

Abstract
Serum immunoglobulin (Ig)M provides the initial response to foreign antigen and plays a regulatory role in subsequent immune response development, accelerating the production of high-affinity IgG. Here we show that mice deficient in serum IgM have an increased propensity to spontaneous autoimmunity as judged by the development with age of serum IgG anti-DNA antibodies and the renal deposition of IgG and complement. They also exhibit augmented anti-DNA IgG production on exposure to lipopolysaccharide. Thus, deficiency in serum IgM leads to diminished responsiveness to foreign antigens but increased responsiveness to self—a paradoxical association reminiscent of that described in humans deficient in complement or IgA.
We wondered whether serum IgM might play an analogous role with regard to the response to self-antigens. However, here—in contrast to the sluggish response to foreign antigens—we find that deficiency in serum IgM actually predisposes to the development of IgG antibodies to autoantigens.
Keywords: immunodeficiency, autoimmunity, B lymphocyte, secretory IgM
Introduction
The first antibody to be produced after initial antigen encounter is of the IgM class. In addition to providing this first line of specific defence, serum IgM also assists subsequent development of the immune response. Thus, while mice deficient in the secretory form of IgM harbor relatively normal numbers of surface IgM+, IgD+ B cells with the serum titers of other Ig isotypes being largely unaffected, they nevertheless exhibit delayed development of specific IgG antibodies to T cell–dependent foreign antigens 1 2. Presumably, antigen–IgM complexes act through the complement system either to facilitate antigen localization to sites suitable for subsequent immune response development or to costimulate B cell activation through the CD21–CD19 receptor complex.
Materials and Methods
The generation of CCB embryonic stem cell lines in which the Cμ secretory tailpiece and the μs polyadenylation site have been deleted on one allele by Lox/Cre-mediated gene targeting has been described previously 2. The litters of μs −/μs − and μs +/μs + mice used in this work are from the F2 generation obtained by intercrossing μs +/μs − heterozygotes generated by breeding embryonic stem cell–derived chimeras (created using C57BL/6 blastocysts) with C57BL/6 females. Mice were housed in a barriered specific pathogen–free facility with sentinel animals from each room screened quarterly for a variety of potential pathogens according to the recommendations of the Federation of European Laboratory Animal Science Associations.
Serum titers of IgG anti–double-stranded (ds)DNA (S1 nuclease treated) were measured as described by Mohan et al. 3 using alkaline phosphatase–conjugated goat anti–mouse IgG (Southern Biotechnology Associates). The assays were calibrated using serum from a 7-mo-old NZB/W mouse, which was arbitrarily assigned a titer of 0.5 U/ml. Total IgG was measured by ELISA calibrating with a monoclonal IgG1 (Sigma Chemical Co.). Sera (diluted 1:20) were also titered by immunohistochemistry for antibody binding to Crithidia lucullia using FITC-conjugated goat anti–mouse IgG (Southern Biotechnology Associates). Antibodies to cardiolipin and myeloperoxidase were monitored by ELISA, and surface plasmon resonance was performed as described previously 4. Glomerular histology was assessed on periodic acid-Schiff–stained sections of kidney fixed in Bouin's fixative and embedded in paraffin. Immunofluorescence microscopy was performed on cryostat sections of kidneys that had been snap-frozen in an N-hexane/dry ice bath using FITC-conjugated polyclonal antibodies to mouse IgG (Sigma Chemical Co.) and mouse C3 (Cappel/ICN Biomedicals). Intensity of fluorescence was graded from 0 to +++ on coded sections.
To monitor LPS induction of autoantibodies, mice (3 mo of age) were immunized intraperitoneally with 30 μg LPS (Escherichia coli strain 0111:B4; Sigma Chemical Co.) four times at weekly intervals, and IgG anti-dsDNA and total IgG levels were measured by ELISA.
Results
To assess the spontaneous production of autoantibodies in mice deficient in serum IgM, litter-matched cohorts of μs −/μs − and μs +/μs + homozygote mice were monitored for the development of serum IgG anti-dsDNA antibodies. 9 out of 30 μs −/μs − mice developed titers of IgG anti-DNA at an age of 12–18 mo that were >3 SEM above the mean titers observed in μs +/μs + controls (Fig. 1 A). None of the 21 control mice showed such elevated titers of autoantibody (Fig. 1 A) nor have such autoantibodies been detected in 1-yr- 4 or 18-mo-old non–gene-targeted mice generated from control F2 breedings (Fig. 1 A). The fact that IgG anti-dsDNA is detected in sera from μs −/μs − mice but not μs +/μs + controls cannot be attributed to any effect of serum IgM antibodies in masking the detection of IgG anti-dsDNA, since a clear difference in IgG anti-dsDNA titers was still evident if the assays were performed using purified serum IgG fractions rather than total serum (Fig. 1 B). The sera from the mice were also screened for autoantibodies by Crithidia immunofluorescence: five mice scored strongly positive, and these were from the animals harboring the highest titer of IgG anti-dsDNA as judged by ELISA.
Figure 1.

(A) Titers of IgG anti-dsDNA and total IgG in the sera of 12–18-mo-old litter-matched μs −/μs − and μs +/μs + mice. A line at 0.24 U/ml marks an IgG anti-dsDNA titer 3 SEM above the average titer of the μs +/μs + controls. The IgG anti-dsDNA titers in sera from 18–20-mo-old mice generated from control (129 × C57BL/6)F2 breedings (labeled F2) are shown for comparison. (B) Titers of IgG anti-dsDNA in the IgG fraction of sera of 12–18-mo-old litter-matched μs −/μs − and μs +/μs + mice. The IgG fractions were obtained by purification on protein G–Sepharose. (C) ELISA titration of autoantibodies in μs −/μs − mice. Open symbols are from μs −/μs − mice, filled symbols from representative μs +/μs + controls. The μs −/μs − sera illustrated harboring anti-dsDNA antibodies are from mice 9128 and 9383; the two sera with elevated anticardiolipin antibodies are from mice 9206 (which also harbors IgG anti-dsDNA; see Table ) and 9484; the mouse harboring antibodies to myeloperoxidase is 9296. (D) Binding kinetics of the anti-DNA antibodies as measured by surface plasmon resonance. Antibody binding to a biotinylated dsDNA oligodeoxyribonucleotide immobilized on a streptavidin chip is depicted in resonance units and was monitored as a function of time. After ∼4 min, residual IgG bound to the chip was identified using a polyclonal anti–mouse IgG antiserum. The dashed line depicts the same experiment performed using serum from a control mouse that did not exhibit IgG anti-dsDNA. (E) Immunofluorescence of Crithidia with serum (diluted 1:20) from μs −/μs − mouse 9383 reveals characteristic staining of the nucleus and kinetoplast.
The quality of the anti-dsDNA antibodies was analyzed by surface plasmon resonance. It is clear that the sera contain a complex mixture of antibodies with different binding characteristics. A minority of the initially bound antibody dissociates rapidly to leave a residue that exhibits strong dsDNA binding, characterized by dissociation half-lives >10 min (Fig. 1 D). This tightly bound antibody can be revealed by developing after several minutes with an anti–mouse IgG antiserum. Sera from control μs +/μs + mice only exhibited the very rapidly dissociating DNA-binding component (which is likely of the IgM isotype). ELISA analysis also revealed that one of the IgM-deficient mice that harbored IgG anti-dsDNA (mouse 9206) also scored positive for anticardiolipin antibodies; furthermore, IgG anticardiolipin and IgG antimyeloperoxidase antibodies were also detected in μs −/μs − mice that had scored negative for IgG anti-dsDNA (Fig. 1 C). In contrast, no mice positive for IgG antibodies to cardiolipin or myeloperoxidase were identified in the control cohort.
The kidneys of the five IgM-deficient mice that contained elevated anti-DNA antibodies as judged by the Crithidia assay as well as the kidneys from eight control mice were scored blindly for histopathological changes. Though overt pathological disease was observed in only 1 mouse (number 9128; see Fig. 2), 6 of the 13 mice examined scored highly for IgG and/or C3 deposition (Table ). Of these six animals, five (four at +++ and one at ++) corresponded to the IgM-deficient mice that had been identified in the Crithidia assay, the kidney staining in three of these mice being both in the capillary wall and the mesangium. The sixth mouse (scoring ++, mesangial staining only) was a μs −/μs + heterozygote with a somewhat elevated IgG anti-DNA titer.
Figure 2.

IgG deposits in the kidneys of μs −/μs − mouse 9128 (and μs +/μs + mouse 9332 as control) detected by immunohistochemistry (original magnification: ×100; 1-s exposure).
Table 1.
IgG and C3 Deposition in Kidney
| Serum IgG anti-dsDNA | Kidney deposition | ||||
|---|---|---|---|---|---|
| Mouse no. | μs | ELISA | Crithidia fluorescence | IgG | C3 |
| U/ml | |||||
| 9116 | −/− | 0.08 | − | + | + |
| 9128 | −/− | 1.41 | + | +++ | +++ |
| 9383 | −/− | 1.74 | + | +++ | ++ |
| 9206 | −/− | 0.68 | + | +++ | + |
| 9397 | −/− | 0.53 | + | ++ | ++ |
| 9444 | −/− | 0.66 | + | +++ | +++ |
| 9461 | −/− | 0.08 | − | + | + |
| 9199 | +/− | 0.24 | − | ++ | ++ |
| 9332 | +/− | 0.10 | − | − | + |
| 9460 | +/− | 0.10 | − | + | + |
| 9288 | +/+ | 0.11 | − | + | + |
| 9338 | +/+ | 0.08 | − | +/− | + |
| 9486 | +/+ | 0.07 | − | +/− | + |
| MRL/lpr | +++ | +++ | |||
| NZB/W | +++ | +++ | |||
Kidney staining was monitored on anonymized samples with scoring ranging from − to +++. The staining, when apparent, was in the mesangium although mice 9128, 9206, and 9383 also revealed IgG deposition in the capillary wall.
As well as monitoring the spontaneous development of autoantibodies, we wished to ascertain whether IgM deficiency facilitated their experimental induction. Repeated injection of bacterial LPS has been shown to lead not only to an increase in serum IgG but also to the development of anti-dsDNA antibodies and the enhancement of autoimmune disease in mouse models 5. Consistent with this, LPS treatment leads to hypergammaglobulinemia in both μs +/μs + and μs −/μs − 3-mo-old mice but it is only among the μs −/μs − animals that we detected enhanced production of IgG anti-dsDNA (Fig. 3).
Figure 3.
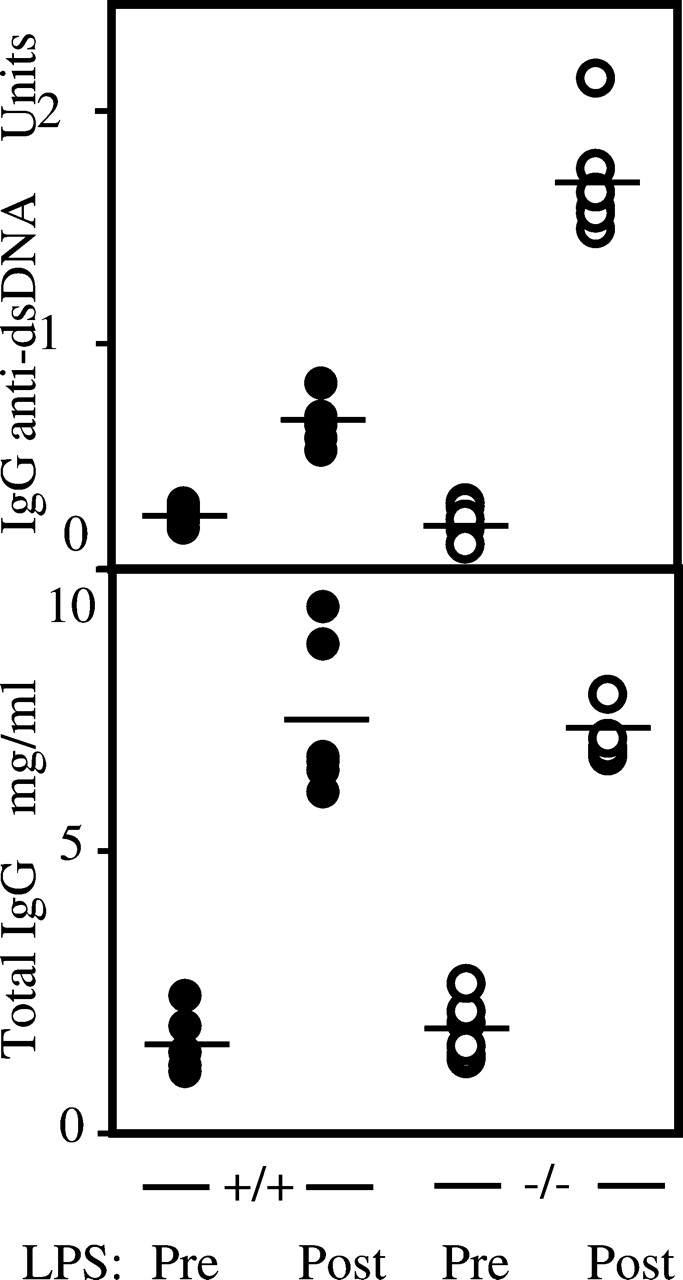
Titers of IgG anti-dsDNA antibodies and total IgG in the sera of 3-mo-old μs −/μs − and μs +/μs + mice before (Pre) and after (Post) administration of LPS.
Discussion
Selective deficiency of serum IgM leads to delayed T cell–dependent IgG responses to foreign antigens while at the same time paradoxically increasing the tendency to develop an IgG response to autoantigens.
The tendency to autoantibody development might be explicable in terms of those perturbations of the immune system in serum IgM–deficient mice that have already been described. Thus, the mice exhibit augmented IgG responses to type 2 T cell–independent (TI-2) antigens 1 2; others have proposed an association between TI-2 type immune responses and the induction of autoantibodies 6. The serum IgM–deficient mice also harbor an increased population of B-1 B cells 1 2; such B cells have been implicated in autoimmunity in some but not other mouse models (see, for example, references 7 and 8).
While selective IgM deficiency in humans has been described only rarely 9 10 11, selective deficiency in IgA in humans is relatively common and is associated with autoimmunity (reviewed in 12, 13). The mechanistic basis of this association is not unknown. Along the lines proposed for association of certain infections with various organ-specific autoimmune diseases, it has been postulated that the autoimmunity might arise as a consequence of the increased number of infections suffered by IgA-deficient patients. A similar logic could be applied to the IgM-deficient mice since they exhibit enhanced sensitivity to microbial challenge 14; however, autoantibody production occurs spontaneously in serum IgM–deficient mice bred in a barrier unit where the infection load is likely to be low. Furthermore, it is far from clear that the association of autoimmunity with IgA deficiency in humans actually reflects a role for the IgA deficiency in triggering the autoimmunity: they could well be independent consequences of a common cause. Indeed, administration of LPS (which enhances autoantibody production in IgM-deficient mice) can trigger both IgA deficiency and autoimmunity in mouse models 15.
We have previously interpreted the delayed maturation of T-dependent immune responses in μs −/μs − mice as reflecting a role for serum IgM, acting through complement, in the handling of foreign antigens 2. This could be enacted by facilitating antigen localization to sites suitable for immune response maturation or by costimulating B cell signaling. Clearly, if IgM were to play an analogous role with regard to the recognition of self-antigens during B cell development, then serum IgM deficiency could lead to a failure to recognize and delete some weakly autoreactive B cell clones from the primary B cell repertoire, thus facilitating development of autoimmunity. Indeed, a predisposition to autoimmunity has already been described in both humans and mice that are genetically deficient in components of the complement cascade 16 17 18, and others have previously interpreted this in terms of a role for complement in the negative selection of autoreactive B cells 17. An alternative model is that the predisposition to autoimmunity in IgM- or C-deficient individuals arises not so much because an altered threshold setting allows the emergence of autoreactive B cell clones but rather as a consequence of alterations to the pathways and kinetics of antigen clearance. This could be a result of the use of Fcγ rather than complement receptors or it could reflect an altered size or site of deposition of immune complexes (for example, see reference 19). This type of explanation, ascribing autoimmunity to an altered pattern of antigen handling, shows parallels to some of the proposals made to explain antinuclear antibodies in mice deficient in serum amyloid P component 20. A further possibility (which follows similar thinking to Stewart et al. 21) is that, in normal mice, some IgM antibodies suppress the expansion of autoreactive IgGs; this balance might then be perturbed by IgM deficiency in serum.
Quite apart from the mechanism by which IgM deficiency leads to autoimmunity, the observations raise the question of whether IgM administration might be useful in treatment of systemic autoimmunity. Nonspecific human polyclonal IgG has been used with variable success to treat patients suffering from diseases of presumed autoimmune pathogenesis. It is possible that supplementing such IgG with IgM may provide a useful adjunct for such therapy. Indeed, polyclonal IgM supplementation of pooled IgG has recently been shown to be effective in a rat nephritis model 22.
Acknowledgments
We thank Facundo Batista for performing the surface plasmon resonance studies, and Theresa Langford and her staff for assistance with animal handling.
A portion of this work was funded by a grant from Lupus UK.
Note added in proof. Since this manuscript was accepted, a paper has appeared (Boes, M., T. Schmidt, K. Linkemann, B.C. Beaudette, A. Marshak-Rothstein, and J. Chen. 2000. Proc. Natl. Acad. Sci. USA. 97:1184–1189) in which the authors come to similar conclusions regarding an increased tendency to autoimmunity in serum IgM–deficient mice.
References
- Boes M, Esau C., Fischer M.B., Schmidt T., Carroll M., Chen J. Enhanced B-1 cell development, but impaired IgG antibody responses in mice deficient in secreted IgM. J. Immunol. 1998;160:4776–4787. [PubMed] [Google Scholar]
- Ehrenstein M.R, O'Keefe T.L., Davies S.L., Neuberger M.S. Targeted gene disruption reveals a role for natural secretory IgM in the maturation of the primary immune response. Proc. Natl. Acad. Sci. USA. 1998;95:10089–10093. doi: 10.1073/pnas.95.17.10089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan C., Shi Y., Laman J.D., Datta S.K. Interaction between CD40 and its ligand gp39 in the development of lupus nephritis. J. Immunol. 1995;154:1470–1480. [PubMed] [Google Scholar]
- O'Keefe T.L., Williams G.T., Batista F.D., Neuberger M.S. Deficiency in CD22, a B cell–specific inhibitory receptor, is sufficient to predispose to development of high affinity autoantibodies. J. Exp. Med. 1999;189:1307–1313. doi: 10.1084/jem.189.8.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallo T., Granholm N.A. Lipopolysaccharide from gram-negative bacteria enhances polyclonal B cell activation and exacerbates nephritis in MRL/lpr mice. Clin. Exp. Immunol. 1990;82:515–521. doi: 10.1111/j.1365-2249.1990.tb05482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehr T., Bachmann M.F., Bucher E., Kalinke U., Di Padova F.E., Lang A.B., Hengartner H., Zinkernagel R.M. Role of repetitive antigen patterns for induction of antibodies against antibodies. J. Exp. Med. 1997;185:1785–1792. doi: 10.1084/jem.185.10.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reap E.A., Sobel E.S., Cohen P.L., Eisenberg R.A. Conventional B cells, not B-1 cells, are responsible for producing autoantibodies in lpr mice. J. Exp. Med. 1993;177:69–78. doi: 10.1084/jem.177.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M., Honjo T. Involvement of B-1 cells in mucosal immunity and autoimmunity. Immunol. Today. 1995;16:534–539. doi: 10.1016/0167-5699(95)80047-6. [DOI] [PubMed] [Google Scholar]
- Yocum M.W., Strong D.M., Chusid M.J., Lakin J.D. Selective immunoglobulin M (IgM) deficiency in two immunodeficient adults with recurrent staphylococcal pyoderma. Am. J. Med. 1976;60:486–494. doi: 10.1016/0002-9343(76)90714-2. [DOI] [PubMed] [Google Scholar]
- Ohno T., Inaba M., Kuribayashi K., Masuda T., Kanoh T., Uchino H. Selective IgM deficiency in adultsphenotypically and functionally altered profiles of peripheral blood lymphocytes. Clin. Exp. Immunol. 1987;68:630–637. [PMC free article] [PubMed] [Google Scholar]
- Guill M.F., Brown D.A., Ochs H.D., Pyun K.H., Moffitt J.E. IgM deficiencyclinical spectrum and immunologic assessment. Ann. Allergy. 1989;62:547–552. [PubMed] [Google Scholar]
- Liblau R.S., Bach J.F. Selective IgA deficiency and autoimmunity. Int. Arch. Allergy Immunol. 1992;99:16–27. doi: 10.1159/000236330. [DOI] [PubMed] [Google Scholar]
- Rankin E.C., Isenberg D.A. IgA deficiency and SLEprevalence in a clinic population and a review of the literature. Lupus. 1997;6:390–394. doi: 10.1177/096120339700600408. [DOI] [PubMed] [Google Scholar]
- Boes M., Prodeus A.P., Schmidt T., Carroll M., Chen J. A critical role of natural immunoglobulin M in immediate defense against systemic bacterial infection. J. Exp. Med. 1998;188:2381–2386. doi: 10.1084/jem.188.12.2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallo T., Granholm N.A. Bacterial lipopolysaccharide induces long lasting IgA deficiency concurrently with features of polyclonal B cell activation in normal and in lupus-prone mice. Clin. Exp. Immunol. 1991;84:134–138. [PMC free article] [PubMed] [Google Scholar]
- Morgan B.P., Walport M.J. Complement deficiency and disease. Immunol. Today. 1991;12:301–306. doi: 10.1016/0167-5699(91)90003-C. [DOI] [PubMed] [Google Scholar]
- Prodeus A.P., Goerg S., Shen L.M., Pozdnyakova O.O., Chu L., Alicot E.M., Goodnow C.C., Carroll M.C. A critical role for complement in maintenance of self-tolerance. Immunity. 1998;9:721–731. doi: 10.1016/s1074-7613(00)80669-x. [DOI] [PubMed] [Google Scholar]
- Botto M., Dell'Agnola C., Bygrave A.E., Thompson E.M., Cok H.T., Petry F., Loos M., Pandolfi P.P., Walport M.J. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat. Genet. 1998;19:56–59. doi: 10.1038/ng0598-56. [DOI] [PubMed] [Google Scholar]
- Mannik M. Mechanisms of tissue deposition of immune complexes J. Rheumatol. 14Suppl. 131987. 35 42 [PubMed] [Google Scholar]
- Bickerstaff M.C.M., Botto M., Hutchinson W.L., Herbert J., Tennent G.A., Bybee A., Mitchell D.A., Cook H.T., Butler P.J.G., Walport M.J., Pepys M.B. Serum amyloid P component controls chromatin degradation and prevents antinuclear autoimmunity. Nat. Med. 1999;6:694–697. doi: 10.1038/9544. [DOI] [PubMed] [Google Scholar]
- Stewart J.J., Agosto H., Litwin S., Welsh J.D., Shlomchik M., Weigert M., Seiden P.E. A solution to the rheumatoid factor paradox. Pathologic rheumatoid factors can be tolerized by competition with natural rheumatoid factors. J. Immunol. 1997;159:1728–1738. [PubMed] [Google Scholar]
- Rieben R., Roos A., Muizert Y., Tinguely C., Gerritsen A.F., Daha M.R. Immunoglobulin M-enriched human intravenous immunoglobulin prevents complement activation in vitro and in vivo in a rat model of acute inflammation. Blood. 1999;93:942–951. [PubMed] [Google Scholar]