

Abstract
Signaling through its widely distributed cell surface receptor, interleukin (IL)-17 enhances the transcription of genes encoding proinflammatory molecules. Although it has been well documented that IL-17 activates the transcription factor nuclear factor (NF)-κB and c-Jun NH2-terminal kinase (JNK), the upstream signaling events are largely unknown. Here we report the requirement of tumor necrosis factor receptor–associated factor (TRAF)6 in IL-17–induced NF-κB and JNK activation. In embryonic fibroblasts (EFs) derived from TRAF6 knockout mice, IL-17 failed to activate the IκB kinases (IKKs) and JNK. Consequently, IL-17–induced IL-6 and intercellular adhesion molecule 1 expression in the TRAF6-deficient cells was abolished. Lack of TRAF6 appeared to be the sole defect responsible for the observed failure to respond to IL-17, because transient transfection of TRAF6 expression plasmid into the TRAF6-deficient cells restored IL-17–induced NF-κB activation in a luciferase reporter assay. Furthermore, the levels of IL-17 receptor (IL-17R) on the TRAF6-deficient EFs were comparable to those on the wild-type control cells. Defect in IL-17 response was not observed in TRAF2-deficient EFs. Moreover, when TRAF6 and IL-17R were coexpressed in 293 cells, TRAF6 coimmunoprecipitated with IL-17R. Together, these results indicate that TRAF6, but not TRAF2, is a crucial component in the IL-17 signaling pathway leading to proinflammatory responses.
Keywords: cytokines, inflammation, signaling, kinases, transcription
Introduction
IL-17, previously termed CTLA-8 (cytotoxic T cell lymphocyte–associated antigen 8; reference 1), is a 20–30-kD protein secreted by activated CD4 cells, mainly Th0 and Th1 cells 2 3. In different cell types, IL-17 induces the expression of a large variety of proinflammatory molecules, including IL-1β, IL-8, inducible nitric oxide synthase, cyclooxigenase-2, and intercellular adhesion molecule (ICAM)-1 3 4 5 6. IL-17 also reportedly induces hematopoietic cytokines such as GM-CSF, leukemia inhibitory factor, and IL-6 7. Thus, IL-17 appears to provide a direct linkage between T cell activation and inflammatory responses. Indeed, IL-17 has been implicated in Th1-mediated inflammatory diseases such as rheumatoid arthritis and organ transplant rejections 7 8 9 10 11.
Like TNF and IL-1, which elicit similar cellular responses, IL-17 also activates the transcription factors nuclear factor (NF)-κB and activator protein (AP)-1, important mediators of gene regulatory activities exhibited by proinflammatory cytokines 6 12 13 14. AP-1 is activated by a phosphorylation event mediated by mitogen-activated protein kinases, including c-Jun NH2-terminal kinase (JNK)-1 15, whereas NF-κB activation is preceded by the activation of IκB kinase-α and -β (IKKs), which are present in most cell types as a heterocomplex 16 17 18 19 20 21. The IKKs phosphorylate two specific serine residues on the IκB proteins, marking them for proteolysis 22 23 24 25 26. The degradation of the inhibitory IκB proteins results in the release and nuclear translocation of the NF-κB proteins, which transcriptionally activate target genes.
TNF and IL-1 signal through different cell surface receptors and utilize different sets of receptor-proximal signaling molecules. TNF induces the trimerization of its type I receptor (the major signaling receptor relative to the type II receptor), which initiates a signaling cascade engaging the adapter TRADD (TNF receptor–associated death domain protein), the serine/threonine kinase RIP (receptor-interacting protein), and TNF receptor–associated factor (TRAF)2 27 28 29. IL-1 induces the heterocomplex formation of two different receptor chains, the type I receptor (IL-1R1) and the receptor accessory protein (IL-1RAcp) 30 31 32. This triggers the signaling events that involve the adapter molecule MyD88, the serine/threonine kinase IRAK (IL-1 receptor–associated kinase), and TRAF6 33 34 35. TRAF2 and TRAF6 mediate the activation of both IKK and JNK 36. Therefore, the TRAF proteins represent a class of adapters that lead distinct upstream signaling pathways for different cytokines to a point of convergence.
The receptor of IL-17 (IL-17R) is a type 1 transmembrane protein with an apparent molecular mass of 130 kD 37. It does not share sequence homology with the receptors of TNF, IL-1, or other cytokines. Little is known about the molecular signaling mechanism of IL-17R. In this study, we used TRAF2- and TRAF6-deficient mouse embryonic fibroblasts (MEFs) to study the role of TRAF molecules in IL-17–induced NF-κB and JNK activation. Our results indicate that TRAF6 is a critical signaling molecule in the IL-17 signaling pathway.
Materials and Methods
Cell Culture and Biological Material.
Primary EFs were gifts from T.W. Mak's laboratory (Amgen Institute/Ontario Cancer Institute, Toronto, Ontario, Canada). The cells were derived from embryos at day 14.5 of gestation as described previously 38 39. EFs and human embryonic kidney (HEK) 293 cells were cultured in high glucose DME (Cellgro) supplemented with 10% certified FBS, 10 mM glutamine, and 50 μg/ml each of streptomycin and penicillin (GIBCO BRL) in a humidified incubator with 5% CO2. Mouse TNF was provided by Genentech; human IL-1β and mouse IL-17 were purchased from Biosource.
Luciferase Reporter Assays.
To reconstitute IL-17 response, TRAF6+/− and TRAF6−/− MEFs were seeded in six-well (35 mm) plates and transfected on the following day with 0.5 μg of luciferase reporter gene controlled by a basic promoter and six NF-κB binding sites, 2 μg of pRSV-β-gal, and 50 ng of either vector DNA or TRAF6 expression plasmid DNA 34 using the Effectene Transfection Reagent (QIAGEN Inc.) according to manufacturer-recommended protocol. 36 h after transfection, the cells were stimulated with TNF (25 g/ml) or IL-17 (100 g/ml) for 8 h. Luciferase activity and β-galactosidase activity was determined with the Luciferase Assay System (Promega Corp.) and chemiluminescent reagents from Tropix, respectively.
Electrophoretic Mobility Assay.
To assay NF-κB–DNA binding activity, 2 × 105 EFs were seeded in 3.5-cm dishes for 24 h. The cells were cultured in serum-free medium for 1 h before the addition of cytokines. At indicated times (see Fig. 1) after cytokine induction, the cells were harvested with PBS containing 1 mM EDTA. Nuclear extract preparation and gel electrophoretic mobility shift assay (EMSA) were performed as described previously 40.
Figure 1.
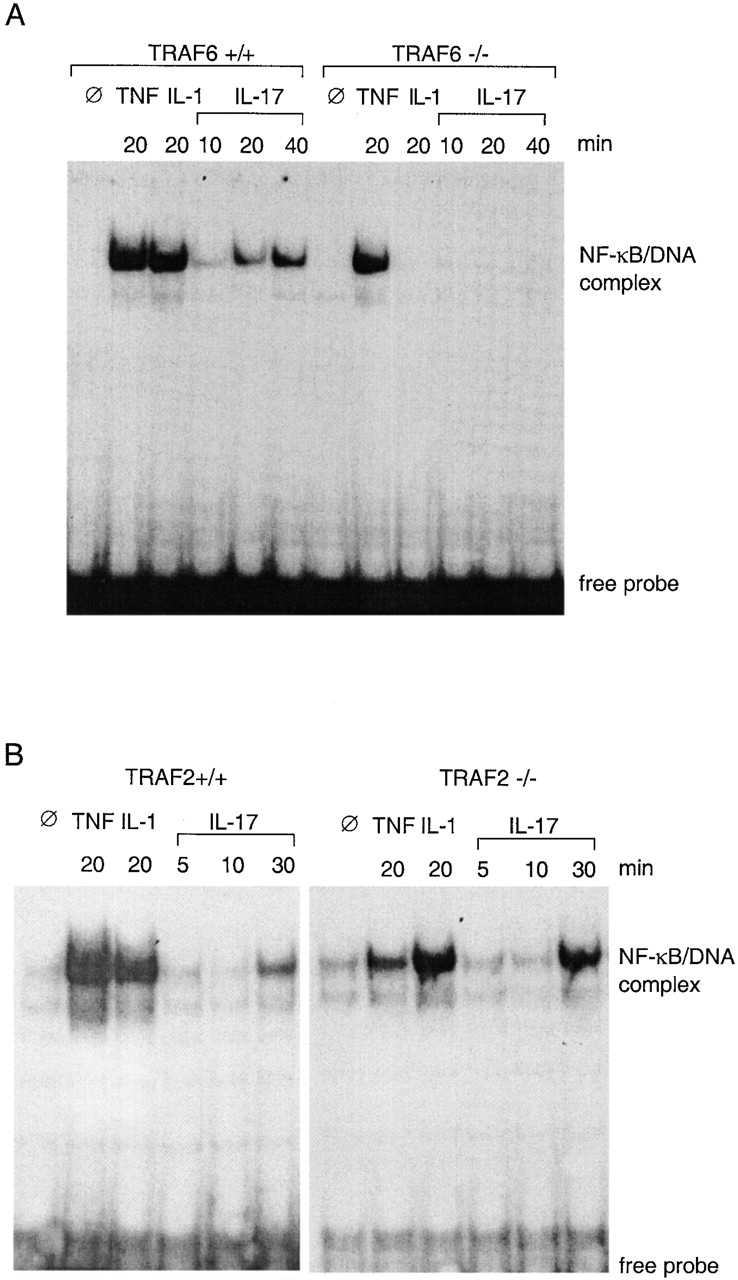
NF-κB activation in MEFs. (A) Lack of NF-κB activation in TRAF6-deficient cells in response to IL-1 and IL-17. Nuclear extracts were prepared from TRAF6+/+ or TRAF6−/− MEFs treated for indicated length of time with 25 ng/ml mTNF, 50 ng/ml IL-1, or 100 ng/ml mIL-17. 6 μg of protein from each sample was tested for ability to bind to a synthetic DNA fragment containing an NF-κB binding site in an EMSA. (B) Normal NF-κB activation in TRAF2-deficient cells in response to IL-17. The same experiments as in A were performed with MEFs derived from TRAF2-deficient mouse embryos and their wild-type littermates.
In Vitro Kinase Assay and Immunoblotting Analysis.
EFs (106) were either left untreated or were treated with the indicated cytokines (see Fig. 2) for 10 min in the absence of serum. Cells were rinsed with cold PBS and lysed on ice for 20 min in lysis buffer (50 mM Hepes, pH 7.6; 125 mM NaCl; 1.5 mM MgCl2; 10% glycerol; 0.5% NP-40; 1 mM EGTA; protease inhibitor cocktail [Boehringer Mannheim]; 20 mM β-glycerophosphate; 1 mM sodium orthovanadate; and 5 mM p-nitro-phenylphosphate). Cell debris was pelleted by centrifugation at 14,000 rpm for 20 min. IKK–NEMO (NF-κB essential modulator) protein complex in the supernatants was immunoprecipitated using anti-NEMO rabbit antiserum and assayed for activity that phosphorylated recombinant IκB-α (amino acids 1–250) as previously described 40. JNK was precipitated with anti–JNK-1 mAb (PharMingen), and the kinase activity was detected in an in vitro kinase assay using recombinant GST–c-Jun 1-79 fusion protein as a substrate (Santa Cruz Biotechnology).
Figure 2.

Kinase activity of the IKK complex and JNK isolated from TRAF6-deficient MEFs. (A) TRAF6-deficient (−/−) and heterozygous EFs (+/−) were treated with 25 ng/ml mTNF, 50 ng/ml IL-1, or 100 ng/ml mIL-17 for 10 min. The IKK complex was immunoprecipitated using anti-NEMO polyclonal antibodies and assayed for activity to phosphorylate IκB-α (amino acids 1–250) (top panel [KA], in vitro kinase assay). Immunoprecipitated materials were separated by SDS-PAGE and immunoblotted with a mixture of antibodies to IKK-α and IKK-β (bottom panel [IB], immunoblot). Molecular mass standards are shown in kilodaltons. (B) JNK-1 was precipitated from wild-type, TRAF2-deficient, and TRAF6-deficient MEFs treated with indicated cytokines and assayed for their activity to phosphorylate recombinant c-Jun (top panel). The amounts of precipitated JNK-1 were determined by immunoblotting (bottom panel).
To detect IKK-α and IKK-β immunoprecipitated with anti-NEMO antiserum, the immunoprecipitates were separated by 10% SDS-PAGE, transferred to nitrocellulose membrane, and immunoblotted with anti–IKK-α and anti–IKK-β polyclonal antibodies (Santa Cruz Biotechnology) as described earlier 40. JNK-1 was detected with an anti–JNK-1 rabbit polyclonal antibody (Santa Cruz Biotechnology).
Expression Plasmids, Transfection, and Coimmunoprecipitation.
Expression plasmids for Flag-tagged CD40 and Flag-tagged IL-1R1 were described elsewhere 33 41. Mammalian expression plasmid for Flag-tagged IL-17R was constructed by inserting PCR-generated cDNA lacking the coding sequence for the signal peptide into expression vector pFlag-CMV-1 (Eastman Kodak Co.). For coprecipitation experiments, 2 × 106 HEK 293 cells were plated on 10-cm dishes and transfected the following day with indicated amounts of expression constructs (see Fig. 5). After 18 h, the cells were collected, washed once with PBS, and lysed for 20 min on ice with lysis buffer. Cell lysates were incubated for 2 h with anti-Flag M2 beads (Sigma Chemical Co.). After extensive washing with lysis buffer, the beads were eluted with 400 μM Flag peptide (Sigma Chemical Co.), separated by SDS-PAGE, and transferred to nitrocellulose membrane. Immunoblot analysis was performed with an anti-Myc or anti-HA (hemagglutinin) mAb (Babco). Expression of transfected constructs was verified by immunoblotting aliquot of cell lysates.
Figure 5.

Interaction of TRAF6 and IL-17R. 293 cells were transfected with indicated amounts of expression plasmid DNA of Flag-tagged IL-1R1, IL-17R, or CD40 together with Myc-tagged TRAF6 or HA-tagged TRAF2 for 18 h. The cells were lysed, and the receptors were immunoprecipitated with anti-Flag beads. Top panel, the Flag immunoprecipitates were resolved by SDS-PAGE and blotted with antibodies to Myc and HA to detect coprecipitating TRAF6 and TRAF2. Center panel, the Flag immunoprecipitates were blotted with Flag antibody to check the amounts of precipitated receptors. Bottom panel, lysates of transfected cells were blotted with both Myc and HA antibodies to determine the expression levels of the TRAF2 and TRAF6 proteins.
Determination of IL-6 Secreted by MEFs.
EFs were seeded at a density of 3 × 104 cells per well in 24-well plates and cultured for 24 h. Cells were left untreated or stimulated with TNF, IL-1, or IL-17 for 15 h. IL-6 concentration of the culture supernatant was determined by ELISA (Endogen) using manufacturer's protocols.
Immunostaining of Cell Surface IL-17R and ICAM-1.
To detect IL-17R, EFs (5 × 105) were first incubated for 1 h with PBS containing goat anti–murine (m)IL-17R antiserum (Santa Cruz Biotechnology) or control serum. After washing twice with PBS, the cells were incubated for 1 h with FITC-conjugated antibodies to goat IgG (Santa Cruz Biotechnology). Flow cytometry was performed using a FACSCalibur™ Analyzer (Becton Dickinson). For detection of ICAM-1, cells were treated for 15 h with 10 ng/ml of TNF, IL-1, or IL-17 or were left untreated. The cells were first incubated with mAb to ICAM-1 (PharMingen) and then stained with FITC-conjugated antibodies to mouse IgG (Santa Cruz Biotechnology).
Results and Discussion
Impairment of IL-17–induced NF-κB Activation in TRAF6-deficient Cells.
EFs obtained from TRAF6−/− embryos or their wild-type littermates were stimulated with IL-17, TNF, or IL-1. NF-κB–DNA binding activity was examined by EMSA. In wild-type cells, NF-κB activation was detected 10 min after IL-17 treatment, followed by a continued increase up to 40 min (Fig. 1 A). Similar results were obtained with heterozygous EF cells (TRAF6+/−; data not shown). However, in TRAF6−/− cells, no NF-κB activation was detected within the time frame examined. In agreement with a previous report, TRAF6−/− cells also failed to respond to IL-1 38. In contrast, TNF-induced NF-κB activation was unaltered by TRAF6 gene disruption, indicating that TRAF6 deficiency only causes defect in response to specific cytokines.
Because TRAF2 has been also implicated in NF-κB activation by certain cytokines 39 42 43 44, we examined if IL-17–induced NF-κB activation was affected by TRAF2 deficiency. Treatment of TRAF2−/− MEFs with either IL-17 or IL-1 induced NF-κB activity comparable to that observed in the wild-type control cells (Fig. 1 B), whereas NF-κB activation induced by 20 min of TNF treatment was significantly reduced. These results indicate that TRAF2 is involved in signal transduction of TNF, as described previously 39, but not in that of IL-1 or IL-17.
Abolishment of IL-17–induced IKK Activation in TRAF6-deficient Cells.
NF-κB activation induced by IL-1, TNF, and LPS entails the activation of IKKs. We investigated whether IL-17–induced NF-κB activation is also mediated by the IKKs, and if so, whether IL-17–induced IKK activation is impaired in the TRAF6 knockout cells. IKK complex in TRAF6+/− or TRAF6−/− EFs untreated or treated with TNF, IL-1, or IL-17 was coimmunoprecipitated with antibodies against NEMO, a stable component in the IKK complex 17 19. Immunoblot analysis indicated that similar amounts of IKK-α and -β were immunoprecipitated from untreated or cytokine-treated TRAF6+/− and TRAF6−/− cells (Fig. 2). The immunoprecipitates were then assayed for activity to phosphorylate recombinant IκB-α (amino acids 1–250) containing IKK substrate, serines 32 and 36. Treatment of TRAF6+/− cells with TNF, IL-1, or IL-17 significantly enhanced the IKK activity, as evidenced by increased phosphorylation of IκB-α, NEMO, and IKKs (Fig. 2 A). On the other hand, IKK activity in the TRAF6−/− cells was enhanced only by TNF and not by IL-1 or IL-17. These results indicate that, like TNF and IL-1, IL-17 activates NF-κB by inducing the catalytic activity of the IKKs, and that IL-17–induced IKK activation involves TRAF6.
Similar results were obtained when JNK activity was measured in TRAF-deficient cells (Fig. 2 B). IL-17– and IL-1–induced JNK activation was abolished in TRAF6 knockout cells but remained intact in TRAF2 knockout cells. In agreement with an earlier report 39, a reduction of TNF-induced JNK activation was observed in TRAF2-deficient cells.
Failure of IL-17 to Induce IL-6 and ICAM-1 Expression in TRAF6-deficient Cells.
To examine if the failure of IL-17 to activate IKK and NF-κB would be translated to its inability to activate target genes, we measured IL-6 secretion by TRAF6-deficient MEFs. Control MEFs responded to TNF, IL-1, and IL-17 with a significant increase in IL-6 secretion (Fig. 3 A). The TRAF6 knockout cells, on the other hand, failed to produce increased levels of IL-6 in response to either IL-17 or IL-1. An increase of IL-6 secretion by the TRAF6-deficient cells was observed after TNF treatment, albeit at a slightly lower level relative to that secreted by control cells. Because TNF induces the release of IL-1, which could contribute to the net increase of IL-6 secretion by TNF-treated cells, this slight reduction in TNF response may be due to a defect in IL-1 response observed in the TRAF6−/− cells.
Figure 3.


Target gene activation in TRAF6-deficient MEFs. (A) Failure of IL-17 to increase IL-6 production by the TRAF6-deficient EFs. Wild-type (+/+) or TRAF6-deficient (−/−) EFs were cultured in the presence of 10 ng/ml mTNF, 10 ng/ml IL-1, or 10 ng/ml mIL-17 for 15 h. IL-6 in cell culture medium was determined by ELISA. Data shown are average values of three independent experiments ± SD. (B) Failure of IL-17 to increase ICAM-1 expression on TRAF6-deficient cells. Wild-type or TRAF6−/− cells were treated with cytokines as in A. Levels of ICAM-1 protein on cell surface were determined by FACS® analysis using mouse mAbs specific to ICAM-1. Dotted line, untreated cells; solid line, cytokine-treated cells.
Similar results were obtained when ICAM-1 cell surface expression was measured by flow cytometry. All three cytokines induced ICAM-1 expression on wild-type MEFs, with the strongest induction observed after TNF treatment (Fig. 3 B). Although TNF-induced increase in ICAM-1 expression remained intact in TRAF6−/− cells, the effects of IL-1 and IL-17 were diminished. These results further support that IL-17–induced response gene activation is mediated by NF-κB and that TRAF-6 is indispensable for the gene regulatory activities of IL-17.
Determinative Effect of TRAF6 Deficiency in IL-17 Response.
To exclude the possibility that the observed unresponsiveness to IL-17 in TRAF6−/− cells might be due to insufficient surface expression of IL-17R, EFs were immunostained with an antiserum against mouse IL-17R and analyzed by flow cytometry. The results indicate that TRAF6+/− and TRAF6−/− cells expressed similar levels of IL-17R (Fig. 4 A).
Figure 4.
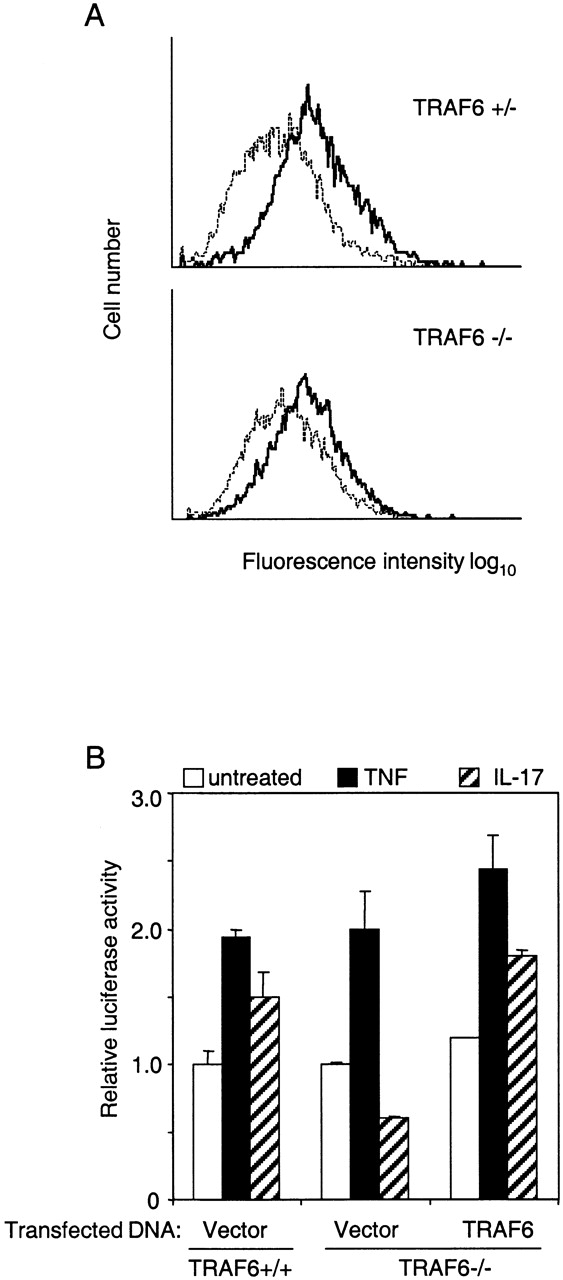
Surface Expression of IL-17R on MEFs and restoration of IL-17 response by transient introduction of TRAF6 protein into TRAF6−/− cells. (A) Determination of IL-17R on TRAF6+/− and TRAF6−/− MEFs by FACS® analysis using polyclonal antibody to mouse IL-17R. Dotted line, mouse-matched isotype IgG; solid line, anti–IL-17R. (B) Reconstitution of IL-17 response. Wild-type or TRAF6-deficient EFs were transfected with RSV-β-gal vector, an NF-κB–dependent luciferase reporter construct, together with 50 ng of vector DNA or TRAF6 expression plasmid. Cells were left untreated (open bars) or stimulated with 25 ng/ml TNF (closed bars) or 100 ng/ml IL-17 (hatched bars) for 8 h. Luciferase activity was determined and normalized based on β-gal activity.
To further eliminate the possibility that failure of TRAF6−/− cells to respond to IL-17 could be caused by defects other than TRAF6 deficiency, we tested if IL-17–induced NF-κB activation could be restored by introducing exogenous TRAF6 into these cells. TRAF6−/− cells were transiently transfected with an NF-κB–driven luciferase report construct together with a TRAF6 expression plasmid or a control vector. Mirroring NF-κB activity measured by EMSA (Fig. 1), IL-17 failed to induce luciferase activity in TRAF6−/− cells, although response to TNF appeared normal (Fig. 4 B). TRAF6−/− cells transfected with TRAF6 expression plasmid responded to IL-17 with an increase of luciferase activity similar to that observed in control cells.
Binding of TRAF6 to IL-17R.
Previous studies indicate that TRAF6 can be recruited to signaling pathways in two modes. It can either bind directly to the receptor intracellular domain, as in the signaling of CD40 and receptor activator of NF-κB 38 45 46 47, or be recruited into the pathways by adapter proteins, as in the signaling of IL-1, IL-18, and LPS 48 49. To determine if TRAF6 interacts with IL-17R, we coexpressed Flag-tagged IL-17R, IL-1R1, or CD40 together with Myc-tagged TRAF6 in 293 cells. In agreement with our earlier finding, TRAF6 did not coimmunoprecipitate with IL-1R1 34. In contrast, TRAF6 coimmunoprecipitated with IL-17R or CD40 (Fig. 5). To determine if this IL-17R–TRAF6 interaction is specific to TRAF6, the same set of Flag-tagged receptors was also coexpressed with HA-tagged TRAF2. TRAF2 was found in the immunoprecipitates of CD40, in agreement with previous reports 47 50, but was not detected in the IL-17R complex. These observations further support the involvement of TRAF6, but not TRAF2, in IL-17 signaling. Because similar amounts of TRAF6 were detected in IL-17R and CD40 complex, TRAF6 may bind to IL-17R directly, although we cannot rule out a remote possibility that this interaction might be mediated by an abundant adapter molecule. The nature of the TRAF6 and IL-17R interaction will be addressed in detail when good antibodies to IL-17R become available.
Acknowledgments
We thank Dave Goeddel, Mike Rothe, and Holger Wesche for helpful discussion; Mark Lomaga, Wen-Chen Yeh, and Tak W. Mak for TRAF2- and TRAF6-deficient MEFs; and Lei Ling for expression vectors.
References
- Rouvier E., Luciani M.F., Mattei M.G., Denizot F., Golstein P. CTLA-8, cloned from an activated T cell, bearing AU-rich messenger RNA instability sequences, and homologous to a herpesvirus saimiri gene. J. Immunol. 1993;150:5445–5456. [PubMed] [Google Scholar]
- Yao Z., Painter S.L., Fanslow W.C., Ulrich D., Macduff B.M., Spriggs M.K., Armitage R.J. Human IL-17a novel cytokine derived from T cells. J. Immunol. 1995;155:5483–5486. [PubMed] [Google Scholar]
- Fossiez F., Djossou O., Chomarat P., Flores-Romo L., Ait-Yahia S., Maat C., Pin J.J., Garrone P., Garcia E., Saeland S. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J. Exp. Med. 1996;183:2593–2603. doi: 10.1084/jem.183.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albanesi C., Cavani A., Girolomoni G. IL-17 is produced by nickel-specific T lymphocytes and regulates ICAM-1 expression and chemokine production in human keratinocytessynergistic or antagonist effects with IFN-gamma and TNF-alpha. J. Immunol. 1999;162:494–502. [PubMed] [Google Scholar]
- Jovanovic D.V., Di Battista J.A., Martel-Pelletier J., Jolicoeur F.C., He Y., Zhang M., Mineau F., Pelletier J.P. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J. Immunol. 1998;160:3513–3521. [PubMed] [Google Scholar]
- Shalom-Barak T., Quach J., Lotz M. Interleukin-17-induced gene expression in articular chondrocytes is associated with activation of mitogen-activated protein kinases and NF-kappaB. J. Biol. Chem. 1998;27:27467–27473. doi: 10.1074/jbc.273.42.27467. [DOI] [PubMed] [Google Scholar]
- Chabaud M., Fossiez F., Taupin J.L., Miossec P. Enhancing effect of IL-17 on IL-1-induced IL-6 and leukemia inhibitory factor production by rheumatoid arthritis synoviocytes and its regulation by Th2 cytokines. J. Immunol. 1998;161:409–414. [PubMed] [Google Scholar]
- Antonysamy M.A., Fanslow W.C., Fu F., Li W., Qian S., Troutt A.B., Thomson A.W. Evidence for a role of IL-17 in alloimmunitya novel IL-17 antagonist promotes heart graft survival. Transplant Proc. 1999;31:93. doi: 10.1016/s0041-1345(98)01453-5. [DOI] [PubMed] [Google Scholar]
- Antonysamy M.A., Fanslow W.C., Fu F., Li W., Qian S., Troutt A.B., Thomson A.W. Evidence for a role of IL-17 in organ allograft rejectionIL-17 promotes the functional differentiation of dendritic cell progenitors. J. Immunol. 1999;162:577–584. [PubMed] [Google Scholar]
- Aarvak T., Chabaud M., Kallberg E., Miossec P., Natvig J.B. Change in the Th1/Th2 phenotype of memory T-cell clones from rheumatoid arthritis synovium. Scand. J. Immunol. 1999;50:1–9. doi: 10.1046/j.1365-3083.1999.00581.x. [DOI] [PubMed] [Google Scholar]
- Kotake S., Udagawa N., Takahashi N., Matsuzaki K., Itoh K., Ishiyama S., Saito S., Inoue K., Kamatani N., Gillespie M.T. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J. Clin. Invest. 1999;103:1345–1352. doi: 10.1172/JCI5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awane M., Andres P.G., Li D.J., Reinecker H.C. NF-kappa B-inducing kinase is a common mediator of IL-17-, TNF-alpha-, and IL-1 beta-induced chemokine promoter activation in intestinal epithelial cells. J. Immunol. 1999;162:5337–5344. [PubMed] [Google Scholar]
- Baeuerle P.A., Henkel T. Function and activation of NF-kappa B in the immune system. Annu. Rev. Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- Barnes P.J., Karin M. Nuclear factor-kappaBa pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 1996;351:127–134. doi: 10.1098/rstb.1996.0008. [DOI] [PubMed] [Google Scholar]
- Zandi E., Chen Y., Karin M. Direct phosphorylation of IkappaB by IKKalpha and IKKbetadiscrimination between free and NF-kappaB-bound substrate. Science. 1998;281:1360–1363. doi: 10.1126/science.281.5381.1360. [DOI] [PubMed] [Google Scholar]
- Yamaoka S., Courtois G., Bessia C., Whiteside S.T., Weil R., Agou F., Kirk H.E., Kay R.J., Israel A. Complementation cloning of NEMO, a component of the IkappaB kinase complex essential for NF-kappaB activation. Cell. 1998;93:1231–1240. doi: 10.1016/s0092-8674(00)81466-x. [DOI] [PubMed] [Google Scholar]
- Woronicz J.D., Gao X., Cao Z., Rothe M., Goeddel D.V. IkappaB kinase-betaNF-kappaB activation and complex formation with IkappaB kinase-alpha and NIK. Science. 1997;278:866–869. doi: 10.1126/science.278.5339.866. [DOI] [PubMed] [Google Scholar]
- Rothwarf D.M., Zandi E., Natoli G., Karin M. IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature. 1998;395:297–300. doi: 10.1038/26261. [DOI] [PubMed] [Google Scholar]
- Regnier C.H., Song H.Y., Gao X., Goeddel D.V., Cao Z., Rothe M. Identification and characterization of an IkappaB kinase. Cell. 1997;90:373–383. doi: 10.1016/s0092-8674(00)80344-x. [DOI] [PubMed] [Google Scholar]
- DiDonato J.A., Hayakawa M., Rothwarf D.M., Zandi E., Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- Thanos D., Maniatis T. NF-kappa Ba lesson in family values. Cell. 1995;80:529–532. doi: 10.1016/0092-8674(95)90506-5. [DOI] [PubMed] [Google Scholar]
- Siebenlist U., Franzoso G., Brown K. Structure, regulation and function of NF-kappa B. Annu. Rev. Cell Biol. 1994;10:405–455. doi: 10.1146/annurev.cb.10.110194.002201. [DOI] [PubMed] [Google Scholar]
- Baldwin A.S., Jr. The NF-kappa B and I kappa B proteinsnew discoveries and insights. Annu. Rev. Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- Baeuerle P.A., Baltimore D. NF-kappa Bten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- Verma I.M., Stevenson J.K., Schwarz E.M., Van Antwerp D., Miyamoto S. Rel/NF-kappa B/I kappa B familyintimate tales of association and dissociation. Genes Dev. 1995;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- Hsu H., Huang J., Shu H.B., Baichwal V., Goeddel D.V. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity. 1996;4:387–396. doi: 10.1016/s1074-7613(00)80252-6. [DOI] [PubMed] [Google Scholar]
- Hsu H., Shu H.B., Pan M.G., Goeddel D.V. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- Kelliher M.A., Grimm S., Ishida Y., Kuo F., Stanger B.Z., Leder P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity. 1998;8:297–303. doi: 10.1016/s1074-7613(00)80535-x. [DOI] [PubMed] [Google Scholar]
- Huang J., Gao X., Li S., Cao Z. Recruitment of IRAK to the interleukin 1 receptor complex requires interleukin 1 receptor accessory protein. Proc. Natl. Acad. Sci. USA. 1997;94:12829–12832. doi: 10.1073/pnas.94.24.12829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korherr C., Hofmeister R., Wesche H., Falk W. A critical role for interleukin-1 receptor accessory protein in interleukin-1 signaling. Eur. J. Immunol. 1997;27:262–267. doi: 10.1002/eji.1830270139. [DOI] [PubMed] [Google Scholar]
- Wesche H., Korherr C., Kracht M., Falk W., Resch K., Martin M.U. The interleukin-1 receptor accessory protein (IL-1RAcP) is essential for IL-1-induced activation of interleukin-1 receptor-associated kinase (IRAK) and stress-activated protein kinases (SAP kinases) J. Biol. Chem. 1997;272:7727–7731. doi: 10.1074/jbc.272.12.7727. [DOI] [PubMed] [Google Scholar]
- Wesche H., Henzel W.J., Shillinglaw W., Li S., Cao Z. MyD88an adapter that recruits IRAK to the IL-1 receptor complex. Immunity. 1997;7:837–847. doi: 10.1016/s1074-7613(00)80402-1. [DOI] [PubMed] [Google Scholar]
- Cao Z., Xiong J., Takeuchi M., Kurama T., Goeddel D.V. TRAF6 is a signal transducer for interleukin-1. Nature. 1996;383:443–446. doi: 10.1038/383443a0. [DOI] [PubMed] [Google Scholar]
- Cao Z., Henzel W.J., Gao X. IRAKa kinase associated with the interleukin-1 receptor. Science. 1996;271:1128–1131. doi: 10.1126/science.271.5252.1128. [DOI] [PubMed] [Google Scholar]
- Baud V., Liu Z.G., Bennett B., Suzuki N., Xia Y., Karin M. Signaling by proinflammatory cytokinesoligomerization of TRAF2 and TRAF6 is sufficient for JNK and IKK activation and target gene induction via an amino-terminal effector domain. Genes Dev. 1999;13:1297–1308. doi: 10.1101/gad.13.10.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z., Spriggs M.K., Derry J.M., Strockbine L., Park L.S., VandenBos T., Zappone J.D., Painter S.L., Armitage R.J. Molecular characterization of the human interleukin (IL)-17 receptor. Cytokine. 1997;9:794–800. doi: 10.1006/cyto.1997.0240. [DOI] [PubMed] [Google Scholar]
- Lomaga M.A., Yeh W.C., Sarosi I., Duncan G.S., Furlonger C., Ho A., Morony S., Capparelli C., Van G., Kaufman S. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999;13:1015–1024. doi: 10.1101/gad.13.8.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh W.C., Shahinian A., Speiser D., Kraunus J., Billia F., Wakeham A., de la Pompa J.L., Ferrick D., Hum B., Iscove N. Early lethality, functional NF-kappaB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity. 1997;7:715–725. doi: 10.1016/s1074-7613(00)80391-x. [DOI] [PubMed] [Google Scholar]
- Tanaka M., Fuentes M.E., Yamaguchi K., Durnin M.H., Dalrymple S.A., Hardy K.L., Goeddel D.V. Embryonic lethality, liver degeneration, and impaired NF-kappa B activation in IKK-beta-deficient mice. Immunity. 1999;10:421–429. doi: 10.1016/s1074-7613(00)80042-4. [DOI] [PubMed] [Google Scholar]
- Hu H.M., O'Rourke K., Boguski M.S., Dixit V.M. A novel RING finger protein interacts with the cytoplasmic domain of CD40. J. Biol. Chem. 1994;269:30069–30072. [PubMed] [Google Scholar]
- Aizawa S., Nakano H., Ishida T., Horie R., Nagai M., Ito K., Yagita H., Okumura K., Inoue J., Watanabe T. Tumor necrosis factor receptor-associated factor (TRAF) 5 and TRAF2 are involved in CD30-mediated NFkappaB activation. J. Biol. Chem. 1997;272:2042–2045. doi: 10.1074/jbc.272.4.2042. [DOI] [PubMed] [Google Scholar]
- Duckett C.S., Gedrich R.W., Gilfillan M.C., Thompson C.B. Induction of nuclear factor kappaB by the CD30 receptor is mediated by TRAF1 and TRAF2. Mol. Cell. Biol. 1997;17:1535–1542. doi: 10.1128/mcb.17.3.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothe M., Sarma V., Dixit V.M., Goeddel D.V. TRAF2-mediated activation of NF-kappa B by TNF receptor 2 and CD40. Science. 1995;269:1424–1427. doi: 10.1126/science.7544915. [DOI] [PubMed] [Google Scholar]
- Darnay B.G., Haridas V., Ni J., Moore P.A., Aggarwal B.B. Characterization of the intracellular domain of receptor activator of NF-kappaB (RANK). Interaction with tumor necrosis factor receptor-associated factors and activation of NF-kappab and c-Jun N-terminal kinase. J. Biol. Chem. 1998;273:20551–20555. doi: 10.1074/jbc.273.32.20551. [DOI] [PubMed] [Google Scholar]
- Ishida T., Mizushima S., Azuma S., Kobayashi N., Tojo T., Suzuki K., Aizawa S., Watanabe T., Mosialos G., Kieff E. Identification of TRAF6, a novel tumor necrosis factor receptor-associated factor protein that mediates signaling from an amino-terminal domain of the CD40 cytoplasmic region. J. Biol. Chem. 1996;271:28745–28748. doi: 10.1074/jbc.271.46.28745. [DOI] [PubMed] [Google Scholar]
- Pullen S.S., Miller H.G., Everdeen D.S., Dang T.T.A., Crute J.J., Kehry M.R. CD40-tumor necrosis factor receptor-associated factor (TRAF) interactionsregulation of CD40 signaling through multiple TRAF binding sites and TRAF hetero-oligomerization. Biochemistry. 1998;37:11836–11845. doi: 10.1021/bi981067q. [DOI] [PubMed] [Google Scholar]
- Kawai T., Adachi O., Ogawa T., Takeda K., Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–122. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- Adachi O., Kawai T., Takeda K., Matsumoto M., Tsutsui H., Sakagami M., Nakanishi K., Akira S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143–150. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- McWhirter S.M., Pullen S.S., Holton J.M., Crute J.J., Kehry M.R., Alber T. Crystallographic analysis of CD40 recognition and signaling by human TRAF2. Proc. Natl. Acad. Sci. USA. 1999;96:8408–8413. doi: 10.1073/pnas.96.15.8408. [DOI] [PMC free article] [PubMed] [Google Scholar]