

Abstract
Reportedly, antiapoptotic Bcl-2 family proteins suppress apoptosis by binding to and inhibiting members of the CED-4 family of caspase activators. To explore this question, we used embryonic stem (ES) cells in which one (−/+) or both (−/−) copies of the gene encoding apoptotic protease activating factor 1 (Apaf-1), a CED-4 homologue, were disrupted by homologous recombination. Stable clones of heterozygous (−/+) and homozygous (−/−) Apaf-1 knockout ES cells that overexpressed Bcl-2 were generated. Withdrawal of serum growth factors or stimulation of heterozygous ES cells with staurosporine (STS), ultraviolet (UV)B irradiation, etoposide (VP16), or cisplatin induced apoptosis followed by cell death (determined by failure to exclude propidium iodide dye). These cell death stimuli also induced activation of several types of caspases and loss of mitochondrial membrane potential (ΔΨ) in heterozygous (+/−) Apaf-1 knockout ES cells. In addition, overexpression of Bcl-2 protected against these events in Apaf-1–expressing ES cells. In contrast, STS, UVB, and VP16 induced little or no caspase activation and apoptosis in homozygous (−/−) Apaf-1 knockout ES cells. Nevertheless, Apaf-1–deficient ES cells subjected to these cell death stimuli or deprived of growth factors did eventually die through a nonapoptotic mechanism associated with loss of ΔΨ. Moreover, Bcl-2 overprotection preserved ΔΨ, reduced the percentage of Apaf-1−/− ES cells undergoing cell death, and increased clonigenic survival. The extent of Bcl-2–mediated cytoprotection was not significantly different for heterozygous (−/+) versus homozygous (−/−) Apaf-1 knockout cells. Furthermore, although Bcl-2 could be readily coimmunoprecipitated with Bax, associations with Apaf-1 were undetectable under conditions where Apaf-1 interactions with procaspase-9 were observed. We conclude that Bcl-2 has cytoprotective functions independent of Apaf-1, preserving mitochondrial function through a caspase-independent mechanism.
Keywords: Bcl-2, apoptotic protease activating factor 1, caspase, apoptosis, mitochondria
Introduction
Many of the mechanisms responsible for apoptosis and programmed cell death are conserved throughout the animal kingdom 1. Members of the caspase family of cell death protease are responsible for the phenomenon of apoptosis in animal cells. When converted from their inactive zymogen forms into active enzymes, these intracellular proteases can cleave a variety of substrates that then produce the characteristic morphological manifestations and associated biochemical changes commonly associated with this form of cell demise.
Genetic analysis of developmental cell death in the nematode, Caenorhabiditis elegans, has revealed two essential cell death genes, ced-3 and ced-4 2. The ced-3 gene encodes a caspase, which has several close homologues in humans and other mammalian species 3. The CED-4 gene product is required for CED-3 activation in C. elegans. CED-4 is an ATP-binding protein that binds the pro-form of CED-3 4, forming an oligomeric complex that results in proteolytic processing and activation of the CED-3 protease 5. Apoptotic protease activating factor 1 (Apaf-1) is a CED-4 homologue that is present in humans, mice, and flies 6 7 8 9 10 11. Similar to CED-4, Apaf-1 binds certain procaspases, forming ATP-dependent oligomeric complexes that result in protease activation 12 13 14 15. Unlike CED-4, however, Apaf-1 contains an additional domain that is not found in the worm protein and that renders Apaf-1 dependent on cytochrome c for its function as a caspase activator. Because cytochrome c is normally found only in mitochondria, activation of Apaf-1 requires release of this protein from these organelles, thus coupling mitochondrial damage to activation of caspases (for a review, see reference 16). Mice and flies with homozygous disruptions of their Apaf-1 genes exhibit defects in developmental cell death in some but not all tissues 7 8 9 10 11. Cells derived from Apaf-1 knockout mice display resistance to certain apoptotic stimuli which are thought to involve mitochondria through cytochrome c release 7.
Bcl-2 family proteins represent an evolutionarily conserved group of cell death regulators, with homologues found in vertebrates and invertebrates. Members of this family include both cell death–inducing and cell death–blocking proteins 17. Though intensively investigated, it remains unclear how Bcl-2 family proteins control cell life and death. Many Bcl-2 family proteins contain a hydrophobic membrane-anchoring domain at their COOH terminus that targets them to the membranes of mitochondria and some other organelles 18 19. Overexpression of mammalian antiapoptotic proteins such as Bcl-2 or Bcl-XL has been shown to protect mitochondria from various cell death stimuli, preserving transmembrane potential (ΔΨ) and preventing release of cytochrome c and other apoptogenic proteins 20 21. Conversely, overexpression of proapoptotic proteins such as Bax or Bak can result in release of cytochrome c from mitochondria and mitochondrial membrane depolarization 22 23 24 25.
In C. elegans, the antiapoptotic Bcl-2 family member, CED-9, suppresses apoptosis by binding CED-4, thereby interfering with CED-4–dependent activation of pro–CED-3 26 27 28. The proapoptotic EGL-1 protein of this nematode dimerizes with CED-9, releasing CED-4 and allowing caspase activation 29. It has been proposed that mammalian Bcl-2 homologues function similarly to their counterparts in C. elegans 17. The antiapoptotic human protein Bcl-XL, for example, reportedly associates with human Apaf-1 and suppresses apoptosis induced by overexpression of Apaf-1 30 31 32. Overexpression of Bcl-2 also may suppress caspase activation downstream of cytochrome c in some circumstances 33 34, consistent with the notion that Bcl-2 can suppress Apaf-1. However, experiments using broad-spectrum caspase-inhibiting compounds have provided evidence that several mammalian Bcl-2 family proteins, including Bcl-2, Bcl-XL, Bax, and Bak, can regulate cytochrome c release and mitochondrial membrane potential independently of caspases 22 23 35 36 37. Moreover, mammalian Bcl-2 family proteins can induce or suppress caspase-independent nonapoptotic cell death (necrosis) under some conditions 22 38, arguing that they operate via mechanisms other than by regulating caspase-activating proteins such as Apaf-1. To address the relation of Apaf-1 to the cytoprotective mechanism by which mammalian Bcl-2 provides cytoprotection, we compared the effects of overexpressing Bcl-2 in murine that which contained or lacked Apaf-1 because targeted ablation of the apaf-1 gene.
Materials and Methods
Plasmid Construction.
cDNAs encoding Apaf-1 or its truncated forms were obtained by PCR based on the published Apaf-1 DNA sequence 6. The full-length Apaf-1 cDNA was cloned into pcI-neo (Promega) with a NH2-terminal Flag tag. Apaf-1 (Trp-Asp [ΔWD]) containing amino acids 1–420 was cloned into pcDNA3-Myc to produce NH2-terminal–tagged Apaf-1 proteins. Plasmids encoding human procaspase-9, procaspase-9 (Cys287Ala), human Bcl-XL, Bcl-2, Bax, and Bax (Iso-Gly-Asp-Glu [ΔIGDE]) have been described 39 40. Some of these were subcloned into the EcoRI and XhoI sites of pcDNA3-myc to produce NH2-terminal tagged proteins. A 0.9-kbp human Bcl-2 cDNA was subcloned into pPGK-neo bpA (gift of H. Baribault, Deltagen, Inc., Menlo Park, CA) in place of neomycin cDNA. pPGK-hygromycin has been described previously 41.
Cell Culture and Transfections.
293T cells were grown in DMEM supplemented with 10% (vol/vol) FCS. 293T cells (3 × 106) seeded in 10-cm dishes were transiently transfected after 0.5 d of culture with various plasmid DNAs using the SuperFect Transfection reagent (Qiagen).
Apaf-1+/−Apaf-1−/− ES cells have been described 7. ES cells were grown in DMEM supplemented with leukemia inhibitory factor, 15% FCS, l-glutamine, and β-mercaptoethanol. pPGK–Bcl-2 and pPGK-hygromycin plasmid DNAs were linearized with NotI or SalI and electroporated into ES cells (V/cm = 300; capacitance = 960 μF). Stably transfected cells were obtained by selection in media containing hygromycin (150 μg/ml) for 2 wk, and clones were isolated using a micromanipulator. Subclones expressing high levels of human Bcl-2 were identified by immunoblotting using an antihuman Bcl-2–specific antibody 42.
Immunoprecipitation and Immunoblot Analysis.
293T cells were transiently transfected with plasmids encoding Bcl-2, Myc–Apaf-1 (ΔWD), Flag–procaspase-9 (C287A), Myc-Bax, Myc-Bax (ΔIGDE) mutant, or various combinations of these plasmids, normalizing for total DNA content. After 2 d, cells were suspended in lysis buffer (10 mM Hepes [pH 7.2], 142.5 mM KCl, 5 mM MgCl2, 1 mM EGTA, 0.2% NP-40) containing a mixture of protease inhibitors (Boehringer). Lysates were precleared by incubation with protein G Sepharose 4B (Zymed Laboratories) without antibody, and proteins were immunoprecipitated using anti-Myc immobilized agarose gel (9E10) or IgG1 control (Santa Cruz Biotechnology, Inc.), fractionated by SDS-PAGE, and then transferred onto nitrocellulose membranes and incubated with anti–Bcl-2 rabbit serum or anti–Flag M2 antibody (Sigma-Aldrich). Antibody detection was accomplished using an enhanced chemiluminescence method (Amersham Pharmacia Biotech). For direct immunoblot analysis of ES cell lysates, 50 μg (total protein) of lysate was prepared from exponentially growing ES cells and was separated in 12% SDS polyacrylamide gels, then blotted and analyzed using anti–Bcl-2 rabbit serum 42 or (as a control) anti–α-tubulin antibody (Sigma-Aldrich).
Assays for Apoptosis, Cell Death, and Clonigenic Survival.
For experiments with ES cells, cell death and apoptosis, respectively, were measured using one- or two-color FACS®-based methods 7. In brief, 2 × 105 ES cells per well were plated in 12-well plates 1 d before replacement of medium with serum-free DMEM, UV irradiation (Stratalinker 2400; Stratagene), or addition to cultures of etoposide (VP16), staurosporine (STS), or cisplatinum (CP; Sigma-Aldrich). At various times thereafter, adherent cells were recovered by trypsinization and pooled with floating cells, then placed on ice in 0.2 ml of PBS containing 10 μg/ml propidium iodide (PI), with or without 0.2 μg/ml of FITC-conjugated annexin V (BD PharMingen). For determination of cell death, the percentage of cells which failed to exclude PI dye was determined by flow cytometry (FACScan™; Becton Dickinson). For apoptosis, the percentage of total recovered cells that were annexin V positive but PI negative (apoptotic) was determined.
Alternatively, cells were recovered from cultures (both floating and adherent), fixed with 3.7% paraformaldehyde-PBS, and stained with 0.1 μg/ml 4,6-diamino-2-phenylindole (DAPI [40]). The percentage of cells with condensed chromatin and fragmented nuclei was determined by UV microscopy. Each experiment was performed in triplicate on several separate days. In addition, the morphology of Apaf-1+/− and Apaf-1−/− ES cells after treatment with apoptotic stimuli was determined by transmission electron microscopy 42.
Release of lactate dehydrogenase (LDH) from dying cells was measured using the medium recovered from cell cultures at various times after withdrawing serum. In brief, to measure released LDH, 0.1 ml aliquots of culture medium were mixed with 10 μl 10 mM NADH, 10 μl of 0.1 M Na-pyruvate (pH 7.4), and 0.88 ml 50 mM Na2HP04 (pH 7.2). The rate of conversion of NADH to NAD+ was determined by spectrophotometry, measuring the decline in absorbance at OD 340 nm at 25°C. To determine cellular LDH activity for normalization of data, cells were recovered from cultures and resuspended in medium containing 0.1% Triton X-100, and LDH activity was measured. Cellular and released LDH activity were then combined to determine total LDH activity in each cell culture. Data for released LDH were normalized relative to total LDH and expressed as a percentage. All measurements were performed in triplicate.
For clonigenic survival assays, ES cells were either treated with STS or deprived of serum. For STS, 106 ES cells were seeded in 6-cm dishes. The next day, 15 μM STS was added to cultures for 6 h. The cells were then washed, recovered by trypsinization, and cultured in triplicate at 104 cells per 35-mm dishes in fresh medium lacking STS. Colony formation was meaured 6 d later by crystal violet staining. Colony-forming efficiency was determined by comparing the ratio of the numbers of colonies formed by cells exposed to STS compared with untreated cells. For serum deprivation, 105 ES cells were seeded into 35-mm dishes in their usual medium containing 15% fetal bovine serum. The next day, the cells were washed three times with medium lacking serum, and then cultured in serum-deficient medium for 9 d. The medium was then replaced with serum-containing medium to allow surviving cells to proliferate and form colonies, which were scored 5 d later. Colony-forming efficiency was calculated as the ratio of the number of colonies arising after serum deprivation compared with the number of cells originally plated (105).
Measurement of Mitochondrial Potential in ES Cells.
To determine the percentage of ES cells experiencing loss of ΔΨ, ES cells were cultured as described above for apoptosis and cell death assays, and then both floating and adherent cells were recovered 1 d after treatment with apoptosis inducers and incubated in DMEM at 37°C with 40 nM 3,3′-dihexyloxacarbocyanine iodide (DiOC6[3]) for 30 min, then analyzed by flow cytometry using the FL1 channel. The percentages of cells with reduced fluorescence indicative of loss of ΔΨ were determined from duplicate samples, and each experiment was repeated at least once.
Caspase Assays.
ES cells were seeded at 3 × 106 cells in 10-cm diameter dishes and treated 1 d later with UV irradiation, anticancer drugs, or serum deprivation as above. At 1 d after treatment of ES cells with apoptosis inducers, cells were recovered into lysis buffer (10 mM Tris-HCl [pH 7.3], 25 mM NaCl, 0.25% Triton X-100, 1 mM EDTA). After centrifugation at 16,000 g for 30 min, the resulting supernatants were adjusted to 1 mg/ml with lysis buffer, and 25 μg total protein was incubated in 100 μl caspase buffer (50 mM Hepes [pH 7.2], 100 mM NaCl, 1 mM EDTA [pH 8.0], 10% sucrose, 0.1% 3[3-cholamidopropyl-dimethylammonio]-1-propanesulfate [CHAPS], and 5 mM dithiothreitol [DTT]) with 100 μM of various fluorigenic substrate peptides, including acetyl-Asp-Glu-Val-Asp-(7-amino-4-trifluoromethyl-coumarin) (Ac-DEVD-AFC), Ac-Tyr-Val-Ala-Asp-AFC (Ac-YVAD-AFC), Ac-Leu-Glu-Val-Asp-AFC (Ac-LEVD-AFC), Ac-Leu-Glu-His-Asp-AFC (Ac-LEHD-AFC), benzyloxycarbonyl-Ile-Glu-Thr-Asp-AFC (z-IETD-AFC), and z-Val-Asp-Val-Ala-Asp-AFC (z-VDVAD-AFC; Calbiochem). Caspase activity was assayed using a fluorimeter plate reader (Fmax™; Molecular Devices) in kinetic mode with excitation and emission wavelengths at 405 and 519 nm, respectively, continuously measuring release of AFC (7-amino-4-trifluoromethyl-coumarin) from substrate peptides as described 43. Data shown represent AFC released after 30 min.
Results
Induction of Cell Death but Not Apoptosis in Apaf-1–deficient ES Cells.
To explore the relevance of Apaf-1 to the mechanism by which Bcl-2 suppresses cell death, we used ES cells in which one or both copies of the gene encoding Apaf-1 were disrupted by homologous recombination 7, thus providing a cellular context in which Apaf-1 was either present or absent. First, the effects of several agents that typically induce apoptosis was tested on Apaf-1+/− and Apaf-1−/− ES cells. Apoptosis was monitored by microscopic examination of fixed cells stained with the DNA-binding fluorochrome DAPI, assessing the percentage of cells that developed nuclear fragmentation and chromatin condensation typical of apoptosis 40. Cell death was determined by failure of cells to exclude the membrane-impermeable dye, PI. In cultures of Apaf-1–expressing ES cells, cytotoxic agents, including the protein kinase inhibitor STS or DNA-damaging anticancer drugs such as etoposide (VP16) or CP (Fig. 1) induced marked increases in both apoptosis (as manifested by nuclear fragmentation and chromatin condensation observed with DAPI staining) and cell death (as revealed by PI dye uptake). In contrast, little or no increase in the percentage of apoptotic cells was induced by these agents in cultures of Apaf-1–deficient ES cells. Nevertheless, an significant increase in the percentage of dead cells (i.e., cells failing to exclude PI dye) was observed in cultures of Apaf-1−/− ES cells after exposure to STS, VP16, and CP. Similar data were obtained when an alternative vital dye was employed, namely trypan blue (not shown). These observations were also confirmed using an assay in which phosphatidylserine surface exposure is detected by annexin V staining of cells, and plasma membrane integrity is simultaneously assessed by PI dye exclusion using two-color FACS® analysis 44. In Apaf-1+/− ES cells, cytotoxic agents induced increases in the percentage of both annexin V–positive/PI-negative (apoptotic) cells and annexin-V–positive/PI-positive (nonapoptotic) cells. In contradistinction, annexin V–positive cells were predominantly PI positive in cultures of Apaf-1−/− ES cells treated with cytotoxic agents (data not presented), suggesting a nonapoptotic demise.
Figure 1.
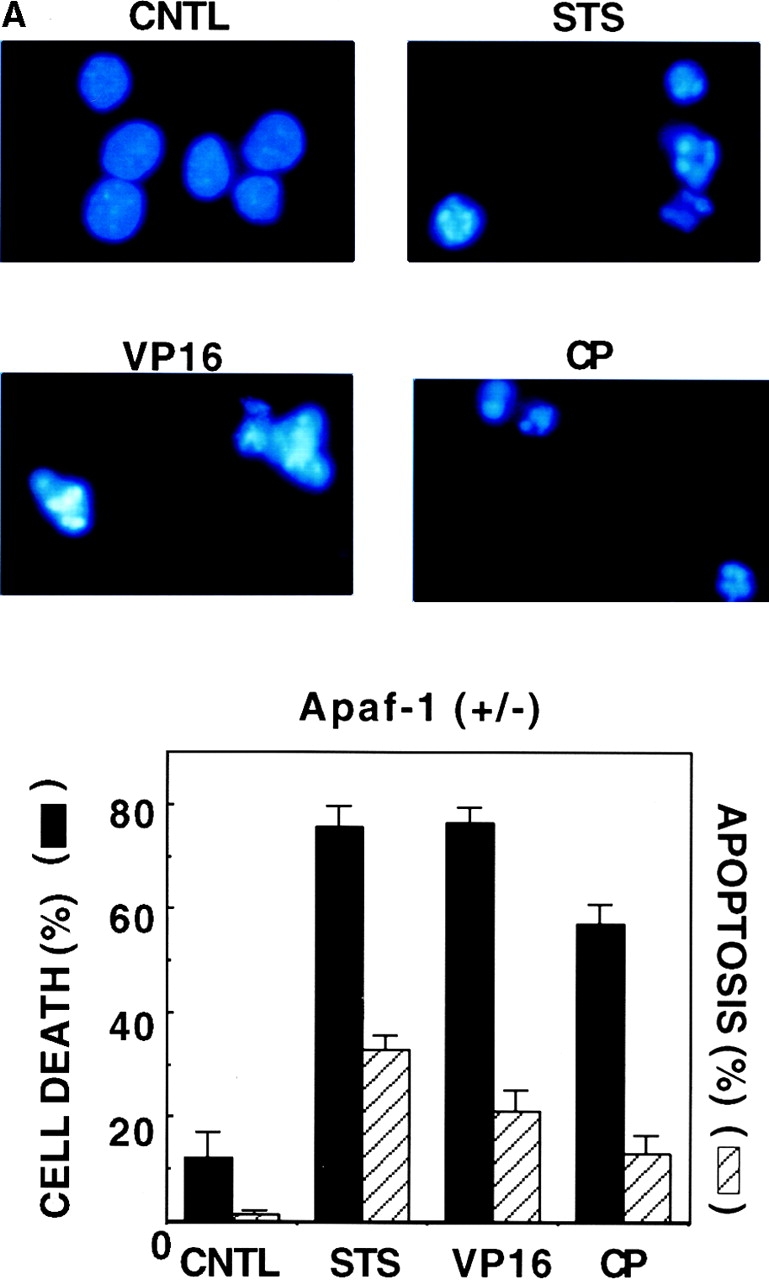
Induction of cell death but not apoptosis in Apaf-1–deficient ES cells. Apaf-1+/− and Apaf-1−/− ES cells were cultured in medium without (control; CNTL) or with cytotoxic agents, including 10 μM STS, 100 μM VP16, or 50 μM CP. After 1–2 d, floating and attached cells were recovered and pooled. Half were fixed and stained with DAPI, and half were placed on ice and then immediately analyzed for PI dye uptake using a flow cytometer. (A and B, top) Representative photomicrographs are presented for fluorescence microscopy analysis of DAPI-stained cells, demonstrating nuclear fragmentation and chromatin condensation typical of apoptosis in cultures of Apaf-1+/− ES cells treated with cytotoxic agents. In contrast, nuclei appear mostly normal in cultures of Apaf-1−/− cells. (A and B, bottom) The percentage of cells with apoptotic nuclear morphology as revealed by DAPI-staining (hatched bars) and the percentage of dead cells as determined by PI dye uptake (black bars) were determined (mean ± SD; n = 3).
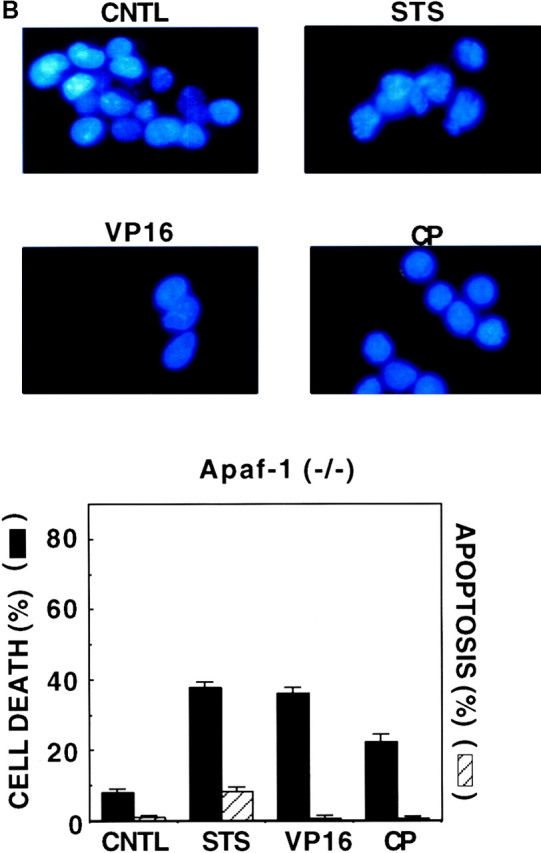
Electron microscopic analysis of Apaf-1 −/− ES cells after exposure to apoptotic stimuli confirmed that these cells did not undergo apoptosis, unlike the typical apoptotic features seen in Apaf-1+/− cells, including chromatin condensation with margination at the nuclear periphery, nuclear fragmentation, plasma membrane blebbing, and apoptotic body formation (Fig. 2). Rather, Apaf-1−/− cells exposed to anticancer drugs or treated with UV irradiation contained intact nuclei with focal rather than peripheral chromatin condensation and had evidence of mitochondrial swelling and distention of the endoplasmic reticulum and Golgi body. Some of these cells had cytosolic vacuolarization, and occasionally rupture of the plasma membrane was also evident. We conclude therefore that Apaf-1–deficient cells treated with anticancer drug or radiation die through a nonapoptotic mechanism.
Figure 2.
Electron microscopic analysis of Apaf-1+/− and Apaf-1−/− ES cells. ES cells were treated without (A and C) or with (B and D) 60 mJ/cm2 UV irradiation, and were fixed 2 d later for electron microscopic analysis. Representative transmission electron micrographs are presented, showing (B) Apaf-1+/− cells with sequence of typical apoptotic features, including plasma membrane blebbing and rupture and fragmented nucleus with highly condensed chromatin (arrow), and (D) Apaf-1−/− cells with intact nucleus containing slightly condensed chromatin, swollen mitochondria, distention of endoplasmic reticulum and Golgi body, and cytosolic vacuolarization. Some of the Apaf-1−/− cells with these morphological features also had evidence of plasma membrane rupture (arrow).
Overexpression of Bcl-2 in Apaf-1+/− and Apaf-1−/− ES Cells.
Using a PGK promoter to drive expression of a Bcl-2–encoding cDNA, stably transfected clones of heterozygous Apaf-1+/− and homozygous Apaf-1−/− ES cells that overexpressed Bcl-2 were generated. Fig. 3 shows results of an immunoblot analysis of lysates prepared from two independent clones of Bcl-2–overexpressing Apaf-1+/− and Apaf-1−/− ES cells, making comparisons with clones of control Apaf-1+/− and Apaf-1−/− ES cells that were electroporated with a control plasmid and subjected to the same hygromycin selection procedure. Note that the ∼26-kD Bcl-2 protein is present at high levels in the expected cell clones (arbitrarily called “A” and “B”) but not in the hygromycin control clones of Apaf-1+/− and Apaf-1−/− ES cells. Probing the same blot with an antibody to α-tubulin confirmed loading of equivalent amounts of total protein for each sample (Fig. 3).
Figure 3.

Immunoblot analysis of transfected ES cells. Stable transfectants of Apaf-1+/− and Apaf-1−/− ES cells were prepared by electroporation of the plasmid pPGK-hygromycin alone or in combination with pPGK–Bcl-2, followed by selection in media containing 150 μg/ml hygromycin for 2 wk. Independent cell clones were generated and analyzed by SDS-PAGE (12% gel)/immunoblotting using lysates containing 50 μg total protein (per lane) and antibodies specific for human Bcl-2 (top) or α-tubulin (bottom). Data shown represent hygromycin control (Hygro) Apaf-1+/− and Apaf-1−/− ES cells compared with two independent Bcl-2–overexpressing clones, termed Bcl-2 (A) and Bcl-2 (B).
From a total of 18 hygromycin-resistant Bcl-2–transfected ES cells lacking Apaf-1, 8 expressed human Bcl-2, whereas the other 10 failed to produce human Bcl-2 protein at levels that were detectable by immunoblotting. From 24 clones of Bcl-2–transfected Apaf-1+/− ES cell clones, 5 expressed Bcl-2. Though the experiments presented below focused on two of the Bcl-2–expressing clones (A and B) each for Apaf-1+/− and Apaf-1−/− ES cells, similar results were obtained with additional clones in pilot experiments. Moreover, cell death experiments using the ES cell clones that failed to express Bcl-2 at immunodetectable levels produced results comparable to the pPGK-hygromycin (control)-transfected ES cells (not shown), consistent with the ineffective expression of the pPGK–Bcl-2 plasmid in those particular stable transfectants.
Bcl-2 Overexpression Protects Apaf-1 Knockout Cells from Death Induced by UV Irradiation and Anticancer Drugs.
To explore the effect of Bcl-2 overexpression in an Apaf-1–deficient cellular context, we contrasted the effects of various cytotoxic stimuli on induction of cell death, as determined by PI dye uptake, in cultures of Apaf-1+/− and Apaf-1−/− ES cells that had been transfected with either hygromycin control or Bcl-2–expression plasmids. Fig. 4 presents the results of experiments in which Apaf-1+/− or Apaf-1−/− ES cells were treated for 1–2 d with UV-irradiation, the topoisomerase inhibitor etoposide (VP16), the DNA-damaging drug CP, or the general kinase inhibitor, STS. In all cases, cell death (failure to exclude PI) was induced in cultures of both Apaf-1+/− and Apaf-1−/− ES cells, although fewer percentages of the Apaf-1–deficient cells died during the same time (Fig. 4 A). Overexpression of Bcl-2 significantly reduced the percentage of cell death induced by UV irradiation, VP16, CP (P < 0.05 by paired t-test), and to some extent STS (P = 0.03–0.08, depending on the particular clone examined) in both Apaf-1+/− and Apaf-1−/− cells. Moreover, this protection afforded by Bcl-2 overexpression in both Apaf-1+/− (not shown) and Apaf-1−/− (Fig. 4 B) ES cells was evident over a range of doses of UV irradiation and concentrations of anticancer drugs. Some quantitative differences in the extent of Bcl-2 cytoprotection in Apaf-1+/− versus Apaf-1−/− cells were noticeable, particularly with VP16, where the percentage inhibition of cell death by Bcl-2 was higher in Apaf-1+/− than in Apaf-1−/− cells (Fig. 4 A). However, the significance of these observations should be interpreted with caution, given that the levels of Bcl-2 protein are not identical in each of the stably transfected ES clones, and considering that subtle differences may exist among clones in terms of how they metabolize or efflux drugs.
Figure 4.

Bcl-2 inhibits death of Apaf-1−/− ES cells afer exposure to UV irradiation and cytotoxic drugs. The Apaf-1+/− and Apaf-1−/− ES cell transfectants were seeded at 2 × 105 cells per well (2-cm) in 12-well plates 1 d before UV irradiation (60–240 mJ/cm2) or addition of 100–300 μM VP16, 50–100 μM CP, or 2–15 μM STS. Floating and adherent cells were recovered after 1 d, and the percentage of dead cells that failed to exclude PI was determined by flow cytometry (mean ± SE; n = 3). (A) Comparison of Apaf-1+/− and Apaf-1−/− ES cells treated with 60 mJ/cm2 UV irradiation for 2 d or with 100 μM VP16, 50 μM CP, or 15 μM STS for 1 d. Cases where cell death was significantly reduced in cultures of Bcl-2 (A) and Bcl-2 (B) cells compared with hygromycin control (Hygro) cells (p ≤ 0.05 as determined by unpaired t-test) are indicated by *. (B) Effects of various doses of UV irradiation or various concentrations of cytotoxic drugs are compared for hygromycin control (Hygro) and Bcl-2–overexpressing Apaf-1–deficient ES cells. Data are representative of multiple experiments.
Though Bcl-2 overexpression reduced cell death induced by UV irradiation, VP16, and CP in both Apaf-1+/− and Apaf-1−/−, it did not prevent cell cycle arrest, as determined by FACS®-based DNA content analysis (not shown). UV irradiation, for example, induced predominantly G1 arrest, whereas VP16 and CP induced predominantly G2/M arrest. Among surviving cells, the percentages of hygromycin control and Bcl-2–overexpressing cells in Go/G1, S, or G2/M phases were not significantly different (data not presented).
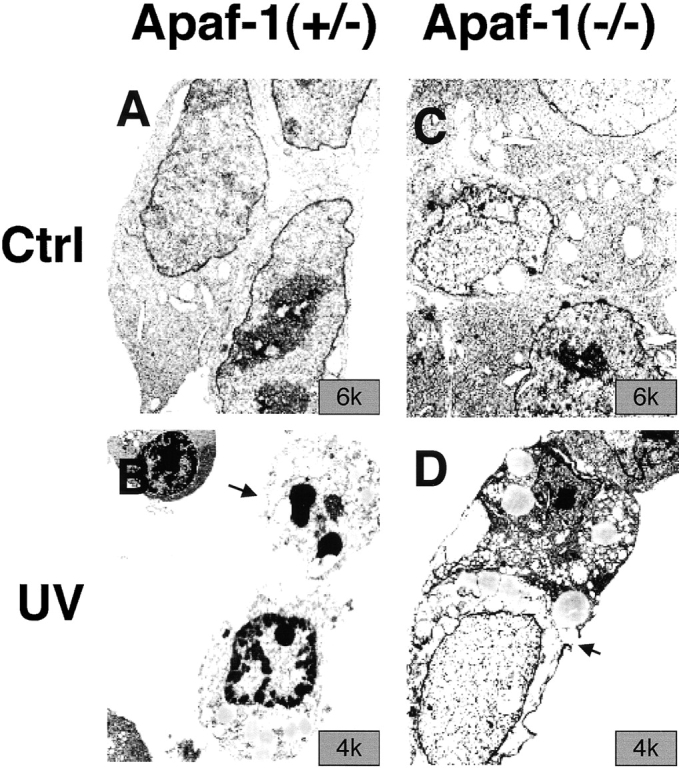
Bcl-2 Overexpression Prolongs Survival of Apaf-1–deficient ES Cells in Absence of Growth Factors.
Gene transfer–mediated overexpression of Bcl-2 has been reported to delay or prevent cell death induced by withdrawal of growth factors from cultures of factor-dependent cells of various lineages (for a review, see reference 45). We therefore asked whether Bcl-2 overexpression in Apaf-1+/− and Apaf-1−/− ES cells could delay cell death induced by serum deprivation. Gene transfer–mediated overexpression of Bcl-2 suppressed cell death induced in cultures of Apaf-1–expressing ES cells, as determined by PI dye exclusion assays (Fig. 5 A). Though death of Apaf-1−/− cells occurred with delayed kinetics relative to Apaf-1+/− ES cells, by 4 d after withdrawal of growth factors, over half the Apaf-1−/− cells failed to exclude PI dye (Fig. 5 A). Moreover, Bcl-2 overexpression prolonged survival of both Apaf-1+/− and Apaf-1−/− ES cells, as measured by PI exclusion.). Similar conclusions were reached when using release of LDH from cells into culture medium as an indicator of cell death (Fig. 5 B) instead of PI dye exclusion. Cell cycle studies (i.e., DNA content analysis) indicated that growth factor–deprived ES–hygromycin control and ES–Bcl-2 cells similarly arrested in Go/G1 before dying, and excluded differences in cell proliferation as an explanation for the differences observed in the relative numbers of surviving cells in cultures deprived of serum growth factors (data not presented).
Figure 5.
Bcl-2 prolongs survival of Apaf-1+/− and Apaf-1−/− ES cells in the absence of growth factors. Stably transfected Apaf-1+/− hygromycin control (Hygro), Bcl-2 (A), and Bcl-2 (B) cells (left) and Apaf-1−/− hygromycin control (Hygro), Bcl-2 (A), and Bcl-2 (B) cells (right) were seeded at 105 cells per 2-cm well. After 12 h, cells attached to plates were washed, and medium was replaced with DMEM lacking FCS. At various times thereafter, (A) both adherent and floating cells were recovered and analyzed by flow cytometry, determining the percentage of viable cells capable of excluding PI (mean ± SE; n = 3), or (B) LDH released into the culture medium by dying cells was measured by enzyme assay (data expressed as percentage relative to total LDH activity present in culture supernatants and cell pellets; cells were lysed with 0.1% Triton X-100; mean ± SE; n = 3).
Bcl-2 Enhances Clonigenic Survival of Apaf-1–deficient ES Cells.
To determine whether the Bcl-2-mediated differences in cell death rates measured in short-term cytotoxicity assays correlate with long–term clonigenic survival, Bcl-2–overexpressing or hygromycin control–transfected Apaf-1−/− ES cells were either exposed to high-dose STS for 6 h or deprived of serum for several days, then washed and placed into complete medium to allow recovery and formation of colonies of growing cells. As shown in Fig. 6, Bcl-2 overexpression increased the clonigenic survival of Apaf-1–deficient ES cells by approximately fivefold after either exposure to STS or deprivation of serum.
Figure 6.

Bcl-2 enhances clonigenic survival of Apaf-1−/− ES cells. Apaf-1–deficient ES cells were either (A) treated with 15 uM STS for 6 h, or (B) deprived of serum for 9 d (−FCS). Cells were washed and cultured for 5–6 d in complete medium containing serum. Cells were then stained with crystal violet, and the number of colonies was counted (mean ± SE; n = 3).
Bcl-2 Protection in Absence of Caspase Activation.
Apoptosis results from activation of caspases. A subgroup of these proteases cleaves the tetrapeptide sequence DEVD (Asp-Glu-Val-Asp 46) and functions as the principal effectors of apoptosis (caspases-3 and -7), whereas upstream, initiator caspases have preferences for other tetrapeptide sequences, such as IETD (Ile-Glu-Thr-Asp; caspases-8 and -10), LEHD (Leu-Glu-His-Asp; caspase-9), LEVD (Leu-Glu-Val-Asp; caspase-4), VDVAD (Val-Asp-Val-Ala-Asp; caspase-2), and YVAD (Tyr-Val-Ala-Asp; caspase-1; references 47, 48). To explore whether caspase activation occurs in Apaf-1+/− and Apaf-1−/− ES cells exposed to apoptogenic stimuli, and to assess whether Bcl-2 overexpression prevents activation of these caspases, we prepared lysates from ES cells at various times after exposure to death stimuli and assayed for active caspases using fluorigenic peptide substrates. As shown in Fig. 7, growth factor withdrawal, UV-irradiation, VP16, and CP all induced the activation of DEVD-cleaving caspases in Apaf-1–containing ES cells. Moreover, Bcl-2 overexpression partially suppressed this caspase activation, as expected 49 50. In contrast, relatively little DEVDase activity was observed in factor-deprived and UV-treated ES cells that lacked Apaf-1, and no caspase activity was detected in these cells after exposure to anticancer drugs VP16 and CP (Fig. 7). Similar observations were made when examining the activities of various initiator caspase activities using fluorigenic tetra- and pentapeptide substrates VDVAD, LEVD, IETD, and LEHD. For example, in lysates prepared from VP16-treated ES cells, VDVAD-, LEVD-, IETD-, and LEHD-cleaving protease activity was present in Apaf-1–containing lysates derived from Apaf-1+/− ES cells but was markedly reduced in lysates from Bcl-2–overexpressing Apaf-1+/− cells (Fig. 7). In contrast, none of these protease activities could be detected in lysates prepared from Apaf-1–deficient ES cells. Studies with the caspase-1 substrate, YVAD, demonstrated no evidence of activation of this type of caspase in either Apaf-1+/− or Apaf-1−/− ES cells (not shown). We conclude therefore that the cytoprotective effects of Bcl-2 in Apaf-1−/− ES cells do not correlate with caspase activity, suggesting a caspase-independent mechanism.
Figure 7.

Measurements of caspase activity in Apaf-1+/− and Apaf-1−/− ES cells. The Apaf-1+/− transfectants hygromycin (Hygro) and Bcl-2 (A) were compared with the Apaf-1−/− ES cell transfectants hygromycin (Hygro) and Bcl-2 (B) with regard to caspase activation. To assay caspase activity, 3 × 106 ES cells were seeded in 10-cm dishes 1 d before treatment. The cells were left untreated or were treated with UV irradiation (240 mJ/cm2), VP16 (100 μM), or CP (100 μM). Floating and adherent cells were collected 24 h later, and lysates were prepared. Alternatively, ES cells were washed and cultured in serum-free medium (−FCS) for 3 d. Caspase activity was measured using lysates containing 25 μg of total cellular protein, monitoring release of AFC from peptide substrates using continuous-reading instruments. Enzyme activity is expressed as the rate of AFC fluorophore released (relative fluorescence units per minute per ng of total protein. (A) Ac-DEVD-AFC was used as the substrate. (B) Various substrate peptides were compared using lysates from VP16-treated cells, including Z-VDVAD-AFC, Ac-LEVD-AFC, Z-IETD-AFC, and Ac-LEHD-AFC. Data are representative of a minimum of two experiments.
Bcl-2 Preserves ΔΨ in Apaf-1− / − ES Cells.
Loss of mitochondrial membrane potential is a concomitant of apoptosis and cell death induced by many stimuli, occurring either upstream or downstream of caspase activation, depending on the particular stimulus employed 51 52. To explore further the mechanism by which Bcl-2 overexpression protects Apaf-1–deficient ES cells, the potential-sensitive dye 3,3′-dihexyloxacarbocyanine iodide DiOC6(3) was used to evaluate changes in ΔΨ after exposure of Apaf-1−/− cells to UV irradiation, VP16, or STS. As expected, loss of ΔΨ was induced in response to these cell death stimuli in hygromycin control cells, as evidence by a two- to threefold increase in the accumulation of cells with reduced DiOC6(3) fluorescence (Fig. 8). In contrast, significantly fewer cells suffered reductions in ΔΨ in cultures of Bcl-2–transfected Apaf-1−/− ES cells (P < 0.05). These results indicate that Bcl-2 can preserve mitochondrial function independently of Apaf-1.
Figure 8.

Bcl-2 preserves mitochondrial ΔΨ in Apaf-1–deficient ES cells. Hygromycin control (Hygro)–transfected or Bcl-2–overexpressing Apaf-1−/− ES cells (clones A and B) were cultured without further treatment or were treated with UV irradiation (240 mJ/cm2), VP16 (100 μM), or STS (10 μM). After 24 h, floating and adherent cells were collected and stained with 40 nM DiOC6(3), followed by FACS® analysis. Loss of ΔΨ was visualized as a reduction in the fluorescence signal in the FL1 channel. (A) Representative FACS® histograms are presented, showing fluorescence intensity (log-scale, x-axis) versus relative cell number (y-axis) for untreated (CNTL) and UV-irradiated hygromycin (Hygro) control cells, Bcl-2 (A), and Bcl-2 (B) cells. (B) The percentages of cells with reduced DiOC6(3) staining are shown (mean ± SE; n = 2 or 3). Statistical significance was assessed by unpaired t-test. *P ≤ 0.05.
Bcl-2 Binds Bax but Does Not Interact with Apaf-1.
Though Bcl-2 protected Apaf-1–deficient ES cells, indicating an Apaf-1–independent mechanism of action, we considered the possibility that Bcl-2 might be capable of operating through both Apaf-1–dependent and –independent mechanisms. Since Bcl-2 homologues associate with Apaf-1 homologues in some lower organisms (e.g., CED-9 binds CED-4 in C. elegans; for a review, see reference 53), we addressed the question of whether Bcl-2 can associate with Apaf-1 using a variety of experimental approaches, including (i) yeast two-hybrid assays, (ii) in vitro protein-binding assays using bacteria-produced glutathione S-transferase–tagged proteins, and (iii) coimmunoprecipitation experiments. Though only results from coimmunoprecipitation experiments are presented here, similar conclusions were reached with all three methods.
Fig. 9 shows representative data from coimmunoprecipitation experiments in which Bcl-2 and Apaf-1 were coexpressed at high levels by transient transfection in 293T cells. For the experiments shown here, an Apaf-1 mutant lacking the COOH-terminal domain containing multiple WD repeats, Apaf-1 (ΔWD), was employed because it accumulated to much higher levels in transfected cells than full-length Apaf-1, and because it retains the domains that would be expected to bind Bcl-2 family proteins 30 54; however, similar conclusions were reached using the full-length Apaf-1 protein (not shown). Although procaspase-9 was readily coimmunoprecipitated with myc epitope–tagged Apaf-1 (ΔWD) protein, Bcl-2 was not (Fig. 9). However, immunoblot analysis of lysates prepared from the transiently transfected cells confirmed production of abundant amounts of the Bcl-2 protein. Moreover, in side-by-side comparisons, Bcl-2 protein was coimmunoprecipitated from 293T cell lysates with Myc-tagged Bax but not with Myc-tagged Apaf-1 (ΔWD), thus confirming that the Bcl-2 protein produced in these experiments was competent to bind other proteins. Though Bcl-2 could be coimmunoprecipitated with Myc-Bax, it did not bind a control Myc-Bax (ΔIGDE) protein, which contains a deletion of four amino acids required for Bcl-2 binding within the Bcl-2 homology (BH)3 domain of Bax 55. Similar results were obtained when using Bcl-XL instead of Bcl-2 (not shown). We conclude therefore that Bcl-2 and Bcl-XL do not bind Apaf-1, at least not under the same conditions where Bcl-2 and Bcl-XL are competent to bind Bax and where Apaf-1 is competent to bind procaspase-9. Bcl-2 and Bcl-xL also failed to suppress apoptosis induced by transient overexpression of Apaf-1 (not shown).
Figure 9.

Bcl-2 binds Bax but does not interact with Apaf-1. 293T cells (3 × 106 cells per 10-cm dish) were transiently transfected with various combinations of plasmids (denoted by + or −), including 7 μg of plasmid DNA encoding Bcl-2 in combination with 4 μg of plasmids encoding either Myc-Bax 1, 8 μg Myc–Apaf-1 (ΔWD) 2, or 4 μg Myc-Bax–ΔIGDE mutant 3. Alternatively, 293T cells were transiently transfected with 7 μg of pFlag–CMV2–Caspase-9 (C287A) and 8 μg of Myc–Apaf-1 (ΔWD)-producing plasmids 4. Cell extracts were prepared 2 d later, and immunoprecipitates were prepared using anti-Myc antibody. Immune complexes (IP) or cell lysates (100 μg total protein) were fractionated by SDS-PAGE and then blotted to nitrocellulose filters, which were incubated with anti–Bcl-2 or anti-Flag (M2) antibodies (top). Alternatively, cell lysates from the four transfections 1 2 3 4 were run directly in gels (100 μg total protein) and analyzed by immunoblotting using anti-Myc antibodies (lower two panels). Control IPs using mouse IgG1 instead of anti-Myc failed to result in precipitation of any of the indicated proteins (not shown), providing an additional specificity control. The positions of the Bcl-2, procaspase-9, Apaf-1 (ΔWD), Bax, and Bax (ΔIGDE) protein are indicated by arrows.
Discussion
Alterations in the expression or function of Bcl-2 family proteins have been associated with many human diseases, including those characterized by excessive cell death (stroke, heart failure, AIDS) and those involving cell accumulation due to insufficient programmed cell death (cancer, autoimmunity 56). Understanding the mechanisms of action of Bcl-2 family proteins is therefore important for devising approaches for eventually manipulating the functions of these proteins for therapeutic benefit. In this report, we have addressed the issue of whether the cytoprotective actions of Bcl-2 require the participation of the CED-4 homologue, Apaf-1.
Using ES cells that lack Apaf-1 because of interruption of both copies of their apaf-1 genes by homologous recombination, we found that Bcl-2 retains cytoprotective activity even in the absence of Apaf-1. Specifically, Bcl-2 overexpression reduced or delayed cell death induced by growth factor deprivation, UV irradiation, and anticancer drugs in Apaf-1−/− ES cells. Furthermore, the extent to which Bcl-2 protected against cell death was similar in both Apaf-1–expressing and Apaf-1–deficient ES cells. These observations thus demonstrate unequivocally that Bcl-2 possesses Apaf-1–independent functions.
It has been debated whether Bcl-2 and its close relatives such as Bcl-XL operate upstream of rather than at the level of Apaf-1. On the one hand, Bcl-2 and Bcl-XL have been shown to block release of cytochrome c from mitochondria 20 21 36, which represents an event upstream of Apaf-1 activation. This ability of Bcl-2 and/or Bcl-XL appears to be caspase independent, further arguing that Apaf-1 is not involved, as the only known activity of Apaf-1 is as an activator of caspases. On the other hand, Bcl-2 overexpression has been shown to block apoptosis downstream of cytochrome c in some scenarios 33 34. Also, Bcl-XL has been reported to associate with Apaf-1 and to suppress apoptosis induced by overexpression of Apaf-1, as well as to prevent Apaf-1 interactions with procaspase-9 30 31 32. However, recent data raise suspicions about the ability of antiapoptotic Bcl-2 family proteins to bind Apaf-1 57, and in our experiments, we were unable to demonstrate an association of Bcl-2 or Bcl-XL with the Apaf-1 protein or fragments of it. Moreover, in contrast to C. elegans CED-4 and CED-9, where membrane-associated CED-9 has been demonstrated to alter the intracellular location of CED-4, pulling this protein to the same membrane locations where CED-9 resides 28, we have been unable to demonstrate an effect of overexpression of Bcl-XL on the intracellular location of Apaf-1 in either resting or apoptosis-stimulated cells (Haraguchi, M. and J.C. Reed, unpublished observations). Thus, although we cannot exclude that an interaction of Bcl-2 or Bcl-XL occurs with Apaf-1 under some unidentified conditions, we have been unable to find any compelling evidence to support this notion. By analogy to other well-known suppressors (e.g., IκB-suppressing nuclear factor κB), if Bcl-2 or Bcl-XL were to function predominantly as Apaf-1 suppressors, then one would expect Bcl-2 and/or Bcl-XL to bind avidly to Apaf-1. This seems not to be the case.
Though Bcl-2 fails to physically or functionally interact with Apaf-1, the CED-9/CED-4 paradigm for explaining the mechanism of Bcl-2 family proteins may still apply. For example, Bcl-2 could interact with other CED-4–like proteins that have perhaps not yet been discovered in humans or mammals. However, in this case, one would still expect the cell death mechanism suppressed by Bcl-2 to involve caspases, since CED-4 homologues operate as caspase activators. In this regard, we found that death of Apaf-1−/− ES cells induced by some anticancer drugs proceeded via a mechanism that resulted in no detectable activation of caspase family proteases, and yet was Bcl-2 suppressible. Moreover, the cell death seen in Apaf-1−/− ES cells did not resemble apoptosis morphologically and was not suppressible by a broad-spectrum caspase inhibitor (benzyloxycarbonyl-Val-Ala-Asp [zVAD] fluoromethyl ketone; Haraguchi, M. and J.C. Reed, unpublished observations). Together, these observations argue against suppression of a CED-4–like protein as the major mechanism by which Bcl-2 opposes cell death.
Mitochondria have been shown to play an important role in both apoptotic and nonapoptotic cell death (for a review, see reference 51). Apaf-1 functions as a coupling device that links mitochondria to the activation of caspases, thereby ensuring an apoptotic demise of cells that have suffered mitochondrial damage. However, if caspases are inhibited, mitochondrial damage can still trigger cell death through nonapoptotic mechanisms. For example, loss of cytochrome c from mitochondria can result not only in apoptosis due to Apaf-1–mediated caspase activation, but it can also cause an arrest of electron transport within the respiratory chain, resulting in generation of membrane-damaging free radicals and cessation of mitochondrial ATP production, culminating in necrosis (for a review, see reference 16). In this regard, we observed that Bcl-2–overexpressing Apaf-1−/− ES cells suffered loss of mitochondrial membrane potential (ΔΨ) when exposed to UV irradiation or anticancer drugs, and that Bcl-2 overexpression reduced the percentage of Apaf-1−/− ES cells that lost ΔΨ. Bcl-2 also preserved mitochondrial oxidative function even in the absence of Apaf-1, as determined by MTT dye reduction assays (not shown). Thus, Apaf-1 is required neither for loss of mitochondrial ΔΨ nor for cell death of ES cells when exposed to UV irradiation or anticancer drugs. Further, Bcl-2 preserves mitochondrial membrane potential via an Apaf-1–independent mechanism, consistent with the idea that Bcl-2 operates upstream of Apaf-1 at the level of the mitochondria, where Bcl-2 predominantly localizes in cells 42 58.
If Bcl-2 controls a cell death checkpoint upstream of Apaf-1, why then does loss of Apaf-1 gene function contribute to neoplastic transformation in collaboration with oncogenes in rodent cell transformation assays 59? Moreover, why is programmed cell death in the developing nervous system and some other locations dependent on Apaf-1 7 8? One possibility is that extramitochondrial pathways for activating Apaf-1 may exist, thus uncoupling mitochondria (the site of Bcl-2 action) from Apaf-1–mediated activation of caspases. Another likely possibility is that Apaf-1 participates in an amplification loop in which Apaf-1–mediated activation of caspases converts what would otherwise be sublethal injury into a lethal hit. In this regard, caspases have been shown to trigger mitochondrial permeability transition, resulting in loss of ΔΨ, mitochondrial swelling, rupture, and release of cytochrome c 60 61. In vivo, circumstances may arise where physiological cell death stimuli induce only a small subpopulation of mitochondria in cells to dump cytochrome c, necessitating that this small proportion of released cytochrome c activates Apaf-1 and secondarily triggers caspase-dependent destruction of additional mitochondria, thereby committing the cell to death. Thus, although Bcl-2 may not directly regulate Apaf-1, functional interactions of Bcl-2 and Apaf-1 are likely to occur within the context of these interdependent cyclical relations involving mitochondria as both initiators and targets in a cell death pathway. Future genetic analysis of Apaf-1 deficiency in combination with Bcl-2 and Bcl-XL gene knockouts in mice will undoubtedly contribute to a greater understanding of the interdependence of Bcl-2 family proteins and Apaf-1 in vivo.
Acknowledgments
We thank R. Cornell for manuscript preparation.
We acknowledge the generous support of the National Institutes of Health (grant GM60554).
Footnotes
Abbreviations used in this paper: Apaf-1, apoptotic protease activating factor 1; CP, cisplatin; ΔΨ, mitochondrial transmembrane potential; ES, embryonic stem; PI, propidium iodide; STS, staurosporine.
References
- Jacobson M.D., Weil M., Raff M.C. Programmed cell death in animal development. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- Yuan J.Y., Horvitz H.R. The Caenorhabditis elegans genes ced-3 and ced-4 act cell autonomously to cause programmed cell death. Dev. Biol. 1990;138:33–41. doi: 10.1016/0012-1606(90)90174-h. [DOI] [PubMed] [Google Scholar]
- Alnemri E.S., Livingston D.J., Nicholson D.W., Salvesen G., Thornberry N.A., Wong W.W., Yuan J. Human ICE/CED-3 protease nomenclature. Cell. 1996;87:171. doi: 10.1016/s0092-8674(00)81334-3. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan A.M., Chaudhary D., O'Rourke K., Koonin E.V., Dixit V.M. Role of CED-4 in the activation of CED-3. Nature. 1997;388:728–729. doi: 10.1038/41913. [DOI] [PubMed] [Google Scholar]
- Yang X., Chang H.Y., Baltimore D. Essential role of CED-4 oligomerization in CED-3 activation and apoptosis. Science. 1998;281:1355–1357. doi: 10.1126/science.281.5381.1355. [DOI] [PubMed] [Google Scholar]
- Zou H., Henzel W.J., Liu X., Lutschg A., Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- Yoshida H., Kong Y.Y., Yoshida R., Elia A.J., Hakem A., Hakem R., Penninger J.M., Mak T.W. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–750. doi: 10.1016/s0092-8674(00)81733-x. [DOI] [PubMed] [Google Scholar]
- Cecconi F., Alvarez-Bolado G., Meyer B.I., Roth K.A., Gruss P. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell. 1998;94:727–737. doi: 10.1016/s0092-8674(00)81732-8. [DOI] [PubMed] [Google Scholar]
- Rodriguez A., Oliver H., Zou H., Chen P., Wang X., Abrams J. Dark is a Drosophila homologue of Apaf-1/CED-4 and functions in an evolutionarily conserved death pathway. Nat. Cell Biol. 1999;1:272–279. doi: 10.1038/12984. [DOI] [PubMed] [Google Scholar]
- Kanuka H., Sawamoto K., Inohara N., Matsuno K., Okano H., Miura M. Control of the cell death pathway by Dapaf-1, a drosophila Apaf-1/CED-4-related caspase activator. Mol. Cell. 1999;4:757–769. doi: 10.1016/s1097-2765(00)80386-x. [DOI] [PubMed] [Google Scholar]
- Zhou L., Song Z., Tittel J., Steller H. HAC-1, a drosophila homolog of APAF-1 and CED-4, functions in developmental and radiation-induced apoptosis. Mol. Cell. 1999;4:745–755. doi: 10.1016/s1097-2765(00)80385-8. [DOI] [PubMed] [Google Scholar]
- Zou H., Li Y., Liu X., Wang X. An APAF-1 cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol. Chem. 1999;274:11549–11556. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]
- Saleh A., Srinivasula S., Acharya S., Fishel R., Alnemri E. Cytochrome c and dATP-mediated oligomerization of Apaf-1 is a prerequisite for procaspase-9 activation. J. Biol. Chem. 1999;274:17941–17945. doi: 10.1074/jbc.274.25.17941. [DOI] [PubMed] [Google Scholar]
- Hu Y., Benedict M.A., Ding L., Nunez G. Role of cytochrome c and dATP/ATP hydrolysis in Apaf-1-mediated caspase-9 activation and apoptosis. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:3586–3595. doi: 10.1093/emboj/18.13.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adrain C., Slee E.A., Harte M.T., Martin S.J. Regulation of apoptotic protease activating factor-1 oligomerization and apoptosis by the WD-40 repeat region. J. Biol. Chem. 1999;274:20855–20860. doi: 10.1074/jbc.274.30.20855. [DOI] [PubMed] [Google Scholar]
- Reed J.C. Cytochrome ccan't live with it; can't live without it. Cell. 1997;91:559–562. doi: 10.1016/s0092-8674(00)80442-0. [DOI] [PubMed] [Google Scholar]
- Adams J.M., Cory S. The Bcl-2 protein familyarbiters of cell survival. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- Tanaka S., Saito K., Reed J.C. Structure-function analysis of the Bcl-2 oncoproteinaddition of a heterologous transmembrane domain to a portion of the Bcl-2 beta protein restores function as a regulator of cell survival. J. Biol. Chem. 1993;268:10920–10926. [PubMed] [Google Scholar]
- Nguyen M., Branton P.E., Walton P.A., Oltvai Z.N., Korsmeyer S.J., Shore G.C. Role of membrane anchor domain of Bcl-2 in suppression of apoptosis caused by E1B-defective adenovirus. J. Biol. Chem. 1994;269:16521–16524. [PubMed] [Google Scholar]
- Yang J., Liu X., Bhalla K., Kim C.N., Ibrado A.M., Cai J., Peng I.-I., Jones D.P., Wang X. Prevention of apoptosis by Bcl-2release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- Kluck R.M., Bossy-Wetzel E., Green D.R., Newmeyer D.D. The release of cytochrome c from mitochondriaa primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- Xiang J., Chao D.T., Korsmeyer S.J. BAX-induced cell death may not require interleukin 1b-converting enzyme-like proteases. Proc. Natl. Acad. Sci. USA. 1996;93:14559–14563. doi: 10.1073/pnas.93.25.14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jürgensmeier J.M., Xie Z., Deveraux Q., Ellerby L., Bredesen D., Reed J.C. Bax directly induces release of cytochrome c from isolated mitochondria. Proc. Natl. Acad. Sci. USA. 1998;95:4997–5002. doi: 10.1073/pnas.95.9.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finucane D.M., Bossy-Wetzel E., Cotter T.G., Green D.R. Bax-induced caspase activation and apoptosis via cytochrome c release from mitochondria is inhibitable by Bcl-XL. J. Biol. Chem. 1999;274:2225–2233. doi: 10.1074/jbc.274.4.2225. [DOI] [PubMed] [Google Scholar]
- Holinger E.P., Chittenden T., Lutz R.J. Bak BH3 Peptides antagonize Bcl-xL function and induce apoptosis through cytochrome c-independent activation of caspases. J. Biol. Chem. 1999;274:13298–13304. doi: 10.1074/jbc.274.19.13298. [DOI] [PubMed] [Google Scholar]
- Spector M.S., Desnoyers S., Heoppner D.J., Hengartner M.O. Interaction between the C. elegans cell-death regulators CED-9 and CED-4. Nature. 1997;385:653–656. doi: 10.1038/385653a0. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan A.M., O'Rourke K., Lane B.R., Dixit V.M. Interaction of CED-4 with CED-3 and CED-9a molecular framework for cell death. Science. 1997;275:1122–1126. doi: 10.1126/science.275.5303.1122. [DOI] [PubMed] [Google Scholar]
- Wu D., Wallen H.D., Nunez G. Interaction and regulation of subcellular localization of CED-4 by CED-9. Science. 1997;275:1126–1129. doi: 10.1126/science.275.5303.1126. [DOI] [PubMed] [Google Scholar]
- Conradt B., Horvitz H. The C. elegans protein EGL-1 is required for programmed cell death and interacts with the Bcl-2-like protein CED-9. Cell. 1998;93:519–529. doi: 10.1016/s0092-8674(00)81182-4. [DOI] [PubMed] [Google Scholar]
- Pan G., O'Rourke K., Dixit V.M. Caspase-9, Bcl-XL, and APAF-1 form a ternary complex. J. Biol. Chem. 1998;273:5841–5845. doi: 10.1074/jbc.273.10.5841. [DOI] [PubMed] [Google Scholar]
- Hu Y., Benedict M.A., Wu D., Inohara N., Nunez G. Bcl-XL interacts with Apaf-1 and inhibits Apaf-1-dependent caspase-9 activation. Proc. Natl. Acad. Sci. USA. 1998;95:4386–4391. doi: 10.1073/pnas.95.8.4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins C., Kim C.N., Fang G., Bhalla K.N. Overexpression of Apaf-1 promotes apoptosis of untreated and paclitaxel- or etoposide-treated HL-60 cells. Cancer Res. 1998;58:4561–4566. [PubMed] [Google Scholar]
- Rossé T., Olivier R., Monney L., Rager M., Conus S., Fellay I., Jansen B., Borner C. Bcl-2 prolongs cell survival after bax-induced release of cytochrome c. Nature. 1998;391:496–499. doi: 10.1038/35160. [DOI] [PubMed] [Google Scholar]
- Zhivotovsky B., Orrenius S., Brustugun O.T., Doskeland S.O. Injected cytochrome c induces apoptosis. Nature. 1998;391:449–450. doi: 10.1038/35060. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E., Newmeyer D.D., Green D.R. Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:37–49. doi: 10.1093/emboj/17.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden M.G., Chandel N.S., Williamson E.K., Schumacker P.T., Thompson C.B. Bcl-XL regulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91:627–637. doi: 10.1016/s0092-8674(00)80450-x. [DOI] [PubMed] [Google Scholar]
- Okuno S., Shimizu S., Ito T., Nomura M., Hamada E., Tsujimoto Y., Matsuda H. Bcl-2 prevents caspase-independent cell death. J. Biol. Chem. 1998;273:34272–34277. doi: 10.1074/jbc.273.51.34272. [DOI] [PubMed] [Google Scholar]
- Kane D.J., Örd T., Anton R., Bredesen D.E. Expression of Bcl-2 inhibits necrotic neural cell death. J. Neurosci. Res. 1995;40:269–275. doi: 10.1002/jnr.490400216. [DOI] [PubMed] [Google Scholar]
- Cardone M.H., Roy N., Stennicke H.R., Salvesen G.S., Franke T.F., Stanbridge E., Frisch S., Reed J.C. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- Zha H., Fisk H.A., Yaffe M.P., Mahajan N., Herman B., Reed J.C. Structure-function comparisons of the proapoptotic protein Bax in yeast and mammalian cells. Mol. Cell. Biol. 1996;16:6494–6508. doi: 10.1128/mcb.16.11.6494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen R.M., Zubiaur M., Neer E.J., Seidman J.G. Embryonic stem cells lacking a functional inhibitory G-protein subunit (alpha i2) produced by gene targeting of both alleles. Proc. Natl. Acad. Sci. USA. 1991;88:7036–7040. doi: 10.1073/pnas.88.16.7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajewski S., Tanaka S., Takayama S., Schibler M.J., Fenton W., Reed J.C. Investigation of the subcellular distribution of the bcl-2 oncoproteinresidence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res. 1993;53:4701–4714. [PubMed] [Google Scholar]
- Deveraux Q.L., Takahashi R., Salvesen G.S., Reed J.C. X-linked IAP is a direct inhibitor of cell death proteases. Nature. 1997;388:300–304. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- Pepper C., Thomas A., Tucker H., Hoy T., Bentley P. Flow cytometric assessment of three different methods for the measurement of in vitro apoptosis. Leuk. Res. 1998;22:439–444. doi: 10.1016/s0145-2126(98)00013-7. [DOI] [PubMed] [Google Scholar]
- Reed J.C. Bcl-2 and the regulation of programmed cell death. J. Cell Biol. 1994;124:1–6. doi: 10.1083/jcb.124.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson D.W., Ali A., Thornberry N.A., Vaillancourt J.P., Ding C.K., Gallant M., Gareau Y., Griffin P.R., Labelle M., Lazebnik Y.A. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- Thornberry N.A., Rano T.A., Peterson E.P., Rasper D.M., Timkey T., Garcia-Calvo M., Houtzager V.M., Nordstrom P.A., Roy S., Vaillancourt J.P. A combinatorial approach defines specificities of members of the caspase family and granzyme B. J. Biol. Chem. 1997;272:17907–17911. doi: 10.1074/jbc.272.29.17907. [DOI] [PubMed] [Google Scholar]
- Salvesen G.S., Dixit V.M. Caspasesintracellular signaling by proteolysis. Cell. 1997;91:443–446. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- Armstrong R.C., Aja T., Xiang J., Gaur S., Krebs J.F., Hoang K., Bai X., Korsmeyer S.J., Karanewsky D.S., Fritz L.C., Tomaselli K.J. Fas-induced activation of the cell death related protease CPP32 is inhibited by Bcl-2 and by ICE family protease inhibitors. J. Biol. Chem. 1996;271:16850–16855. doi: 10.1074/jbc.271.28.16850. [DOI] [PubMed] [Google Scholar]
- Boulakia C.A., Chen G., Ng F.W., Teodoro J.G., Branton P.E., Nicholson D.W., Poirier G.G., Shore G.C. Bcl-2 and adenovirus E1B 19 kDA protein prevent E1A-induced processing of CPP32 and cleavage of poly(ADP-ribose) polymerase. Oncogene. 1996;12:529–536. [PubMed] [Google Scholar]
- Green D.R., Reed J.C. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Green D.R., Kroemer G. The central executioners of apoptosiscaspases or mitochondria? Trends Cell Biol. 1998;8:267–271. doi: 10.1016/s0962-8924(98)01273-2. [DOI] [PubMed] [Google Scholar]
- Hengartner M.O. Apoptosis. Death cycle and Swiss army knives. Nature. 1998;391:441–442. doi: 10.1038/35036. [DOI] [PubMed] [Google Scholar]
- Chaudhary D., O'Rourke K., Chinnaiyan A.M., Dixit V.M. The death inhibitory molecules CED-9 and CED-4L use a common mechanism to inhibit the CED-3 death protease. J. Biol. Chem. 1998;273:17708–17712. doi: 10.1074/jbc.273.28.17708. [DOI] [PubMed] [Google Scholar]
- Zha H., Aime-Sempe C., Sato T., Reed J.C. Pro-apoptotic protein Bax heterodimerizes with Bcl-2 and homodimerizes with Bax via a novel domain (BH3) distinct from BH1 and BH2. J. Biol. Chem. 1996;271:7440–7444. doi: 10.1074/jbc.271.13.7440. [DOI] [PubMed] [Google Scholar]
- Reed J.C. Mechanisms of Bcl-2 family protein function and dysfunction in health and disease. Behring Inst. Mitt. 1996;97:72–100. [PubMed] [Google Scholar]
- Moriishi K., Huang D.C., Cory S., Adams J.M. Bcl-2 family members do not inhibit apoptosis by binding the caspase activator Apaf-1. Proc. Natl. Acad. Sci. USA. 1999;96:9683–9688. doi: 10.1073/pnas.96.17.9683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockenbery D.M., Zutter M., Hickey W., Nahm M., Korsmeyer S.J. Bcl-2 protein is topographically restricted in tissues characterized by apoptotic cell death. Proc. Natl. Acad. Sci. USA. 1991;88:6961–6965. doi: 10.1073/pnas.88.16.6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soengas M.S., Alarcon R.M., Yoshida H., Giaccia A., Hakem R., Mak T., Lowe S.W. Apaf-1 and caspase-9 in p53-dependent apoptosis and tumor inhibition. Science. 1999;284:156–159. doi: 10.1126/science.284.5411.156. [DOI] [PubMed] [Google Scholar]
- Marzo I., Brenner C., Zamzami N., Susin S.A., Beutner G., Brdiczka D., Xie Z.H., Reed J.C., Kroemer G. The permeability transition pore complexa target for apoptosis regulation by caspases and Bcl-2-related proteins. J. Exp. Med. 1998;187:1261–1271. doi: 10.1084/jem.187.8.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzo I., Susin S.A., Petit P.X., Ravagnan L., Brenner C., Larochette N., Zamzami N., Kroemer G. Caspases disrupt mitochondrial membrane barrier function. FEBS Lett. 1998;427:198–202. doi: 10.1016/s0014-5793(98)00424-4. [DOI] [PubMed] [Google Scholar]