

Abstract
We have previously identified two antiviral cytokines (interferon [IFN]-γ and IFN-α/β) that downregulate hepatitis B virus (HBV) replication in the liver of transgenic mice. The cytokine-inducible downstream events that inhibit HBV replication have not been identified. One possible factor is nitric oxide (NO), a pleiotropic free radical with antiviral activity that is produced in the liver by the inducible NO synthase (iNOS). To examine the role of NO in our model, we crossed transgenic mice that replicate HBV with mice that lack a functional iNOS. Importantly, iNOS-deficient mice were almost completely resistant to the noncytopathic inhibitory effect of HBV-specific cytotoxic T lymphocytes on viral replication, an effect that we have shown previously to depend on the intrahepatic induction of IFN-γ. Conversely, iNOS-deficient mice were not resistant to the antiviral effect of IFN-α/β induced by either polyinosinic-polycytidylic acid complex or by lymphocytic choriomeningitis virus (LCMV) infection. These results indicate that NO mediates the antiviral activity of IFN-γ, whereas the antiviral activity of IFN-α/β is NO independent. We also compared the relative sensitivity of LCMV to control by NO in these animals. Interestingly, LCMV replicated to higher levels in the liver of iNOS-deficient mice than control mice, indicating that NO controls LCMV replication in the liver, as well as HBV.
Keywords: nitric oxide, cytokine, liver, hepatitis B virus, lymphocytic choriomeningitis virus
Introduction
We have shown previously that hepatitis B virus (HBV)-specific CTLs can abolish viral replication in the hepatocytes of HBV transgenic mice by noncytopathic mechanisms that are mediated by cytokines such as IFN-γ 1 2 3. In addition, we have shown that IFN-α/β inhibits HBV replication in these animals after polyinosinic-polycytidylic acid complex (poly-I/C) injection 3 or during unrelated hepatotropic virus infections, including lymphocytic choriomeningitis virus (LCMV [4]). Recent results obtained in chimpanzees acutely infected with HBV indicate that similar cytokine-dependent noncytopathic antiviral events contribute to viral clearance during HBV infection 5.
As IFN-α/β and IFN-γ presumably do not represent the final mediators in this system, downstream factors induced by the cytokines such as RNase L, double-stranded RNA-dependent protein kinase 6, and nitric oxide (NO) probably mediate their antiviral effects. In this study, we examined the role of NO as a potential candidate in this process. NO is a highly unstable product of L-arginine metabolism that exerts its functions in a paracrine and autocrine fashion 7. NO is made by distinct categories of NO synthase (iNOS) isoforms 7. The constitutive forms are present in different cell types (including endothelial and neuronal cells) and synthesize low amounts of NO 7. The inducible forms are present in macrophages and hepatocytes and synthesize high amounts of NO upon activation by cytokines such as IFN-γ, TNF-α, and IL-1α 7. NO is a mediator of several physiological functions, including vasodilatation, inhibition of platelet aggregation, and neuronal communication 7.
It is well known that the inducible production of NO by macrophages inhibits the growth of many pathogens, including bacteria, fungi, and parasites 7. Similarly, NO inhibits herpes simplex virus, ectromelia virus, and vaccinia virus replication in vitro 7 8. Thus, it is possible that NO may mediate the antiviral activity of IFN-γ and/or IFN-α/β in our system. To test this hypothesis, we crossed transgenic mice that replicate HBV with mice that are genetically iNOS-deficient, and we monitored the contribution of NO to the antiviral effects of cytokines induced in the liver by HBV-specific CTLs, poly-I/C, or LCMV infection in these animals. In addition, we monitored the ability of NO to inhibit LCMV replication in these animals in order to compare the relative sensitivity of HBV and LCMV to NO.
Materials and Methods
Mice.
The HBV transgenic mouse lineage 1.3.46 (inbred B10.D2) used in this study (official designation, Tg[HBV 1.3 genome]Chi46) has been described previously 9. These mice replicate HBV at high levels in the liver without any evidence of cytopathology. Lineage 1.3.46 was crossed with mice that lack a functional iNOS allele (iNOS−/− [10]). The knockout mice were provided by Drs. John Mudgett (Merck Research Laboratories, Rahway, NJ), and John MacMicking and Carl Nathan (Cornell University Medical College, New York, NY). Heterozygous mice from lineage 1.3.46 were crossed with iNOS−/− mice to yield progeny that were screened for hepatitis B e antigen (HBeAg) in the serum using commercially available reagents from Abbott Laboratories. HBeAg-positive progeny were screened for homozygosity of the null mutation by PCR exactly as described 10, and homozygosity of the H-2d class I molecule (the restriction element used by the hepatitis B surface antigen [HBsAg]-specific CTLs used in this study; see below) was screened by FACS® analysis as described 11. HBV transgenic mice that were homozygous for the H-2d class I molecule and either homozygous or heterozygous for the null mutation were matched for age (8–10 wk), sex (male), and levels of HBeAg in their serum before experimental manipulations. All animals were housed in pathogen-free rooms under strict barrier conditions.
Injection of HBsAg-specific CTLs.
A HBsAg-specific, H-2d restricted, CD8+ CTL line was derived from spleen cells of nontransgenic B10.D2 mice immunized with 50 μg of plasmid pCMV-S2/S as described 11. The CTL line was maintained by weekly restimulation with irradiated P815 cells that stably express the HBV large envelope protein (ayw subtype) containing HBsAg, as described previously 12. 5 d after the last stimulation, the cells were washed, counted, suspended in HBSS containing 2% FCS, and injected intravenously into HBV transgenic mice. 2 d after injection, mice were killed, and their livers were harvested for histological and histochemical analyses, or they were snap frozen in liquid nitrogen and stored at −80°C for subsequent molecular analyses (see below).
LCMV Isolates and Infection of Mice.
Clone 2.2 of the WE isolate of LCMV used in this study has been described previously 4. Stocks of virus were prepared by growth on baby hamster kidney cells. All virus stocks were free of mycoplasma contamination as determined by Hoechst staining of cells growing in antibiotic-free medium 48 h after virus infection. The titers of the LCMV stocks, and also infectious virus titers in murine tissues, were determined by plaque assay on Vero cells as described 4. Adult male mice (8-10-wk old) were infected by single intravenous inoculation (2 × 106 PFU) of LCMV WE clone 2.2 per mouse and killed either 24 h or 7 d after infection, when their livers were processed exactly as described for the CTL-injected animals.
Poly-I/C Treatment.
Mice were injected intravenously with a single dose (200 μg per mouse) of poly-I/C (Sigma Chemical Co.) and killed 24 h later. Their livers were also processed exactly as described for the CTL-injected animals.
Tissue DNA and RNA Analyses.
Frozen liver tissue was mechanically pulverized under liquid nitrogen, and total genomic DNA and RNA were isolated for Southern and Northern blot analyses exactly as described previously 9. Nylon membranes were analyzed for HBV DNA, HBV RNA, LCMV RNA, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and 2′5′-oligoadenylate synthetase (2′5′-OAS) as described 4. Quantitation of cytokine, T lymphocyte, and macrophage marker mRNAs was performed by RNase protection assay exactly as described previously 2. The iNOS probe (provided by Iain Campbell, The Scripps Research Institute, La Jolla, CA) used in the RNase protection has been described previously 13. The relative abundance of specific DNA and RNA molecules was quantitated by PhosphorImaging analysis using the Optiquant™ image analysis software (Packard).
In Situ Hybridization.
This procedure was carried out exactly as described 9. The 33P-labeled antisense RNA probe used in this study was prepared by transcription from the T7 promoter of a plasmid containing the full-length cDNA for iNOS (provided by Iain Campbell) as described previously 13.
Biochemical and Histological Analyses.
The extent of hepatocellular injury was monitored by measuring serum alanine aminotransferase (sALT) activity at multiple time points after treatment with saline, CTLs, IFN-α, poly-I/C, or infection with LCMV. sALT activity was measured in a Paramax™ chemical analyzer (Baxter Diagnostics) exactly as described previously 9. For histological analysis, liver tissue samples were fixed in 10% zinc-buffered formalin (Anatech), embedded in paraffin, sectioned (3 μm), and stained with hematoxylin and eosin exactly as described elsewhere 9.
Results and Discussion
HBV Replicates at High Levels in the Liver of iNOS-deficient Mice.
HBV transgenic mice from lineage 1.3.46 were crossed with iNOS−/− mice. Groups (six mice each) of age- (8–10 wk), sex- (male), and serum HBeAg–matched animals that were either heterozygous (+/−) or homozygous (−/−) for the iNOS null mutation were killed, and their livers were harvested. After extraction, total hepatic RNA and DNA were pooled in each group and analyzed for HBV gene expression and replication by Northern and Southern blot analyses.
As shown in Fig. 1, iNOS−/− mice replicate HBV in the liver at levels that are almost twofold higher than the respective heterozygous control littermates (as measured by PhosphorImaging analysis; data not shown), whereas the intrahepatic levels of HBV RNA, including the pregenomic 3.5-kb RNA, were very similar. Actually, the pregenomic RNA content was slightly lower (0.92-fold) in iNOS−/− mice. These results suggest that NO inhibits HBV replication at a step(s) in the viral life cycle after pregenomic RNA accumulation. Although the differences are relatively small (less than twofold), the higher content of HBV replicative forms in the liver of iNOS−/− mice suggests that basal levels of NO may regulate HBV replication to some degree in the uninflamed liver. Heterozygous and homozygous iNOS-deficient mice have been monitored histologically for over 1 yr without observing pathological changes in any organ, especially the liver (not shown). Lack of inflammation in these livers was also underscored by the absence of sALT elevation (Fig. 1, bottom).
Figure 1.
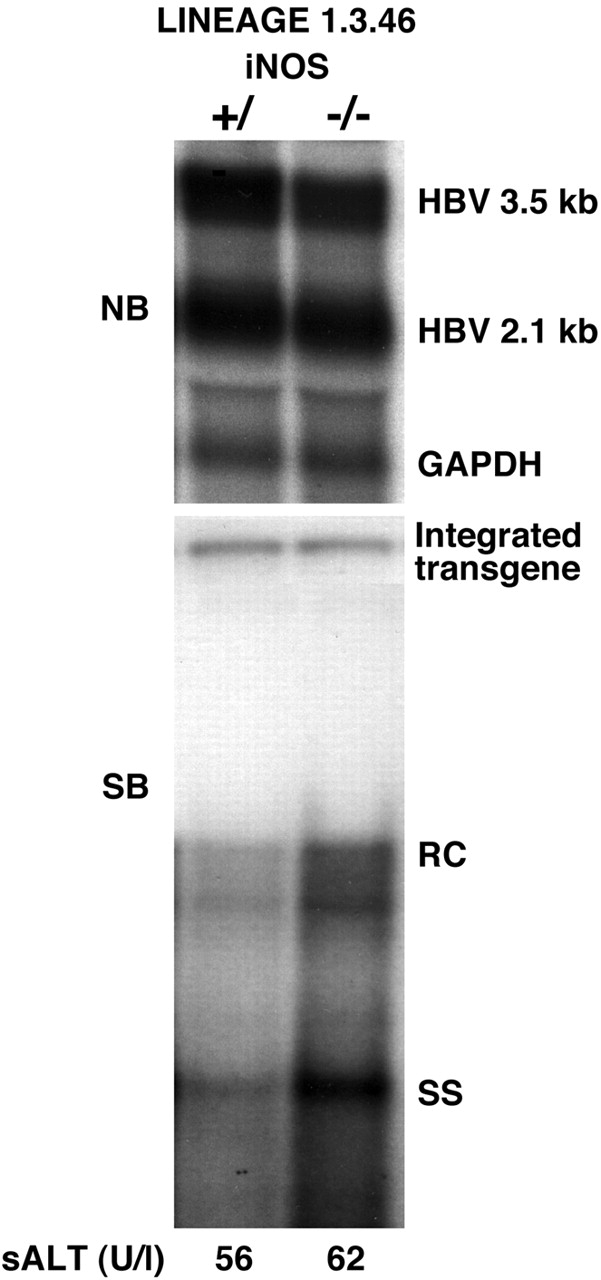
Hepatic HBV replication in iNOS−/− mice. Six age- (8–10 wk), sex- (male), and serum HBeAg–matched mice that were either heterozygous (+/−) or homozygous (−/−) for the indicated null mutation were killed, and the livers were harvested. After extraction, total hepatic RNA and DNA were pooled in each group and analyzed for HBV gene expression and replication by Northern (NB) and Southern blot (SB) analyses. The membranes were hybridized with 32P-labeled HBV- and GAPDH-specific DNA probes. Southern blot analysis was performed with 30 μg of total hepatic DNA. All DNA samples were RNase treated before gel electrophoresis. Bands corresponding to the integrated transgene, relaxed circular (RC), and single-stranded (SS) linear HBV DNA replicative forms are indicated. The integrated transgene can be used to normalize the amount of DNA bound to the membrane. The filter was hybridized with a 32P-labeled HBV-specific DNA probe. The mean sALT activity, measured at the time of autopsy, is indicated (bottom) for each group and is expressed in U/liter (U/l).
NO Mediates Most of the Antiviral Activity of IFN-γ Produced by HBsAg-specific CTLs.
We have shown previously that the noncytopathic antiviral effect of CTLs is blocked by a cocktail of antibodies to IFN-γ and TNF-α 2. We have also shown recently that the amount of IFN-γ produced by passively transferred CTLs is sufficient to inhibit HBV replication in the liver of transgenic mice, indicating that TNF-α and other cytokines, including IFN-α/β, are not required for the antiviral activity of CTLs 3. To monitor the CTL-dependent antiviral activity in the liver of iNOS−/− mice, six age-, sex-, and serum HBeAg–matched transgenic mice that were either heterozygous or homozygous for the iNOS null mutation were injected intravenously with 2.5 × 107 lymphocytes derived from a CD8+ HBsAg-specific CTL line. Mice were bled and killed, livers were harvested 2 d later, and results were compared with those observed in livers pooled from 10 age-, sex-, and serum HBeAg–matched transgenic littermates injected with saline (NaCl).
As shown in Fig. 2 for three representative mice per group, iNOS mRNA was induced in the liver of iNOS+/− mice after CTL injection, and this was associated with a profound inhibition of HBV DNA replicative forms. In contrast, iNOS mRNA was not induced in the liver of iNOS−/− mice, and this was associated with persistence of HBV DNA replicative forms (Fig. 2). The lack of inhibition of HBV replication in iNOS−/− mice was not due to a defect in the entry and/or activation of CTLs into the liver, as CD8, CD3, IFN-γ, and TNF-α mRNA were induced in the liver of these animals at levels that were even higher than those observed in the liver of iNOS+/− mice (Fig. 2). As IFN-γ produced by transferred CTLs is sufficient to inhibit HBV replication 3, these results demonstrate that NO mediates most of the antiviral activity of IFN-γ in our system. To determine which cells produce NO in the CTL-injected livers, the hepatic content of iNOS mRNA was also analyzed by in situ hybridization analysis in wild-type animals 2 d after CTL injection. As shown in Fig. 3, most iNOS mRNA was detected in lymphomononuclear inflammatory cells (arrowheads), indicating that they, rather than hepatocytes, represent the major source of NO in the CTL-injected livers. Along with the increased inflammatory infiltrate, the severity of liver disease (monitored by sALT) was also significantly higher in iNOS−/− mice compared with heterozygous controls (Fig. 2, bottom). These results suggest that NO may have inhibited the severity of liver disease by inhibiting the recruitment of the transferred CTLs into the liver, in keeping with reports that NO inhibits leukocyte adhesion to endothelial cells. 14 15 16.
Figure 2.
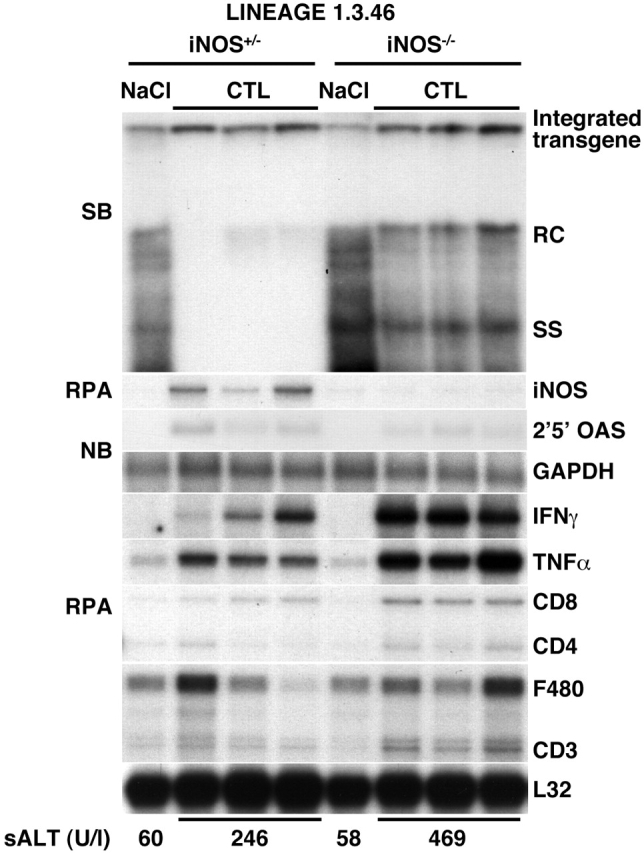
NO mediates most of the antiviral activity of IFN-γ produced by HBsAg-specific CTLs. Age-, sex-, and serum HBeAg–matched transgenic mice from the same groups of mice described in the legend to Fig. 1 were injected intravenously with 2.5 × 107 CTLs and killed 2 d later. Total hepatic RNA and DNA were analyzed for HBV gene expression and replication and for the message of 2′5′-OAS and GAPDH as described in the legend to Fig. 1. Total hepatic RNA (10 μg) from the same mice was also analyzed by RNase protection assay (RPA) for the expression of iNOS, IFN-γ, and TNF-α transcripts and for the expression of CD3, CD4, CD8, and F480, as indicated. The mRNA encoding the ribosomal protein L32 was used to normalize the amount of RNA loaded in each lane. Results were compared with those observed in livers pooled from six age-, sex-, and serum HBeAg–matched transgenic saline controls (NaCl). The mean sALT activity, measured at the time of autopsy, is indicated (bottom) for each group and is expressed in U/liter (U/l). SB, Southern blot; NB, Northern blot; RC, relaxed circular; SS, single stranded.
Figure 3.

Intrahepatic distribution of iNOS mRNA after adoptive transfer of CTLs. Livers from two wild-type animals (A, B and C, D) that were injected 2 d earlier with CTLs were analyzed for the expression of iNOS mRNA by in situ hybridization using a specific 33P-labeled iNOS riboprobe. Note that most iNOS RNA is contained in lymphomononuclear inflammatory cells (arrowheads), whereas no iNOS RNA is detectable in the hepatocytes (hematoxylin and eosin; original magnification: ×600). d. 2, day 2.
NO Does Not Mediate the Antiviral Activity of IFN-α/β Induced by Poly-I/C or LCMV Infection.
We have shown previously that poly-I/C inhibits HBV replication by inducing the production of IFN-α/β in the liver 3. To monitor the antiviral activity of poly-I/C in the animals described above, three age-, sex-, and serum HBeAg–matched transgenic mice that were either heterozygous or homozygous for the iNOS null mutation were injected intravenously with poly-I/C and killed 24 h after injection. As shown in Fig. 4, a strong induction of 2′5′-OAS (a known IFN-α/β–responsive gene) was observed in the liver of iNOS+/− and iNOS−/− mice, and this coincided with a profound inhibition of HBV replication in both group of animals. Importantly, poly-I/C did not induce iNOS mRNA in the iNOS+/− controls in which HBV replication was profoundly reduced (Fig. 4). Collectively, these results demonstrate that NO does not mediate the antiviral activity of IFN-α/β. No liver disease was observed histologically (not shown) or biochemically (Fig. 4, bottom) in poly-I/C–injected animals; accordingly, their livers showed no induction of IFN-γ, T cell, and macrophage marker RNA (Fig. 4).
Figure 4.
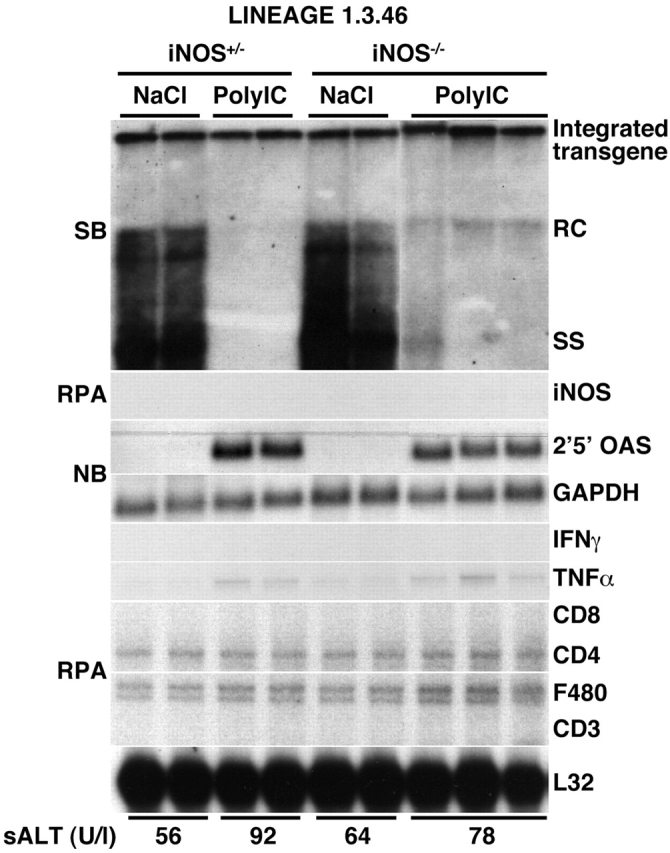
NO does not mediate the antiviral activity of IFN-α/β induced by poly-I/C. Age-, sex-, and serum HBeAg–matched transgenic mice from the same groups of mice described in the legend to Fig. 1 were killed 24 h after a single injection of poly-I/C (200 μg per mouse). Total hepatic RNA and DNA were analyzed for HBV gene expression and replication and for the message of iNOS, 2′5′-OAS, IFN-γ, TNF-α, T cell (CD8, CD4, CD3), and macrophage (F480) markers exactly as described in the legends to Fig. 1 and Fig. 2. Results were compared with those observed in livers of two age-, sex-, and serum HBeAg–matched littermates that were injected with saline (NaCl). The mean sALT activity, measured at the time of autopsy, is indicated (bottom) for each group and is expressed in U/liter (U/l). SB, Southern blot; NB, Northern blot; RC, relaxed circular; SS, single stranded; RPA, RNase protection assay.
We have shown previously that HBV replication is also inhibited by IFN-α/β induced in the liver during LCMV infection 3 4. In this study, we showed that the hepatic induction of IFN-α/β on day 1 after LCMV infection coincided with a profound decrease in HBV replication in iNOS−/− mice (Fig. 5), reiterating that NO is not involved in the IFN-α/β–dependent inhibition of HBV replication. HBV replication was still abolished at day 7 after LCMV infection, when the hepatic induction of IFN-α/β was substantially decreased, and IFN-γ was strongly induced (Fig. 5). The fact that HBV replication did not reappear in the liver of iNOS−/− mice (in which NO mediates most of the antiviral activity of IFN-γ) is not surprising for the following reasons: (a) IFN-α/β is still strongly induced between days 1 and 5 after acute LCMV infection 4; (b) the reappearance of HBV replication after cessation of intrahepatic cytokine induction is a relatively slow process that takes about 1 wk 17; and (c) IFN-γ or TNF-α, both of which are strongly induced at this time point, could trigger iNOS-independent antiviral events within the hepatocyte.
Figure 5.

Relative sensitivity of HBV and LCMV to the antiviral effects of NO. Age-, sex-, and serum HBeAg–matched transgenic mice from the same groups of mice described in the legend to Fig. 1 were infected with LCMV WE clone 2.2 (2 × 106 PFU per mouse) and killed either 24 h or 7 d after infection. Total hepatic RNA and DNA were analyzed for HBV gene expression and replication and for the message of iNOS, 2′5′-OAS, IFN-γ, TNF-α, T cell (CD8, CD4, CD3), and macrophage (F480) markers exactly as described in the legend to Fig. 1. Northern blot (NB) membranes were also hybridized with a 32P-labeled LCMV-specific DNA probe (top). The results of the plaque assay (PFU/g) of individual liver tissues are indicated for the day 7 time point. The PFU results for the day 1 time point are expressed as mean values. Results were compared with those observed in livers pooled from six age-, sex-, and serum HBeAg–matched transgenic saline-injected controls (NaCl). The mean sALT activity, measured at the time of autopsy, is indicated (bottom) for each group and is expressed in U/liter (U/l). ND, not detectable; SB, Southern blot; RC, relaxed circular; SS, single stranded; RPA, RNase protection assay.
LCMV Is also Susceptible to the Antiviral Effects of NO.
To determine the sensitivity of LCMV to NO, we monitored the content of LCMV RNA in the liver of iNOS−/− mice after an acute LCMV infection. No difference in the hepatic content of LCMV RNA was observed between iNOS−/− and iNOS+/− mice at day 1 after infection (Fig. 5, top), in keeping with the fact that iNOS mRNA was not induced at this time point (Fig. 5). In contrast, IFN-α/β (monitored by the increase in the message for 2′5′-OAS) was strongly induced in the liver of both groups of animals at this time point (Fig. 5). This indicates that NO does not mediate the ability of IFN-α/β to inhibit LCMV replication that we and others have reported previously 3 18 19.
Importantly, iNOS−/− mice showed about fourfold higher hepatic levels (calculated by PhosphorImaging analysis as mean values) of LCMV RNA on day 7 after infection, compared with iNOS+/− control mice in which iNOS mRNA was strongly induced (Fig. 5, top). Similarly, higher levels of viral titers (analyzed by plaque assay, Fig. 5) were detected in these animals. At this time point, the LCMV-specific T cell response is known to be maximal in the liver 4, and as expected, CD8, CD4, CD3, F480, IFN-γ, and TNF-α mRNAs were strongly induced in both groups of animals (Fig. 5). These results demonstrate that LCMV is probably susceptible to the antiviral effects of NO, like HBV. The fact that the iNOS-associated inhibition of LCMV replication observed on day 7 was associated with a strong intrahepatic induction of IFN-γ suggests that NO may have contributed to the known capacity of this cytokine to control LCMV replication 3 20 21. Despite similar content of cytokine, T cell, and macrophage marker RNA, on day 7 after infection, there was a relative high degree of variation in viral titers and LCMV RNA in different individual iNOS+/− mice. A similar degree of variation in LCMV titer and RNA levels were found in four additional iNOS+/− mice from a different experiment. The reason for this variability remains to be determined.
In summary, this study shows that NO mediates the ability of IFN-γ, but not IFN-α/β, to inhibit HBV replication. The results also show that NO may also control LCMV replication in the liver by an IFN-α/β–independent mechanism that is probably triggered by IFN-γ. Finally, we have shown that the absence of NO increases the severity of liver disease observed after passive transfer of HBV-specific CTLs or after infection with LCMV, suggesting a protective role for this mediator. It is possible that by inhibiting viral replication, NO may reduce the expression of viral antigens in the cell, thereby diminishing the severity of the immune-mediated liver disease. Future research intended to elucidate this protective mechanism as well as the mechanisms responsible for the antiviral activity of NO against HBV and LCMV is clearly warranted.
Acknowledgments
We thank John Mudgett, John MacMicking, and Carl Nathan for providing iNOS knockout mice; Persephone Borrow and Michael Oldstone for providing the clone WE 2.2 of LCMV (work supported by National Institutes of Health grant AI09484); Iain Campbell for providing the iNOS probe and Monte Hobbs for providing the cytokine gene and T cell marker probe sets used in the RNase Protection assays; Rick Koch, Christina Whitten, and Margie Chadwell for excellent technical assistance; and Jennifer Newmann for help with manuscript preparation.
This work was supported by grants AI40696 (to L.G. Guidotti) and CA40489 (to F.V. Chisari) from the National Institutes of Health. This is manuscript number 12743-MEM from The Scripps Research Institute.
References
- Guidotti L.G., Chisari F.V. Cytokine-induced viral purging—role in viral pathogenesis. Curr. Opin. Microbiol. 1999;2:388–391. doi: 10.1016/s1369-5274(99)80068-x. [DOI] [PubMed] [Google Scholar]
- Guidotti L.G., Ishikawa T., Hobbs M.V., Matzke B., Schreiber R., Chisari F.V. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity. 1996;4:25–36. doi: 10.1016/s1074-7613(00)80295-2. [DOI] [PubMed] [Google Scholar]
- McClary H., Koch R., Chisari F.V., Guidotti L.G. Relative sensitivity of hepatitis B virus and other hepatotropic viruses to the antiviral effects of cytokines. J. Virol. 2000;74:2255–2264. doi: 10.1128/jvi.74.5.2255-2264.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidotti L.G., Borrow P., Hobbs M.V., Matzke B., Gresser I., Oldstone M.B., Chisari F.V. Viral cross talkintracellular inactivation of the hepatitis B virus during an unrelated viral infection of the liver. Proc. Natl. Acad. Sci. USA. 1996;93:4589–4594. doi: 10.1073/pnas.93.10.4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidotti L.G., Rochford R., Chung L., Shapiro M., Purcell R., Chisari F.V. Viral clearance without destruction of infected cells during acute HBV infection. Science. 1999;284:825–829. doi: 10.1126/science.284.5415.825. [DOI] [PubMed] [Google Scholar]
- Kalvakolanu D.V., Borden E.C. An overview of the interferon systemsignal transduction and mechanisms of action. Cancer Invest. 1996;14:25–53. doi: 10.3109/07357909609018435. [DOI] [PubMed] [Google Scholar]
- Lowenstein C.J., Dinerman J.L., Snyder S.H. Nitric oxidea physiologic messenger. Ann. Intern. Med. 1994;120:227–237. doi: 10.7326/0003-4819-120-3-199402010-00009. [DOI] [PubMed] [Google Scholar]
- Reiss C.S., Komatsu T. Does nitric oxide play a critical role in viral infections? J. Virol. 1998;72:4547–4551. doi: 10.1128/jvi.72.6.4547-4551.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidotti L.G., Matzke B., Schaller H., Chisari F.V. High level hepatitis B virus replication in transgenic mice. J. Virol. 1995;69:6158–6169. doi: 10.1128/jvi.69.10.6158-6169.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMicking J.D., Nathan C., Hom G., Chartrain N., Fletcher D.S., Trumbauer M., Stevens K., Xie Q.-W., Sokol K., Hutchinson N. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. 1995;81:641–650. doi: 10.1016/0092-8674(95)90085-3. [DOI] [PubMed] [Google Scholar]
- Shimizu Y., Guidotti L.G., Fowler P., Chisari F.V. Dendritic cell immunization breaks cytotoxic T lymphocyte tolerance in hepatitis B virus transgenic mice. J. Immunol. 1998;161:4520–4529. [PubMed] [Google Scholar]
- Ando K., Moriyama T., Guidotti L.G., Wirth S., Schreiber R.D., Schlicht H.J., Huang S., Chisari F.V. Mechanisms of class I restricted immunopathology. A transgenic mouse model of fulminant hepatitis. J. Exp. Med. 1993;178:1541–1554. doi: 10.1084/jem.178.5.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell I.L., Samimi A., Chiang C.S. Expression of the inducible nitric oxide synthase. Correlation with neuropathology and clinical features in mice with lymphocytic choriomeningitis. J. Immunol. 1994;153:3622–3629. [PubMed] [Google Scholar]
- Kanwar S., Kubes P. Nitric oxide is an antiadhesive molecule for leukocytes. New Horiz. 1995;3:93–104. [PubMed] [Google Scholar]
- Hickey M.J., Sharkey K.A., Sihota E.G., Reinhardt P.H., Macmicking J.D., Nathan C., Kubes P. Inducible nitric oxide synthase-deficient mice have enhanced leukocyte-endothelium interactions in endotoxemia. FASEB J. 1997;11:955–964. doi: 10.1096/fasebj.11.12.9337148. [DOI] [PubMed] [Google Scholar]
- Hickey M.J., Kubes P. Role of nitric oxide in regulation of leucocyte-endothelial cell interactions. Exp. Physiol. 1997;82:339–348. doi: 10.1113/expphysiol.1997.sp004029. [DOI] [PubMed] [Google Scholar]
- Cavanaugh V.J., Guidotti L.G., Chisari F.V. Interleukin-12 inhibits hepatitis B virus replication in HBV transgenic mice. J.Virol. 1997;71:3236–3243. doi: 10.1128/jvi.71.4.3236-3243.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskophidis D., Battegay M., Bruendler M.-A., Laine E., Gresser I., Zinkernagel R. Resistance of lymphocytic choriomeningitis virus to alpha/beta interferon and gamma interferon. J. Virol. 1994;68:1951–1955. doi: 10.1128/jvi.68.3.1951-1955.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller U., Steinhoff U., Reis L.F., Hemmi S., Pavlovic J., Zinkernagel R.M., Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- Leist T.P., Eppler M., Zinkernagel R.M. Enhanced virus replication and inhibition of lymphocytic choriomeningitis virus disease in anti-gamma interferon-treated mice. J. Virol. 1989;63:2813–2819. doi: 10.1128/jvi.63.6.2813-2819.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wille A., Gessner A., Lother H., Lehmann-Grube F. Mechanism of recovery from acute virus infection. VIII. Treatment of lymphocytic choriomeningitis virus-infected mice with anti-interferon-gamma monoclonal antibody blocks generation of virus-specific cytotoxic T lymphocytes and virus elimination. Eur. J. Immunol. 1989;19:1283–1288. doi: 10.1002/eji.1830190720. [DOI] [PubMed] [Google Scholar]