

Abstract
We have directly compared the efficacy of two immunotherapeutic strategies for the treatment of cancer: “vaccination” of tumor-bearing mice with genetically modified dendritic cells (DCs), and vaccination with genetically modified tumor cells. Using several different preexisting tumor models that make use of B16F10 melanoma cells expressing a target tumor antigen (human melanoma-associated gene [MAGE]-1), we found that vaccination with bone marrow–derived DCs engineered to express MAGE-1 via adenoviral-mediated gene transfer led to a dramatic decrease in the number of metastases in a lung metastasis model, and led to prolonged survival and some long-term cures in a subcutaneous preexisting tumor model. In contrast, vaccination with granulocyte/macrophage colony-stimulating factor (GM-CSF)–transduced tumor cells, previously shown to induce potent antitumor immunity in standard tumor challenge assays, led to a decreased therapeutic effect in the metastasis model and no effect in the subcutaneous tumor model. Further engineering of DCs to express either GM-CSF, tumor necrosis factor α, or CD40 ligand via retroviral-mediated gene transfer, led to a significantly increased therapeutic effect in the subcutaneous tumor model. The immunological mechanism, as shown for GM-CSF–transduced DCs, involves MAGE-1–specific CD4+ and CD8+ T cells. Expression of GM-CSF by DCs led to enhanced cytotoxic T lymphocyte activity, potentially mediated by increased numbers of DCs in draining lymph nodes. Our results suggest that clinical studies involving the vaccination with genetically modified DCs may be warranted.
Keywords: tumor immunology, gene therapy, retrovirus, adenovirus, B16F10
Introduction
Successful treatment of malignant diseases by the enhancement of antitumor immunity has been a long-standing goal of immunological research. Over the past decade, advances in our understanding of the role that specific cytokines and other gene products play in determining the outcome of an immune response have particularly rekindled optimism that immunotherapeutic approaches to the treatment of cancer will ultimately succeed. Among the many different immunotherapeutic strategies that are currently being evaluated, therapeutic vaccination with genetically modified tumor cells and vaccination with dendritic cells (DCs) manipulated to present specific tumor antigens have attracted particular attention 1 2. In the case of studies involving the use of gene-modified tumor cells as therapeutic vaccines, a large number of different gene products have been shown to possess the ability to stimulate antitumor immunity 3 4 5 6 7 8. Interestingly, however, in studies that directly compare the relative immunostimulatory activities of different gene products in nonimmunogenic tumor challenge models, one specific gene product, GM-CSF, appears to possess particularly potent activity 9. Early clinical studies suggest that GM-CSF may have significant biological activity in patients as well 10. Although the mechanism by which GM-CSF mediates antitumor immunity is still not completely understood, a variety of evidence suggests that secretion of cytokine local to the injected tumor cells results in the accumulation of large numbers of DCs, and may augment the antigen presentation functions of the cells.
In parallel to studies with cytokine-expressing tumor cells, various groups have also evaluated the direct use of DCs as therapeutic cancer vaccines. Early studies demonstrated that murine DCs pulsed with peptides or protein can protect mice against a lethal challenge with tumor cells 11 12 13. More recently, a variety of preparations of DCs have been shown to stimulate antitumor immunity, including DCs loaded with peptide 14 or tumor cell extracts 15, fused with tumor cells 16, transduced with RNA isolated from tumor tissues 17 18, or transduced by viral vectors 19 20.
Because the immunotherapeutic strategies involving GM-CSF–expressing tumor cells and DCs both appear to potently stimulate antitumor immunity and likely act through a common immunologic mechanism, we were interested in determining the relative efficacy of the two strategies in a direct comparative study. Here we report a side by side comparison of the antitumor activity of genetically modified DCs engineered to express a target tumor antigen versus cytokine-expressing tumor cells, and examine the effects of further genetic modifications of transduced DCs on their therapeutic efficacy.
Materials and Methods
Animals and Cell Lines.
Specific pathogen-free, age-matched (8–10 wk old) female C57BL/6 mice and congenic CD4−/− and CD8−/− mice were obtained from The Jackson Laboratory. B16–melanoma-associated gene (MAGE)-1 is a subclone of B16F10 cells stably transduced with a retrovirus encoding the human MAGE-1 gene 21. B16-MAGE-1 was further transduced with the retroviral vectors CMMP-GM-CSF, SFG-IL12 22, or SFG-CD80 to yield B16M-GM-CSF, B16M-IL12, and B16M-CD80, respectively. B16M-CD40L was generated by transduction of B16M cells with MMP-CD40L (CD40L cDNA; provided by R. Geha, Children's Hospital, Boston, MA). Embryonic fibroblast (EF) cells were obtained from congenic mouse embryos and retrovirally transduced with SFG-MAGE-1. Tumor cell lines were maintained in DME (GIBCO BRL) supplemented with 10% (vol/vol) heat-inactivated FCS, 2 mmol/liter glutamine, 50 U/ml penicillin and streptomycin (Gibco BRL). NIH3T3 cells (CRL 1658; American Type Culture Collection) were grown in DME containing 10% (vol/vol) calf serum (Sigma-Aldrich) and 50 U/ml penicillin and streptomycin. The packaging cell line 293GPG was grown in DME supplemented with 10% (vol/vol) heat-inactivated FCS, 2 mmol/liter glutamine, 50 U/ml penicillin and streptomycin (GIBCO BRL), tetracycline (1 μg/ml), puromycin (2 μg/ml), and G418 (0.3 mg/ml) (all from Sigma-Aldrich).
Generation of DCs from Murine Bone Marrow Cells.
Bone marrow was flushed from the long bones of the hind limbs of 10–14-wk-old female C57BL/6 mice. Upon depletion of erythrocytes, the cells were cultured in DC medium (DMEM supplemented with 10% inactivated FCS, 2 mmol/liter glutamine, 10 mmol Hepes-buffered saline, 100 mmol nonessential amino acids, 100 U/ml penicillin, 100 μg/ml streptomycine, 5 × 10−5 μM 2-ME (GIBCO BRL), 500 U/ml recombinant murine GM-CSF, and 500 U/ml murin IL-4 (both from Sigma-Aldrich) 23. On day 7 of culture, released mature nonadherent cells with the typical features of DCs were used for in vivo studies. For phenotypic analysis, the expression of the cell surface markers CD11c, CD80, CD86, MHC class II, and intercellular adhesion molecule 1 was tested using commercially available mAbs and appropriate isotype control antibodies (all from BD PharMingen). More than 90% of the cells showed high expression of these markers while they were negative for expression of CD3, B220, and GR-1. In some experiments, the capacity of DCs to initiate a mixed lymphocyte reaction was also confirmed.
Transduction of DCs with Retroviral and Adenoviral Constructs.
For studies involving retroviral-mediated gene transfer, CMMP, a novel derivative of MFG, was used. In this vector, the murine leukemia virus (MLV) LTRs are replaced with the corresponding myeloproliferative sarcoma virus LTRs and the normal MLV tRNA primer binding site is replaced by a glutamine tRNA primer binding site. In experiments requiring coexpression of a gene product and a reporter gene (green fluorescent protein [GFP]), bicistronic CMMP viruses were created by insertion of the internal ribosomal entry site (IRES)-GFP sequence from the plasmid MSCV2.2-IRES-GFP (gift from B. Sha, University of California, San Francisco, CA) downstream of the site for insertion of sequences in CMMP. Concentrated vesicular stomatitis virus G protein (VSV-G)-pseudotyped retroviral particles were generated via transient transfection of the 293GPG packaging cell line and ultracentrifugation as described previously 24. For retroviral transductions, 10 μl of high titer viral particles (5 × 108 to 5 × 109/ml) was added to 1 ml of DC cultures on days 2 and 4 (in the presence of 8 μg/ml polybrene). The DCs were precooled, incubated with the virus at 4°C for 3–4 h, and then transferred to 37°C incubators.
For studies involving adenoviral gene transfer, replication-deficient E1/E3-deleted adenoviral vectors were used, in which transcription of the desired coding sequence was driven by a CMV promoter 25. DCs were infected by exposure to adenoviral particles (multiplicity of infection 100) for 3–4 h at 37°C immediately before use. Expression of human MAGE-1 was confirmed by immunoblotting with the MAGE-1–specific mAb MA-454 (gift of L. Old, Ludwig Institute for Cancer Research, New York Branch at Memorial Sloan-Kettering Cancer Center, New York, NY) 26. 48 h after transduction with recombinant adenoviral particles DCs were lysed, and 10 μg of protein extract, as determined by a modified Lowry assay (detergent-compatible protein assay; Bio-Rad), was subjected to SDS-gel electrophoresis (12% polyacrylamide gel) in reducing conditions and blotted onto polyvinylidine difluoride membrane (Bio-Rad). Equal protein loading was confirmed by Coomassie staining.
Tumor Studies.
Tumor cells were treated with trypsin, washed twice in HBSS (GIBCO BRL), and injected in 0.2–0.5 ml of HBSS either subcutaneously (solid tumor model) or intravenously in the tail vein (lung metastasis model). In the solid tumor model, the development of tumors was monitored every 3–4 d. The mice were killed when the sum of the two perpendicular tumor diameters reached 30 mm, or when ulcerating lesions were detected. Statistical survival differences were evaluated with the Wilcoxon rank sum test, using JMP3.1.5 software (SAS Institute). In the short-term experiments, tumor size was measured every 2 d and the tumor volume was estimated using the following formula: (short diameter)2 × long diameter × 0.52.
Generation of CTL Cultures and IFN-γ Release Assay.
Animals were immunized with 106 DCs administered subcutaneously into hind limbs and forearms. 4 d later, regional LNs were harvested and T cells were purified by negative depletion of B cells and APCs using magnetic beads (pan IgG Dynabeads; Dynal). T cells were then restimulated with irradiated B16M-CD80 cells (16 Gy) at a cellular ratio of 20:1 in the presence of recombinant human IL-2 (20 U/ml; Sigma-Aldrich). After 5 d, 2 × 105 viable T cells were incubated with 5 × 104 target cells in 96-well plates for 24 h. The IFN-γ concentration in the supernatant was determined by ELISA, using commercially available reagents (Endogen).
In Situ Analysis of DCs in LNs.
To study the homing of DCs into draining LNs, CMMP-GFP and CMMP-GM-CSF-IRES-GFP–transduced DCs were sorted for green fluorescence in a Becton Dickinson FACScan™ and additionally stained with 5-(and-6)-([{4chloromethyl}benzoyl]amino)tetramethylrhodamine (CMTMR) in the case of GM-CSF–secreting DCs, and 5-chloromethylfluorescein diacetate (CMFDA) in the case of CMMP-GFP–transduced cells (both from Molecular Probes) according to the manufacturer's guidelines. Immediately before subcutaneous injection, both populations were mixed at a ratio of 1:1. 1.25 × 106 cells were then injected subcutaneously at four sites into the calves and forearms, and regional LNs were dissected after 24 and 48 h, respectively. The LNs were embedded into OCT Compound 4583 (Sakura) and frozen in liquid nitrogen. 10-μm cryosections of individual LNs were fixed in 3.7% formaldehyde and visualized with an MRC-1024 laser scanning confocal imaging system (Bio-Rad). Excitation wavelengths were 488 and 522 nm, respectively, using emission filters of 522 and 598 nm. Cytological preparations of the DC mixture were also evaluated to confirm the ratio of red and green cells. Both colors could be well differentiated despite the common underlying green fluorescence of GFP. For the in situ analysis, green and red cells were counted independently on each section. LN sections with ≥5 fluorescent cells were included in the scoring. The ratio of green to red cells was determined on 10–20 sections/LN.
Results
Establishment of Tumor Model for Evaluation of DC Vaccines.
To generate a tumor model suitable for vaccination studies involving genetically modified DCs, murine melanoma B16F10 cells were genetically engineered to express the human MAGE-1, an embryonal antigen that has been shown to be an immunological target in patients with melanoma 27 via retroviral infection 21. One particular clonal cell line expressing MAGE-1 (termed B16-MAGE-1), which showed identical in vivo growth characteristics to unmodified B16F10 cells, was chosen for use in tumor challenge and preexisting tumor studies. To generate DCs expressing MAGE-1, DCs were prepared from murine bone marrow cells by culture of the cells in the presence of GM-CSF and IL-4, as described previously 23, and subsequently infected with an adenoviral vector encoding MAGE-1 (see Materials and Methods). A majority of cells were transduced by adenoviral vector infection, as evaluated with a lacZ-encoding recombinant adenovirus (Ad) (data not shown).
The ability of MAGE-1–expressing DCs to induce antitumor immunity against MAGE-1–expressing B16F10 cells was first evaluated in a standard tumor challenge model. Groups of eight mice were first injected with either 2 × 105 MAGE-1–expressing DC cells, 2 × 105 MAGE-1–expressing EFs (EF-MAGE-1 cells), or 107 particles of Ad-MAGE (an adenoviral vector encoding MAGE-1) subcutaneous in the abdominal wall. Then, they were challenged with either 106 B16-MAGE-1 or the parental B16F10 cells 12 d later. As shown in Fig. 1, vaccination with MAGE-1–expressing DCs protected a significant number of the mice against challenge with B16-MAGE-1, but did not protect against challenge with the parental B16F10 cells, which do not express MAGE-1. These results indicate that, as expected, the vaccination effect of MAGE-1–expressing DCs is antigen specific. Animals injected with either EF-MAGE-1 cells or Ad-MAGE-1 showed some protection against challenge with B16-MAGE-1 cells as well, albeit weaker than that elicited by MAGE-1–expressing DCs.
Figure 1.

DC-Ad-MAGE-1 cells induce specific antitumor immunity. Groups of eight mice were first immunized with 2 × 105 DCs, EFs retrovirally transduced to express MAGE-1, or 107 adenoviral particles by subcutaneous injection. After 12 d, the mice were subcutaneously injected with 106 B16 wild-type (open symbols) or B16-MAGE-1 (filled symbols) tumor cells.
Comparative Analysis of the Efficacy of Genetically Modified DCs and Tumor Cells in Preexisting Tumor Models.
To compare the relative efficacy of genetically modified DCs and cytokine-expressing tumor cells, we employed two different preexisting tumor models. In the first model, 5 × 105 viable B16-MAGE-1 cells were injected intravenously into normal C57BL/6 mice. Subsequently, the mice were “vaccinated” with either 106 MAGE-1–expressing DC cells, GFP-expressing DCs, GM-CSF–expressing B16-MAGE-1 cells, or IL-12–expressing B16-MAGE-1 cells on days 3 and 7 after injection of tumor. 24 d after injection of tumor, animals were killed, and macroscopically visible lung metastases were counted. IL-12–expressing cells were chosen for study based on reports by others that IL-12 stimulates antitumor activity in preexisting tumor models 28 29 30. As shown in Fig. 2 A, vaccination with either MAGE-1–expressing DCs or cytokine–transduced B16 cells led to a dramatic reduction in the number of pulmonary metastases. Injection with either MAGE-1–expressing DCs or IL-12–expressing tumor cells led to a significantly greater reduction in metastases than injection with GM-CSF–expressing tumor cells. Antitumor immunity against pulmonary metastases may not necessarily reflect the pathophysiological mechanisms that govern the elimination of solid tumors implanted under the skin. Therefore, we also evaluated the relative efficacy of DC-based vaccines in a subcutaneous tumor model and included also CD40L–transduced tumor cells in these studies. 5 × 105 B16-MAGE-1 cells were injected subcutaneously in C57BL/6 animals, and the mice were subsequently injected with transduced DCs or tumor cells (106) one time 5 d later. As shown in Fig. 2 B, 10% of the tumor bearing mice treated with MAGE-1–expressing DCs showed long-term survival without clinical evidence of tumor-growth >90 d after tumor inoculation (P < 0.001). In contrast, animals treated with either GM-CSF–, CD40L–, or IL-12–expressing B16M cells succumbed to growing tumors. As expected, GFP-expressing DCs provided no therapeutic effect. To address the question of whether the increased efficacy of DC-based vaccines may be due to higher expression levels of the tumor antigen MAGE-1, we determined the relative amount of MAGE-1 in DCs transduced with adenoviruses and retrovirally transduced tumor cells. Equal protein amounts of cell lysates (10 μg) from transduced DCs and tumor cells were subjected to immunoblotting with the human MAGE-1–specific mAb MA454. As shown in Fig. 2 C, despite their increased potency as tumor cell vaccines, the expression level of MAGE-1 in DCs was inferior to that in B16-derived tumor cells.
Figure 2.
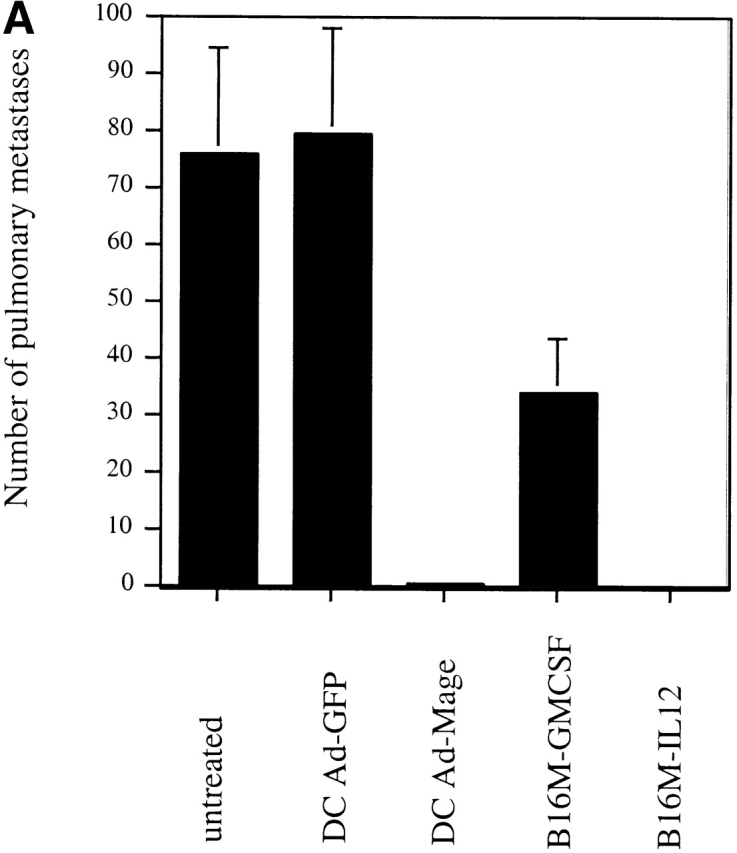


(A) Comparative analysis of DCs and cytokine-transduced tumor cells in preexisting pulmonary metastasis model. Animals were injected with 5 × 106 tumor cells by tail vein injection. DCs and cytokine-transduced tumor cell vaccines (5 × 105 cells) were given on days 3 and 7 by subcutaneous injection. All animals (n = 4/group) were killed on day 24 and the number of macroscopically visible metastases was determined. Error bars represent SE. (B) Comparative analysis of DCs and cytokine-transduced tumor cells in preexisting subcutaneous tumor model. Groups of 10 mice were treated with a single dose of Ad-transduced DCs (106) or irradiated cytokine-transduced tumor cells (both via subcutaneous administration) on day 5 after subcutaneous inoculation of 5 × 105 B16-MAGE-1 tumor cells. (C) MAGE-1 detection in transduced tumor cells and DCs. 10 μg of cell extract was loaded onto a 12% polyacrylamide gel and separated by electrophoresis. The 46-kD MAGE-1 protein was detected by the mAb MA454 (arrow).
Transduction with Cytokine-encoding Retroviruses Further Enhances the Potency of DCs.
To determine whether further genetic modifications of DCs might improve their efficacy as therapeutic vaccines, we evaluated the effect of expressing three gene products known to influence the development and/or function of DCs: GM-CSF, TNF-α, and CD40L. For these studies, retroviral vectors were generated which encoded the individual gene products. In each vector, a reporter gene, GFP, was also included and expressed through the use of IRES sequences in order to permit the normalization of transduction efficiencies obtained with the different cytokine-encoding viruses. An additional virus that only encoded GFP was also generated for use as a control. Using an optimized method for transduction (see Materials and Methods), ∼20–50% of DCs could be routinely transduced (Fig. 3 A). In the case of DCs engineered to express either GM-CSF or TNF-α, ELISA assays indicated the expression of ∼500 pg GM-CSF and 1,000 pg TNF-α per 106 cells/d, respectively.
Figure 3.





Retroviral transduction of DCs enhances their efficacy as therapeutic tumor vaccines. (A) FACS® profile of DCs transduced with the retrovirus CMMP-GFP. The fluorescence is compared with nontransduced DCs. (B) Mice with preexisting B16-MAGE-1 tumors (n = 5/group) were immunized on day 5 after tumor inoculation with 106 DC subcutaneously. Error bars represent the SE of the mean. (C–E) Kaplan-Meier survival graphs showing long-term tumor-free survival. Groups of 10 mice were treated with a single dose of 106 DCs (in E, 1 group received 3 injections in weekly intervals) or irradiated cytokine-transduced tumor cell vaccines (106) 5 d after subcutaneous injection of 5 × 105 B16-MAGE-1 tumor cells. P values are determined by statistical comparison against the survival of mice treated with DC-Ad-MAGE-1 cells.
Fig. 3b Fig. c Fig. d Fig. e, shows the results of four independent experiments in which the therapeutic efficacy of different genetically modified DC preparations was compared. In each experiment, the DCs used for vaccination were retrovirally transduced either with a control virus (CMMP-GFP), or with a retrovirus encoding a cytokine before transduction with Ad-MAGE-1. DCs transduced by the different bicistronic viruses were normalized for the percentage of GFP-expressing cells, and equal numbers of GFP+ cells were used for vaccination. In a first experiment, the relative efficacy of retrovirally transduced DCs was evaluated by measuring the tumor progression after a single subcutaneous administration of 106 DCs. In comparison to GFP-transduced control DCs, a significant effect of DCs that were engineered to express GM-CSF, TNF-α, or CD40L was noted (Fig. 3 B). The following experiments show Kaplan-Meier graphs and reflect long-term survival. In some experiments, additional groups of mice were immunized with the cytokine-transduced B16-MAGE-1 cells to provide additional points of comparison. As shown in each independent experiment, vaccination with DCs expressing only MAGE-1 (and GFP, the reporter gene product) led to the long-term tumor-free survival of 10–20% of the animals (Fig. 3c Fig. d Fig. e). As shown above in Fig. 2 B, vaccination with GM-CSF–transduced B16M cells provided no therapeutic efficacy in this preexisting tumor model, and successful vaccination was dependent on MAGE-1 expression by DCs. In contrast to vaccination with only MAGE-1–expressing DCs, DCs that additionally expressed GM-CSF, CD40L, or TNF-α appeared to significantly improve the therapeutic efficacy of the vaccination. Whereas the long-term survival benefit of GM-CSF–transduced DCs reached statistical significance (DC-GM-CSF versus DC-GFP, P = 0.008, Fig. 3 C), the improved survival curve of mice treated with CD40L- and TNF-α–transduced DCs only reached statistical significance if the data from two experiments were pooled (e.g., experiments shown in Fig. 3c and Fig. d; P ≤ 0.05). It remains to be determined from larger studies whether one of the three cytokines will prove to be superior to the others.
Not surprisingly, the long-term tumor-free survival of vaccinated animals also appeared to depend on the frequency of vaccinations and the numbers of DCs administered. Best results were seen in mice that received either repetitive doses (Fig. 3 E) or increased numbers of transduced DCs (data not shown). In these experiments, up to 70% of animals showed long-term tumor-free survival. Furthermore, the effect of GM-CSF was dependent on the antigen presentation of MAGE-1 in DCs, as the administration of DCs transduced with GM-CSF and GFP did not result in prolonged survival (not shown).
The Antitumor Effect of GM-CSF–transduced DCs Is Mediated by Antigen-specific T Cells.
To further dissect the immunologic mechanisms involved in the DC-mediated antitumor response, we employed a T cell-deficient in vivo system as well as in vitro studies of antigen-specific CTLs. First, mice deficient for either CD4+ or CD8+ subsets of T lymphocytes were inoculated with B16-MAGE-1 tumor cells and subsequently immunized with GM-CSF–transduced DCs. As shown in Fig. 4 A, the tumors in both CD4−/− and CD8−/− deficient animals grew as fast as the tumors in C57BL/6 wild-type mice immunized with control transduced DCs, indicating that the antitumor immunity induced by GM-CSF–transduced DCs is mediated both by CD4+ and CD8+ T cells. Second, we analyzed MAGE-1–specific T cells after immunization with various DC vaccines. Upon subcutaneous administration of GM-CSF– and GFP-transduced DCs, T cells were purified from draining LNs and restimulated in vitro using B16-MAGE-1-CD80 cells, according to a previously published protocol 31. The cells were then reincubated with B16 wild-type cells or B16-MAGE-1 cells. We measured the concentration of INF-γ as an indicator for CTL activity. Although control DCs gave rise to some CTL activity (potentially due to in vitro priming by residual DCs), DC-Ad-MAGE-1 clearly induced a MAGE-specific CTL response (Fig. 4 B). This effect was significantly stronger in animals immunized with GM-CSF–transduced DCs, indicating that DCs retrovirally transduced with GM-CSF–encoding constructs induce a stronger antigen-specific T cell response. On histological examination of hematoxylin and eosin–stained tumor sections, we noted an increased inflammatory response and tumor cell necrosis upon vaccination with DC-CMMP-GFP-Ad-MAGE-1, which was even more pronounced in animals that received DCs engineered to secrete GM-CSF (data not shown).
Figure 4.


(A) Antitumor immunity of transduced DCs is mediated by CD4 and CD8 cells. 5 × 105 tumor cells were subcutaneously injected in CD4−/−, CD8−/−, and congenic wild-type mice (n = 5/group). A single dose of DCs (106) was given on day 5 after the inoculation of tumor cells. (B) Enhanced cytotoxic T cell activity after immunization with GM-CSF–transduced DCs. After vaccination with genetically transduced DCs, T cells were purified from draining LNs, restimulated in vitro, and incubated with B16 wild-type (white bars) or B16-MAGE-1 (black bars) target cells in triplicates. IFN-γ was measured in the supernatant by ELISA.
Effects of GM-CSF Expression on In Vivo Properties of Genetically Modified DCs.
Based on the observed effects of GM-CSF expression on the therapeutic efficacy of genetically modified DCs, we next asked whether the expression of GM-CSF influenced the behavior of the cells in vivo in any discernible way. We reasoned that the increased efficacy of GM-CSF-transduced DCs might be mediated by increased survival of those cells that produce their growth factor in an autocrine fashion. Therefore, we quantified the number of GM-CSF–transduced DCs in situ. To account for potential confounding variations between different animals, we used a mixture of GM-CSF– and GFP-transduced DCs so that the number of GFP control transduced DCs served as an internal standard. For these studies, DCs expressing either no cytokine (CMMP-GFP–transduced) or GM-CSF (CMMP-GM-CSF-IRES-GFP–transduced) were first sorted for expression of GFP. GM-CSF–transduced DCs were then stained with the CellTracker™ dye CMTMR (red fluorescence). CMMP-GFP–transduced DCs were labeled with the CellTracker™ dye CMFDA (green fluorescence) to assure higher fluorescence intensity and an appropriate labeling control. Both populations of cells were mixed to equal ratios immediately before their subcutaneous injection. After 24 and 48 h, the animals were killed, and the ratio of GM-CSF– to GFP-transduced DCs was determined in the draining LNs. This ratio was compared with the ratio of the cells before injection by confocal microscopy. As shown in Fig. 5, we detected a proportionally higher number of GM-CSF–transduced DCs in all LNs examined. Similar results were seen when GM-CSF–transduced cells were labeled with the green dye and GFP-transduced cells were stained in red, excluding the possibility of dye artifacts. This finding is consistent with the hypothesis that DCs that secrete their growth factor GM-CSF either home more efficiently to draining LNs, continue to divide in vivo, or generally persist longer in vivo.
Figure 5.

GM-CSF–transduced DCs give rise to higher cell counts in draining LNs. DCs were retrovirally transduced with CMMP-GFP or CMMP-GM-CSF-IRES-GFP and sorted for GFP+ expression. CMMP-GFP–transduced cells were additionally stained with the green CMFDA CellTracker™ dye; CMMP-GM-CSF-IRES-GFP+ cells were stained with the red fluorescent CMTMR CellTracker™ dye. Both populations were mixed immediately before subcutaneous injection. Animals were killed after 24 and 48 h and the ratio of green to red cells was determined in three draining LNs. The error bars indicate the SD of 10–20 sections/LN.
Discussion
In light of the many promising immunotherapeutic strategies that have been evaluated in preclinical studies over the past few years, there has been an increasing need to define the most promising candidate therapies for clinical studies. The studies described here provide an important comparison of two commonly used strategies for inducing antitumor immunity which appear to have a related mechanism of action. The major conclusion from our studies is that DCs engineered to express a surrogate tumor antigen elicit a significantly stronger antitumor immunity than tumor cells that coexpress that same antigen and one of several immunostimulatory cytokines. Moreover, we have shown that it is possible to further enhance the immunotherapeutic potential of DCs by additional genetic modifications.
Previous comparative analyses of DC-mediated T cell priming have included a determination of the relative efficiency of presenting antigen on DCs via conventional pulsing of cells with protein or peptide versus expression of the antigen through the use of vaccinia or Ad vector 32, as well as assessments of the relative efficacy of transfected DCs versus transfected fibroblasts or vaccination with naked DNA 33. In the most relevant comparison to the studies reported here, Ashley et al. compared the efficiency of immunostimulation by DCs pulsed with either tumor extracts or tumor RNA to vaccination by GM-CSF–modified tumor cells in an intracranial tumor challenge model 18. While no significant differences were observed in their studies, it is likely that this is due to the use of a less stringent tumor challenge model than those used in the current study, as GM-CSF–expressing cells possessed readily detectable activity in their model. In both our own published 9 and unpublished studies (our unpublished data), we have found that GM-CSF possesses relatively little activity in most preexisting tumor models in which the vaccine is administered >3 d after tumor inoculation.
There have also been a limited number of recent reports that the engineered expression of specific gene products by DCs, such as IL-7 34 and lymphotactin 35, can improve their immunostimulatory function. For our studies, we chose to evaluate the activity of several gene products, GM-CSF, TNF-α, and CD40L, known to be critically involved in the differentiation, activation, and maturation of DCs. For example, GM-CSF has been identified as a critically important growth factor for DCs 23 and seems to be important in maintaining the efficient presentation of soluble antigen 36. Members of the TNF family are known to activate DCs. TNF-α contributes to both DC maturation 37 and activation 38. CD40L has equally been shown to promote DC maturation from human hematopoietic progenitor cells 39 40 41. Signaling via CD40 upregulates the expression of costimulatory molecules 39 42 and may be the key event that enables the DCs to stimulate T killer cells 43 44 45. Our finding that the engineered expression of either of the three molecules tested led to significantly improved biological activity suggests that the optimal in vivo behavior of DCs may be quite dependent on a pattern of normal endogenous gene expression not easily maintained after ex vivo manipulation of the cells. This notion is consistent with a variety of studies by others that suggest that optimal DC function can be highly dependent on the precise methodology used for their generation and expansion 46 47. While the precise explanation for the enhanced immunogenicity of GM-CSF–transduced DCs (or cells transduced by TNF-α or CD40L viruses) remains to be determined, our dye-tracking experiments indicate that GM-CSF expression results in more transduced DCs reaching the LN. This could be due to several mechanisms, including direct effects upon the in vivo migration characteristics of injected DCs 48 and/or their resistance against apoptosis 49 50. Future experiments will address these hypotheses.
Unfortunately, a critical issue that cannot be easily answered by any preclinical studies, including those presented here, is whether a general statement can be made about the relative efficacy of antigen-specific vaccination strategies, such as vaccination with antigen–expressing DCs, and strategies that aim to mount an immune response against a diversity of antigens, as is the goal of vaccination with genetically modified tumor cells. Furthermore, we are aware that our data are based on the introduction of a xenogeneic protein and that it may be even more difficult to induce antitumor immunity against autologous antigens. However, in light of the current studies and the promising clinical studies involving the use of cytokine-expressing tumor cells, parallel clinical studies that evaluate the relative efficacy of transduced tumor cell vaccine and genetically modified DCs would appear to be warranted. In addition, our studies suggest that it will be important to directly test in patients whether DCs genetically modified to express both a target tumor antigen and a specific cytokine, such as GM-CSF, will show improved biological activity relative to strategies involving DCs prepared in other ways.
Acknowledgments
We thank Peggy Russell, Steve Jungles, Li Zhang, and Michelle Lowi for expert technical help, the members of the Mulligan laboratory for fruitful discussions, and G. Dranoff for his critical review of the manuscript.
C. Klein was supported by the Deutsche Forschungsgemeinschaft (DFG Kl 2-1), and is a Scholar of the American Society of Hematology. This work was supported by the Howard Hughes Medical Institute.
Footnotes
Abbreviations used in this paper: Ad, adenovirus; DC, dendritic cell; EF, embryonic fibroblast; GFP, green fluorescent protein; IRES, internal ribosomal entry site; MAGE, melanoma-associated gene.
References
- Dranoff G., Mulligan R.C. Gene transfer as cancer therapy. Adv. Immunol. 1995;58:417–454. doi: 10.1016/s0065-2776(08)60624-0. [DOI] [PubMed] [Google Scholar]
- Schuler G., Steinman R.M. Dendritic cells as adjuvants for immune-mediated resistance to tumors. J. Exp. Med. 1997;186:1183–1187. doi: 10.1084/jem.186.8.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepper R.I., Pattengale P.K., Leder P. Murine interleukin-4 displays potent anti-tumor activity in vivo. Cell. 1989;57:503–512. doi: 10.1016/0092-8674(89)90925-2. [DOI] [PubMed] [Google Scholar]
- Golumbek P.T., Lazenby A.J., Levitsky H.I., Jaffee L.M., Karasuyama H., Baker M., Pardoll D.M. Treatment of established renal cancer by tumor cells engineered to secrete interleukin-4. Science. 1991;254:713–716. doi: 10.1126/science.1948050. [DOI] [PubMed] [Google Scholar]
- Fearon E.R., Pardoll D.M., Itaya T., Golumbek P., Levitsky H.I., Simons J.W., Karasuyama H., Vogelstein B., Frost P. Interleukin-2 production by tumor cells bypasses T helper function in the generation of an antitumor response. Cell. 1990;60:397–403. doi: 10.1016/0092-8674(90)90591-2. [DOI] [PubMed] [Google Scholar]
- Watanabe Y., Kuribayashi K., Miyatake S., Nishihara K., Nakayama E., Taniyama T., Sakata T. Exogenous expression of mouse interferon gamma cDNA in mouse neuroblastoma C1300 cells results in reduced tumorigenicity by augmented anti-tumor immunity. Proc. Natl. Acad. Sci. USA. 1989;86:9456–9460. doi: 10.1073/pnas.86.23.9456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng M.N., Park B.H., Koeppen H.K., Tracey K.J., Fendly B.M., Schreiber H. Long-term inhibition of tumor growth by tumor necrosis factor in the absence of cachexia or T-cell immunity. Proc. Natl. Acad. Sci. USA. 1991;88:3535–3539. doi: 10.1073/pnas.88.9.3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock H., Dorsch M., Diamantstein T., Blankenstein T. Interleukin 7 induces CD4+ T cell–dependent tumor rejection. J. Exp. Med. 1991;174:1291–1298. doi: 10.1084/jem.174.6.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranoff G., Jaffee E., Lazenby A., Golumbek P., Levitsky H., Brose K., Jackson V., Hamada H., Pardoll D., Mulligan R. Vaccination with irradiated tumor cells engineered to secrete murine GM-CSF stimulates potent, specific, and long-lasting immunity. Proc. Natl. Acad. Sci. USA. 1993;90:3539–3543. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soiffer R., Lynch T., Mihm M., Jung K., Rhuda C., Schmollinger J.C., Hodi F.S., Liebster L., Lam P., Mentzer S. Vaccination with irradiated autologous melanoma cells engineered to secrete human granulocyte-macrophage colony-stimulating factor generates potent antitumor immunity in patients with metastatic melanoma. Proc. Natl. Acad. Sci. USA. 1998;95:13141–13146. doi: 10.1073/pnas.95.22.13141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celluzzi C.M., Mayordomo J.I., Storkus W.J., Lotze M.T., Falo L.D., Jr. Peptide-pulsed dendritic cells induce antigen-specific CTL-mediated protective tumor immunity. J. Exp. Med. 1996;183:283–287. doi: 10.1084/jem.183.1.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel L., Mayordomo J.I., Tjandrawan T., DeLeo A.B., Clarke M.R., Lotze M.T., Storkus W.J. Therapy of murine tumors with tumor peptide–pulsed dendritic cellsdependence on T cells, B7 costimulation, and T helper cell 1–associated cytokines. J. Exp. Med. 1996;183:87–97. doi: 10.1084/jem.183.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paglia P., Chiodoni C., Rodolfo M., Colombo M.P. Murine dendritic cells loaded in vitro with soluble protein prime cytotoxic T lymphocytes against tumor antigen in vivo. J. Exp. Med. 1996;183:317–322. doi: 10.1084/jem.183.1.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayordomo J.I., Zorina T., Storkus W.J., Zitvogel L., Celluzzi C., Falo L.D., Melief C.J., Ildstad S.T., Kast W.M., Deleo A.B. Bone marrow-derived dendritic cells pulsed with synthetic tumour peptides elicit protective and therapeutic antitumour immunity. Nat. Med. 1995;1:1297–1302. doi: 10.1038/nm1295-1297. [DOI] [PubMed] [Google Scholar]
- Fields R.C., Shimizu K., Mule J.J. Murine dendritic cells pulsed with whole tumor lysates mediate potent antitumor immune responses in vitro and in vivo. Proc. Natl. Acad. Sci. USA. 1998;95:9482–9487. doi: 10.1073/pnas.95.16.9482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong J., Chen D., Kashiwaba M., Kufe D. Induction of antitumor activity by immunization with fusions of dendritic and carcinoma cells. Nat. Med. 1997;3:558–561. doi: 10.1038/nm0597-558. [DOI] [PubMed] [Google Scholar]
- Boczkowski D., Nair S.K., Snyder D., Gilboa E. Dendritic cells pulsed with RNA are potent antigen-presenting cells in vitro and in vivo. J. Exp. Med. 1996;184:465–472. doi: 10.1084/jem.184.2.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley D.M., Faiola B., Nair S., Hale L.P., Bigner D.D., Gilboa E. Bone marrow–generated dendritic cells pulsed with tumor extracts or tumor RNA induce antitumor immunity against central nervous system tumors. J. Exp. Med. 1997;186:1177–1182. doi: 10.1084/jem.186.7.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W., Kong H.L., Carpenter H., Torii H., Granstein R., Rafii S., Moore M.A., Crystal R.G. Dendritic cells genetically modified with an adenovirus vector encoding the cDNA for a model antigen induce protective and therapeutic antitumor immunity. J. Exp. Med. 1997;186:1247–1256. doi: 10.1084/jem.186.8.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Specht J.M., Wang G., Do M.T., Lam J.S., Royal R.E., Reeves M.E., Rosenberg S.A., Hwu P. Dendritic cells retrovirally transduced with a model antigen gene are therapeutically effective against established pulmonary metastases. J. Exp. Med. 1997;186:1213–1221. doi: 10.1084/jem.186.8.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueler H., Mulligan R.C. Induction of antigen-specific tumor immunity by genetic and cellular vaccines against MAGEenhanced tumor protection by coexpression of granulocyte-macrophage colony-stimulating factor and B7-1. Mol. Med. 1996;2:545–555. [PMC free article] [PubMed] [Google Scholar]
- Lieschke G.J., Rao P.K., Gately M.K., Mulligan R.C. Bioactive murine and human interleukin-12 fusion proteins which retain antitumor activity in vivo. Nat. Biotechnol. 1997;15:35–40. doi: 10.1038/nbt0197-35. [DOI] [PubMed] [Google Scholar]
- Inaba K., Inaba M., Romani N., Aya H., Deguchi M., Ikehara S., Muramatsu S., Steinman R.M. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ory D.S., Neugeboren B.A., Mulligan R.C. A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc. Natl. Acad. Sci. USA. 1996;93:11400–11406. doi: 10.1073/pnas.93.21.11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy S., Kitamura M., Harris-Stansil T., Dai Y., Phipps M.L. Construction of adenovirus vectors through Cre-lox recombination. J. Virol. 1997;71:1842–1849. doi: 10.1128/jvi.71.3.1842-1849.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.T., Stockert E., Chen Y., Garin-Chesa P., Rettig W.J., van der Bruggen P., Boon T., Old L.J. Identification of the MAGE-1 gene product by monoclonal and polyclonal antibodies. Proc. Natl. Acad. Sci. USA. 1994;91:1004–1008. doi: 10.1073/pnas.91.3.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Bruggen P., Traversari C., Chomez P., Lurquin C., De Plaen E., Van den Eynde B., Knuth A., Boon T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254:1643–1647. doi: 10.1126/science.1840703. [DOI] [PubMed] [Google Scholar]
- Tahara H., Zeh H.J., III, Storkus W.J., Pappo I., Watkins S.C., Gubler U., Wolf S.F., Robbins P.D., Lotze M.T. Fibroblasts genetically engineered to secrete interleukin 12 can suppress tumor growth and induce antitumor immunity to a murine melanoma in vivo. Cancer Res. 1994;54:182–189. [PubMed] [Google Scholar]
- Zitvogel L., Tahara H., Robbins P.D., Storkus W.J., Clarke M.R., Nalesnik M.A., Lotze M.T. Cancer immunotherapy of established tumors with IL-12. Effective delivery by genetically engineered fibroblasts. J. Immunol. 1995;155:1393–1403. [PubMed] [Google Scholar]
- Nanni P., Rossi I., De Giovanni C., Landuzzi L., Nicoletti G., Stoppacciaro A., Parenza M., Colombo M.P., Lollini P.L. Interleukin 12 gene therapy of MHC-negative murine melanoma metastases. Cancer Res. 1998;58:1225–1230. [PubMed] [Google Scholar]
- van Elsas A., Hurwitz A.A., Allison J.P. Combination immunotherapy of B16 melanoma using anti–cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)–producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J. Exp. Med. 1999;190:355–366. doi: 10.1084/jem.190.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brossart P., Goldrath A.W., Butz E.A., Martin S., Bevan M.J. Virus-mediated delivery of antigenic epitopes into dendritic cells as a means to induce CTL. J. Immunol. 1997;158:3270–3276. [PubMed] [Google Scholar]
- Timares L., Takashima A., Johnston S.A. Quantitative analysis of the immunopotency of genetically transfected dendritic cells. Proc. Natl. Acad. Sci. USA. 1998;95:13147–13152. doi: 10.1073/pnas.95.22.13147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann J., Aicher A., Qin Z., Cayeux Z., Daemen K., Blankenstein T., Dorken B., Pezzutto A. Retroviral interleukin-7 gene transfer into human dendritic cells enhances T cell activation. Gene. Ther. 1998;5:264–271. doi: 10.1038/sj.gt.3300568. [DOI] [PubMed] [Google Scholar]
- Cao X., Zhang W., He L., Xie Z., Ma S., Tao Q., Yu Y., Hamada H., Wang J. Lymphotactin gene-modified bone marrow dendritic cells act as more potent adjuvants for peptide delivery to induce specific antitumor immunity. J. Immunol. 1998;161:6238–6244. [PubMed] [Google Scholar]
- Sallusto F., Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J. Exp. Med. 1994;179:1109–1118. doi: 10.1084/jem.179.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F., Cella M., Danieli C., Lanzavecchia A. Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartmentdownregulation by cytokines and bacterial products. J. Exp. Med. 1995;182:389–400. doi: 10.1084/jem.182.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieser C., Bock G., Klocker H., Bartsch G., Thurnher M. Prostaglandin E2 and tumor necrosis factor alpha cooperate to activate human dendritic cellssynergistic activation of interleukin 12 production. J. Exp. Med. 1997;186:1603–1608. doi: 10.1084/jem.186.9.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caux C., Massacrier C., Vanbervliet B., Dubois B., Van Kooten C., Durand I., Banchereau J. Activation of human dendritic cells through CD40 cross-linking. J. Exp. Med. 1994;180:1263–1272. doi: 10.1084/jem.180.4.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Romo L., Bjorck P., Duvert V., van Kooten C., Saeland S., Banchereau J. CD40 ligation on human cord blood CD34+ hematopoietic progenitors induces their proliferation and differentiation into functional dendritic cells. J. Exp. Med. 1997;185:341–349. doi: 10.1084/jem.185.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brossart P., Grunebach F., Stuhler G., Reichardt V.L., Mohle R., Kanz L., Brugger W. Generation of functional human dendritic cells from adherent peripheral blood monocytes by CD40 ligation in the absence of granulocyte-macrophage colony-stimulating factor. Blood. 1998;92:4238–4247. [PubMed] [Google Scholar]
- Cella M., Scheidegger D., Palmer-Lehmann K., Lane P., Lanzavecchia A., Alber G. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacityT–T help via APC activation. J. Exp. Med. 1996;184:747–752. doi: 10.1084/jem.184.2.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridge J.P., Di Rosa F., Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 1998;393:474–478. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- Bennett S.R., Carbone F.R., Karamalis F., Flavell R.A., Miller J.F., Heath W.R. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 1998;393:478–480. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- Schoenberger S.P., Toes R.E., van der Voort E.I., Offringa R., Melief C.J. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- Chen B., Shi Y., Smith J.D., Choi D., Geiger J.D., Mule J.J. The role of tumor necrosis factor alpha in modulating the quantity of peripheral blood-derived, cytokine-driven human dendritic cells and its role in enhancing the quality of dendritic cell function in presenting soluble antigens to CD4+ T cells in vitro. Blood. 1998;91:4652–4661. [PubMed] [Google Scholar]
- Labeur M.S., Roters B., Pers B., Mehling A., Luger T.A., Schwarz T., Grabbe S. Generation of tumor immunity by bone marrow-derived dendritic cells correlates with dendritic cell maturation stage. J. Immunol. 1999;162:168–175. [PubMed] [Google Scholar]
- Cumberbatch M., Dearman R.J., Kimber I. Langerhans cells require signals from both tumour necrosis factor-alpha and interleukin-1 beta for migration. Immunology. 1997;92:388–395. doi: 10.1046/j.1365-2567.1997.00360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludewig B., Graf D., Gelderblom H.R., Becker Y., Kroczek R.A., Pauli G. Spontaneous apoptosis of dendritic cells is efficiently inhibited by TRAP (CD40-ligand) and TNF-alpha, but strongly enhanced by interleukin-10. Eur. J. Immunol. 1995;25:1943–1950. doi: 10.1002/eji.1830250722. [DOI] [PubMed] [Google Scholar]
- Bjorck P., Banchereau J., Flores-Romo L. CD40 ligation counteracts Fas-induced apoptosis of human dendritic cells. Int. Immunol. 1997;9:365–372. doi: 10.1093/intimm/9.3.365. [DOI] [PubMed] [Google Scholar]