

Abstract
The immune system, despite its complexity, is maintained at a relative steady state. Mechanisms involved in maintaining lymphocyte homeostasis are poorly understood; however, recent availability of transgenic (Tg) and knockout mouse models with altered balance of lymphocyte cell populations suggest that cytokines play a major role in maintaining lymphocyte homeostasis. We show here that transforming growth factor (TGF)-β plays a critical role in maintaining CD8+ T cell homeostasis in a Tg mouse model that specifically overexpresses a dominant negative TGF-β II receptor (DNRII) on T cells. DNRII T cell Tg mice develop a CD8+ T cell lymphoproliferative disorder resulting in the massive expansion of the lymphoid organs. These CD8+ T cells are phenotypically “naive” except for the upregulation of the cell surface molecule CD44, a molecule usually associated with memory T cells. Despite their dominance in the peripheral lymphoid organs, CD8+ T cells appear to develop normally in the thymus, suggesting that TGF-β exerts its homeostatic control in the peripheral immune system.
Keywords: lymphoproliferative disorder, T lymphocyte subsets, TCR repertoire, T cell transformation, thymocyte development
Introduction
TGF-β is a member of a large family of secreted proteins involved in cell growth and differentiation (for review see reference 1). This protein family, known as the TGF-β superfamily, consists of many structurally conserved members that function by several distinct mechanisms dependent on the species, cell type, or cell environment involved.
Mammalian TGF-β is itself a multifunctional subfamily that consists of three isoforms (TGF-β1–3) that have been shown to have similar functions in vitro but appear to have unique functions in vivo based on tissue expression and activity 2. In the immune system, TGF-β has been shown to have two major effects, a proinflammatory effect and an immunosuppressive effect 3. Both of these effects are elicited by all three isoforms and are regulated by multiple control mechanisms, including regulation of cell adhesion 4 5 6, modulation of macrophage function 7 8 9, and regulation of lymphocyte proliferation and differentiation 10 11 12.
In the absence of TGF-β1, mice develop a massive multifocal inflammatory disease, suggesting a major role for TGF-β1 in immune regulation 13 14. Further analysis has shown that the disease consists of an initial inflammatory response followed by an autoimmune manifestation 15 16 17. Dissection of these two phenomena has been possible by crossing TGF-β1 knockout (KO) mice to other immune-deficient mice. TGF-β1 KO mice crossed to SCID mice result in mice with no inflammatory lesions or indications of autoimmunity 15, implying a role for B and T cells. Similarly, if TGF-β1 KO mice are crossed to MHC class II–deficient mice, they no longer develop an inflammatory or autoimmune-type disease, implicating an MHC class II–restricted T cell component in the disease 18. However, because of the multiple effects of TGF-β, it is difficult to characterize the actions of TGF-β as specific to T cells in the murine KO models.
Regulation of homeostasis in the immune system is complex. Lymphocytes are maintained at relatively constant numbers and ratios when no foreign antigens are detected. Rapid expansion of lymphocyte number follows antigen encounter, with eventual return to preimmune stimulation numbers and ratios after antigen removal. Antigen type and route of entry, as well as previous antigen encounters, determine which populations of lymphocytes expand and differentiate and which ones remain quiescent 19 20 21 22. Much is known about the activation, differentiation, and expansion of lymphocytes involved in such antigen responses, but little is known about the maintenance of lymphocyte homeostasis before and after the response.
Recent studies using transgenic (Tg) and KO mice have shown that a multitude of these mice have defects in lymphocyte homeostasis. Many of the defects in these mice involve cytokines or their signaling pathways 23 24 25 26. Defects in the common cytokine receptor γ (present in IL-2, IL-4, IL-7, IL-9, and IL-15 receptors) or its signaling pathway result in an overexpansion of CD4+ T cells 23 24. Likewise, a CD4+ Th cell imbalance occurs in mice defective for the interferon regulatory factor 1 and the IL-2 gene 27 28. A second category of genetically altered mice exhibiting lymphocyte homeostatic defects involves genes important in negative regulation. Fas, Fas ligand, and CTLA-4 are examples of genes that result in a severe lymphoproliferative disease when defective in mice 29 30.
Interestingly, TGF-β is both a negative regulator of the immune system and a cytokine; however, its role in lymphocyte homeostasis has been difficult to analyze because of its diverse effects on multiple organ systems. To better study the role of TGF-β in T cells and its effect in vivo, we have generated a Tg mouse that expresses a dominant negative TGF-β type II receptor (DNRII) specifically on T cells 31 32 using a human CD2 promoter/enhancer 33. This model is distinct from the TGF-β1 KO model; since it would be expected to have a interruption in signaling from all three isoforms of TGF-β, it would not be expected to interfere with stem cell development 34, and it would not influence endothelial cell adhesiveness 5. Rather, a T cell–specific, DNRII Tg mouse allows direct investigation of the role of TGF-β on T cell number and function in vivo.
Mice expressing a DNRII transgene on their T cells develop a CD8+ T cell hyperproliferation that dominates the lymphocyte population by 2–9 mo of age. These CD8+ T cells express multiple TCR V-β families, suggesting a polyclonal expansion of CD8+ T cells; however, over time oligoclonal populations of TCR V-β families appear in most mice. In contrast to the TGF-β1 KO mice, this expansion of T cells is limited to the CD8+ T cell population and does not involve CD4+ T cells or B cell populations, nor does it have an inflammatory component. These results suggest that TGF-β plays a critical role in maintaining CD8+ T cell homeostasis.
Materials and Methods
Gene Constructs.
A 0.7-kb fragment containing the DNRII with a 5′ influenza hemagglutinin (HA) tag cloned into pcDNRIIA3 (Invitrogen Corp.) was removed with HindIII (5′) and XhoI (3′). It was inserted into Bluescript SKII (Stratagene Inc.) at the 5′ HindIII and 3′ XhoI sites. The DNRII transgene fragment was then removed from Bluescript using XhoI (5′) and XbaI (3′) and then reinserted into Bluescript SKII at the 5′ SmaI and 3′ XbaI sites. The DNRII transgene was then removed with EcoRI and cloned into the CD2 minigene 33 at the EcoRI site. The construct was removed from the vector using a 5′ SalI site and a 3′ NotI site and purified over a sucrose gradient for injection.
Mice.
DNRII Tg mice were generated and housed at the Science Applications International Corp. Tg core facility in Frederick, MD according to National Institutes of Health (NIH) animal guidelines. All Tg mice were generated and maintained with C57BL/6 strain mice.
Cell Populations.
Splenocytes, LN cells, and thymocytes were prepared as previously described 35. T cell populations were purified from splenocytes by either magnetic bead separation (Miltenyi Biotec) or passage over a T cell column (Biotex Labs.), followed by magnetic bead separation. Purity of the T cell populations was established by flow cytometric analysis and was always >85%. Cells were maintained in complete culture medium (CCM): RPMI 1640 (Life Technologies, Inc.) with 1 mM sodium pyruvate (Life Technologies), 100 mM nonessential amino acids (Life Technologies), 100 U/ml penicillin plus 100 mg/ml streptomycin (Life Technologies), 50 mM 2-ME (Fisher Scientific Co.), 2 mM l-glutamine (Life Technologies), and 10% FCS (Hyclone). Cells were incubated at 37°C with 5% CO2/95% air.
Cell line TG1124 was generated from Tg founder T12H1 by incubation of splenocytes with Con A (2.5 μg/ml; Sigma Chemical Co.) in CCM. A stable, long-term cell line, TG1124, emerged from this culture and was carried by successive passes in CCM alone.
Flow Cytometry.
Biotin-, FITC-, and PE-conjugated mAbs against mouse CD8 (Calbiochem Corp.), CD4, CD25, CD44, CD45Rb, CD69, and TCR V-β panel (PharMingen) were used to stain cells from thymus, LN, and spleen. Anti-HA (Boehringer Mannheim) was used at 1 μg/ml. Biotin-conjugated (1 μg/ml) Abs were incubated in a total volume of 20 μl for 20 min on ice, washed once in FACS media (FM; HBSS [GIBCO BRL] plus 1% BSA [Sigma Chemical Co.] plus 1% azide [wt/vol; Sigma Chemical Co.]). A secondary reagent, streptavidin R–PE (Caltag Labs.) used at 1:40 dilution was used in conjunction with biotin-modified reagent. An Fc-specific Ab, 2.4G2, was used to block Fc binding. Secondary reagent was added in 20 μl of total volume, incubated for 15 min on ice, washed in FM twice, and then resuspended in 0.3 ml of FM. Stained cells were analyzed by FACScan™ (Becton Dickinson) as previously reported 35. Data are presented on log scale as either histogram or dot plot.
Semiquantitative Reverse Transcriptase–PCR.
T Cells were isolated using magnetic cell sorting using MACS microbeads (Miltenyi Biotec). T cells were separated from non-T cells by positive selection using anti-Thy 1.2–conjugated microbeads or were sequentially purified by positive selection using anti-CD8–conjugated microbeads followed by positive selection using anti-CD4–conjugated microbeads. In both procedures, the non-T cell fraction was the final eluate and gave similar results with both methods. RNA was prepared by the Trizol® method (Life Technologies). cDNA was prepared using the Superscript® choice system (Life Technologies), and PCR was performed as previously described 35, except conditions were optimized to generate PCR products in a linear range. Southern hybridization was performed on the PCR products using specific 32P-labeled internal oligonucleotides. Radioactivity was detected using a PhosphorImaging screen (Eastman Kodak Co.) with a Storm 860 analyzer (Molecular Dynamics). Data were analyzed using ImageQuant® software (Molecular Dynamics).
Cross-linking Assay.
Equal amounts of total protein from either non-Tg splenocytes or TGF-β T cell lines were used as previously described 36. In brief, 125I-labeled TGF-β1 was incubated with either C57BL/6 splenocytes or DNRII T cell line TG1124 in the absence or presence of cold TGF-β1. After cross-linking, lysates were immunoprecipitated with either anti–TGF-β type II receptor, directed at the cytoplasmic tail, or anti-HA mAb. Immunoprecipitates were run on a polyacrylamide gel and analyzed as previously described 36.
Cell Cycle Analysis.
Splenocytes were harvested as previously described, washed in FM, and stained with anti-CD4 FITC or anti-CD8 FITC mAb. Stained cells were washed twice in cold FM and fixed with a 0.5% formaldehyde solution. Cells were treated with a citrate hypotonic solution containing propidium iodide to stain for DNA 37. Cells were analyzed on a FACScan™ (Becton Dickinson) and analyzed using ModFit LT® (Becton Dickinson) cell cycle analysis software as previously described 37.
Results
Generation of DNRII Tg Mouse.
To target the DNRII transgene to the T cell lineage, a truncated TGF-β type II receptor containing an HA tag sequence (reference 36; Fig. 1 A) was inserted into the human CD2 minigene construct. Expression of the DNRII transgene was evaluated by an 125I-labeled TGF-β1 binding assay, demonstrating surface expression of DNRII capable of binding its ligand in T cells of Tg mice (Fig. 1 B). Transgene expression was also confirmed by semiquantitative reverse transcriptase (RT)-PCR (Fig. 1C and Fig. D) and Northern blot analysis (data not shown). No appreciable amount of DNRII transgene mRNA was detected in T cell–depleted populations, as determined by RT-PCR (Fig. 1 C). Both CD4+ and CD8+ T cells expressed comparable amounts of DNRII transgene mRNA, as determined by RT-PCR (Fig. 1 D).
Figure 1.
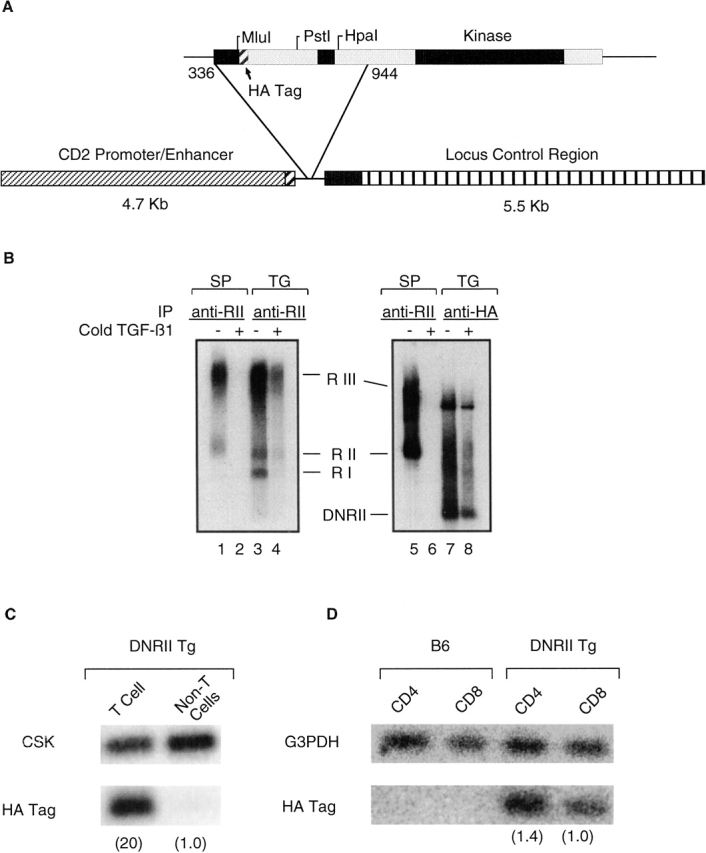
Construction and expression of the DNRII transgene. (A) Schematic representation of the expression vector with transgene. (B) Association of TGF-β1 with DNRII. Either splenocytes (SP) from C57BL/6 mice or TG1124 cells (TG) were affinity labeled with 125I–TGF-β1 in the absence (−) or presence (+) of cold TGF-β1. Cell lysates were subjected to immunoprecipitation by either anti–TGF-β II receptor, specific for cytoplasmic tail of endogenous TGF-β II, or anti-HA tag, specific for DNRII. Immunoprecipitation of receptors was analyzed by SDS-PAGE and autoradiography. (C) Semiquantitative RT-PCR to establish tissue-specific expression of DNRII transgene. CSK, a ubiquitously expressed kinase, was used to establish relative expression of the transgene in T cell and non-T cell populations (number in parentheses). (D) Semiquantitative RT-PCR to establish expression within T cell subsets. Expression of G3PDH was used to calculate relative expression between CD4+ and CD8+ T cell subsets (number in parentheses).
Seven Tg lines (T12I2, TI2El, T12E2, T12G2, Tl2H1, T12I3, and T12#9) were identified by Southern blot analysis; five transmitted the transgene to offspring. Offspring that were Tg for the DNRII gene were born at the expected Mendelian frequency and appeared healthy at birth. However, over time all seven Tg founder mice and their offspring developed a severe lymphoproliferative disorder resulting in enlarged spleen (2–20×) and LNs (2–50×), with moderate infiltration of lymphocytes into other organs. The severity of the disease phenotype varied among founders and correlated with transgene copy number. Founder T12Hl had the most severe phenotype as well as the highest transgene copy number (five to six copies) among the six transmitting founders (data not shown), with a marked infiltration of all lymphoid organs with small lymphocytes (Fig. 2). Other organs, such as the liver, heart, and lungs, had moderate infiltration of similar small lymphocytes with minimal inflammation (Fig. 2). Founder Tl2H1 died at 3 mo of age but was able to transmit the transgene to one female Fl mouse. The female T12H1 offspring also died at 3 mo of age with a phenotype similar to that of the founder.
Figure 2.
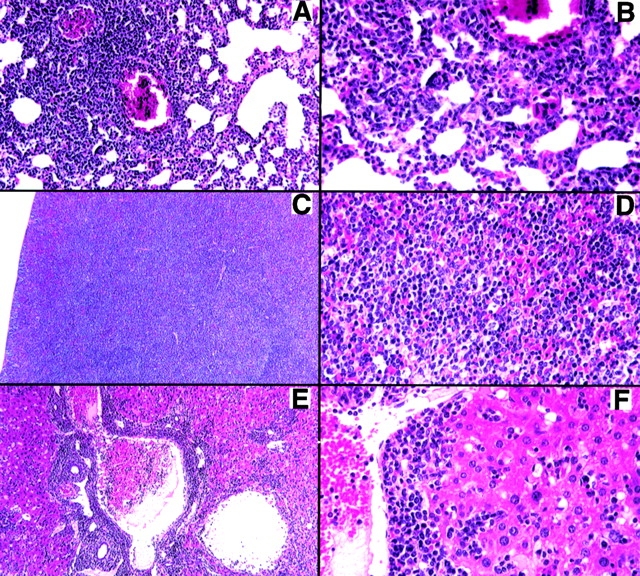
Small lymphocyte infiltration in DNRII Tg founder T12H1. (A) Extensive perivascular infiltration of small lymphocytes into the lung with little inflammation. (B) Higher magnification showing lymphoid cells in alveolar walls. (C) Spleen containing large white pulp areas with disruption of normal architecture. (D) Higher magnification showing a uniform population of lymphoid cells. (E) Liver with extensive perivascular and periportal lymphoid infiltration, with many lymphoid cells present in the blood but little inflammation. (F) Higher magnification showing infiltration around the blood vessel and in the sinusoids. Magnifications: A, 75×; C and E, 50×; B, D, and F, 150×.
Founders T12I2 and T12E1 had a similar phenotype to T12H1, except for a slower onset. Founder T12E1 was found to have a single integration site containing two to four copies of the transgene (data not shown). Founder T12I2 was found to have two independent integration sites, each with one to two copies of the transgene (data not shown). T12I2 offspring containing either of the two integrated transgenes were phenotypically similar, with each having a slower onset of the phenotype than the founder.
Phenotypic Analysis of DNRII Tg Mice.
Analysis of cell surface phenotype of splenocytes and LN cells from founder TI2H1 revealed a 10-fold increase in the number of CD8+ T cells as compared with control (Fig. 3), suggesting that the lymphoproliferative disorder was based solely on a CD8+ T cell expansion. In concordance with this observation, absolute cell numbers of CD4+ T cells and B cells were only slightly decreased when compared with control (Fig. 3). All DNRII Tg founders and their offspring exhibited a similar increase in CD8+ cell numbers resulting in a severe CD4/8 ratio alteration (data not shown), suggesting that the phenotype was integration site independent.
Figure 3.

Flow cytometric analysis of B and T cells in DNRII Tg mice. Splenocytes from 12-wk-old founder T12H1 (right panels) or an aged-matched C57BL/6 mouse (left panels) were analyzed for expression of T cell markers, CD4 and CD8 (top panels), and B cell markers, B220 and MHC class II (CII) molecules (bottom panels). Absolute cell numbers ×10−6 shown in parentheses. At least 10,000 events were collected for each group.
Further T cell surface marker phenotype analysis was performed to identify the activation status of the CD8+ T cells. Lymphocytes from DNRII Tg mice were stained for CD44, CD69, CD25, CD45Rb, and CD62L. Normal naive CD8+ T cells express low levels of CD44, CD69, and CD25 and high levels of CD45Rb and CD62L. DNRII Tg CD8+ T cells had a striking increase in CD44 levels (Fig. 4) yet unchanged levels of CD25, CD62L, CD69, and CD45Rb (Fig. 4). Increased levels of CD69 have been detected in a small percentage of the DNRII Tg mice studied, all of which have had the most severe progression of the lymphoproliferative disorder (data not shown).
Figure 4.
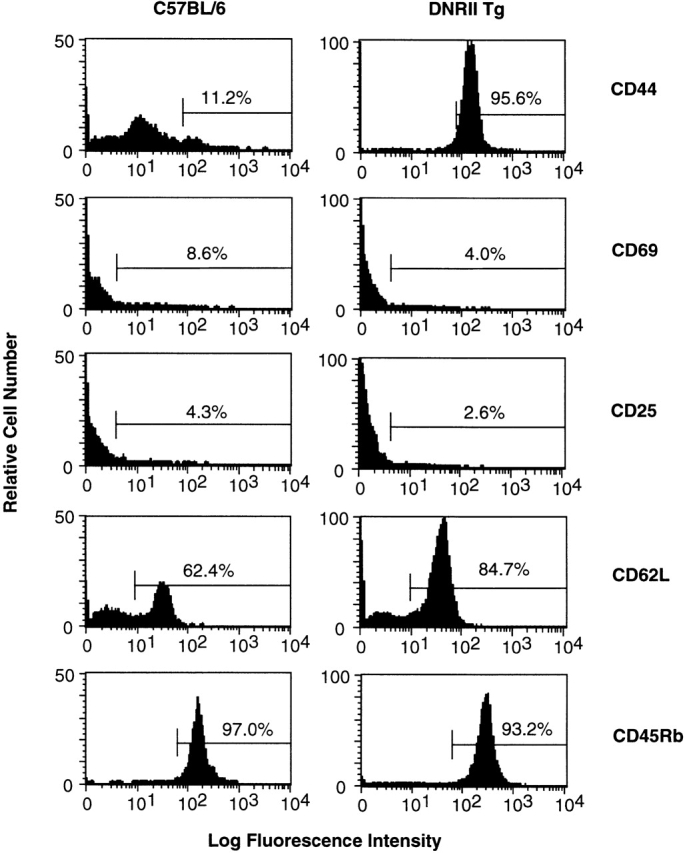
T cell activation marker expression on surfaces of DNRII Tg CD8+ T cells. LN T cells from 27-wk-old non-Tg littermate mouse (left histograms) or DNRII Tg mouse I2A1 (right histograms) were stained with Abs specific for a panel of T cell activation/memory markers. At least 10,000 events were collected for each group.
Time Course Analysis of Phenotypic Changes.
CD8+ T cell changes were consistent in all of the DNRII Tg founder lines studied, with each progressing in a similar manner toward a CD8+ T cell dominance. To better characterize this progression, a time course analysis of cell surface marker expression was performed. The CD44hi phenotype was the earliest observed abnormality, with high CD44 levels occurring on CD8+ T cells by 6–12 wk of age (Fig. 5 A). Similar time points in slower-progressing DNRII Tg lines (T12I2) indicate that the Tg mice have normal percentages of CD44hi cells at 4–6 wk, establishing that initial CD8+ T cell populations are normal with respect to CD44 phenotype (data not shown). No changes were observed in the cell surface phenotype of the DNRII Tg CD4+ T cells (data not shown). Between 12 and 24 wk of age, a second major change occurred. The percentage of CD8+ T cells began to rise, causing a significant decrease in the CD4/CD8 ratio (Fig. 5 A). This decreased CD4/CD8 ratio was mostly a result of an absolute increase in CD8+ T cells, not a decrease in CD4+ T cells (Fig. 3).
Figure 5.
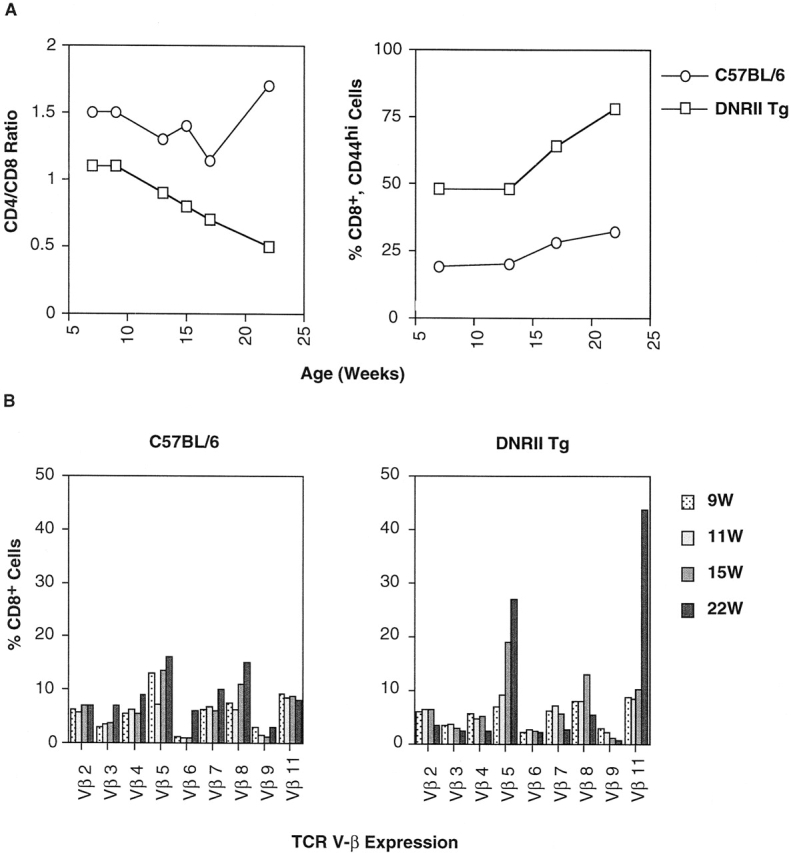
Time course analysis of CD8+ peripheral T cells in DNRII Tg mice. (A) PBLs from founder line E1A3 (□) or non-Tg littermate mice (○) were analyzed by flow cytometry for T cell markers CD4 and CD8 (left; expressed as CD4/CD8 ratio) and CD8+CD44hi surface marker (right) over time. (B) TCR V-β repertoire was measured by a panel of TCR V-β–specific mAbs. Percentage of CD8+ PBLs expressing a specific TCR V-β receptor was plotted over time. At least 10,000 events were collected per time point.
To address the issue of monoclonal or polyclonal expansion, changes in the TCR V-β repertoire were measured using a panel of mAbs. These data demonstrate that the CD8+ T cell repertoire was being skewed from a polyclonal to an oligoclonal repertoire, containing several TCR V-β families (Fig. 5 B). Finally, in late onset of the lymphoproliferative disorder, one to two TCR V-β family usually dominated between 6 and 12 mo of age. However, no one TCR V-β family dominated across individual mice, suggesting that the TCR V-β expansion was not due to a single environmental antigen (data not shown).
Thymocyte Development in DNRII Tg Mice.
This gross overpopulation of the lymphoid organs in DNRII Tg mice can be explained by an increase in production of CD8+ T cells from the thymus, decreased cell death of CD8+ T cells, or an expansion of CD8+ T cells in the periphery. The thymi of DNRII Tg mice, despite a dominance of CD8+ T cells in the periphery, have a normal appearance and normal subpopulation phenotype (Fig. 6 A). Although increased thymic export of CD8+ cells is a formal possibility, this observation rather suggested that the expansion of CD8+ cells was due to a lack of cell death, a profound proliferation in the periphery, or a combination of both.
Figure 6.
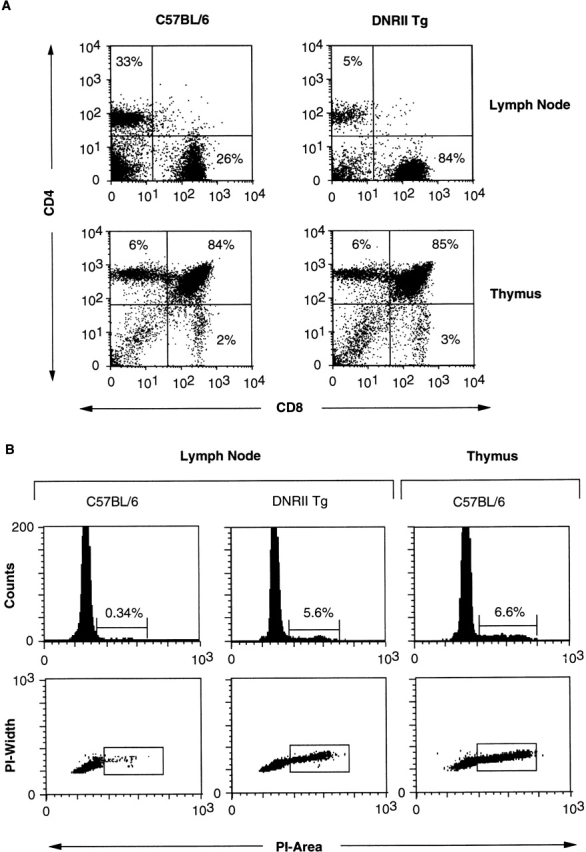
Thymic development and cell cycle analysis of DNRII Tg T cells. (A) LN cells (top panels) or thymocytes (bottom panels) from 27-wk-old DNRII Tg line I2A1 (right panels) or littermate control mice (left panels) were stained with anti-CD4 and -CD8 mAbs and were detected by flow cytometric analysis. (B) CD8+ LN T cells (four leftmost panels) or thymocytes (right panels) from 19-wk-old DNRII Tg line I2C2 (center panels) or littermate control mice (left and right panels) were stained with propidium iodide (PI). Percentage of cells in cycle (S/M/G2) is indicated as percentage of incorporation of PI as detected by flow cytometric analysis. Thymocytes (right panels) were used as a positive control for the detection of cycling cells. At least 10,000 events were collected for each group.
Cell Cycle Analysis of CD8+ T Cell Populations.
To differentiate between these possibilities, cell cycle analysis of CD8+ T cells from the DNRII Tg mice was performed. It was found that whereas normal lymphocytes from C57BL/6 mice (Fig. 6 B) had very few cells in cycle, DNRII Tg CD8+ T cells resembled the cell cycle status of C57BL/6 (Fig. 6 B) or DNRII Tg (data not shown) thymocytes, with 5–8% of cells in cycle. These data suggest that DNRII Tg CD8+ T cells are actively dividing, which could account for their overpopulation in the periphery of DNRII Tg mice. No increased resistance to apoptosis was detected in Tg CD8+ T cells when compared with normal T cells when incubated for various time periods in vitro or subjected to cytokine withdraw after activation (data not shown), suggesting that CD8+ T cell proliferation rather than resistance to cell death is responsible for the CD8+ T cell expansion found in DNRII Tg mice.
Discussion
The immune system is maintained at a steady state, with distinct populations of cells held at a relatively constant number and ratio. This homeostatic mechanism is especially apparent within T lymphocyte subpopulations, where a constant CD4/CD8 ratio is maintained. Studies have shown that this constant CD4/CD8 ratio is maintained even when T cells varying in initial CD4/CD8 ratios are used to reconstitute athymic mice 38 39. In addition, it appears that naive and memory T cells, based on cell surface expression of the CD44+ marker, are also strictly interrelated, with neither population able to fully reconstitute T cell numbers by itself 40. Together, these data suggest that interactions between T lymphocyte subpopulations are strictly regulated.
DNRII-expressing T cells alter this strict T cell homeostatic process by allowing a massive expansion of CD8+ CD44hi cells in the periphery. Absolute numbers of CD4+ T cells, as well as B cells, remained unchanged or slightly decreased over time; however, slight increases in the number of CD4+ cells in cycle have been detected, indicating that CD4+ T cells may be affected by the transgene. The CD8+ T cell expansion is directly proportional to the dose of TGF-β DNRII expression in the Tg line, as demonstrated by varying rates of CD8+ T cell expansion by different Tg founder lines. Similar dose-dependent effects have been seen in other TGF-β DNRII Tg mice 41.
These results are quite distinct from those for TGF-β1 KO mice, which develop a severe inflammatory disease with diverse infiltrating cell populations. These differences between the two mouse models may be explained by the differences in the tissues affected by the genetic alterations. Whereas the TGF-β DNRII transgene directly affects only T lymphocytes, the TGF-β1 KO alteration affects many tissues, including tissues vital to the survival of the mouse 13 14. In addition, TGF-β1 gene inactivation affects only TGF-β1 signaling, whereas the TGF-β DNRII transgene affects signaling from all three isoforms of TGF-β, which may contribute to differences seen between the two mouse models.
Differential expression of the transgene in CD4+ and CD8+ T cells does not appear to be a likely explanation for the discrepancies, as RT-PCR analysis demonstrated equal amounts of transgene mRNA in both populations. In addition, other Tg mice using the same expression vector have had equal expression in CD4+ and CD8+ T cells 33 42 43. Similarly, all founder mice, regardless of transgene copy number, had the same phenotype, which would indicate that high transgene expression, which was capable of causing massive CD8+ T cell expansion by 8 wk of age, was not sufficient to cause a major change in the CD4+ T cell population.
Differential control of CD4+ and CD8+ T cell homeostasis has been observed repeatedly in recent gene KO models. Most strikingly, defects in cytokines binding to receptors containing the common cytokine receptor γ chain have altered lymphocyte homeostasis dramatically 23 24 28 44 45 46. However, many changes that occur in these mice can be attributed to abnormal thymic development. One exception appears to be the IL-2R complex that shows normal thymic development when disrupted but abnormal expansion of CD4+ cells in the periphery over time 28 46 47. Similarly, CTLA-4 KO mice appear to have normal T cell development in the thymus, but these mice rapidly develop a T cell lymphoproliferative disease in the periphery characterized by an activated T cell phenotype that appears to be mediated by CD4+ T cells 30 48 49.
Thymic development appears to be normal in the TGF-β DNRII Tg mouse, despite a gross overpopulation of CD8+ T cells in the LNs and spleen. This type of CD8+ T cell expansion has been infrequently observed in other Tg or KO mouse models but is commonly observed with viral infections. Massive CD8+ T cell expansion has been observed with lymphocytic choriomeningitis, murine CMV, pichinde virus, and vaccinia virus and has been associated with CMV, Epstein-Barr virus, and Kawasaki disease in humans, which is thought to be caused by an infectious agent 50 51 52 53 54 55. Like virally induced CD8+ T cell expansions, CD8+ T cells in TGF-β DNRII Tg mice are associated with an oligoclonal expansion with a CD44hi phenotype. However, unlike virally induced CD8+ T cells, CD8+ T cells in TGF-β DNRII Tg mice do not express IL-2Rα, which could be upregulated by an antigen-specific release of IL-2 not found in TGF-β DNRII Tg mice (data not shown). Alternatively, expression of certain T cell activation markers could have been transiently altered 56 57.
Similar differential stimulation of CD8+ T cells over CD4+ T cells has also been observed with low doses of LPS in vivo 58. These responses, as well as the CD8+ T cell expansion seen in viral infections, are thought to be induced by type I IFN (IFN-I) secreted by APCs 59. This idea is supported by the results of Tough et al. 60 showing an increase in CD8+CD44hi T cells in mice injected with IFN-I. CD8+ T cells in these mice are CD69lo CD25−, suggesting that the T cells expanding in TGF-β DNRII Tg mice might be induced by a similar mechanism.
If similar mechanisms are involved, one could hypothesize that TGF-β normally acts as a negative regulator of CD8+ T cell expansion in an unchallenged immune system. When encountering antigen capable of stimulating an IFN-I response, TGF-β negative regulation is reversed, allowing CD8+ T cells to enter the cell cycle. This would result in an increased pool of available CD8+ T cells capable of interacting with the foreign antigen.
Several recent reports support this hypothesis. One report describes a direct inhibitory link between the IFN-γ signaling pathway and the TGF-β signaling pathway 61. As IFN-γ is also induced by many of the same agents that induce IFN-I, as well as by IFN-I itself, the presence of IFN-I could reverse the negative regulation of TGF-β, allowing CD8+ T cell expansion. A second report demonstrates that IFN-I can stimulate IL-15 production, which is capable of selectively expanding a CD8+CD44hi T cell population in vivo 45. CD8+ T cell homeostasis may thus involve a complex influence of many cytokines, with TGF-β acting as a major negative regulator.
Acknowledgments
We thank R. Hodes and M. Kuehn for helpful discussion and advice on the manuscript, J. Ward for analysis of histology and helpful discussion, S. Poer for excellent technical support, and S. Sharrow and the Experimental Immunology Branch FACS lab for helpful discussion and reagents.
P.J. Lucas is supported by an NIH Intramural Research Training Award fellowship.
Footnotes
Abbreviations used in this paper: CCM, complete culture medium; DNRII, dominant negative TGF-β type II receptor; FM, FACS media; HA, hemagglutinin; KO, knockout; RT, reverse transcriptase; Tg, transgenic.
References
- Kingsley D.M. The TGF-β superfamilynew members, new receptors, and new genetic tests of function in different organisms. Genes Dev. 1994;8:133–146. doi: 10.1101/gad.8.2.133. [DOI] [PubMed] [Google Scholar]
- Letterio J.J., Roberts A.B. Transforming growth factor-beta1-deficient miceidentification of isoform-specific activities in vivo. J. Leukoc. Biol. 1996;59:769–774. doi: 10.1002/jlb.59.6.769. [DOI] [PubMed] [Google Scholar]
- Ruscetti F., Varesio L., Ochoa A., Ortaldo J. Pleiotropic effects of transforming growth factor-beta on cells of the immune system. Ann. NY Acad. Sci. 1993;685:488–500. doi: 10.1111/j.1749-6632.1993.tb35911.x. [DOI] [PubMed] [Google Scholar]
- Wahl S.M., Allen J.B., Weeks B.S., Wong H.L., Klotman P.E. Transforming growth factor beta enhances integrin expression and type IV collagenase secretion in human monocytes. Proc. Natl. Acad. Sci. USA. 1993;90:4577–4581. doi: 10.1073/pnas.90.10.4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble J.R., Khew-Goodall Y., Vadas M.A. Transforming growth factor-beta inhibits E-selectin expression on human endothelial cells. J. Immunol. 1993;150:4494–4503. [PubMed] [Google Scholar]
- Hines K.L., Kulkarni A.B., McCarthy J.B., Tian H., Ward J.M., Christ M., McCartney-Francis N.L., Furcht L.T., Karlsson S., Wahl S.M. Synthetic fibronectin peptides interrupt inflammatory cell infiltration in transforming growth factor beta 1 knockout mice. Proc. Natl. Acad. Sci. USA. 1994;91:5187–5191. doi: 10.1073/pnas.91.11.5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutolo M., Sulli A., Barone A., Seriolo B., Accardo S. Macrophages, synovial tissue and rheumatoid arthritis. Clin. Exp. Rheumatol. 1993;11:331–339. [PubMed] [Google Scholar]
- Fontana A., Constam D.B., Frei K., Malipiero U., Pfister H.W. Modulation of the immune response by transforming growth factor beta. Int. Arch. Allergy Immunol. 1992;99:1–7. doi: 10.1159/000236328. [DOI] [PubMed] [Google Scholar]
- Vodovotz Y., Bogdan C., Paik J., Xie Q.W., Nathan C. Mechanisms of suppression of macrophage nitric oxide release by transforming growth factor beta. J. Exp. Med. 1993;178:605–613. doi: 10.1084/jem.178.2.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Young S.L. Requirement of transforming growth factor-beta (TGF-beta) type II receptor for TGF-beta-induced proliferation and growth inhibition. J. Biol. Chem. 1996;271:2369–2372. doi: 10.1074/jbc.271.5.2369. [DOI] [PubMed] [Google Scholar]
- Lowrance J.H., O'Sullivan F.X., Caver T.E., Waegell W., Gresham H.D. Spontaneous elaboration of transforming growth factor beta suppresses host defense against bacterial infection in autoimmune MRL/lpr mice. J. Exp. Med. 1994;180:1693–1703. doi: 10.1084/jem.180.5.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young M.R., Wright M.A., Matthews J.P., Malik I., Prechel M. Suppression of T cell proliferation by tumor-induced granulocyte-macrophage progenitor cells producing transforming growth factor-β and nitric oxide. J. Immunol. 1996;156:1916–1922. [PubMed] [Google Scholar]
- Shull M.M., Ormsby I., Kier A.B., Pawlowski S., Diebold R.J., Yin M., Allen R., Sidman C., Proetzel G., Calvin D. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni A.B., Huh C.G., Becker D., Geiser A., Lyght M., Flanders K.C., Roberts A.B., Sporn M.B., Ward J.M., Karlsson S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold R.J., Eis M.J., Yin M., Ormsby I., Boivin G.P., Darrow B.J., Saffitz J.E., Doetschman T. Early-onset multifocal inflammation in the transforming growth factor beta 1-null mouse is lymphocyte mediated. Proc. Natl. Acad. Sci. USA. 1995;92:12215–12219. doi: 10.1073/pnas.92.26.12215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koevary S.B. Prevention of diabetes in BB/Wor rats by injection of peritoneal exudate cells cultured in the presence of transforming growth factor beta (TGF-beta) and islet cells. Diabetes Res. 1994;27:1–14. [PubMed] [Google Scholar]
- Dang H., Geiser A.G., Letterio J.J., Nakabayashi T., Kong L., Fernandes G., Talal N. SLE-like autoantibodies and Sjogren's syndrome-like lymphoproliferation in TGF-beta knockout mice. J. Immunol. 1995;155:3205–3212. [PubMed] [Google Scholar]
- Letterio J.J., Geiser A.G., Kulkarni A.B., Dang H., Kong L., Nakabayashi T., Mackall C.L., Gress R.E., Roberts A.B. Autoimmunity associated with TGF-beta1-deficiency in mice is dependent on MHC class II antigen expression. J. Clin. Invest. 1996;98:2109–2119. doi: 10.1172/JCI119017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler S., Paiha S., Winkler H., Graninger W., Marberger M., Steiner G.E. Microfilarial clearance in loiasis involves elevation of Th1 and Th2 products and emergence of a specific pattern of T-cell populations. Parasite Immunol. 1996;18:479–482. doi: 10.1111/j.1365-3024.1996.tb01032.x. [DOI] [PubMed] [Google Scholar]
- Morris M.M., Dyson H., Baker D., Harbige L.S., Fazakerley J.K., Amor S. Characterization of the cellular and cytokine response in the central nervous system following Semliki Forest virus infection. J. Neuroimmunol. 1997;74:185–197. doi: 10.1016/s0165-5728(96)00786-2. [DOI] [PubMed] [Google Scholar]
- Kanagawa O., Vaupel B.A., Gayama S., Koehler G., Kopf M. Resistance of mice deficient in IL-4 to retrovirus-induced immunodeficiency syndrome (MAIDS) Science. 262 1993. 240 242[see comments] [DOI] [PubMed] [Google Scholar]
- Guler M.L., Gorham J.D., Hsieh C.S., Mackey A.J., Steen R.G., Dietrich W.F., Murphy K.M. Genetic susceptibility to LeishmaniaIL-12 responsiveness in TH1 cell development Science. 271 1996. 984 987[see comments] [DOI] [PubMed] [Google Scholar]
- Nakajima H., Shores E.W., Noguchi M., Leonard W.J. The common cytokine receptor gamma chain plays an essential role in regulating lymphoid homeostasis. J. Exp. Med. 1997;185:189–195. doi: 10.1084/jem.185.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn S.J., Forbush K.A., Nguyen N., Witthuhn B., Nosaka T., Ihle J.N., Perlmutter R.M. Requirement for Jak3 in mature T cellsits role in regulation of T cell homeostasis. J. Immunol. 1998;160:2130–2138. [PubMed] [Google Scholar]
- Lodolce J.P., Boone D.L., Chai S., Swain R.E., Dassopoulos T., Trettin S., Ma A. IL-15 receptor maintains lymphoid homeostasis by supporting lymphocyte homing and proliferation. Immunity. 1998;9:669–676. doi: 10.1016/s1074-7613(00)80664-0. [DOI] [PubMed] [Google Scholar]
- McElligott D.L., Phillips J.A., Stillman C.A., Koch R.J., Mosier D.E., Hobbs M.V. CD4+ T cells from IRF-1-deficient mice exhibit altered patterns of cytokine expression and cell subset homeostasis. J. Immunol. 1997;159:4180–4186. [PubMed] [Google Scholar]
- Matsuyama T., Kimura T., Kitagawa M., Pfeffer K., Kawakami T., Watanabe N., Kundig T.M., Amakawa R., Kishihara K., Wakeham A. Targeted disruption of IRF-1 or IRF-2 results in abnormal type I IFN gene induction and aberrant lymphocyte development. Cell. 1993;75:83–97. [PubMed] [Google Scholar]
- Sadlack B., Lohler J., Schorle H., Klebb G., Haber H., Sickel E., Noelle R.J., Horak I. Generalized autoimmune disease in interleukin-2-deficient mice is triggered by an uncontrolled activation and proliferation of CD4+ T cells. Eur. J. Immunol. 1995;25:3053–3059. doi: 10.1002/eji.1830251111. [DOI] [PubMed] [Google Scholar]
- Senju S., Negishi I., Motoyama N., Wang F., Nakayama K.I., Nakayama K., Lucas P.J., Hatakeyama S., Zhang Q., Yonehara S. Functional significance of the Fas molecule in naive lymphocytes. Int. Immunol. 1996;8:423–431. doi: 10.1093/intimm/8.3.423. [DOI] [PubMed] [Google Scholar]
- Chambers C.A., Sullivan T.J., Allison J.P. Lymphoproliferation in CTLA-4-deficient mice is mediated by costimulation-dependent activation of CD4+ T cells. Immunity. 1997;7:885–895. doi: 10.1016/s1074-7613(00)80406-9. [DOI] [PubMed] [Google Scholar]
- Ebner R., Chen R.H., Shum L., Lawler S., Zioncheck T.F., Lee A., Lopez A.R., Derynck R. Cloning of a type I TGF-beta receptor and its effect on TGF-beta binding to the type II receptor. Science. 1993;260:1344–1348. doi: 10.1126/science.8388127. [DOI] [PubMed] [Google Scholar]
- Chen R.H., Ebner R., Derynck R. Inactivation of the type II receptor reveals two receptor pathways for the diverse TGF-beta activities. Science. 1993;260:1335–1338. doi: 10.1126/science.8388126. [DOI] [PubMed] [Google Scholar]
- Zhumabekov T., Corbella P., Tolaini M., Kioussis D. Improved version of a human CD2 minigene based vector for T cell-specific expression in transgenic mice. J. Immunol. Methods. 1995;185:133–140. doi: 10.1016/0022-1759(95)00124-s. [DOI] [PubMed] [Google Scholar]
- Waegell W.O., Higley H.R., Kincade P.W., Dasch J.R. Growth acceleration and stem cell expansion in Dexter-type cultures by neutralization of TGF-beta. Exp. Hematol. 1994;22:1051–1057. [PubMed] [Google Scholar]
- Lucas P.J., Negishi I., Nakayama K., Fields L.E., Loh D.Y. Naive CD28-deficient T cells can initiate but not sustain an in vitro antigen-specific immune response. J. Immunol. 1995;154:5757–5768. [PubMed] [Google Scholar]
- Park K., Kim S.-J., Bang Y.-J., Park J.-G., Kim N.K., Roberts A.A., Sporn M.B. Genetic changes in the transforming growth factor β (TGF-β) type II receptor gene in human gastric cancer cellscorrelation with sensitivity to growth inhibition by TGF-β. Proc. Natl. Acad. Sci. USA. 1994;91:8772–8776. doi: 10.1073/pnas.91.19.8772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahama Y., Letterio J.J., Suzuki H., Farr A.G., Singer A. Early progression of thymocytes along the CD4/CD8 developmental pathway is regulated by a subset of thymic epithelial cells expressing transforming growth factor beta. J. Exp. Med. 1994;179:1495–1506. doi: 10.1084/jem.179.5.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha B., Dautigny N., Pereira P. Peripheral T lymphocytesexpansion potential and homeostatic regulation of pool sizes and CD4/CD8 ratios in vivo. Eur. J. Immunol. 1989;19:905–911. doi: 10.1002/eji.1830190518. [DOI] [PubMed] [Google Scholar]
- Tanchot C., Rosado M.M., Agenes F., Freitas A.A., Rocha B. Lymphocyte homeostasis. Semin. Immunol. 1997;9:331–337. doi: 10.1006/smim.1997.0090. [DOI] [PubMed] [Google Scholar]
- Tanchot C., Rocha B. The peripheral T cell repertoireindependent homeostatic regulation of virgin and activated CD8+ T cell pools. Eur. J. Immunol. 1995;25:2127–2136. doi: 10.1002/eji.1830250802. [DOI] [PubMed] [Google Scholar]
- Wang X.J., Greenhalgh D.A., Bickenbach J.R., Jiang A., Bundman D.S., Krieg T., Derynck R., Roop D.R. Expression of a dominant-negative type II transforming growth factor beta (TGF-beta) receptor in the epidermis of transgenic mice blocks TGF-beta-mediated growth inhibition. Proc. Natl. Acad. Sci. USA. 1997;94:2386–2391. doi: 10.1073/pnas.94.6.2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspinall R. Age-associated thymic atrophy in the mouse is due to a deficiency affecting rearrangement of the TCR during intrathymic T cell development. J. Immunol. 1997;158:3037–3045. [PubMed] [Google Scholar]
- Greaves D.R., Wilson F.D., Lang G., Kioussis D. Human CD2 3′-flanking sequences confer high-level, T cell-specific, position-independent gene expression in transgenic mice. Cell. 1989;56:979–986. doi: 10.1016/0092-8674(89)90631-4. [DOI] [PubMed] [Google Scholar]
- Maraskovsky E., Teepe M., Morrissey P.J., Braddy S., Miller R.E., Lynch D.H., Peschon J.J. Impaired survival and proliferation in IL-7 receptor-deficient peripheral T cells. J. Immunol. 1996;157:5315–5323. [PubMed] [Google Scholar]
- Zhang X., Sun S., Hwang I., Tough D.F., Sprent J. Potent and selective stimulation of memory-phenotype CD8+ T cells in vivo by IL-15. Immunity. 1998;8:591–599. doi: 10.1016/s1074-7613(00)80564-6. [DOI] [PubMed] [Google Scholar]
- Willerford D.M., Chen J., Ferry J.A., Davidson L., Ma A., Alt F.W. Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity. 1995;3:521–530. doi: 10.1016/1074-7613(95)90180-9. [DOI] [PubMed] [Google Scholar]
- Suzuki H., Kundig T.M., Furlonger C., Wakeham A., Timms E., Matsuyama T., Schmits R., Simard J.J., Ohashi P.S., Griesser H. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor beta. Science. 1995;268:1472–1476. doi: 10.1126/science.7770771. [DOI] [PubMed] [Google Scholar]
- Waterhouse P., Marengere L.E., Mittrucker H.W., Mak T.W. CTLA-4, a negative regulator of T-lymphocyte activation. Immunol. Rev. 1996;153:183–207. doi: 10.1111/j.1600-065x.1996.tb00925.x. [DOI] [PubMed] [Google Scholar]
- Waterhouse P., Bachmann M.F., Penninger J.M., Ohashi P.S., Mak T.W. Normal thymic selection, normal viability and decreased lymphoproliferation in T cell receptor-transgenic CTLA-4-deficient mice. Eur. J. Immunol. 1997;27:1887–1892. doi: 10.1002/eji.1830270811. [DOI] [PubMed] [Google Scholar]
- Yang H.Y., Dundon P.L., Nahill S.R., Welsh R.M. Virus-induced polyclonal cytotoxic T lymphocyte stimulation. J. Immunol. 1989;142:1710–1718. [PubMed] [Google Scholar]
- Buchmeier M.J., Welsh R.M., Dutko F.J., Oldstone M.B. The virology and immunobiology of lymphocytic choriomeningitis virus infection. Adv. Immunol. 1980;30:275–331. doi: 10.1016/s0065-2776(08)60197-2. [DOI] [PubMed] [Google Scholar]
- Rubin R.H., Carney W.P., Schooley R.T., Colvin R.B., Burton R.C., Hoffman R.A., Hansen W.P., Cosimi A.B., Russell P.S., Hirsch M.S. The effect of infection on T lymphocyte subpopulationsa preliminary report. Int. J. Immunopharmacol. 1981;3:307–312. doi: 10.1016/0192-0561(81)90024-2. [DOI] [PubMed] [Google Scholar]
- Oldstone M.B., Buchmeier M.J., Doyle M.V., Tishon A. Virus-induced immune complex diseasespecific anti-viral antibody and C1q binding material in the circulation during persistent lymphocytic choriomeningitis virus infection. J. Immunol. 1980;124:831–838. [PubMed] [Google Scholar]
- Butz E.A., Bevan M.J. Massive expansion of antigen-specific CD8+ T cells during an acute virus infection. Immunity. 1998;8:167–175. doi: 10.1016/s1074-7613(00)80469-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi I.H., Chwae Y.J., Shim W.S., Kim D.S., Kwon D.H., Kim J.D., Kim S.J. Clonal expansion of CD8+ T cells in Kawasaki disease. J. Immunol. 1997;159:481–486. [PubMed] [Google Scholar]
- Topham D.J., Tripp R.A., Hamilton-Easton A.M., Sarawar S.R., Doherty P.C. Quantitative analysis of the influenza virus-specific CD4+ T cell memory in the absence of B cells and Ig. J. Immunol. 1996;157:2947–2952. [PubMed] [Google Scholar]
- Zimmerman C., Brduscha-Riem K., Blaser C., Zinkernagel R.M., Pircher H. Visualization, characterization, and turnover of CD8+ memory T cells in virus-infected hosts. J. Exp. Med. 1996;183:1367–1375. doi: 10.1084/jem.183.4.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tough D.F., Sun S., Sprent J. T cell stimulation in vivo by lipopolysaccharide (LPS) J. Exp. Med. 1997;185:2089–2094. doi: 10.1084/jem.185.12.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprent J., Tough D.F. Lymphocyte life-span and memory. Science. 1994;265:1395–1400. doi: 10.1126/science.8073282. [DOI] [PubMed] [Google Scholar]
- Tough D.F., Borrow P., Sprent J. Induction of bystander T cell proliferation by viruses and type I interferon in vivo Science. 272 1996. 1947 1950[see comments] [DOI] [PubMed] [Google Scholar]
- Ulloa L., Doody J., Massague J. Inhibition of transforming growth factor-beta/SMAD signalling by the interferon-gamma/STAT pathway. Nature. 1999;397:710–713. doi: 10.1038/17826. [DOI] [PubMed] [Google Scholar]