for the indicated BM and splenic populations. The data are representative of two experiments. All symbols have error bars even though in some instances (i.e., Basal migration) the error bars are smaller than the symbol.
CXCR5 expression increases, paralleling BCA-1 responsiveness, as B cells exit the BM and progress through the transitional stages into recirculating μδ+ follicular B cells. Few immature BM B cells expressed significant levels of CXCR5 (Fig. 5 D, first column), but a majority of Transition 1 recent emigrants expressed CXCR5 (Fig. 5 D, second column), and expression became more uniform and more intense on Transition 2 and μδ+ follicular B cells (Fig. 5 D, third and fourth columns). CCR6 shows a different temporal pattern of acquisition compared with CXCR5. CCR6 is undetectable on any BM B cell population (Fig. 4 C, first four columns) including immature BM B cells and the Transition 1 B cells to which they give rise (Fig. 5 C, first and second columns, respectively). In contrast, all Transition 2 and recirculating μδ+ follicular B cells display uniform CCR6 expression (Fig. 5 C, third and fourth columns, respectively).
Chemotaxis Profiles of B Cells Occupying Distinct Microenvironmental Niches within Lymphoid Tissues: Follicular, Splenic MZ, and GC Cells.
B cells occupying distinct anatomical sites in lymphoid tissue can be distinguished by their antigenic phenotypes 31. Follicular B cells are B220hi CD21intCD23int and are sIgDhi and sIgMlo (μδ1). In comparison, splenic MZ B cells are B220+CD21hiCD23lo/− and have low to zero IgD but high levels of IgM on their surface (Fig. 6 A). In stimulated lymphoid tissues, such as PPs, B cells in GCs are identified as B220+ cells that have higher level expression of terminal galactosyl residues that are recognized by the plant lectin peanut agglutinin (PNAhi) compared with the surrounding B220+PNAlo follicular mantle subset (Fig. 6 B 32 33 34). In addition, GC B cells are μδ− compared with the follicular mantle μδ+ B cells. In the experiments described here, we analyzed the migration properties of cells in each of these regions individually.
Figure 6.

Flow cytometric separation of anatomically distinct B cell subsets. Unfractioned splenic lymphocytes (A) and PP lymphocytes (B) were prepared and stained with B220-APC, CD3ε-biotin/SAv-PerCP, CD23-PE, and CD21-FITC for spleen or B220-APC, CD3ε-biotin/SAv-PerCP, and PNA-FITC for PPs. The data shown have been gated on the B220+CD3ε− population.
Splenic follicular (B220hiCD21intCD23int) and MZ (B220+CD21hiCD23lo/−) B cells migrate with similar efficiency to SLC, SDF-1α, and BCA-1 (Fig. 7 B). In contrast to IgM+ immature BM B cells and Transition 1 B cells (Fig. 5 A), follicular B cells also migrate significantly to MIP-3α (Fig. 7 A). MZ splenocytes also respond to MIP-3α, but substantially less efficiently (Fig. 7 A). Follicular B cells are also found in peripheral LNs, mesenteric LNs, and PPs (also called follicular mantle B cells if GCs are present); however, MZ B cells are far less numerous in these organs compared with spleen 35. As expected, LN (data not shown) and PP (Fig. 7E and Fig. F) follicular B cell subsets displayed very similar chemotactic fingerprints to those of their splenic counterparts. The only difference observed was that the MIP-3α responses of splenic follicular B cells were less pronounced in the corresponding PP (Fig. 7 E) and LN (data not shown but similar to PP) populations. Chemotaxis profiles of CD3ε+B220− T cells found in the corresponding organs are shown for comparison.
Figure 7.
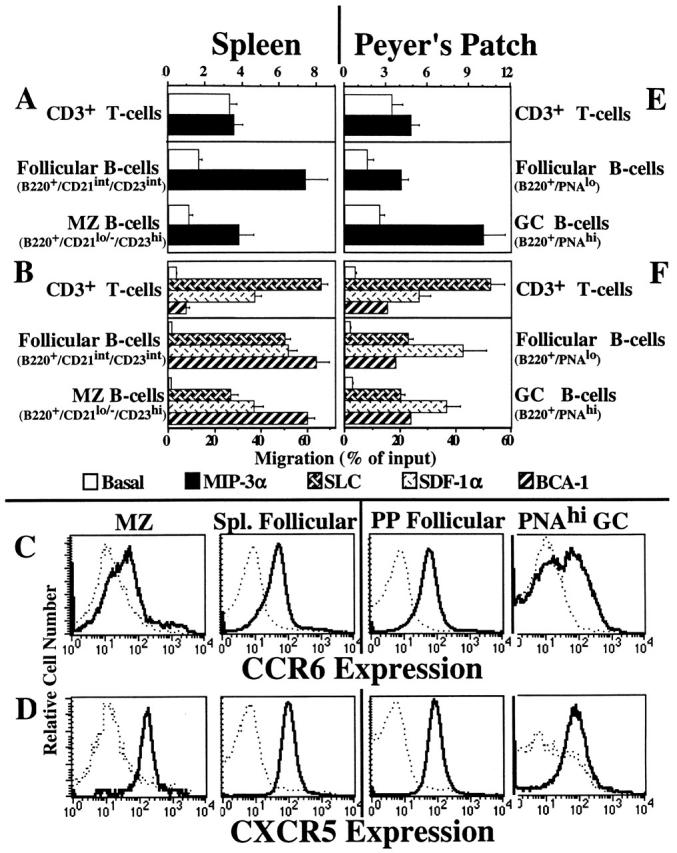
Chemotactic responses of follicular, MZ, and GC B cells. (A and B) Unfractioned splenic lymphocytes were added to inserts that were placed in wells containing 100 nM MIP-3α (A), or 100 nM SLC, 50 nM SDF-1α, or 500 nM BCA-1 (B). Responding cells were harvested, stained with the mAbs, and gated on the follicular and MZ regions shown in Fig. 6 A. The data are the mean ± SEM from three to eight experiments. (C) Control mAb staining (dashed line) and anti-CCR6 staining (bold line) are shown for the splenic CD21hiCD23−/lo MZ (first column), CD21intCD23int splenic follicular B cells (second column), B220+PNAlo PP follicular mantle (third column), and B220+PNAhi PP GC B cells (fourth column). The data are representative of two experiments. (D) Control Ab staining (dashed line) and anti-CXCR5 staining (bold line) are shown for the indicated splenic and PP populations described in C. The data are representative of two experiments. (E and F) Unfractioned PP lymphocytes were prepared and added to inserts that were placed in wells containing 100 nM MIP-3α (E) or 100 nM SLC, 50 nM SDF-1α, or 500 nM BCA-1 (F). Responding cells were harvested, stained with the mAbs, and gated on the follicular mantle and GC regions shown in Fig. 6 B. The data are the mean ± SEM from two to four experiments.
The expression of CCR6 and CXCR5 correlates with the functional responses to MIP-3α and BCA-1 by splenic B cells. In Fig. 7 C (second column), staining for CCR6+ reveals a sharp, uniform peak of expression on CD21int CD23int follicular B cells, whereas CD21hiCD23−/lo MZ B cells display broader and dimmer CCR6 expression levels (Fig. 7 C, first column). Fig. 7 D shows a sharp, uniform peak of CXCR5 expression on both CD21intCD23int follicular (second column) and CD21hiCD23−/lo MZ (first column) B cells. The levels of CCR6 and CXCR5 on peripheral LN and PP Region 1 B cells are identical to the levels observed on splenic follicular B cells (data not shown). Similar CXCR5 expression by follicular and MZ B cells has been observed by Cyster et al. 22. No detectable CXCR3 expression was observed on splenic or PP B cells (data not shown).
We studied GC B cells in PPs, secondary lymphoid organs of the gut wall, which are chronically stimulated by their constant exposure to food-borne antigens and the intestinal microflora. PP GC B cells (B220+PNAhi) responded to SLC, SDF-1α, and BCA-1 as well as the unactivated follicular mantle B cell population (B220+PNAlo), with a slight reduction in the SDF-1α response (Fig. 7 F). Interestingly, GC B cells in the PPs of C57BL/6 mice (the principle strain studied here) displayed somewhat enhanced chemotaxis to MIP-3α (Fig. 7 E). In contrast, GC B cells in the PPs of Balb/c mice displayed significantly reduced MIP-3α responses compared with their follicular mantle counterparts (data not shown). In spite of their relatively efficient migration to MIP-3α, many C57BL/6 GC B cells lacked CCR6 expression (Fig. 7 C, fourth column), with only a small number displaying high levels comparable to PP follicular B cells (Fig. 7 C, third column). PP GC B cells displayed uniform levels of CXCR5 expression (Fig. 7 D, fourth column) that were similar in magnitude to splenic follicular (Fig. 7 D, second column) and PP follicular mantle (Fig. 7 D, third column) B cells. Similar chemotaxis and receptor expression data were obtained when μδ− staining was used to discriminate PP follicular mantle and GC B cells (data not shown).
Chemotaxis Profiles of Extrafollicular Peritoneal Cavity B Cells.
A unique type of B cell is found primarily in the peritoneal and pleural cavities of the mouse. B1 or CD5+Ly-1+ B cells are part of the preimmune repertoire and may arise via an alternative developmental pathway. They are self-renewing 1 and are associated with autoimmune diseases and B cell leukemias and lymphomas 10. Peritoneal B1 B cells are B220lo and most are CD5+ and CD43+ 1. The conventional B cells (termed B2) in the peritoneal cavity have higher levels of B220 and are CD5− and CD43−. Both types of B cells from the peritoneal cavity had zero to very low basal migration compared with any of the other B cell populations we tested (Fig. 8A and Fig. B). Both B1 and B2 B cells responded to SLC, SDF-1α, and BCA-1 equally well, though to a lesser degree than splenic B cells (compare Fig. 8 B with Fig. 7 B), and their response to MIP-3α was reduced compared with splenic B cells (compare Fig. 8 A with Fig. 7 A). CCR6 expression was demonstrated on both B1 and B2 cells, although background staining of B1 B cells was relatively high (Fig. 8 C). Both B cell populations expressed high levels of the BCA-1 receptor CXCR5 (Fig. 8 D). Cyster et al. has also demonstrated CXCR5 expression on peritoneal cavity B1 B cells 22.
Figure 8.

B1 (CD5+) B cells respond to lymphoid tissue chemokines but not significantly to MIP-3α. (A and B) Unfractioned peritoneal cavity lymphocytes were prepared and added to inserts that were placed in wells containing 100 nM MIP-3α (A) or 100 nM SLC, 50 nM SDF-1α, or 500 nM BCA-1 (B). Responding cells were harvested and stained with B220-APC, CD3ε-biotin/SAv-PerCP, CD5-PE, and CD43-FITC. The data are the mean ± SEM from two to four experiments. (C) Control mAb staining (dashed line) and anti-CCR6 staining (bold line) are shown for the indicated peritoneal cavity populations. The data are representative of two experiments. (D) Control Ab staining (dashed line) and anti-CXCR5 staining (bold line) are shown for the indicated peritoneal cavity populations. The data are representative of two experiments.
Discussion
A central tenet of current models of leukocyte trafficking is that coordinated regulation of adhesive and migratory responses controls cellular positioning or homing in vivo 36. In turn, this homing determines the leukocyte's cellular and microenvironmental interactions, and therefore its fate and function. For example, developmental switches in homing receptor and vascular addressin expression have been implicated in the maturation of peripheral lymphoid tissue during the perinatal period 37 and in developmental and antigen-induced transitions in T cells 38 39. We show here that developing B lineage cells undergo dramatic alterations in their chemotactic responses to different chemokines, alterations that may help define particular stages of B cell maturation and may regulate their cellular and microenvironmental interactions, development, and/or function. We will discuss our findings in relation to (a) the major changes that occur during B lineage development, including the unique expression of a TECK response during an early B lineage stage and a dramatic switch to peripheral secondary lymphoid organ–expressed chemokine responses associated with B cell export from the marrow compartment, and (b) the patterns of chemokine responses displayed by B cells occupying different microenvironmental niches in the periphery.
We first addressed the chemokine responses of B cells at their earliest committed stage of development in the adult BM. B cell genesis derives from dividing hematopoietic stem cells (HSCs) that differentiate into progenitor cells, which in turn give rise to various hematopoietic lineages. The first identifiable BM cells committed to the B lineage are Fraction A1/A2 (AA4.1+NK1.1− cells within Fraction A 25 26 40); these cells still have their Ig genes in the germline state but display the B lineage marker B220. Surprisingly, we found that a significant fraction of AA4.1+NK1.1− Fraction A cells, unlike B lineage cells at later stages of development, migrate in response to RANTES, JE (mouse MCP-1), MIG, and TECK. These chemokines attract monocytes (RANTES and JE) and subsets of T cells (RANTES, JE, MIG, TECK) in the periphery, but have no activity on mature B cells.
Limiting dilution analysis of B220−HSA−AA4.1+CD4+ multipotent progenitors demonstrated that a large number of colonies derived from single cells gave rise to progeny colonies containing both B cells and macrophages, suggesting a close developmental relationship between monocytes and B cells 40. These data, and the observation of bipotential precursors of B cells and macrophages in the fetal liver 41, may explain the low level responses of AA4.1+ NK1.1− Fraction A cells to seemingly monocytic chemokines if both cell types arose from a common precursor. Interestingly, a recent report has shown that CXCR3 is expressed on leukemic B cells from all patients with chronic lymphocytic leukemia and is found on a subset of other B cell leukemias and lymphomas 42. In other studies, we have found that the JE/MCP-1 response in Fraction A cells is absent in CCR2-deficient mice, further confirming the association of functional responses with specific chemokine receptor expression (Bowman, E., unpublished results). However, the lack of significant response by RANTES-, JE-, and MIG-responding cells in secondary pro-B CFU assays (Fig. 2) lessens the emphasis we can place on these chemokine responses in the scheme of B cell development. Other non-B lineage cells within the Fraction A gate including AA4.1−NK1.1+ immature NK precursors 26 43 migrate very efficiently to these chemokines, and the responses seen in the AA4.1+NK1.1− population may be due to contaminating non-B lineage cells that fall within the flow cytometry gates we have set. AA4.1+ Fraction A cells do not express consistently detectable levels of CXCR3 (Fig. 4 A) even though they consistently migrated to MIG (Fig. 1 B). Either this population expresses another undescribed MIG receptor or they may express levels of CXCR3 that, while functional, are much lower than those on CXCR3+ AA4.1− Fraction A cells, levels beneath our detection limits. Preliminary studies suggest that earlier HSCs, like later stage B lineage cells shown here, are also unresponsive to these chemokines (data not shown).
TECK was the only chemokine that could effectively migrate BM cells that would give rise to pro-B CFUs in the presence of IL-7 and Flt3-L (Fig. 2), but not IL-7 alone (data not shown). IL-7 alone stimulates the proliferation of late pro-B cells and early pre-B cells, but early pro-B cells, pre-B cells, and immature B cells are unresponsive 23. Flt3-L can not stimulate the proliferation of any B lineage population alone, but Flt3-L synergizes with IL-7 to increase the growth of Lineage−/Sca-1+ BM cells (a heterogeneous population of cells containing B cell progenitors before they express B220 24) and the B cell progenitors among the B220loCD43+HSA−/lo Fraction A population 23. These data combined suggests that TECK selectively attracts very early B220+ B lineage progenitors 23 and/or B220− BM cells with potential to develop into B220+ B cells 24, as these cell types require both IL-7 and Flt3-L for growth into pro-B colonies.
It is attractive to postulate that responsiveness to TECK (and potentially RANTES and JE) by B lineage progenitors may help target them into supportive specialized niches appropriate to their developmental stage, and that downregulation of this (these) response with subsequent maturation would then allow their progression to different marrow microenvironments. However, B lineage progenitors may respond to these chemokines not only as chemotactic agents, but also as growth promoting and/or arresting cytokines. Several chemokines have been demonstrated to have potent effects on the growth of BM progenitors (especially myeloid progenitors) 44 45 46 47 48 and a feedback system has been hypothesized whereby chemokines produced in the periphery control the further production of different leukocyte lineages. Therefore, B lineage progenitor responses to these chemokines may play a role in the total B cell output by the BM by regulating the total number of B lineage cells that are allowed to develop.
As B cells progress through their developmental program, there is a dramatic switch in their chemotactic profiles. AA4.1+NK1.1− Fraction A cells convert into pro-B cells (Fraction B and C) with increasing D-JH but not V-D-JH rearrangement 25. Concomitant with Ig gene recombination, these cells lose their response to TECK and acquire responsiveness to BCA-1. Pro-B cells progress into pre-B cells (Fraction D) with increasing V-D-JH and V-Jκ rearrangement 25. These cells lose the BCA-1 response present in pro-B cells, but still possess the same small response to the CCR7 ligands exhibited by both AA4.1+ NK1.1− Fraction A cells and pro-B cells. Pre-B cells differentiate into immature B cells (Fraction E) with fully rearranged Ig-encoded DNA, completing the BM stage of B cell development 25. These sIgM+ immature B cells migrate better to the CCR7 ligands SLC and MIP-3β than their pre-B cell precursors, and reacquire responsiveness to BCA-1. CXCR5 expression mirrors BCA-1 responsiveness, with a minor proportion of pro-B cells and immature B cells expressing detectable levels of the receptor and migrating to BCA-1. Thus, developing B cells undergo a variety of developmental alterations, from the on/off response to TECK (and potentially RANTES, JE, and MIG), the on/off/on again response to BCA-1, and the progressive increases in responses to the secondary lymphoid homing–associated chemokines, SLC and MIP-3β. Similar conclusions concerning CCR7 ligands have been made in the human system by Kim et al. 49, who demonstrated that the CCR7 ligands SLC and MIP-3β induce the migration of late stage B cell progenitors (pre–B II, immature, and mature B cells), but not early pro/pre–B I cells.
Interestingly, B lineage cells at all stages of development migrate with similar efficiency to SDF-1α, at least when assayed at the optimal chemotactic concentrations used here. This finding of uniform SDF-1α responsiveness is similar to that of Kim et al. 49. However, it contrasts with the results by D'Apuzzo et al. 50; these authors report that early B lineage cells from mouse BM migrate much better to SDF-1α than do more developed B cells, and postulated that SDF-1α selectively attracts and confines early B cell precursors within the BM. The basis for this difference is unclear. Our results and those of Kim et al. also raise the possibility that apparent effects of SDF-1α and CXCR4 on B cell retention in the BM may reflect secondary or indirect effects, rather than a direct SDF-1α–mediated retention mechanism. One scenario would be that stromal-produced SDF-1α stimulates hematopoietic non-B cells in the BM to produce the factor required to retain B cells in BM until their development is complete, at which time the B cells would lose responsiveness to this factor and exit the BM. Reconstitution of lethally irradiated mice with CXCR4−/− fetal liver cells (as performed by Ma et al. 51) would leave both the hypothetical non-B cell and B lineage cells incapable of responding to stromal-produced SDF-1α. In this indirect manner, B lineage cells would not be retained in the BM, but this would be due to the lack of action of SDF-1α on the non-B cell, which then prevent expression of the pro-retention factor, instead of a direct SDF-1α–B cell interaction. Also unclear is the significance of the increased SDF-1α responsiveness in B cells after exit to the periphery, since CXCR4−/− lymphocytes migrate to secondary lymphoid organs and repopulate their niches efficiently 51.
Though sIgM+ immature BM B cells respond to SLC, MIP-3β, SDF-1α, and BCA-1, the magnitude of the responses by B cells increases substantially after their migration to the spleen (i.e., as transitioning recent emigrants). These heightened responses are further increased after progression of the splenic Transition 1 B cells into Transition 2 B cells. No further increase in responsiveness to these chemokines is seen as Transition 2 B cells convert into the naive, recirculating μδ+ B cells. In the case of BCA-1 responses, the increased chemotaxis is associated with an increase in both the frequency and intensity of CXCR5 expression (Fig. 5B and Fig. D).
BCA-1 is preferentially expressed by dendritic cells in B cell follicles 14. Along with its receptor, CXCR5, it has been postulated to control B cell entry into the follicular environment 14 15 52. Consistent with studies of CXCR5 expression on human B cells by Forster et al. 52, we observed that peripheral B cells migrate better to BCA-1 than immature BM B cells, supporting an important role for these molecules in peripheral B cell function. Moreover, we have found that (a) immature BM B cells respond relatively poorly to BCA-1; (b) there is an increased BCA-1 response in splenic transitioning populations; (c) the recirculating pool of splenic B cells possesses the most robust response to BCA-1 measured; and (d) CXCR5 expression and responsiveness to BCA-1 characterize all the major peripheral B cell subsets, including MZ B cells and B1 B cells—cells that are not associated with the follicular microenvironment. Together, these results indicate that CXCR5 expression and BCA-1 responsiveness are not, in and of themselves, sufficient to confer follicular localization to B cells. It is important to consider this conclusion in light of studies that demonstrated that CXCR5-deficient B cells display selective follicular localization defects in PPs and spleen (but not in most peripheral LNs, where follicular homing was normal 52).Taken together with our finding of CXCR5 expression and BCA-1 responsiveness by B cells that are normally excluded from the follicular compartment, these data suggest that follicular localization may be under complex (combinatorial) control as a function of tissue site (and other factors), so that a contribution of CXCR5 is essential in PPs and spleen, but is accessory or redundant in most normal LNs. BCA-1 may have other important functions in peripheral B cell biology as well.
The increasing responsiveness to SLC and MIP-3β during B cell maturation in and after emigration from the BM is of particular interest because these chemokines signal through a common receptor, CCR7, and have been implicated in directing peripheral lymphocyte homing. SLC but not MIP-3β can also signal through CXCR3 53, but this receptor is not expressed by peripheral B cells. SLC is made and displayed by high endothelial venule (HEV) cells 54 55, can trigger rapid shear-resistant integrin-dependent arrest of rolling lymphocytes 16 56, and has been implicated in the recognition and arrest of circulating lymphocytes on HEVs in PPs and LNs 55 57. SLC and MIP-3β are also expressed by stromal dendritic cells in T cell zones of all secondary lymphoid organs 54. Although recent in situ studies show that B cells do not require SLC or MIP-3β signaling to arrest on HEVs in vivo 55, the ability of all B cell subsets to respond to these chemokines may nevertheless permit or facilitate their entry into lymphoid organs from the blood, perhaps at the level of diapedesis as evidenced by the poor homing of CCR7−/− B cells into LNs and PPs compared with wild-type B cells 58. Interestingly, SLC is also expressed by lymphatic endothelium and has been implicated in lymphatic entry of activated dendritic cells. It may play a similar role for lymphatic recruitment of activated or memory lymphocytes in extralymphoid tissues. Thus, the upregulation of MIP-3β and SLC responses in association with B cell export to the periphery may facilitate mature B cell migration and circulation through secondary lymphoid organs at multiple levels.
Importantly, all mature peripheral B cell populations, including follicular and MZ B cells as well as GC and B1 B cells, respond equally well to SLC and MIP-3β, as they do to SDF-1α and BCA-1. Therefore, although these chemokine responses may be critical to B cell trafficking in the periphery, they appear unlikely to be responsible for the specialized homing properties that target B cell subsets to distinct microenvironments.
B cell responses to MIP-3α were unique, in that detectable chemotaxis to MIP-3α was limited to a subset of peripheral mature B cells. No migration to MIP-3α and no CCR6 expression were observed in any BM B cell population, including the most developed sIgM+ immature BM B cell. Recently emigrated Transition 1 splenic B cells also lacked CCR6 and did not respond to MIP-3α, but Transition 2 B cells and splenic follicular μδ+ B cells are uniformly CCR6+ and migrate consistently (albeit at lower efficiency than the other peripheral chemokines tested). Although only a subset of PP GC B cells display CCR6, GC B cells respond well to MIP-3α, suggesting that the CCR6+ GC B cells have an enhanced migratory response to this chemokine.
Our finding of significant MIP-3α responses in these mature B cell population conflicts with the conclusions by Liao et al. 59, who were unable to generate a calcium flux with MIP-3α in human B cells in spite of low level uniform expression of CCR6, as revealed by mAb staining. They proposed that B cells are unresponsive to MIP-3α. However, our chemotaxis data and parallel studies with human lymphocytes (Campbell, J.J., unpublished data) clearly demonstrate that B cells migrate to MIP-3α. Moreover, in recent studies, Tanaka et al. confirm our findings that PP B cells migrate to MIP-3α and report an efficiency of MIP-3α–induced chemotaxis similar to that observed here 20.
MIP-3α is primarily expressed by epithelial cells, especially during inflammation or on epithelium associated with lymphoepithelial organs such as PPs. For example, in Northern blot analysis it is reportedly absent or only weakly expressed in resting spleen and LNs, but it is detectable in inflamed tonsils, intestine, and appendices (lymphoid tissues intimately associated with epithelial surfaces 20 60 61 62). In situ hybridization studies reveal high level expression by antigen-recruiting epithelium in the lymphoepithelial tonsils and PPs 20 61. MIP-3α expression by such epithelium has been postulated to help recruit antigen-presenting dendritic cells, and it may also help define sites of B cell recruitment to epithelial-associated lymphoid organs, including PPs and also bronchus-associated lymphoid tissue. Such a concept might provide teleological explanation for the association of MIP-3α responses and CCR6 expression with follicle-homing B cell populations.
There are only minor differences in the chemotactic responses between resting follicular mantle and antigen-reactive GC B cells to the chemokines studied here. This contrasts with the results of Bleul et al. 63, who showed that human tonsillar GC (CD38+) B cells were unable to respond to SDF-1α even though they express the SDF-1α receptor CXCR4. We have extended their studies and examined the chemokine responsiveness of human tonsil B cells to MIP-3α, SLC, and BCA-1 in addition to SDF-1α. In each case, human tonsil GC B cells failed to respond to any of the chemokines tested, whereas tonsillar non-GC CD38− B cells responded robustly (Campbell, J.J., unpublished observation). This discrepancy in functional responses may be due to a species difference between mice and humans, or alternatively may reflect differences in GCs from different lymphoid organs. In this context, it is relevant that we observed consistent significant strain-dependent differences in the GC B cell responses to MIP-3α, suggesting genetic variability in GC responses.
Receptor expression alone has not proven an adequate parameter to predict a cell's chemotactic potential. Forster et al. reported that human tonsil GC CD38+ B cells express CXCR5 on their surface 52 and Bleul et al. reported that human tonsil CD38+ B cells express CXCR4 63 even though these cells were unresponsive to the chemokines BCA-1 and SDF-1α (see above paragraph). We conclude that the functional measurement of chemotaxis is the only way to determine the chemotactic response profile for any given cell population. By assessing the correlation between function and surface expression of known receptors (as presented here for CCR6, CXCR3, and CXCR5), a secondary question can be investigated: Which receptors mediate the distinctive chemotactic fingerprints of cells? However, potential involvement of unidentified receptors (as yet) must always be kept in mind.
Both B1 and B2 B cells from the mouse peritoneal cavity respond to SLC, SDF-1α, and BCA-1 with equal efficiency (albeit not as well as splenic follicular B cells). However, unlike splenic B cells, there was no response of B1 B cells and very little response of B2 B cells to MIP-3α even though both cell types expressed CCR6. It was surprising that B1 B cells, which are not found in great quantities in secondary lymphoid organs, can nevertheless respond to the follicle-associated chemokine BCA-1 and the HEV-associated chemokine SLC. If B1 B cells do arise through a unique developmental program 9 10, that program still enables them to respond to all the same chemokines (except MIP-3α) to which more conventional B cells respond. As it seems unlikely that these lymphoid organ chemokines play a role in recruiting cells into the peritoneal or other serosal cavities, they may function in allowing B1 B cells to migrate or circulate through lymphoid tissues themselves.
In conclusion, we have uncovered a dynamic regulation of the chemotactic responses of B cells as they progress through development (Fig. 9). Responses to secondary lymphoid chemokines increase progressively during B cell differentiation in the BM and are upregulated even further in cells that have progressed into fully mature recirculating peripheral B cells. The remarkable similarity in chemotactic responses to the peripheral chemokines by different mature peripheral B cell subsets (follicular, MZ, GC, and B1 B cells) was unexpected in light of their unique patterns of microenvironmental homing. The only exception was the response to MIP-3α, which was mainly detected in follicle-associated μδ+ and GC B cells. We conclude that these chemokines and their receptors likely act in combination with other chemoattractants, signaling, and adhesion molecules to control the microenvironmental homing and the localization of distinct B cell subsets in the periphery. In contrast to the similar chemotactic fingerprints of microenvironmentally defined peripheral B cell populations, developing BM B cells display striking, stage-specific chemokine response profiles. The dramatic switches in chemokine responses in maturing B cells suggest that chemoattractant responsiveness may play an important role in B cell development.
Figure 9.

Schematic depiction of the acquisition and loss of chemokine responsiveness as B cells develop in the BM and migrate to the periphery.
Acknowledgments
We thank J. Cyster, E. O'Hara, C. Kim, J. Kim, M. Williams, and K. Youngman for critical reading of the manuscript.
This work was supported by National Institutes of Health grants, an Award from the Department of Veterans Affairs (to E.C. Butcher), and by the FACS® Core Facility of the Stanford Digestive Diseases Center under National Institutes of Health grant DK38707. E.P. Bowman was supported by U.S. Public Health Service grant 5T32CA09151 and was a recipient of an Arthritis Foundation Postdoctoral Fellowship. J.J. Campbell was supported by National Institutes of Health Cancer Etiology, Prevention, Detection, and Diagnosis grant 5T32CA090302, National Institutes of Health Individual National Research Service Award 1F32AI08930, and was a recipient of an Arthritis Foundation Postdoctoral Fellowship. R.R. Hardy was supported by National Institutes of Health grant AI26782.
Footnotes
Abbreviations used in this paper: APC, allophycocyanin; BCA-1, B cell–attracting chemokine 1; BCS, bovine calf serum; BLC, B lymphocyte chemoattractant; BM, bone marrow; CCR, CC chemokine receptor; CXCR, CXC chemokine receptor; GC, germinal center; Flt3-L, flt3 ligand; HSA, heat stable antigen; HSC, hematopoietic stem cell; HEV, high endothelial venule; LARC, liver and activation-regulated chemokine; MCP, monocyte chemoattractant protein; MIG, monokine induced by IFN-γ; MIP, macrophage inflammatory protein; MZ, marginal zone; PerCP, peridinine chlorophyll protein; PNA, peanut agglutinin; PP, Peyer's patch; RANTES, regulated upon activation, normal T cell expressed and secreted; SDF, stromal cell–derived factor; sIg, surface Ig; SLC, secondary lymphoid tissue chemoattractant; SAv, streptavidin; TARC, thymus and activation-regulated chemokine; TECK, thymus-expressed chemokine.
References
- Goodnow C.C., Cyster J.G., Hartley S.B., Bell S.E., Cooke M.P., Healy J.I., Akkaraju S., Rathmell J.C., Pogue S.L., Shokat K.P. Self-tolerance checkpoints in B lymphocyte development. Adv. Immunol. 1995;59:279–368. doi: 10.1016/s0065-2776(08)60633-1. [DOI] [PubMed] [Google Scholar]
- Humphrey J.H. Tolerogenic or immunogenic activity of hapten-conjugated polysaccharides correlated with cellular localization. Eur. J. Immunol. 1981;11:212–220. doi: 10.1002/eji.1830110310. [DOI] [PubMed] [Google Scholar]
- Lane P.J., Gray D., Oldfield S., MacLennan I.C. Differences in the recruitment of virgin B cells into antibody responses to thymus-dependent and thymus-independent type-2 antigens. Eur. J. Immunol. 1986;16:1569–1575. doi: 10.1002/eji.1830161216. [DOI] [PubMed] [Google Scholar]
- Oliver A.M., Martin F., Gartland G.L., Carter R.H., Kearney J.F. Marginal zone B cells exhibit unique activation, proliferative and immunoglobulin secretory responses. Eur. J. Immunol. 1997;27:2366–2374. doi: 10.1002/eji.1830270935. [DOI] [PubMed] [Google Scholar]
- Liu Y.J., Oldfield S., MacLennan I.C. Memory B cells in T cell-dependent antibody responses colonize the splenic marginal zones. Eur. J. Immunol. 1988;18:355–362. doi: 10.1002/eji.1830180306. [DOI] [PubMed] [Google Scholar]
- Groeneveld P.H., Erich T., Kraal G. In vivo effects of LPS on B lymphocyte subpopulations. Migration of marginal zone-lymphocytes and IgD-blast formation in the mouse spleen. Immunobiology. 1985;170:402–411. doi: 10.1016/S0171-2985(85)80064-4. [DOI] [PubMed] [Google Scholar]
- Kelsoe G. Life and death in germinal centers (redux) Immunity. 1996;4:107–111. doi: 10.1016/s1074-7613(00)80675-5. [DOI] [PubMed] [Google Scholar]
- Klinman N.R. The cellular origins of memory B cells. Semin. Immunol. 1997;9:241–247. doi: 10.1006/smim.1997.0075. [DOI] [PubMed] [Google Scholar]
- Duchosal M.A. B-cell development and differentiation Semin. Hematol. 34Suppl.1997. 2 12 [PubMed] [Google Scholar]
- Kearney J.F., Won W.J., Benedict C., Moratz C., Zimmer P., Oliver A., Martin F., Shu F. B cell development in mice. Int. Rev. Immunol. 1997;15:207–241. doi: 10.3109/08830189709068177. [DOI] [PubMed] [Google Scholar]
- Zlotnik A., Morales J., Hedrick J.A. Recent advances in chemokines and chemokine receptors. Crit. Rev. Immunol. 1999;19:1–47. [PubMed] [Google Scholar]
- Foxman E.F., Campbell J.J., Butcher E.C. Multistep navigation and the combinatorial control of leukocyte chemotaxis. J. Cell Biol. 1997;139:1349–1360. doi: 10.1083/jcb.139.5.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hromas R., Kim C.H., Klemsz M., Krathwohl M., Fife K., Cooper S., Schnizlein-Bick C., Broxmeyer H.E. Isolation and characterization of Exodus-2, a novel C-C chemokine with a unique 37-amino acid carboxyl-terminal extension. J. Immunol. 1997;159:2554–2558. [PubMed] [Google Scholar]
- Gunn M.D., Ngo V.N., Ansel K.M., Ekland E.H., Cyster J.G., Williams L.T. A B-cell-homing chemokine made in lymphoid follicles activates Burkitt's lymphoma receptor-1. Nature. 1998;391:799–803. doi: 10.1038/35876. [DOI] [PubMed] [Google Scholar]
- Legler D.F., Loetscher M., Roos R.S., Clark-Lewis I., Baggiolini M., Moser B. B cell-attracting chemokine 1, a human CXC chemokine expressed in lymphoid tissues, selectively attracts B lymphocytes via BLR1/CXCR5. J. Exp. Med. 1998;187:655–660. doi: 10.1084/jem.187.4.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell J.J., Bowman E.P., Murphy K., Youngman K.R., Siani M.A., Thompson D.A., Wu L., Zlotnik A., Butcher E.C. 6-C-kine (SLC), a lymphocyte adhesion-triggering chemokine expressed by high endothelium, is an agonists for the MIP-3beta receptor CCR7. J. Cell Biol. 1998;141:1053–1059. doi: 10.1083/jcb.141.4.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo V.N., Tang H.L., Cyster J.G. Epstein-Barr virus–induced molecule 1 ligand chemokine is expressed by dendritic cells in lymphoid tissues and strongly attracts naive T cells and activated B cells. J. Exp. Med. 1998;188:181–191. doi: 10.1084/jem.188.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C.H., Pelus L.M., White J.R., Applebaum E., Johanson K., Broxmeyer H.E. CK beta-11/macrophage inflammatory protein-3 beta/EBI1-ligand chemokine is an efficacious chemoattractant for T and B cells. J. Immunol. 1998;160:2418–2424. [PubMed] [Google Scholar]
- Nagira M., Imai T., Yoshida R., Takagi S., Iwasaki M., Baba M., Tabira Y., Akagi J., Nomiyama H., Yoshie O. A lymphocyte-specific CC chemokine, secondary lymphoid tissue chemokine (SLC), is a highly efficient chemoattractant for B cells and activated T cells. Eur. J. Immunol. 1998;28:1516–1523. doi: 10.1002/(SICI)1521-4141(199805)28:05<1516::AID-IMMU1516>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Tanaka Y., Imai T., Baba M., Ishikawa I., Uehira M., Nomiyama H., Yoshie O. Selective expression of liver and activation-regulated chemokine (LARC) in intestinal epithelium in mice and humans. Eur. J. Immunol. 1999;29:633–642. doi: 10.1002/(SICI)1521-4141(199902)29:02<633::AID-IMMU633>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Campbell J.J., Qin S., Bacon K.B., Mackay C.R., Butcher E.C. Biology of chemokine and classical chemoattractant receptorsdifferential requirements for adhesion-triggering versus chemotactic responses in lymphoid cells. J. Cell Biol. 1996;134:255–266. doi: 10.1083/jcb.134.1.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyster J., Ngo V.N., Ekland E.H., Gunn M.D., Sedgwick J.D., Ansel K.M. Chemokines and B-cell homing to follicles. Curr. Top. Microbiol. Immunol. 1999;246:87–92. doi: 10.1007/978-3-642-60162-0_11. [DOI] [PubMed] [Google Scholar]
- Hunte B.E., Hudak S., Campbell D., Xu Y., Rennick D. flk2/flt3 ligand is a potent cofactor for the growth of primitive B cell progenitors. J. Immunol. 1996;156:489–496. [PubMed] [Google Scholar]
- Veiby O.P., Lyman S.D., Jacobsen S.E. Combined signaling through interleukin-7 receptors and flt3 but not c-kit potently and selectively promotes B-cell commitment and differentiation from uncommitted murine bone marrow progenitor cells. Blood. 1996;88:1256–1265. [PubMed] [Google Scholar]
- Hardy R.R., Carmack C.E., Shinton S.A., Kemp J.D., Hayakawa K. Resolution and characterization of pro-B and pre–pro-B cell stages in normal mouse bone marrow. J. Exp. Med. 1991;173:1213–1225. doi: 10.1084/jem.173.5.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.S., Wasserman R., Hayakawa K., Hardy R.R. Identification of the earliest B lineage stage in mouse bone marrow. Immunity. 1996;5:527–535. doi: 10.1016/s1074-7613(00)80268-x. [DOI] [PubMed] [Google Scholar]
- Forster R., Mattis A.E., Kremmer E., Wolf E., Brem G., Lipp M. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell. 1996;87:1037–1047. doi: 10.1016/s0092-8674(00)81798-5. [DOI] [PubMed] [Google Scholar]
- Pevzner V., Wolf I., Burgstahler R., Forster R., Lipp M. Regulation of expression of chemokine receptor BLR1/CXCR5 during B cell maturation. Curr. Top. Microbiol. Immunol. 1999;246:79–84. doi: 10.1007/978-3-642-60162-0_10. [DOI] [PubMed] [Google Scholar]
- Allman D.M., Ferguson S.E., Cancro M.P. Peripheral B cell maturation. I. Immature peripheral B cells in adults are heat-stable antigenhi and exhibit unique signaling characteristics. J. Immunol. 1992;149:2533–2540. [PubMed] [Google Scholar]
- Loder F., Mutschler B., Ray R.J., Paige C.J., Sideras P., Torres R., Lamers M.C., Carsetti R. B cell development in the spleen takes place in discrete steps and is determined by the quality of B cell receptor–derived signals. J. Exp. Med. 1999;190:75–89. doi: 10.1084/jem.190.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroese F.G., Butcher E.C., Lalor P.A., Stall A.M., Herzenberg L.A. The rat B cell systemthe anatomical localization of flow cytometry-defined B cell subpopulations. Eur. J. Immunol. 1990;20:1527–1534. doi: 10.1002/eji.1830200718. [DOI] [PubMed] [Google Scholar]
- Rose M.L., Birbeck M.S., Wallis V.J., Forrester J.A., Davies A.J. Peanut lectin binding properties of germinal centres of mouse lymphoid tissue. Nature. 1980;284:364–366. doi: 10.1038/284364a0. [DOI] [PubMed] [Google Scholar]
- Butcher E.C., Rouse R.V., Coffman R.L., Nottenburg C.N., Hardy R.R., Weissman I.L. Surface phenotype of Peyer's patch germinal center cellsimplications for the role of germinal centers in B cell differentiation. J. Immunol. 1982;129:2698–2707. [PubMed] [Google Scholar]
- Kraal G., Weissman I.L., Butcher E.C. Germinal centre B cellsantigen specificity and changes in heavy chain class expression. Nature. 1982;298:377–379. doi: 10.1038/298377a0. [DOI] [PubMed] [Google Scholar]
- Hardy R.R., Hayakawa K., Haaijman J., Herzenberg L.A. B-cell subpopulations identified by two-colour fluorescence analysis. Nature. 1982;297:589–591. doi: 10.1038/297589a0. [DOI] [PubMed] [Google Scholar]
- Butcher E.C., Picker L.J. Lymphocyte homing and homeostasis. Science. 1996;272:60–66. doi: 10.1126/science.272.5258.60. [DOI] [PubMed] [Google Scholar]
- Mebius R.E., Streeter P.R., Michie S., Butcher E.C., Weissman I.L. A developmental switch in lymphocyte homing receptor and endothelial vascular addressin expression regulates lymphocyte homing and permits CD4+ CD3− cells to colonize lymph nodes. Proc. Natl. Acad. Sci. USA. 1996;93:11019–11024. doi: 10.1073/pnas.93.20.11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman I.L. Developmental switches in the immune system. Cell. 1994;76:207–218. doi: 10.1016/0092-8674(94)90329-8. [DOI] [PubMed] [Google Scholar]
- Butcher E.C., Williams M., Youngman K., Rott L., Briskin M. Lymphocyte trafficking and regional immunity. Adv. Immunol. 1999;72:209–253. doi: 10.1016/s0065-2776(08)60022-x. [DOI] [PubMed] [Google Scholar]
- Allman D., Li J., Hardy R.R. Commitment to the B lymphoid lineage occurs before DH-JH recombination. J. Exp. Med. 1999;189:735–740. doi: 10.1084/jem.189.4.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumano A., Paige C.J., Iscove N.N., Brady G. Bipotential precursors of B cells and macrophages in murine fetal liver. Nature. 1992;356:612–615. doi: 10.1038/356612a0. [DOI] [PubMed] [Google Scholar]
- Trentin L., Agostini C., Facco M., Piazza F., Perin A., Siviero M., Gurrieri C., Galvan S., Adami F., Zambello R., Semenzato G. The chemokine receptor CXCR3 is expressed on malignant B cells and mediates chemotaxis. J. Clin. Invest. 1999;104:115–121. doi: 10.1172/JCI7335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolink A., ten Boekel E., Melchers F., Fearon D.T., Krop I., Andersson J. A subpopulation of B220+ cells in murine bone marrow does not express CD19 and contains natural killer cell progenitors. J. Exp. Med. 1996;183:187–194. doi: 10.1084/jem.183.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn B.S., Jang I.K., Broxmeyer H.E., Cooper S., Jenkins N.A., Gilbert D.J., Copeland N.G., Elick T.A., Fraser M.J., Jr., Kwon B.S. A novel chemokine, macrophage inflammatory protein-related protein-2, inhibits colony formation of bone marrow myeloid progenitors. J. Immunol. 1995;155:2661–2667. [PubMed] [Google Scholar]
- Broxmeyer H.E., Cooper S., Cacalano G., Hague N.L., Bailish E., Moore M.W. Involvement of interleukin (IL)-8 receptor in negative regulation of myeloid progenitor cells in vivoevidence from mice lacking the murine IL-8 receptor homologue. J. Exp. Med. 1996;184:1825–1832. doi: 10.1084/jem.184.5.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel V.P., Kreider B.L., Li Y., Li H., Leung K., Salcedo T., Nardelli B., Pippalla V., Gentz S., Thotakura R. Molecular and functional characterization of two novel human C-C chemokines as inhibitors of two distinct classes of myeloid progenitors. J. Exp. Med. 1997;185:1163–1172. doi: 10.1084/jem.185.7.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn B.S., Zhang S.M., Broxmeyer H.E., Cooper S., Antol K., Fraser M., Jr., Kwon B.S. Characterization of CKbeta8 and CKbeta8-1two alternatively spliced forms of human beta-chemokine, chemoattractants for neutrophils, monocytes, and lymphocytes, and potent agonists at CC chemokine receptor 1. Blood. 1998;91:3118–3126. [PubMed] [Google Scholar]
- Quackenbush E.J., Wershil B.K., Aguirre V., Gutierrez-Ramos J.C. Eotaxin modulates myelopoiesis and mast cell development from embryonic hematopoietic progenitors. Blood. 1998;92:1887–1897. [PubMed] [Google Scholar]
- Kim C.H., Pelus L.M., Appelbaum E., Johanson K., Anzai N., Broxmeyer H.E. CCR7 ligands, SLC/6Ckine/Exodus2/TCA4 and CKbeta-11/MIP-3beta/ELC, are chemoattractants for CD56+CD16− NK cells and late stage lymphoid progenitors. Cell. Immunol. 1999;193:226–235. doi: 10.1006/cimm.1999.1483. [DOI] [PubMed] [Google Scholar]
- D'Apuzzo M., Rolink A., Loetscher M., Hoxie J.A., Clark-Lewis I., Melchers F., Baggiolini M., Moser B. The chemokine SDF-1, stromal cell-derived factor 1, attracts early stage B cell precursors via the chemokine receptor CXCR4. Eur. J. Immunol. 1997;27:1788–1793. doi: 10.1002/eji.1830270729. [DOI] [PubMed] [Google Scholar]
- Ma Q., Jones D., Springer T.A. The chemokine receptor CXCR4 is required for the retention of B lineage and granulocytic precursors within the bone marrow microenvironment. Immunity. 1999;10:463–471. doi: 10.1016/s1074-7613(00)80046-1. [DOI] [PubMed] [Google Scholar]
- Forster R., Emrich T., Kremmer E., Lipp M. Expression of the G-protein–coupled receptor BLR1 defines mature, recirculating B cells and a subset of T-helper memory cells. Blood. 1994;84:830–840. [PubMed] [Google Scholar]
- Soto H., Wang W., Strieter R.M., Copeland N.G., Gilbert D.J., Jenkins N.A., Hedrick J., Zlotnik A. The CC chemokine 6Ckine binds the CXC chemokine receptor CXCR3. Proc. Natl. Acad. Sci. USA. 1998;95:8205–8210. doi: 10.1073/pnas.95.14.8205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn M.D., Tangemann K., Tam C., Cyster J.G., Rosen S.D., Williams L.T. A chemokine expressed in lymphoid high endothelial venules promotes the adhesion and chemotaxis of naive T lymphocytes. Proc. Natl. Acad. Sci. USA. 1998;95:258–263. doi: 10.1073/pnas.95.1.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnock R.A., Campbell J.J., Dorf M.E., McEvoy L.M., Butcher E.C. The role of chemokines in the microenvironmental control of T versus B cell arrest in Peyer's patch high endothelial venules. J. Exp. Med. 2000;191:77–88. doi: 10.1084/jem.191.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachynski R.K., Wu S.W., Gunn M.D., Erle D.J. Secondary lymphoid-tissue chemokine (SLC) stimulates integrin alpha 4 beta 7-mediated adhesion of lymphocytes to mucosal addressin cell adhesion molecule-1 (MAdCAM-1) under flow. J. Immunol. 1998;161:952–956. [PubMed] [Google Scholar]
- Stein J.V., Rot A., Luo Y., Narasimhaswamy M., Nakano H., Gunn M.D., Matsuzawa A., Quackenbush E.J., Dorf M.E., von Andrian U.H. The CC chemokine thymus-derived chemotactic agent 4 (TCA-4, secondary lymphoid tissue chemokine, 6Ckine, exodus-2) triggers lymphocyte function–associated antigen 1–mediated arrest of rolling T lymphocytes in peripheral lymph node high endothelial venules. J. Exp. Med. 2000;191:61–76. doi: 10.1084/jem.191.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster R., Schubel A., Breitfeld D., Kremmer E., Renner-Muller I., Wolf E., Lipp M. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99:23–33. doi: 10.1016/s0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- Liao F., Rabin R.L., Smith C.S., Sharma G., Nutman T.B., Farber J.M. CC-chemokine receptor 6 is expressed on diverse memory subsets of T cells and determines responsiveness to macrophage inflammatory protein 3 alpha. J. Immunol. 1999;162:186–194. [PubMed] [Google Scholar]
- Rossi D.L., Vicari A.P., Franz-Bacon K., McClanahan T.K., Zlotnik A. Identification through bioinformatics of two new macrophage proinflammatory human chemokinesMIP-3alpha and MIP-3beta. J. Immunol. 1997;158:1033–1036. [PubMed] [Google Scholar]
- Dieu M.C., Vanbervliet B., Vicari A., Bridon J.M., Oldham E., Ait-Yahia S., Briere F., Zlotnik A., Lebecque S., Caux C. Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J. Exp. Med. 1998;188:373–386. doi: 10.1084/jem.188.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varona R., Zaballos A., Gutierrez J., Martin P., Roncal F., Albar J.P., Ardavin C., Marquez G. Molecular cloning, functional characterization and mRNA expression analysis of the murine chemokine receptor CCR6 and its specific ligand MIP-3alpha. FEBS Lett. 1998;440:188–194. doi: 10.1016/s0014-5793(98)01450-1. [DOI] [PubMed] [Google Scholar]
- Bleul C.C., Schultze J.L., Springer T.A. B lymphocyte chemotaxis regulated in association with microanatomic localization, differentiation state, and B cell receptor engagement. J. Exp. Med. 1998;187:753–762. doi: 10.1084/jem.187.5.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenh C.H., Cox M.A., Kaminski H., Zhang M., Byrnes H., Fine J., Lundell D., Chou C.C., Narula S.K., Zavodny P.J. Cutting edgespecies specificity of the CC chemokine 6Ckine signaling through the CXC chemokine receptor CXCR3: human 6Ckine is not a ligand for the human or mouse CXCR3 receptors. J. Immunol. 1999;162:3765–3769. [PubMed] [Google Scholar]