

Abstract
The serine/threonine kinase protein kinase B (PKB)/Akt mediates cell survival in a variety of systems. We have generated transgenic mice expressing a constitutively active form of PKB (gag-PKB) to examine the effects of PKB activity on T lymphocyte survival. Thymocytes and mature T cells overexpressing gag-PKB displayed increased active PKB, enhanced viability in culture, and resistance to a variety of apoptotic stimuli. PKB activity prolonged the survival of CD4+CD8+ double positive (DP) thymocytes in fetal thymic organ culture, but was unable to prevent antigen-induced clonal deletion of thymocytes expressing the major histocompatibility complex class I–restricted P14 T cell receptor (TCR). In mature T lymphocytes, PKB can be activated in response to TCR stimulation, and peptide-antigen–specific proliferation is enhanced in T cells expressing the gag-PKB transgene. Both thymocytes and T cells overexpressing gag-PKB displayed elevated levels of the antiapoptotic molecule Bcl-XL. In addition, the activation of peripheral T cells led to enhanced nuclear factor (NF)-κB activation via accelerated degradation of the NF-κB inhibitory protein IκBα. Our data highlight a physiological role for PKB in promoting survival of DP thymocytes and mature T cells, and provide evidence for the direct association of three major survival molecules (PKB, Bcl-XL, and NF-κB) in vivo in T lymphocytes.
Keywords: PKB/Akt, Bcl-XL, apoptosis, thymocyte selection, NF-κB
Introduction
The induction of survival versus apoptosis is a central issue during T cell development and activation. The differential regulation of thymocyte survival versus death through TCR-mediated selection signals plays a key role in establishing a functional mature T cell repertoire 1. In mature T cells, several studies have demonstrated that signals from MHC molecules are required for the survival of resting T cells 2 3 4. In addition, the induction of apoptosis is strictly regulated after antigenic triggering 5. However, during T cell activation, the T cell costimulatory molecule CD28 is believed to contribute to survival signals 6 7 8.
Several molecules have been identified that play key roles in regulating apoptosis in T cells 9. Important advances in understanding T cell apoptosis have come through the study of Bcl-2 family members. Bcl-2–related proteins function to promote either cell survival (such as Bcl-2 and Bcl-XL) or cell death (such as Bax and BAD). Several studies have demonstrated that overexpression of Bcl-2 10 11 or Bcl-XL 12 13 in thymocytes confers resistance to spontaneous apoptosis and apoptosis induced by a variety of death stimuli. Conversely, overexpression of death-promoting proteins Bax 14 or BAD 15 in thymocytes accelerates cell death in response to apoptosis-inducing stimuli. However, signaling pathways that control expression of these molecules in T cells are not well understood.
Recent studies have identified an important role for a phosphatidylinositol 3-kinase (PI3K)1-mediated signaling pathway in the suppression of apoptosis. The serine/threonine kinase protein kinase Bα (PKB/Akt), a downstream target of PI3K activity, is emerging as a key molecule involved in regulating cell survival in a variety of models 16. Activated PKB has been demonstrated to play a role in growth factor– and cytokine-mediated survival, and protects cells from apoptosis induced by a variety of stimuli, including UV irradiation, c-myc overexpression, Fas-mediated cell death, and matrix detachment 17. PI3K-mediated phosphorylation of phosphatidylinositol functions to recruit PKB and the protein kinases PDK-1 and PDK-2 to the plasma membrane, allowing for direct phosphorylation of PKB by these kinases. Phosphorylation on Thr-308 and Ser-473 is required for the full activation of PKB-α, which then dissociates from the plasma membrane and phosphorylates a variety of substrates 18.
The mechanisms by which PKB promotes cell survival remain controversial, although recent data suggest that PKB may interact directly with several components of cell death machinery. PKB has been shown to antagonize the proapoptotic functions of the Bcl-2–related molecule BAD 19 20 and caspase 9 21 through direct phosphorylation. Alternatively, PKB may alter gene transcription upon binding of survival factors through the regulation of transcription factors 22 23 24 25. PKB is able to inhibit the function of members of the Forkhead family of transcription factors via direct phosphorylation, resulting in their nuclear exclusion. In addition, PKB has recently been implicated as a positive regulator of nuclear factor (NF)-κB activation in fibroblasts 26, embryonic kidney cells 27, and Jurkat cells 28. PKB has been shown to transactivate IκB kinase α (IKKα), which leads to IκB degradation via phosphorylation and nuclear translocation of NF-κB 26 27.
Although the role of PKB as a general mediator of cell survival has been well documented, the function of PKB in T cells is unknown. To investigate the role of PKB activity in primary T cells, we generated mice expressing a constitutively active form of PKB (gag-PKB) in the T cell lineage. In this study, we demonstrate that overexpressed, active PKB can promote CD4+CD8+ double positive (DP) thymocyte survival and mature T cell survival. We also show that PKB can be activated downstream of the TCR in peripheral T cells and link PKB activation with the NF-κB survival pathway in T cells. Finally, we provide evidence that PKB regulates expression of the antiapoptotic molecule Bcl-XL. Our results support a model of PKB-mediated survival through the regulation of the antiapoptotic molecules NF-κB and Bcl-XL in T cells.
Materials and Methods
Generation of Transgenic Mice.
Construction of the gag-PKB cDNA has been described previously 29. To target gag-PKB to the T cell lineage, gag-PKB cDNA was inserted into EcoRI and SmaI sites of the human CD2 minigene cassette 30. The hCD2-gag-PKB insert was linearized and microinjected into fertilized eggs from (CBA/J × B6)F2 animals. Two founder lines, 4-1 and 5-4, were backcrossed to C57BL/6J mice for at least three generations before characterization. The presence of the gag-PKB transgene was detected by Southern blot using an α-32P–radiolabeled gag-PKB–specific probe and EcoRI-digested tail DNA from 3–4-wk-old mice. Alternatively, mice were genotyped via PCR using tail DNA, sense primer 5′-CCTTGAACCTCCTCGTTCGAC-3′, and antisense primer 5′-GATGTACTCAC CTCGCTTGTG-3′.
Mice.
C57BL/6J mice were obtained from The Jackson Laboratory. TCR transgenic mice were generated previously using α and β chains isolated from CTL clone P14, which recognizes the lymphocytic choriomeningitis virus (LCMV) glycoprotein (peptide p33-41) in the context of H-2Db 31. Mice homozygous for the P14 TCR transgene (327 line) were used in timed matings with heterozygous gag-PKB mice for fetal thymic organ culture (FTOC). All mice were maintained in a specific pathogen-free environment at the Ontario Cancer Institute according to institutional guidelines.
Cell Preparation.
Single cell suspensions of lymphoid organs were generated by gently pressing organs through sterile wire mesh. Cells were washed once in cold HBSS and resuspended in IMDM, supplemented with 10% heat-inactivated FCS (GIBCO BRL), 50 μM β-mercaptoethanol, 2 mM glutamine, and 0.1% penicillin/streptomycin. CD4+ and CD8+ splenic T cells were isolated via magnetic cell sorting using MACS® separation columns (Miltenyi Biotec). Splenic preparations were enriched >90% for CD4+ and CD8+ T cells as determined by flow cytometry.
Reagents and Antibodies.
Dexamethasone and 7-amino-actinomycin D (7AAD) were purchased from Sigma-Aldrich. Recombinant hCD8-mFasL fusion protein 32 and hamster anti–mouse CD3 antibody were supplied by Amgen. Purified anti-CD28 (37.51) was purchased from BD PharMingen. The LCMV glycoprotein peptide p33 (KAVYNFATM) and the adenovirus peptide AV (SGPSNTPPEI) were synthesized and purified as described previously 33. The following mAbs were used for flow cytometry: PE-conjugated anti–mouse CD4, FITC-conjugated anti-CD8, biotinylated anti–heat-stable antigen (HSA) (M1/69), biotinylated anti-Vα2 (B20.1), and FITC-conjugated anti–Bcl-2 (3F11) (BD PharMingen). Biotinylated antibodies were detected with streptavidin–Red 670 (GIBCO BRL).
Apoptosis Assays.
For analysis of spontaneous cell death, thymocytes or magnetically sorted CD4+ and CD8+ splenic T cells were suspended at 106 cells per ml in IMDM supplemented with 5% heat-inactivated FCS, 2 mM l-glutamine, 50 μM β-mercaptoethanol, and antibiotics, and cultured in 24-well tissue culture plates. Samples were harvested at various time points, and cell viability was determined by trypan blue exclusion. CD4 versus CD8 thymocyte profiles were assessed over time by flow cytometry. Thymocytes were stained with PE-conjugated anti-CD4, FITC-conjugated anti-CD8, 7AAD, and CD4 versus CD8 thymocyte profiles were determined for 7AAD− populations 34.
For analysis of apoptotic cell death, thymocytes and sorted T cells were plated as above and subjected to apoptotic treatment as indicated. Cell viability was determined by trypan blue exclusion or by FACS® analysis using Annexin V-FITC and propidium iodide (PI) staining according to the manufacturer's instructions (R&D Systems).
FTOC.
Timed matings were established between female mice homozygous for the P14 transgenic TCR (327 line) and male mice heterozygous for the gag-PKB transgene, and fetal thymic lobes were isolated from day 16.5 embryos. PCR analysis was performed on DNA isolated from individual embryos to detect the presence or absence of the gag-PKB transgene. The fetal thymic lobes were separated and transferred onto 0.8-μM nucleopore polycarbonate filters (Costar) and cultured for 6 d at 37°C, 5% CO2 in complete medium (IMDM supplemented with 10% heat-inactivated FCS, 2 mM l-glutamine, 50 μM β-mercaptoethanol, and antibiotics). In addition, thymic lobes were cultured in the presence or absence of exogenous p33 peptide (10−6 M). Media and peptides were changed daily. Thymocytes were harvested by passing thymic lobes through a nylon mesh, stained with mAbs at 4°C, and analyzed via flow cytometry.
T Cell Proliferation Assays.
Splenocytes (5 × 104) from P14 TCR transgenic or P14 TCR/gag-PKB double transgenic mice were cultured with 105 C57BL/6 spleen cells prepulsed with peptide in triplicate in 96-well flat-bottomed plates. Cultures were pulsed with 1 μCi/well [3H]thymidine (Dupont) for the final 8 h of culture and harvested onto glass fiber filters. [3H]Thymidine uptake was measured using a scintillation counter (Topcount; Canberra Packard).
Western Blot Analysis.
Single cell suspensions were lysed by incubation on ice in Gentle Soft Buffer (10 mM NaCl, 20 mM Pipes [pH 7.4], 0.5% NP-40, 5 mM EDTA, 5 μg/ml leupeptin, 1 mM benzamidine, 0.5 mM NaF, and 100 μM Na3VO4) for 20 min. Lysates were cleared by centrifugation and supernatants were normalized for total protein by Bradford assay (Bio-Rad). Proteins were resolved by 12.5% SDS-PAGE, electroblotted to polyvinylidene difluoride membrane (Costar), blocked in 5% Tris-buffered saline, 0.05% Tween 20, and probed with primary antibodies. Anti-PKB, antiphospho S473 PKB, and anti-IκBα antibodies were purchased from New England Biolabs, Inc. Mouse monoclonal anti–Bcl-XL (H-5) and goat polyclonal anti-BAD (C-20) were from Santa Cruz Biotechnology, Inc. Rabbit polyclonal anti–Bcl-2 was a gift from Dr. David Andrews (McMaster University, Hamilton, Ontario). Antiactin antibodies were purchased from Sigma-Aldrich. After incubation with horseradish peroxidase–conjugated goat anti–rabbit antibody (Amersham Pharmacia Biotech) or goat anti–mouse antibody (Santa Cruz Biotechnology, Inc.), bound immunoglobulins were detected using enhanced chemiluminescence (Amersham Pharmacia Biotech) according to the manufacturer's directions.
Electrophoretic Mobility Shift Assay.
Nuclear extracts were harvested according to protocols described previously 35. In brief, 6 × 106 purified T cells, either untreated or stimulated for 4 h with various treatments, were washed twice with PBS and resuspended in 250 μl of buffer A (10 mM Hepes [pH 7.8], 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM dithiothreitol, and 1 mM PMSF). After incubation on ice for 5 min, NP-40 was added to a final concentration of 0.6%. After centrifugation, cytoplasmic proteins were removed and the pelleted nuclei were resuspended in 25 μl buffer C (20 mM Hepes [pH 7.9], 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol, and 1 mM PMSF). After 30 min agitation at 4°C, the samples were centrifuged and supernatants, containing nuclear proteins, were transferred to a fresh vial. Equal amounts of nuclear protein, as determined using the Bio-Rad protein assay, were incubated with an end-labeled, double-stranded NF-κB–specific oligonucleotide probe containing two tandemly positioned NF-κB–binding sites (5′-ATC AGG GAC TTT CCG CTG GGG ACT TTC CG-3′ and 5′-CGG AAA GTC CCC AGC GGA AAG TCC CTG AT-3′). The reaction was performed in a total of 20 μl of binding buffer (5 mM Hepes [pH 7.8], 50 mM KCl, 0.5 mM dithiothreitol, 1 μg poly [dI-dC], and 10% glycerol) for 20 min at room temperature. After incubation, samples were fractionated on a 5% polyacrylamide gel and visualized by autoradiography.
Results
Generation of Transgenic Mice Expressing gag-PKB in Thymocytes and Peripheral T Cells.
To study the effect of PKB on T cell development and peripheral function, we generated transgenic mice overexpressing a constitutively active form of PKB (gag-PKB) in T lymphocytes under control of the CD2 promoter and locus control region (LCR) 30. The gag-PKB–encoded protein is equivalent to the oncogenic form of PKB, v-akt, and consists of three parts: an in frame fusion of the viral gag protein, a 21–amino acid peptide linker derived primarily from the 5′ untranslated region of the endogenous PKB gene locus, and PKB (Fig. 1 A) 36. An NH2-terminal glycine residue of the gag portion of the chimeric protein serves as a site for myristylation 37 38. This myristyl group preferentially targets the gag-PKB fusion protein to the cell membrane where it becomes constitutively activated by upstream kinases 39. Two individual founder lines were established, B6/PKB5-4 and B6/PKB4-1, and the latter was predominantly used in this study. As the active form of PKB is oncogenic 40, thymocytes and T cells from mice were screened for evidence of lymphomagenesis via hematoxylin and eosin histological staining or by flow cytometry using Vβ-specific antibodies to detect monoclonal expansion of T lymphocytes (data not shown). No evidence of lymphoma was detected in mice up to 5 mo of age. All mice used in this study were 6–16 wk of age.
Figure 1.



T cells from transgenic mice express activated PKB. (A) Illustration of the gag-PKB transgene. A 2.3-kb cDNA fragment containing the coding sequence of gag-PKB was inserted into the EcoRI and SmaI sites of the human CD2 minigene. The CD2 promoter, polyadenylation signal (Poly A), and enhancer/locus control region (LCR) are indicated. The construct was linearized at the KpnI and XbaI sites before microinjection. (B) gag-PKB T cells express transgenic gag-PKB mRNA. DNase-treated total RNA isolated from gag-PKB transgenic (B6/PKB) and nontransgenic (B6) LN T cells was subjected to RT-PCR using gag-PKB–specific primers. PCR was performed on the RNA to control for DNA contamination. In the right lane, gag-PKB cDNA was amplified as a positive control for the PCR reaction. RT-PCR products were analyzed on a 1% agarose gel stained with ethidium bromide. (C) Transgenic gag-PKB T cells display elevated levels of PKB protein and increased levels of PKB phosphorylation. Expression of PKB was assessed in thymus, LN, and purified CD4+ and CD8+ T cells from wild-type mice (B6) and gag-PKB transgenic mice from two independent founder lines (B6/PKB5-4 and B6/PKB4-1) by Western blot analysis with a polyclonal anti-PKB antibody. Phosphorylated PKB was detected for the same samples using a polyclonal antibody specific for PKB phosphorylated at Ser-473. Lysates from 2 × 106 cells per tissue sample were loaded per lane after normalization for protein content via the Bradford assay.
gag-PKB mRNA expression was measured by reverse transcription (RT)-PCR on total cellular RNA prepared from LNs of transgenic mice or littermate controls. Primers specific for the gag-PKB construct were used to detect specific expression of the chimeric mRNA and not endogenous PKB mRNA. RT-PCR using RNA from transgenic (B6/PKB) or nontransgenic (B6) lymphocytes showed expression of gag-PKB mRNA in transgenic lymphocytes, and RT minus controls confirmed that there was no contaminating genomic DNA in these samples (Fig. 1 B). Thus, gag-PKB mRNA is produced in lymphocytes of the transgenic mice.
The expression of gag-PKB and its phosphorylation status were determined by Western blot analysis of cell extracts prepared from thymocytes and peripheral T cells of gag-PKB transgenic mice. Studies have demonstrated that the phosphorylation status of PKB, as detected using an antibody specific for PKB phosphorylated at Ser-473, correlates with its kinase activity in vitro 41. Lysates from the thymus, LN, and purified peripheral T cells from nontransgenic animals (B6) and two transgenic founder lines (B6/PKB5-4 and B6/PKB4-1, respectively) were probed with anti-PKB or anti–P-S473 PKB antibodies (Fig. 1 C). PKB levels were elevated in the T cells of mice expressing the gag-PKB transgene (Fig. 1 C, top). In addition, PKB was hyperphosphorylated in the transgenic mice compared with nontransgenic controls (Fig. 1 C, middle). Protein levels of the p85 regulatory subunit of PI3K, the upstream activator of PKB, were similar in control and transgenic lines, demonstrating equal protein loading (Fig. 1 C, bottom). Similar results were obtained using whole cell extracts from splenocytes (data not shown). The size of the transgenic protein detected on the Western blot was much smaller (∼55 kD) than the expected gag-PKB fusion protein (∼90 kD). This phenomenon has been observed in transfection studies using the gag-PKB construct, and was suggested to be due to an internal translation start site in the gag-PKB mRNA 42. However, there is evidence that viral gag polyproteins encode their own cleavage sites during retroviral packaging events 43. As gag-PKB mRNA is produced, it is possible that the gag sequence is cleaved from the chimeric protein after activation, possibly in an autolytic fashion. Overexpressed PKB lacking the gag sequence would not be targeted to the plasma membrane, and would not be expected to exist in a hyperphosphorylated state. Together, these analyses show that we have established lines of transgenic mice that express elevated levels of PKB in the T cell lineage, and despite the possible loss of the gag sequence, the kinase is phosphorylated and activated well above endogenous levels in the T cells of these mice.
PKB Prevents Thymocyte Death in Response to Various Apoptotic Stimuli.
To assay survival of gag-PKB transgenic thymocytes in response to apoptotic stimuli, thymocytes were treated with various death agonists and cell viability was measured over time. Transgenic (B6/PKB5-4 and B6/PKB4-1) and nontransgenic (B6) thymocytes were treated with γ radiation (250 or 500 rads), the glucocorticoid dexamethasone (0.1 or 1 μM), or recombinant hCD8-mFasL fusion protein (50 or 100 ng/ml), a physiological activator of Fas 32, and the proportion of viable cells relative to untreated controls was determined over time via trypan blue exclusion (Fig. 2 A). In all conditions, thymocytes expressing the gag-PKB transgene showed enhanced viability over nontransgenic controls. In particular, pronounced differences in viability (more than two- to threefold) were observed between gag-PKB transgenic and control thymocytes at the higher apoptotic treatments tested (500 rads, 1 μM dexamethasone, and 100 ng/ml hCD8-mFasL; Fig. 2 A). The resistance to Fas-induced killing in transgenic thymocytes was not due to differences in Fas receptor expression, as the levels of Fas expression were similar in both sets of thymocytes (data not shown). The transgenic thymocytes were also resistant to treatment with UV radiation and etoposide compared with nontransgenic cells (data not shown). Similar results were obtained using Annexin V and PI staining (data not shown), indicators of early and late stages of programmed cell death, respectively. Together these data support a role for PKB in regulating T cell survival.
Figure 2.

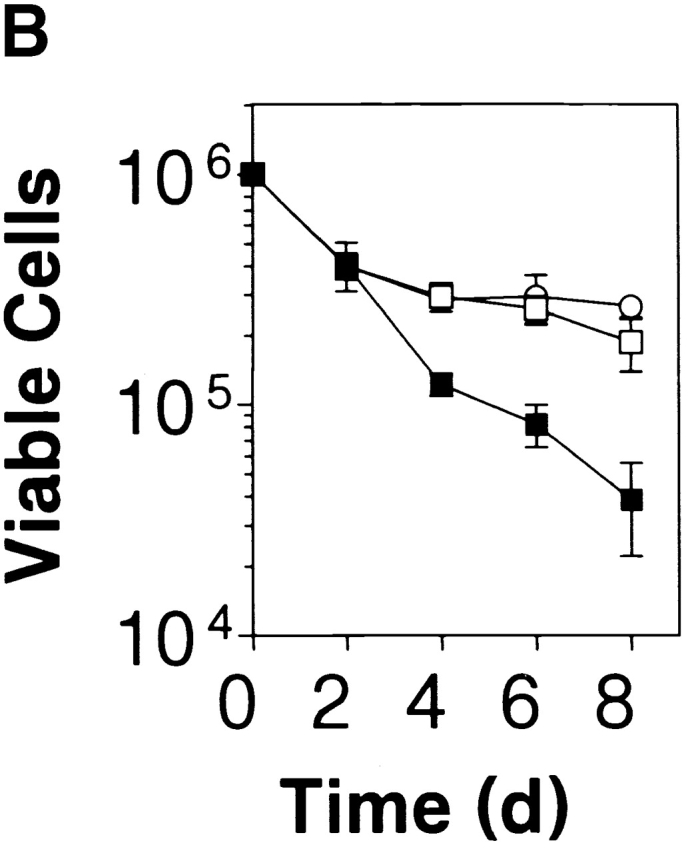

PKB kinase activity protects thymocytes from various apoptosis-inducing stimuli and spontaneous apoptosis in vitro. (A) Transgenic gag-PKB thymocytes are resistant to γ irradiation, dexamethasone, and anti-Fas–induced apoptosis. Thymocytes from control mice (B6, ▪) or mice expressing the gag-PKB transgene (B6/PKB5-4, □; or B6/PKB4-1, ○) were cultured at 106 cells/ml after exposure to γ radiation or incubation in the presence of dexamethasone or recombinant CD8-FasL fusion protein. Thymocyte viability was determined by trypan blue exclusion at various time points after apoptotic treatment. Viability is expressed normalized to untreated thymocytes and in triplicate (±SEM). The data are representative of four independent experiments. (B) PKB activity promotes survival in thymocytes. Thymocytes from transgenic mice (B6/PKB5-4, □; or B6/PKB4-1, ○) or nontransgenic littermate controls (B6, ▪) were cultured in 24-well tissue culture plates at 106 cells/ml in IMDM supplemented with 5% heat-inactivated FCS, 2 mM l-glutamine, and 50 μM β-mercaptoethanol. Cell viability was determined in triplicate by trypan blue exclusion over 8 d of culture at 37°C. The error bars represent the SEM expressed as cell numbers for each triplicate value. These data are representative of six independent experiments. (C) PKB activity selectively protects CD4+CD8+ DP thymocytes from spontaneous apoptosis. Thymocytes from B6/PKB and B6 mice were cultured as in B and harvested after 0, 4, or 8 d of culture. Cells were stained with FITC-conjugated anti-CD8α, PE-conjugated anti-CD4, and 7AAD, and analyzed via flow cytometry. Viable thymocytes were determined by negative staining with 7AAD. The mean percentage of DP thymocytes at each time point is indicated. The presented data are representative of three separate experiments with two mice per experiment.
PKB Protects DP Thymocytes from Spontaneous Death In Vitro.
To assess the effect of activated PKB on thymocyte survival, thymocytes from gag-PKB transgenic (B6/PKB5-4 or B6/PKB4-1) and control littermates (B6) were placed in culture in IMDM medium supplemented with 5% FCS, and viability was assessed at various time points by trypan blue exclusion. Expression of gag-PKB in thymocytes significantly improved their rate of survival over time compared with nontransgenic littermate controls (Fig. 2 B). Viability was similar between the two groups after 2 d in culture, but gag-PKB thymocytes exhibited a marked increase in survival after this time. Approximately 27 ± 3% of thymocytes from B6/PKB4-1 mice were still viable by day 8 of culture, compared with 3.8 ± 1.7% for control thymocytes (Fig. 2 B). These results strongly suggest that PKB can promote extended thymocyte survival in vitro.
Because PKB is expressed, phosphorylated in thymocytes (Fig. 1 C), and acts to regulate thymocyte survival (Fig. 2 B), we determined which thymocyte subpopulations displayed enhanced survival in the presence of activated PKB. Thymocytes from transgenic and control mice were cultured for 8 d in IMDM plus 5% FCS, and viable cells from various time points were assessed for CD4 and CD8 coreceptor expression by flow cytometry. There was a gradual decrease in DP thymocyte survival from 84 to 7% after 8 d in culture in control thymocytes, whereas gag-PKB transgenic DP thymocytes showed a modest decrease from 80 to 61% over the same time period (Fig. 2 C). In terms of absolute cell numbers, this corresponds to a decrease in nontransgenic DP thymocytes from 8.4 ± 0.15 × 105 to 2.7 ± 1.1 × 103, compared with a decrease from 8.0 ± 0.2 × 105 to 1.6 ± 0.2 × 105 for gag-PKB transgenic DP thymocytes. Thus, overexpression of activated PKB confers enhanced survival to thymocytes and appears to selectively regulate the survival of DP thymocytes.
PKB Alters the Survival of DP Thymocytes in FTOC.
The development of a functional T cell repertoire is dependent on the selective survival and programmed death of thymocytes during positive and negative selection in the thymus. As the expression of activated PKB appears to enhance thymocyte survival in culture and decreases their sensitivity to apoptosis, we examined whether PKB may play a role during selection events in the thymus. Flow cytometric analysis of thymocyte subpopulations in adult mice aged 8–14 wk showed no apparent difference in the number or proportion of DP, single positive (SP) CD4+, and SP CD8+ thymocytes between the gag-PKB transgenic mice and nontransgenic littermates (data not shown). In addition, there appeared to be no significant difference in thymus cellularity in these animals. These experiments suggest that expression of gag-PKB does not dramatically alter thymocyte selection.
To further investigate the effect of PKB on thymocyte selection, FTOCs were done using the P14 TCR transgenic mouse model. P14 TCR transgenic mice express receptors specific for the LCMV glycoprotein peptide (amino acids 33–41) presented in the context of H-2Db 31. Homozygous TCR transgenic mice were bred with mice heterozygous for the gag-PKB transgene, and day 16 fetal thymic lobes were harvested and genotyped for the gag-PKB transgene by PCR. After 6 d in culture, P14 or P14/PKB fetal thymic lobes were analyzed by three-color flow cytometry with mAbs against CD4, CD8, and Vα2 to follow the transgenic TCR (Fig. 3 A). P14 thymocytes appeared to display increased positive selection of CD8+ thymocytes compared with P14/PKB thymocytes. However, as shown in Fig. 3 B, P14 thymic lobes expressing the gag-PKB transgene showed an increase in cellularity. An average of 45.0 ± 4.4 × 104 (n = 6) thymocytes were recovered from P14/PKB thymic lobes, compared with 22.0 ± 3.8 × 104 (n = 4) thymocytes for P14 control lobes.
Figure 3.


Active PKB enhances CD4+CD8+ DP thymocyte survival in FTOC. FTOCs from P14 TCR transgenic mice with (P14/PKB, hatched bars) or without (P14, black bars) the gag-PKB transgene were cultured and stained with FITC-conjugated anti-CD8, PE-conjugated anti-CD4, and biotinylated anti-Vα2 or HSA antibodies. (A) Dot plots show CD4 and CD8 expression of thymocytes isolated from P14 and P14/PKB fetal thymic lobes after 6 d in culture. The percentage of cells from each thymocyte subpopulation is indicated. Histograms display the Vα2 expression levels of mature (HSAlo) CD8+ thymocytes. The data shown are representative of three independent experiments. (B) Total cell numbers from P14 (n = 4) and P14/PKB (n = 6) fetal thymic lobes are described in A.
In terms of cellularity, there was no significant difference in the total number of CD8+ cells generated in P14 thymic lobes with or without expression of the gag-PKB transgene. An average of 12.6 ± 3.0 × 104 (n = 6) CD8+ cells were recovered from P14/PKB lobes, compared with 9.0 ± 1.7 × 104 (n = 4) cells for P14 lobes (Fig. 3 B). In addition, Vα2 staining of the CD8+ compartment in P14 versus P14/PKB thymic lobes suggested that there was no difference in TCR density associated with the maturation of these CD8+ cells (Fig. 3 A). These data are consistent with the idea that PKB activity does not significantly alter the positive selection of thymocytes; rather, active PKB enhances the viability of CD4+ CD8+ DP thymocytes in thymic organ culture.
PKB Does Not Prevent Peptide-induced Negative Selection of Thymocytes in FTOC.
To test whether thymocyte negative selection could be altered by active PKB, P14 and P14/PKB thymic lobes were cultured in the presence of 10−6 M LCMV glycoprotein peptide p33, a concentration known to induce deletion of P14 TCR transgenic thymocytes in FTOC 44. Negative selection in the P14 TCR transgenic mouse model is defined by a loss of Vα2+ TCR transgenic thymocytes and an overall decrease in thymus cellularity. 6 d of culture in the presence of p33 peptide resulted in the deletion of CD4+CD8+ DP and CD8+ SP thymocytes expressing the Vα2 chain in P14 thymic lobes regardless of gag-PKB expression (Fig. 4 A). However, while P14 lobes cultured with 10−6 M LCMV glycoprotein p33 showed a substantial decrease in DP thymocytes, P14/PKB lobes maintained a higher proportion of DP thymocytes as observed by flow cytometry (23.7 ± 6.9%, n = 4, compared with 53.6 ± 9%, n = 6). Despite increased numbers, these DP thymocytes did not express high levels of Vα2. It is possible that DP thymocytes expressing gag-PKB downregulate the transgenic TCR through a receptor editing process in response to p33, resulting in a Vα2lo phenotype. In terms of cellularity, both P14 and P14/PKB thymic lobes cultured with p33 peptide showed a decrease in total CD4+CD8+ DP thymocyte populations (Fig. 4 B). These data show that negative selection may not be as efficient in the presence of active PKB, consistent with the role of PKB in enhancing DP survival. However, negative selection of TCR transgenic thymocytes is not abrogated in the presence of active PKB.
Figure 4.

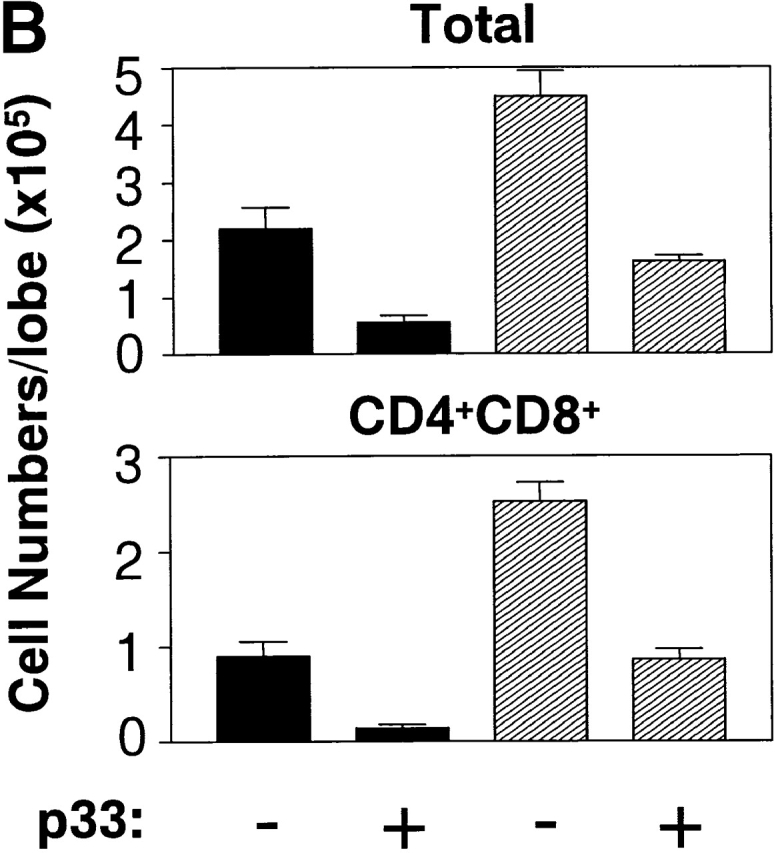
Overexpression of gag-PKB does not prevent thymocyte negative selection in FTOC. FTOCs from P14 TCR transgenic mice with (P14/PKB) or without (P14) the gag-PKB transgene were cultured in the presence or absence of 10−6 M p33 peptide and stained with FITC-conjugated anti-CD8, PE-conjugated anti-CD4, and biotinylated anti-Vα2 or HSA antibodies. (A) The percentage of cells from each thymocyte subpopulation is indicated. Histograms display the Vα2 expression levels on immature (HSAhi) CD4+CD8+ DP thymocytes and mature (HSAlo) CD8+ thymocytes. (B) Total thymocyte and CD4+CD8+ DP thymocyte cell numbers from P14 (black bars) and P14/PKB lobes (hatched bars) after incubation in the presence or absence of p33 peptide. Values shown represent the mean ± SEM for four (P14) and six (P14/PKB) thymic lobes. These data are representative of two independent experiments.
Active PKB Leads to Increased Expression of Bcl-XL in Thymocytes.
Several studies have identified roles for antiapoptotic molecules of the Bcl-2 family during lymphocyte development. Bcl-2 expression is upregulated during positive selection 45 46, whereas Bcl-XL is essential for the survival of DP thymocytes 47 48. To investigate the role of PKB in regulating endogenous levels of these molecules in vivo, thymocytes from B6/PKB and B6 control mice were lysed and Bcl-2 and Bcl-XL protein levels were examined by Western blot (Fig. 5). There was no difference in Bcl-2 protein levels in the thymus of these mice as determined by Western blot (Fig. 5, bottom) or by flow cytometry (data not shown), suggesting that PKB does not affect Bcl-2 expression in thymocytes. However, endogenous levels of Bcl-XL were increased in gag-PKB transgenic mice compared with negative littermate control mice (Fig. 5, top). It should be noted that Bcl-XL was readily detected in control thymocytes from nontransgenic animals; however, Bcl-XL expression in gag-PKB transgenic thymocytes was considerably higher relative to the controls. These results suggest that PKB influences the level of Bcl-XL protein in developing thymocytes.
Figure 5.

Elevated Bcl-XL protein levels are detected in gag-PKB transgenic thymocytes. Cell lysates from 5 × 106 transgenic (B6/PKB) or nontransgenic (B6) thymocytes were resolved by 10% SDS-PAGE and analyzed via Western blotting. Blots were probed with antibodies specific for Bcl-XL (top) and Bcl-2 (bottom).
To determine whether RNA levels were increased, semiquantitative RT-PCR analysis was performed on total cellular RNA isolated from gag-PKB transgenic or nontransgenic thymocytes. When normalized to β-actin levels, Bcl-XL mRNA levels were not significantly different between gag-PKB thymocytes and nontransgenic controls (data not shown). These results suggest that PKB does not regulate Bcl-XL expression in thymocytes by modulating its transcription. Together, these studies show that active PKB enhances Bcl-XL levels, which corresponds to the enhanced survival of DP thymocytes observed in vitro.
gag-PKB Confers Resistance to Apoptotic Stimuli and Spontaneous Apoptosis in Peripheral T Lymphocytes.
To investigate the effects of gag-PKB expression on mature T cell survival, sorted CD4+ and CD8+ splenic T cells from gag-PKB transgenic (B6/PKB5-4 or B6/PKB4-1) or nontransgenic animals were exposed to γ radiation or treated with various concentrations of dexamethasone. As measured by viability dye exclusion, cultured T cells from gag-PKB transgenic mice showed marked reduction in the kinetics of cell death relative to nontransgenic littermates in response to dexamethasone and γ radiation (Fig. 6 A). At 24 h, 53% of transgenic T cells subjected to 250 rads of γ radiation were apoptotic compared with 77% for control T cells. Likewise, only 46% of T cells from gag-PKB transgenic mice were apoptotic in the presence of 0.1 μM dexamethasone compared with 80% of control T cells at 24 h. It should be noted that expression of gag-PKB confers a 1.5–3-fold increase in survival relative to untreated controls, which is less than that observed in both Bcl-2 and Bcl-XL transgenic animals 12. It is possible that the differences in apoptosis to various stimuli may be greater but remain masked due to the enhanced survival of the untreated gag-PKB transgenic population. Collectively, these observations indicate that PKB kinase activity can promote cell survival in peripheral T cells in response to various apoptotic stimuli.
Figure 6.
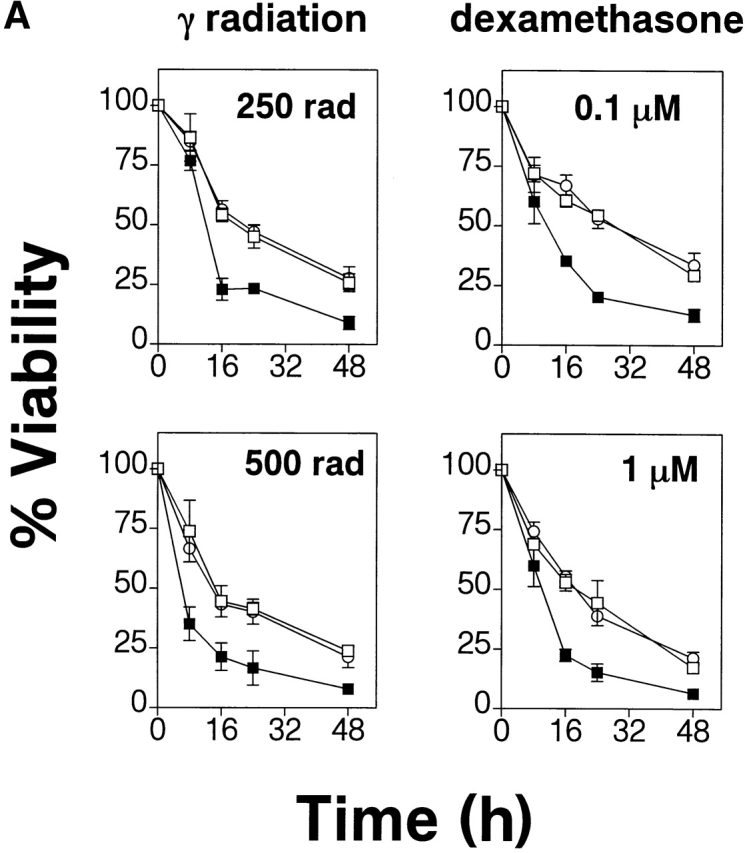
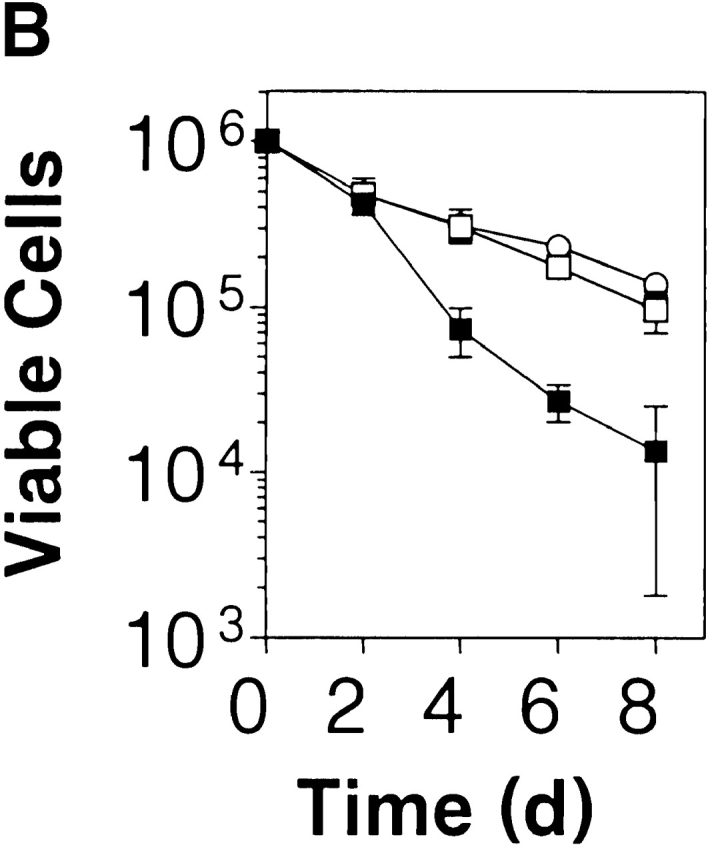
PKB kinase activity protects mature T cells from apoptosis-inducing stimuli and spontaneous cell death in vitro. (A) Transgenic gag-PKB T cells are resistant to γ irradiation and dexamethasone-induced apoptosis. Purified CD4+ and CD8+ splenic T cells from mice expressing the gag-PKB transgene (B6/PKB5-4, □; or B6/PKB4-1, ○) or littermate controls (B6, ▪) were cultured at 106 cells/ml after exposure to γ radiation or dexamethasone. Viability was determined by trypan blue exclusion up to 48 h after treatment. Viability is expressed normalized to untreated T cells and in triplicate (±SEM). The data are representative of four independent experiments. (B) PKB kinase activity enhances T cell survival. Purified CD4+ and CD8+ splenic T cells were cultured at 106 cells/ml for 8 d at 37°C. Cell viability was determined in triplicate by trypan blue exclusion. The error bars represent the SEM expressed as cell numbers for each triplicate value. These data are representative of four independent experiments.
To address the question of whether PKB activity can regulate basal T cell survival, we assessed the ability of peripheral T cells to resist spontaneous apoptosis in culture. CD4+ and CD8+ peripheral T cells isolated from the spleen of gag-PKB transgenic mice displayed increased survival in culture over time compared with nontransgenic controls (Fig. 6 B). After 4 d in culture, ∼31% of B6/PKB splenic T cells were viable, whereas 7% of nontransgenic splenic T cells were alive (Fig. 6 B). This trend was observed at up to 8 d of culture in vitro. Thus, expression of PKB kinase activity can provide signals necessary for the regulation of survival in mature T cells.
TCR-specific PKB Activation in Mature T Cells.
Peripheral T lymphocytes remain in a quiescent state until their antigen-specific TCR is triggered by peptide antigen bound to MHC molecules on the surface of an APC. As ligation of the TCR initiates a series of signal transduction events resulting in T cell proliferation and differentiation, we examined whether PKB was activated in a TCR-dependent fashion in naive T cells. As shown in Fig. 7 A, activation of peripheral T cells with anti-CD3 alone or anti-CD3 plus anti-CD28 led to a substantial increase in PKB activation relative to untreated cells (NT). Activated PKB was detected as early as 10 min after TCR ligation and continued to remain high after 6 h (data not shown). In addition, the failure of these stimulatory antibodies to activate PKB in cells pretreated with the PI3K inhibitor wortmannin 49 indicated that TCR-mediated activation of PKB was dependent on PI3K activity. These results establish that PKB represents a physiologically relevant target downstream of the TCR in primary T cells.
Figure 7.
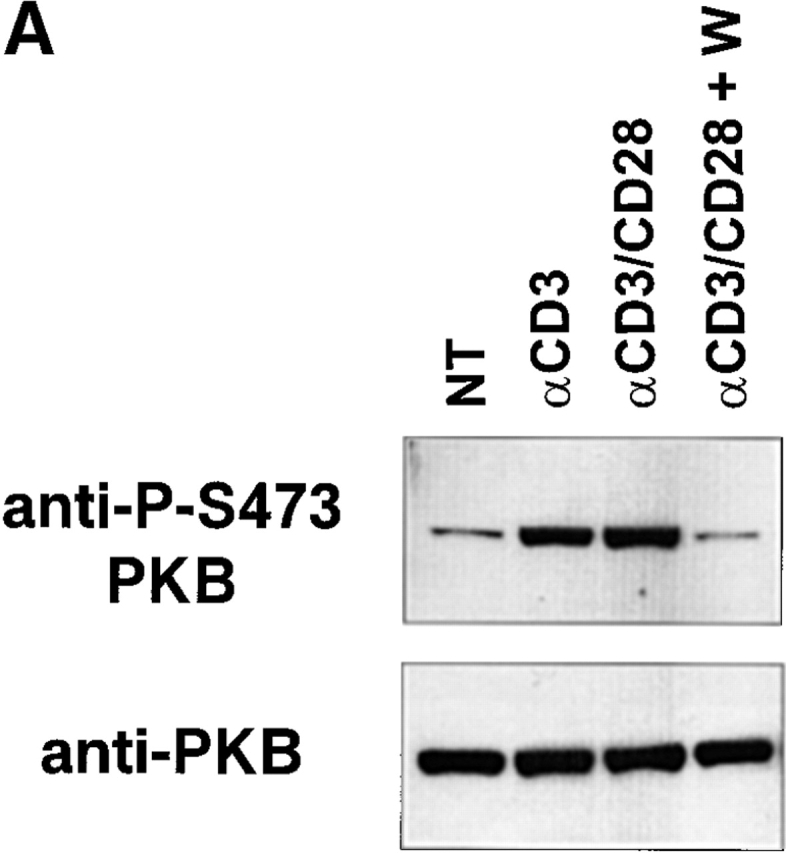

PKB is activated after TCR ligation and enhances antigen-specific proliferation. (A) PKB is activated in T cells in a TCR- and PI3K-dependent fashion. Purified T cells (5 × 106) from C57BL/6J animals were left untreated (NT) or stimulated with 10 μg/ml anti-CD3 antibody (αCD3), or anti-CD3 plus 2 μg/ml anti-CD28 antibody for 2 h at 37°C. W, pretreatment of cells with wortmannin (100 nM) for 30 min before stimulation. PKB activity was assayed by Western blot with antibodies specific for PKB phosphorylated on Ser-473. Endogenous PKB levels were measured using anti-PKB antibodies. (B) P14 TCR transgenic thymocytes expressing gag-PKB display enhanced antigen-specific proliferation over time. Splenocytes from P14 TCR transgenic (P14, •) or P14 TCR/gag-PKB double transgenic (P14/PKB, ○) animals were cultured for up to 5 d in the presence of APCs pre-pulsed with 10−5 M p33 peptide, and pulsed with [3H]thymidine at the indicated time points. To standardize for different experiments, scintillation counts obtained were normalized and expressed as a percentage of maximal proliferation (where maximal proliferation equals 100%). Mean proliferative values for each time point are indicated.
To investigate the functional consequences of PKB activation downstream of the TCR, we analyzed antigen-specific T cell proliferation using TCR transgenic mice. Splenocytes from P14 TCR transgenic and P14/PKB double transgenic animals were cultured in the presence of APCs preincubated with p33 peptide, and proliferation was assessed over time. While levels of proliferation remained similar in both groups at 48 h, T cells expressing the gag-PKB transgene displayed enhanced proliferation relative to P14 control lymphocytes up to 120 h after activation (Fig. 7 B). Proliferation against an irrelevant control peptide (adenovirus peptide AV) was <4% of maximal proliferation for all time points, regardless of gag-PKB transgene expression (data not shown). These data suggest that PKB activity can alter antigen-specific T cell proliferation.
Active PKB Leads to Increased Bcl-XL Levels but Not Phosphorylated BAD in Mature T Cells.
Expression of the antiapoptotic proteins Bcl-2 and Bcl-XL is tightly regulated in mature T cells. Bcl-2 expression is high in mature peripheral T cells and remains high after activation 50, whereas Bcl-XL is upregulated only upon activation and has been proposed to confer transient survival to activated T cells 6. As transgenic thymocytes expressing gag-PKB upregulate Bcl-XL protein levels, we investigated whether PKB activity could promote discordant expression of Bcl-XL in mature T cells. Purified T lymphocytes from B6/PKB and B6 control mice were lysed and Bcl-XL and Bcl-2 protein levels were examined by Western blot (Fig. 8 A). Endogenous levels of Bcl-XL were increased in transgenic mice (B6/PKB) relative to negative littermate control mice (B6) (Fig. 8, top), although not as dramatically as the levels observed in thymocytes. There was no difference in Bcl-2 protein levels in the T cells of these mice (Fig. 8, middle), suggesting that PKB does not affect Bcl-2 expression in mature T cells. These results indicate that PKB influences the level of Bcl-XL protein in mature T cells.
Figure 8.
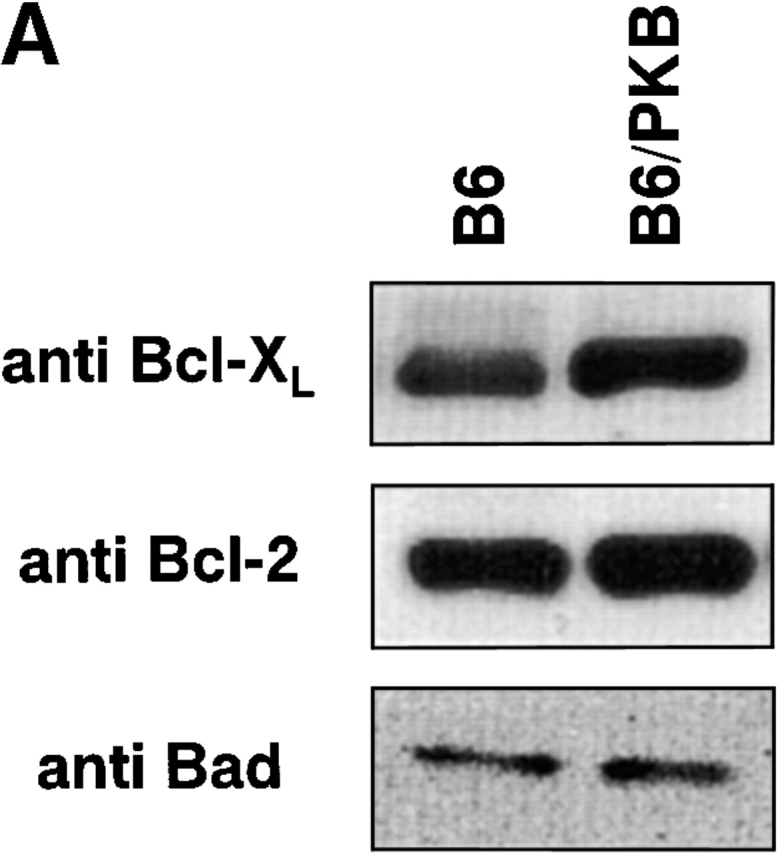


Mediators of survival in gag-PKB transgenic T cells. (A) Increased Bcl-XL levels, but no phosphorylated BAD, can be detected in gag-PKB transgenic T cells. Cell lysates from transgenic (B6/PKB) or nontransgenic (B6) T cells were resolved by 10% SDS-PAGE and analyzed by Western blot. Cell lysates from 5 × 106 thymocytes were probed with antibodies specific for Bcl-XL (top) and Bcl-2 (middle). Lysates from 2 × 107 thymocytes were probed with antibodies against BAD (bottom). (B) PKB activity enhances NF-κB activation in T cells. Primary T cells isolated from LNs and spleens of gag-PKB transgenic (B6/PKB) and control (B6) mice were left untreated (NT) or treated with the indicated stimuli for 4 h. Nuclear extracts were incubated with a radiolabeled probe containing NF-κB binding sites, and NF-κB activation was determined using a gel mobility shift assay as described in Materials and Methods. Bands corresponding to specific NF-κB–DNA complexes and nonspecific binding are indicated. (C) PKB activity enhances IκBα degradation in response to TCR stimulation. T cells purified from gag-PKB transgenic (B6/PKB) or control (B6) mice were pretreated with 50 μg/ml cyclohexamide for 15 min, followed by stimulation with 10 μg/ml anti-CD3 and 2 μg/ml anti-CD28 antibodies for the times indicated. Whole cell lysates were analyzed for IκBα protein levels by Western blot. The blots were stripped and probed with an antibody to β-actin to assess for equal protein loading. This figure is representative of two independent experiments.
Recent studies have identified BAD, a proapoptotic molecule of the Bcl-2 family, as being an important mediator of cell death 15 19 20. PKB has been shown to promote survival by directly phosphorylating BAD at Ser-136, which favors the dissociation of BAD from Bcl-2/Bcl-XL and permits its association with 14-3-3 51. Thus, PKB activity is hypothesized to antagonize BAD-mediated repression of Bcl-2 and Bcl-XL. To determine the effect of PKB kinase activity on BAD phosphorylation in T lymphocytes, we examined endogenous levels of BAD in gag-PKB transgenic and nontransgenic T cells via Western blot analysis (Fig. 8 A, bottom). BAD was detected in low amounts in samples containing 2 × 107 T cells, consistent with findings that BAD is not highly expressed in resting T cells 15. Previous studies have indicated that hyperphosphorylation of BAD causes a decrease in mobility, and that phosphorylated BAD can be detected as a protein doublet via Western blot 51. Endogenous BAD was detected as a single BAD protein band in both transgenic (B6/PKB) and nontransgenic (B6) T cells (Fig. 8 A, bottom). These data suggest that endogenous BAD is not normally phosphorylated in T cells, and that expression of an activated allele of PKB does not lead to phosphorylation of BAD in vivo.
PKB Activity Induces NF-κB Activation and IκB Degradation in Primary T Cells.
Recent studies have shown that PKB can activate the transcription factor NF-κB and promote survival 26 27 28. To determine whether PKB kinase activity influences survival via NF-κB activity in T cells, the following experiments were done. T cells from gag-PKB transgenic (B6/PKB) or nontransgenic (B6) littermates were treated with various stimuli and NF-κB activation was determined by gel mobility shift assays (Fig. 8 B). Nuclear NF-κB was absent in untreated T cells (NT) from both sets of mice, suggesting that expression of activated PKB itself was insufficient to induce NF-κB activation (Fig. 8 B). By contrast, nuclear levels of NF-κB site binding complexes were elevated dramatically in gag-PKB transgenic T cells relative to controls when stimulated with antibodies against CD3 and CD28, or PMA and ionomycin. Interestingly, significant levels of NF-κB activation were also detected when gag-PKB transgenic T cells were treated with anti-CD28 antibodies alone. These data indicate that PKB may enhance NF-κB–mediated gene transcription driven by costimulatory signals through CD28. More importantly, these data demonstrate that activated PKB can facilitate the signaling pathway downstream of the TCR that leads to NF-κB activation.
NF-κB activity is normally inhibited in resting T cells through sequestration by inhibitory proteins known as IκBs. After T cell activation, IκB proteins are rapidly degraded through ubiquitin-mediated proteolysis, permitting nuclear translocation of NF-κB and induction of gene transcription 52. To investigate the mechanism of PKB-mediated NF-κB activation in T cells, we examined the kinetics of IκBα degradation via Western blot. T cells from transgenic (B6/PKB) and nontransgenic (B6) animals were treated with cyclohexamide to prevent NF-κB–mediated reexpression of IκB, and then stimulated with antibodies specific for CD3 and CD28. As seen in Fig. 8 C, T cells expressing the gag-PKB transgene displayed accelerated loss of IκB over time. Significant decreases in IκBα were observed 1 h after stimulation, with complete loss of IκBα protein at 2 h. By contrast, nontransgenic T cells maintained high levels of IκBα at 1 h, and only began to show a decrease in IκBα levels 2 h after activation. Collectively, these results clearly demonstrate that PKB activity can increase NF-κB activation in stimulated T cells, and that this regulation is promoted at the checkpoint of IκBα degradation.
Discussion
PKB Inhibits T Cell Apoptosis: Association with Bcl-XL.
Recent work suggests that mammalian cells have developed several different apoptotic pathways that become activated in a cell type– and stimulus-dependent fashion. Our studies demonstrate that one of the mechanisms by which PKB antagonizes apoptosis involves the antiapoptotic molecule Bcl-XL. Previous studies have shown that Bcl-XL can suppress different pathways of apoptotic death in T lymphocytes. Overexpression of Bcl-XL prevented dexamethasone- and γ irradiation–induced death in thymocytes and T cells 12 13. Apoptotic signaling downstream of Fas can be blocked in T cells by Bcl-XL in some models 6 53 54 but not in other lymphoid cell lines 55 56 57, indicating that the ability of Bcl-XL to block Fas-mediated cell death may be restricted to certain cell types.
We have observed that T lymphocytes expressing active PKB exhibit significantly increased rates of survival after induction of DNA damage (γ irradiation) or steroid-induced apoptosis (dexamethasone). Immature thymocytes expressing gag-PKB also show increased resistance to apoptosis through stimulation of the Fas pathway. These data are consistent with recent findings showing impaired Fas-mediated apoptosis when COS7 cells are cotransfected with gag-PKB and Fas constructs 42. Further evidence suggests that the mechanism of PKB-mediated survival overlaps with Bcl-XL pathways, as transgenic expression of both gag-PKB and Bcl-XL enhances the survival of DP thymocytes 48. Accordingly, increased levels of Bcl-XL protein were observed in gag-PKB transgenic mice. Collectively, the gag-PKB mouse phenotype resembles that of Bcl-XL transgenic mice 12 13 and contrasts that of Bcl-X gene–deficient mice 47 48, suggesting that these two molecules work in concert to oppose apoptosis in T cells.
Role of PKB during Thymocyte Selection.
Current research suggests the importance of two survival signals during thymocyte development: a basal survival signal mediated by Bcl-XL that maintains the CD4+CD8+ DP thymocyte pool 47 48, and a second signal induced upon receipt of positive selection signals through the TCR that results in the upregulation of Bcl-2 45 58. In our studies, DP thymocytes expressing the gag-PKB transgene displayed a selective survival advantage over nontransgenic controls. In this respect, activated PKB may enhance Bcl-XL levels to ensure the survival of DP thymocytes that audition for positive or negative selection. Although it is well documented that PKB acts downstream of PI3K, the signals that activate PI3K during thymocyte development remain unknown.
Our studies have also shown that activated PKB is unable to block antigen-induced negative selection. This is consistent with past work that has demonstrated that Bcl-XL does not prevent self-antigen–induced clonal deletion 13. Together, these results demonstrate that PKB kinase activity acts at the level of DP thymocyte survival, and that PI3K/PKB signaling does not play a role in the apoptotic pathway activated by TCR-mediated negative selection signals.
Mechanisms of PKB-mediated Survival: The Good and the BAD.
PKB may elicit its protective phenotype in T lymphocytes through a variety of mechanisms. We have demonstrated a novel correlation between PKB activity and Bcl-XL levels, and suggest that PKB promotes T cell survival through enhanced Bcl-XL expression. In other studies, Bcl-XL has been shown to interact with caspase 9 and apoptotic protease-activating factor 1 (Apaf1), resulting in the inhibition of caspase activation 59 60. Increased levels of Bcl-XL induced by PKB kinase activity may form inhibitory complexes with caspase 9, Apaf1, and cytochrome c after apoptotic insult, delaying caspase activation and slowing the progression of cell death in T cells. PKB-mediated regulation of Bcl-XL may also antagonize the apoptotic effects of Fas. Ligation of Fas by its cognate ligand or through antibody stimulation results in the recruitment of several proteins to form the death-inducing signaling complex 61, and leads to caspase 8 activation 62. Bcl-XL has been shown to act downstream of death-inducing signaling complex formation and caspase 8 activation to impede Fas-mediated death signals 54. This may be one way that PKB acts to antagonize Fas-mediated death signals in thymocytes.
PI3K activity has been shown to induce Bcl-XL expression at both the mRNA and protein levels in Baf-3 cells 63, implying that the PI3K/PKB pathway may regulate transcription of this antiapoptotic molecule. However, PKB kinase activity does not regulate expression of Bcl-XL at the transcriptional level in our transgenic mouse model. Sequence analysis confirms that Bcl-XL lacks traditional PKB phosphorylation consensus sites (RXRXXS/T) 64, suggesting that PKB is unlikely to regulate Bcl-XL by direct phosphorylation. However, relatively few downstream substrates of PKB have been identified to date, giving rise to the possibility of intermediary molecules that control PKB-mediated Bcl-XL expression.
To date, one of the links between PKB and the cellular apoptotic machinery has been through the ability of PKB to phosphorylate the proapoptotic molecule BAD 19 20. Work by Mok et al. 15 indicates that transgenic overexpression of BAD in thymocytes renders them hypersensitive towards dexamethasone, γ radiation, and Fas-induced death, possibly due to sequestration of endogenous Bcl-XL. We show that BAD expression in normal T lymphocytes is extremely low, and in contrast to other studies, our data suggest that BAD is not phosphorylated in vivo by activated PKB. This is in accordance with other models suggesting that PKB-mediated phosphorylation of BAD is not the only pathway by which PKB counters apoptotic death 65 66. BAD is not ubiquitously expressed, and cells that do not express BAD are still protected from apoptosis via PKB signaling 67. This appears to be the case in primary T cells; past results show that BAD is not highly expressed in T cells and induced only in response to apoptotic stimuli 15. The effector molecules that promote survival downstream of PKB signals are likely cell and stimulus specific. Taking these findings into account, it is unlikely that BAD functions as a relevant physiological substrate for PKB in T cells.
Integration of PKB Signaling with the NF-κB Pathway.
Another key pathway associated with cell survival is signaling via the transcription factor NF-κB 68. In this study, we provide the first evidence that PKB can be activated by TCR cross-linking in mature T cells and that PKB influences NF-κB activation in primary T cells in response to TCR stimulation. In T lymphocytes, NF-κB activation is induced through engagement of the TCR 69. Mobility shift assays performed using nuclear fractions from T cells treated with anti-CD3 and anti-CD28 antibodies clearly demonstrated that NF-κB activity was significantly increased in T cells expressing gag-PKB, indicating that active PKB mediates this effect in vivo. Interestingly, nuclear translocation of NF-κB was induced in transgenic T cells through CD28 stimulation alone. This evidence, coupled with the fact that PKB is activated downstream of CD28 in T lymphocytes 70, implicates PKB as a mediator of NF-κB activation downstream of CD28 costimulation. Whereas CD3-mediated NF-κB activation is low in gag-PKB transgenic T cells, stimulation with anti-CD3 and anti-CD28 antibodies leads to a synergistic increase in NF-κB nuclear translocation that is greater than that observed with either treatment alone. Past studies indicate that costimulatory signals can enhance TCR-mediated NF-κB activation 71, and our data suggest that PKB plays a key role in this process.
PKB-mediated regulation of NF-κB in T cells can be influenced by IκB degradation 52. The kinetics of IκBα degradation were accelerated in gag-PKB transgenic mice relative to controls, suggesting that PKB kinase activity influences NF-κB nuclear translocation by regulating levels of IκB. Recent reports have suggested that PKB can phosphorylate one of the regulatory IκB kinases, IKKα, through phosphorylation at a PKB phosphorylation consensus sequence (Thr-23) located at its NH2 terminus 27. Moreover, PKB-mediated NF-κB activity is impaired in Jurkat cells cotransfected with PKB and kinase-deficient IKK molecules 28. Thus, in our model, PKB may promote enhanced NF-κB transcriptional activity in response to TCR triggering and costimulation through the activation of IKK. It is important to note that the expression of active PKB does not lead to increased basal levels of NF-κB activity, consistent with similar results observed in Jurkat cells 28. This points to a mechanism by which PKB does not act alone, but requires the activation of other molecules downstream of TCR-mediated signals to promote IKK activation, IκB degradation, and enhanced NF-κB activity.
The ability of PKB to induce activation of NF-κB adds an additional level of complexity to its function as a cytoprotective agent. Our data, and those of others, suggest that PKB may not simply function as an antagonist of proapoptotic molecules; rather, PKB may promote cell survival, including protection from apoptotic stimuli such as Fas ligand or γ-irradiation, through the induced expression of NF-κB–regulated survival genes such as c-IAP1 72, c-IAP2 73, or Bfl-1/A1 74. Previous work suggests that PI3K/PKB regulates survival of Baf-3 cells via Bcl-XL 63. Indeed, Bcl-XL has been identified as an NF-κB target gene, but the exact role NF-κB plays in its regulation remains controversial. NF-κB has been shown to prevent Fas-induced death in B cells through the upregulation of Bcl-XL 75, but has also been demonstrated to promote apoptosis in thymocytes by downregulating Bcl-XL expression 76. Exactly how PKB-mediated NF-κB activation functions in T cells remains to be elucidated.
Concluding Remarks.
Expression of active PKB in T lymphocytes has provided key insights into the regulation of T cell survival. It has provided an important link to a previously undefined signaling pathway regulating Bcl-XL expression. In addition, experiments with mature T cells demonstrate that T cell activation via the TCR and costimulatory molecules enhances T cell survival via PKB and NF-κB. Our findings demonstrate that PKB-mediated survival is multifaceted; it is apparent that PKB signaling exerts its antiapoptotic effects in T lymphocytes through a number of ways. However, more research is needed to fully understand the intricate regulation of T cell survival versus apoptosis and the role that PKB plays in this process.
Acknowledgments
We would like to thank Ann Marie Walsh for generation of transgenic animals. We thank Drs. Mark Bray and Sudha Arya for supplying recombinant hCD8-mFasL fusion protein and anti-CD3 antibody, Dr. David Andrews for his generous gift of anti–Bcl-2 antibody, Arsen Zakarian for technical assistance, and Dr. Razqallah Hakem for expert advice.
This work was supported by the Medical Research Council of Canada (to P.S. Ohashi and J.R. Woodgett). R.G. Jones was supported by the Natural Science and Engineering Council of Canada and the Medical Research Council. J.R. Woodgett is a Medical Research Council Senior Scientist and an International Scholar of the Howard Hughes Medical Institute. P.S. Ohashi is a Medical Research Council Scientist.
Footnotes
Abbreviations used in this paper: 7AAD, 7-amino-actinomycin D; DISC, death-inducing signaling complex; DP, double positive; FTOC, fetal thymic organ culture; HSA, heat-stable antigen; LCMV, lymphocytic choriomeningitis virus; NF, nuclear factor; PI, propidium iodide; PI3K, phosphatidylinositol 3-kinase; PKB, protein kinase B; RT, reverse transcription; SP, single positive.
R.G. Jones and M. Parsons contributed equally to this work.
References
- Sebzda E., Mariathasan S., Ohteki T., Jones R., Bachmann M.F., Ohashi P.S. Selection of the T cell repertoire. Annu. Rev. Immunol. 1999;17:829–874. doi: 10.1146/annurev.immunol.17.1.829. [DOI] [PubMed] [Google Scholar]
- Takeda S., Rodewald H.-R., Arakawa H., Bluethmann H., Shimizu T. MHC class II molecules are not required for survival of newly generated CD4+ T cells, but affect their long-term life span. Immunity. 1996;5:217–228. doi: 10.1016/s1074-7613(00)80317-9. [DOI] [PubMed] [Google Scholar]
- Tanchot C., Lemonnier F.A., Pérarnau B., Freitas A.A., Rocha B. Differential requirements for survival and proliferation of CD8 naive or memory T cells. Science. 1997;276:2057–2062. doi: 10.1126/science.276.5321.2057. [DOI] [PubMed] [Google Scholar]
- Kirberg J., Berns A., von Boehmer H. Peripheral T cell survival requires continual ligation of the T cell receptor to major histocompatibility complex–encoded molecules. J. Exp. Med. 1997;186:1269–1275. doi: 10.1084/jem.186.8.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenardo M.J., Chan F.K.M., Hornung F., McFarland H., Siegel R., Wang J., Zheng L. Mature T lymphocyte apoptosis-immune regulation in a dynamic and unpredictable environment. Annu. Rev. Immunol. 1999;17:221–253. doi: 10.1146/annurev.immunol.17.1.221. [DOI] [PubMed] [Google Scholar]
- Boise L.H., Minn A.J., Noel P.J., June C.H., Accavitti M.A., Lindsten T., Thompson C.B. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-xL . Immunity. 1995;3:87–98. doi: 10.1016/1074-7613(95)90161-2. [DOI] [PubMed] [Google Scholar]
- Radvanyi L., Shi Y., Vaziri H., Sharma A., Dhala R., Mills G.B., Miller R.G. CD28 costimulation inhibits TCR-induced apoptosis during a primary T cell response. J. Immunol. 1996;156:1788–1798. [PubMed] [Google Scholar]
- Vella A.T., Mitchell T., Groth B., Linsley P.S., Green J.M., Thompson C.B., Kappler J.W., Marrack P. CD28 engagement and proinflammatory cytokines contribute to T cell expansion and long-term survival in vivo. J. Immunol. 1997;158:4714–4720. [PubMed] [Google Scholar]
- Rathmell J.C., Thompson C.B. The central effectors of cell death in the immune system. Annu. Rev. Immunol. 1999;17:781–828. doi: 10.1146/annurev.immunol.17.1.781. [DOI] [PubMed] [Google Scholar]
- Sentman C.L., Shutter J.R., Hockenbery D., Kanagawa O., Korsmeyer S.J. Bcl-2 inhibits multiple forms of apoptosis but not negative selection in thymocytes. Cell. 1991;67:879–888. doi: 10.1016/0092-8674(91)90361-2. [DOI] [PubMed] [Google Scholar]
- Strasser A., Harris A.W., Cory S. Bcl-2 transgene inhibits T cell death and perturbs thymic self-censorship. Cell. 1991;67:889–899. doi: 10.1016/0092-8674(91)90362-3. [DOI] [PubMed] [Google Scholar]
- Chao D.T., Linette G.P., Boise L.H., White L.S., Thompson C.B. Bcl-XL and Bcl-2 repress a common pathway of cell death. J. Exp. Med. 1995;182:821–828. doi: 10.1084/jem.182.3.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillot D.A., Merino R., Nunez G. Bcl-XL displays restricted distribution during T cell development and inhibits multiple forms of apoptosis but not clonal deletion in transgenic mice. J. Exp. Med. 1995;182:1973–1983. doi: 10.1084/jem.182.6.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams O., Norton T., Halligey M., Kioussis D., Brady H.J.M. The action of Bax and Bcl-2 on T cell selection. J. Exp. Med. 1998;188:1125–1133. doi: 10.1084/jem.188.6.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok C., Gil-Gomez G., Williams O., Coles M., Taga S., Tolaini M., Norton T., Kioussis D., Brady H.J.M. Bad can act as a key regulator of T cell apoptosis and T cell development. J. Exp. Med. 1999;189:575–586. doi: 10.1084/jem.189.3.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffer P.J., Jin J., Woodgett J.R. Protein kinase B (c-Akt)a multifunctional mediator of phosphatidylinositol 3-kinase activation. Biochem. J. 1998;335:1–13. doi: 10.1042/bj3350001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S.R., Brunet A., Greenberg M.E. Cellular survivala play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- Alessi D.R., Cohen P. Mechanism of activation and function of protein kinase B. Curr. Opin. Gen. Dev. 1998;8:55–62. doi: 10.1016/s0959-437x(98)80062-2. [DOI] [PubMed] [Google Scholar]
- Datta S.R., Dudek H., Tao X., Masters S., Fu H., Gotoh Y., Greenberg M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- del Peso L., Gonzalez-Garcia M., Page C., Herrera R., Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- Cardone M.H., Roy N., Stennicke H.R., Salvesen G.S., Franke T.F., Stanbridge E., Frisch S., Reed J.C. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- Paradis S., Ruvkun G. Caenorhabitis elegans Akt/PKB transduces insulin receptor-like signals from AGE-1 PI3 kinase to the DAF-16 transcription factor. Genes Dev. 1998;12:2488–2498. doi: 10.1101/gad.12.16.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A., Bonni A., Zigmond M.J., Lin M.Z., Juo P., Hu L.S., Anderson M.J., Arden K.C., Blenis J., Greenberg M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Biggs W.H., III, Meisenhelder J., Hunter T., Cavenee W.K., Arden K.C. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc. Natl. Acad. Sci. USA. 1999;96:7421–7426. doi: 10.1073/pnas.96.13.7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops G.J.P., de Ruiter N.D., de Vries-Smits A.M.M., Powell D.R., Bos J.L., Burgering B.M. Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature. 1999;398:630–634. doi: 10.1038/19328. [DOI] [PubMed] [Google Scholar]
- Romashkova J.A., Makarov S.S. NF-κB is a target of Akt in anti-apoptotic PDGF signalling. Nature. 1999;401:86–89. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
- Ozes O.N., Mayo L.D., Gustin J.A., Pfeffer S.R., Pfeffer L.M., Donner D.B. NF-κB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- Kane L.P., Shapiro V.S., Stokoe D., Weiss A. Induction of NF-κB by the Akt/PKB kinase. Curr. Biol. 1999;9:601–604. doi: 10.1016/s0960-9822(99)80265-6. [DOI] [PubMed] [Google Scholar]
- Burgering B.M., Coffer P.J. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature. 1995;376:599–602. doi: 10.1038/376599a0. [DOI] [PubMed] [Google Scholar]
- Zhumabekov T., Corbella P., Tolaini M., Kioussis D. Improved version of a human CD2 minigene based vector for T cell-specific expression in transgenic mice. J. Immunol. Methods. 1995;185:133–140. doi: 10.1016/0022-1759(95)00124-s. [DOI] [PubMed] [Google Scholar]
- Pircher H., Bürki K., Lang R., Hengartner H., Zinkernagel R. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature. 1989;342:559–561. doi: 10.1038/342559a0. [DOI] [PubMed] [Google Scholar]
- Kayagaki N., Yamaguchi N., Nagao F., Matsuo S., Maeda H., Okumura K., Yagita H. Polymorphism of murine Fas ligand that affects the biological activity. Proc. Natl. Acad. Sci. USA. 1997;94:3914–3919. doi: 10.1073/pnas.94.8.3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebzda E., Kündig T.M., Thomson C.T., Aoki K., Mak S.Y., Mayer J., Zamborelli T.M., Nathenson S., Ohashi P.S. Mature T cell reactivity altered by a peptide agonist that induces positive selection. J. Exp. Med. 1996;183:1093–1104. doi: 10.1084/jem.183.3.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid I., Uittenbogaart C.H., Giorgi J.V. Sensitive method for measuring apoptosis and cell surface phenotype in human thymocytes by flow cytometry. Cytometry. 1994;15:12–20. doi: 10.1002/cyto.990150104. [DOI] [PubMed] [Google Scholar]
- Pfeffer K., Matsuyama T., Kündig T.M., Wakeham A., Kishihara K., Shahinian A., Wiegmann K., Ohashi P.S., Krönke M., Mak T.W. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell. 1993;73:456–467. doi: 10.1016/0092-8674(93)90134-c. [DOI] [PubMed] [Google Scholar]
- Bellacosa A., Testa J.R., Staal S.P., Tsichlis P.N. A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-like region. Science. 1991;254:274–277. doi: 10.1126/science.254.5029.274. [DOI] [PubMed] [Google Scholar]
- Henderson L.E., Krutzsch H.C., Oroszlan S. Myristyl amino-terminal acylation of murine retrovirus proteinsan unusual post-translational proteins modification. Proc. Natl. Acad. Sci. USA. 1983;80:339–343. doi: 10.1073/pnas.80.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz A.M., Oroszlan S. In vivo modification of retroviral gag gene-encoded polyproteins by myristic acid. J. Virol. 1983;46:355–361. doi: 10.1128/jvi.46.2.355-361.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed N.N., Franke T.F., Bellacosa A., Datta K., Gonzalez-Portal M.E., Taguchi T., Testa J.R., Tsichlis P.N. The proteins encoded by c-akt and v-akt differ in post-translational modification, subcellular localization and oncogenic potential. Oncogene. 1993;8:1957–1963. [PubMed] [Google Scholar]
- Staal S.P., Hartley J.W. Thymic lymphoma induction by the AKT8 murine retrovirus. J. Exp. Med. 1988;167:1259–1264. doi: 10.1084/jem.167.3.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambolic V., Suzuki A., de la Pompa J.L., Brothers G.M., Mirtsos C., Sasaki T., Ruland J., Penninger J.M., Siderovski D.P., Mak T.W. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- Hausler P., Papoff G., Eramo A., Reif K., Cantrell D., Ruberti G. Protection of CD95-mediated apoptosis by activation of phosphatidylinositide 3-kinase and protein kinase B. Eur. J. Immunol. 1998;28:57–69. doi: 10.1002/(SICI)1521-4141(199801)28:01<57::AID-IMMU57>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Konvalinka J., Heuser A.M., Hruskova-Heidingsfeldova O., Vogt V.M., Sedlacek J., Strop P., Krausslich H.G. Proteolytic processing of particle-associated retroviral polyproteins by homologous and heterologous viral proteinases. Eur. J. Biochem. 1995;228:191–198. doi: 10.1111/j.1432-1033.1995.tb20249.x. [DOI] [PubMed] [Google Scholar]
- Sebzda E., Wallace V.A., Mayer J., Yeung R.S.M., Mak T.W., Ohashi P.S. Positive and negative thymocyte selection induced by different concentrations of a single peptide. Science. 1994;263:1615–1618. doi: 10.1126/science.8128249. [DOI] [PubMed] [Google Scholar]
- Linette G.P., Grusby M.J., Hedrick S.M., Hansen T.H., Glimcher L.H., Korsmeyer S.J. BcI-2 is upregulated at the CD4+ CD8+ stage during positive selection and promotes thymocyte differentiation at several control points. Immunity. 1994;1:197–205. doi: 10.1016/1074-7613(94)90098-1. [DOI] [PubMed] [Google Scholar]
- Tao W., Teh S.-J., Melhado I., Jirik F., Korsmeyer S.J., Teh H.-S. The T cell receptor repertoire of CD4-8+ thymocytes is altered by overexpression of the BCL-2 protooncogene in the thymus. J. Exp. Med. 1994;179:145–153. doi: 10.1084/jem.179.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motoyama N., Wang F., Roth K.A., Sawa H., Nakayama K., Negishi I., Senju S., Zhang Q., Fujii S., Loh D.Y. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science. 1995;267:1506–1510. doi: 10.1126/science.7878471. [DOI] [PubMed] [Google Scholar]
- Ma A., Pena J.C., Chang B., Margosian E., Davidson L., Alt F.W., Thompson C.B. Bclx regulates the survival of double-positive thymocytes. Proc. Natl. Acad. Sci. USA. 1995;92:4763–4767. doi: 10.1073/pnas.92.11.4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada T., Sakuma L., Fukui Y., Hazeki O., Ui M. Blockage of chemotactic peptide-induced stimulation of neutrophils by wortmannin as a result of selective inhibition of phosphatidylinositol 3-kinase. J. Biol. Chem. 1994;269:3563–3567. [PubMed] [Google Scholar]
- Veis D.J., Sentman C.L., Bach E.A., Korsmeyer S.J. Expression of the bcl-2 protein in murine and human thymocytes and in peripheral T lymphocytes. J. Immunol. 1993;151:2546–2554. [PubMed] [Google Scholar]
- Zha J., Harada H., Yang E., Jockel J., Korsmeyer S.J. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-XL . Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- Baldwin A.S., Jr. The NF-κB and IκB proteinsnew discoveries and insights. Annu. Rev. Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- Peter M.E., Kischkel F.C., Scheuerpflug C.G., Medema J.P., Debatin K.-M., Krammer P.H. Resistance of cultured peripheral T cells towards activation-induced cell death involves a lack of recruitment of FLICE (MACH/caspase 8) to the CD95 death-inducing signaling complex. Eur. J. Immunol. 1997;27:1207–1212. doi: 10.1002/eji.1830270523. [DOI] [PubMed] [Google Scholar]
- Medema J.P., Scaffidi C., Krammer P.H., Peter M.E. Bcl-xL acts downstream of caspase-8 activation by the CD95 death-inducing signaling complex. J. Biol. Chem. 1998;273:3388–3393. doi: 10.1074/jbc.273.6.3388. [DOI] [PubMed] [Google Scholar]
- Strasser A., Harris A.W., Huang D.C.S., Krammer P.H., Cory S. Bcl-2 and Fas/APO-1 regulate distinct pathways to lymphocyte apoptosis. EMBO (Eur. Mol. Biol. Organ.) J. 1995;14:6136–6147. doi: 10.1002/j.1460-2075.1995.tb00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi C., Fulda S., Srinivasan A., Friesen C., Li F., Tomaselli K.J., Debatin K.-M., Krammer P.H., Peter M.E. Two CD95 (APO-1/Fas) signaling pathways. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:1675–1687. doi: 10.1093/emboj/17.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi C., Schmitz I., Zha J., Korsmeyer S.J., Krammer P.H., Peter M.E. Differential modulation of apoptosis sensitivity in CD95 type I and type II cells. J. Biol. Chem. 1999;274:22532–22538. doi: 10.1074/jbc.274.32.22532. [DOI] [PubMed] [Google Scholar]
- Gratiot-Deans J., Merino R., Nunez G., Turka L.A. Bcl-2 expression during T-cell developmentearly loss and late return occur at specific stages of commitment to differentiation and survival. Proc. Natl. Acad. Sci. USA. 1994;91:10685–10689. doi: 10.1073/pnas.91.22.10685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y., Benedict M.A., Wu D., Inohara N., Nunez G. Bcl-XL interacts with Apaf-1 and inhibits Apaf-1-dependent caspase-9 activation. Proc. Natl. Acad. Sci. USA. 1998;95:4386–4391. doi: 10.1073/pnas.95.8.4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan G., O'Rourke K., Dixit V.M. Caspase-9, Bcl-XL, and Apaf-1 form a ternary complex. J. Biol. Chem. 1998;273:5841–5845. doi: 10.1074/jbc.273.10.5841. [DOI] [PubMed] [Google Scholar]
- Muzio M., Chinnaiyan A.M., Koschkel F.C., O'Rourke K., Shevchenko A., Ni J., Scaffidi C., Bretz J.D., Zhang M., Gentz R. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- Kischkel F.C., Hellbardt S., Behrmann I., Germer M., Pawlita M., Krammer P.H., Peter M.E. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO (Eur. Mol. Biol. Organ.) J. 1995;14:5579–5588. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leverrier Y., Thomas J., Mathieu A., Low W., Blanquier B., Marvel J. Role of P13-kinase in Bcl-X induction and apoptosis inhibition mediated by IL-3 or IGF-1 in Baf-3 cells. Cell Death Differ. 1999;6:290–296. doi: 10.1038/sj.cdd.4400492. [DOI] [PubMed] [Google Scholar]
- Alessi D.R., Caudwell F.B., Andjelkovic M., Hemmings B.A., Cohen P. Molecular basis for the substrate specificity of protein kinase B; comparison with MAPKAP kinase-1 and p70 S6 kinase. FEBS Lett. 1996;399:333–338. doi: 10.1016/s0014-5793(96)01370-1. [DOI] [PubMed] [Google Scholar]
- Downward J. How BAD phosphorylation is good for survival. Nat. Cell Biol. 1999;1:E33–E35. doi: 10.1038/10026. [DOI] [PubMed] [Google Scholar]
- Khwaja A. Akt is more than just a Bad kinase. Nature. 1999;401:33–34. doi: 10.1038/43354. [DOI] [PubMed] [Google Scholar]
- Kitada S., Krajewska M., Zhang X., Scudiero D., Zapata J.M., Wang H.G., Shabiak A., Tudor G., Krajewski S., Myers T.G. Expression and location of pro-apoptotic Bcl-2 family protein BAD in normal human tissues and tumor cell lines. Am. J. Pathol. 1998;152:51–61. [PMC free article] [PubMed] [Google Scholar]
- Sonenshein G.E. Rel/NF-κB transcription factors and the control of apoptosis. Semin. Cancer Biol. 1997;8:113–119. doi: 10.1006/scbi.1997.0062. [DOI] [PubMed] [Google Scholar]
- Jamieson C., McCaffrey P.G., Roa A., Sen R. Physiologic activation of T cells via the T cell receptor induces NF-kappa B. J. Immunol. 1991;147:416–420. [PubMed] [Google Scholar]
- Parry R.V., Reif K., Smith G., Sanson D.M., Hemmings B.A., Ward S.G. Ligation of the T cell co-stimulatory receptor CD28 activates the serine-threonine protein kinase protein kinase B. Eur. J. Immunol. 1997;27:2495–2501. doi: 10.1002/eji.1830271006. [DOI] [PubMed] [Google Scholar]
- Costello R., Lipcey C., Algarte M., Cerdan C., Baeuerle P.A., Olive D., Imbert J. Activation of primary human T-lymphocytes through CD2 plus CD28 molecules induces long-term nuclear expression of NF-kappa B. Cell Growth Differ. 1993;1993:329–339. [PubMed] [Google Scholar]
- Chu Z.L., McKinsey T.A., Liu L., Gentry J.J., Malim M.H., Ballard D.W. Suppression of tumor necrosis factor-induced cell death by inhibitor of apoptosis c-IAP2 is under NF-kappaB control. Proc. Natl. Acad. Sci. USA. 1997;94:10057–10062. doi: 10.1073/pnas.94.19.10057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.Y., Mayo M.W., Korneluk R.G., Goeddel D.V., Baldwin A.S., Jr. NF-kappaB antiapoptosisinduction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- Zong W.-X., Edelstein L.C., Chen C., Bash J., Gelinas C. The prosurvival Bcl-2 homolog Bfl-1/A1 is a direct transcriptional target of NF-κB that blocks TNFα-induced apoptosis. Genes Dev. 1999;13:382–387. doi: 10.1101/gad.13.4.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.H., Dadgostar H., Cheng Q., Shu J., Cheng G. NF-κB-mediated up-regulation of Bcl-x and Bfl-1/A1 is required for CD40 survival signaling in B lymphocytes. Proc. Natl. Acad. Sci. USA. 1999;96:9136–9141. doi: 10.1073/pnas.96.16.9136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hettmann T., DiDonato J., Karin M., Leiden J.M. An essential role for nuclear factor κB in promoting double positive thymocyte apoptosis. J. Exp. Med. 1999;189:145–158. doi: 10.1084/jem.189.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]