

Abstract
Cytotoxic T lymphocytes (CTLs) destroy target cells through a mechanism involving the exocytosis of cytolytic granule components including granzyme B (grB) and perforin, which have been shown to induce apoptosis through caspase activation. However, grB has also been linked with caspase-independent disruption of mitochondrial function. We show here that cytochrome c release requires the direct proteolytic cleavage of Bid by grB to generate a 14-kD grB-truncated product (gtBid) that translocates to mitochondria. In turn, gtBid recruits Bax to mitochondria through a caspase-independent mechanism where it becomes integrated into the membrane and induces cytochrome c release. Our results provide evidence for a new pathway by which CTLs inflict damage and explain the caspase-independent mechanism of mitochondrial dysfunction.
Keywords: cytotoxic T lymphocyte, granzyme B, Bcl-2, Bid, Bax
Introduction
CTLs are cells which participate in adaptive immune responses such as the elimination of virus-infected and tumor cells, as well as transplant rejection 1 2 3 4. The destruction of target cells is believed to involve either engagement of the Fas receptor or granule exocytosis. Fas ligation and oligomerization lead to the formation of the death-inducing signaling complex (DISC), which begins with recruitment of Fas-associated death domain (FADD)/mediator of receptor-induced toxicity (MORT)1 to the cytoplasmic death domains of Fas 5. FADD acts as an adapter molecule between the Fas receptor and the prodomain of caspase-8 6 7, a member of the caspase family of cysteine proteinases (cysteinyl aspases). These enzymes exist in cells as inactive zymogens but are proteolytically cleaved following aspartate residues to become activated in response to an apoptotic stimulus (for reviews, see references 8 9 10). The recruitment of caspase-8 to the DISC brings identical molecules into close proximity and allows for catalytic activation. In cells where caspase-8 is efficiently recruited to the DISC, activated caspase-8 directly acts on caspase-3 11 to initiate the effector stages of the apoptotic program, including chromatin condensation, DNA fragmentation, membrane alterations, and cytoskeletal rearrangements 12 13 14 15 16. However, in cells such as Jurkat, where recruitment of capsase-8 to the DISC is relatively inefficient, indirect activation of caspase-3 mediated through a mitochondrial pathway takes precedence. This occurs through proteolytic activation of Bid, a proapoptotic member of the Bcl-2 family 17 18 19. Caspase-8 cleavage of 26-kD human Bid following Asp59 generates a COOH-terminal cleavage product of 15 kD 17 18 20. Truncated (t)Bid translocates to mitochondria where it causes the permeabilization of the outer mitochondrial membrane through a poorly defined mechanism leading to the release of cytochrome c from the intermembrane space 17 18 19. Once released to the cytoplasm, cytochrome c acts as a cofactor in conjunction with Apaf-1, procaspase-9, and ATP/dATP to induce activation of caspase-9 and subsequently caspase-3 21 22.
The second mechanism used by CTLs to kill targets involves the calcium-dependent exocytosis of CTL-derived granule proteins and their uptake by the target cell. Among the granule components are the pore-forming protein perforin and several granule proteinases or granzymes, which gain entry into the target cell through a poorly defined mechanism (for reviews, see references 1, 2 3 4). Granzyme B (grB), the prototypic member of the granzymes, is a serine proteinase with a unique specificity among mammalian serine proteinases for cleavage on the carboxyl side of aspartate residues 23. Several caspases contain cleavage sites potentially recognized by grB 24 25, implying that grB may activate caspases in the target cell after uptake. Indeed, caspases, including caspase-3 (CPP32/Apopain/Yama), caspase-6 (Mch2), caspase-7 (Mch3/CMH-1/ICE-LAP3), caspase-8 (FLICE/MACH/Mch5), capsase-9 (ICE-LAP6/Mch6), and caspase-10 (Mch4), are cleaved by grB in vitro 6 26 27 28 29 30 31 32 33 34. In addition, processed caspases-1 (ICE), -3, -7, and -8 are found in whole cells after treatment with purified grB and perforin or a replication-deficient adenovirus (Ad), or in response to treatment with human CTLs 29 35 36 37 38.
GrB can proteolytically cleave caspase-3 both in vitro and in whole cells in the presence of the broad-spectrum caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone (zVAD-fmk [26, 36, 37]). This implies that grB directly activates caspase-3, leading to apoptosis. In addition, it was recently shown that treatment of targets with grB and Ad or perforin, or whole CTLs, leads to mitochondrial disruptions through a caspase-independent mechanism 39 40. These data suggest that grB can also induce necrotic cell death without caspase activation. However, it was not known whether grB acted directly on mitochondria or indirectly through the proteolytic activation or inactivation of a cellular substrate(s).
We show here that Bid is required for the release of cytochrome c in response to grB. Bid is known to be cleaved by grB to generate a 14-kD grB-truncated Bid fragment (herein designated gtBid) that is distinct from tBid 18 35. Here we present evidence that, once generated, gtBid translocates to mitochondria where it recruits the proapoptotic Bcl-2 family member, Bax, to the mitochondrial membrane. Bax becomes integrated into the membrane to induce release of cytochrome c through a caspase-independent mechanism. Importantly, Bcl-2 does not inhibit Bid cleavage and translocation to mitochondria but does prevent Bax insertion and cytochrome c release after treatment with grB.
Materials and Methods
Cell Culture and Reagents.
The human T cell lymphoma line Jurkat (American Type Culture Collection) was grown in RPMI (Life Technologies) supplemented with 10% fetal bovine serum (Hyclone) and 100 μM 2-mercaptoethanol (RHFM). GrB was purified as described previously 41. Stable transfected cells were prepared as follows. The Bcl-2 gene was subcloned into the XhoI site of the eukaryotic expression vector BMGneo 42. Jurkat cells were stably transfected with 10 μg of NotI linearized DNA by electroporation (250 V, 250 μF). Stably transfected cells were selected in RHFM containing 1 mg/ml of G418 (Life Technologies) and cloned by limiting dilution. After selection, cells were routinely grown in RHFM containing 800 μg/ml of G418. Jack Gauldie (McMaster University, Hamilton, Ontario, Canada) provided replication-deficient Ad (type 5). zVAD-fmk was from Kamiya Biochemicals. Anti–human Fas IgM antibody (clone CH11) was from Upstate Biotechnology. Monoclonal anti–human cytochrome c antibody (clone 7H8.2C12) was from BD PharMingen. Rabbit polyclonal anti-Bax antiserum (N-20) was from Santa Cruz Biotechnology, Inc. The goat anti–mouse and goat anti–rabbit horseradish peroxidase (HRP)-conjugated secondary antibodies were from Bio-Rad Laboratories. The rabbit anti–mouse Bid antiserum and the pET-15b vector containing His-tagged human Bid were provided by Xiaodong Wang (Howard Hughes Medical Institute, Dallas, TX). Sigma-Aldrich provided all other reagents unless otherwise stated.
Subcellular Fractionation.
Subcellular fractionation of Jurkat cells was performed as described 43. In brief, 1–6 × 108 cells were collected for 10 min at 2,000 g (3,500 rpm in a Sorvall GSA rotor) and washed once with PBS. The pellet was washed once with buffer A (20 mM morpholino propane sulfonic acid [MOPS], pH 7.4, 100 mM sucrose, 1 mM EGTA) and then resuspended in a volume of buffer B (20 mM MOPS, pH 7.4, 100 mM sucrose, 1 mM EGTA, 5% Percoll, and 191 μg/ml digitonin) giving a final cell density of 2 × 107 cells/ml. After a 15-min incubation on ice with occasional stirring, the cells were spun at 2,500 g (4,500 rpm in a Sorvall SS-34 rotor) for 10 min at 4°C. The pellet, containing nuclei and cell debris, was discarded. The supernatant was further fractionated by centrifugation at 15,000 g (11,500 rpm in a Sorvall SS-34 rotor) for 15 min at 4°C. The mitochondrial fraction, a loose fluffy layer at the bottom of the tube, was collected, washed three times with buffer A, and then resuspended in buffer A. The supernatant was spun at 100,000 g (39,000 rpm in a Beckman 70Ti rotor) for 1 h at 4°C. The S-100 cytosolic fraction is herein referred to as the cytosol. Protein concentrations were determined using a bicinchoninic acid (BCA) kit (Pierce Chemical Co.).
Expression and Purification of Recombinant Bid.
His-tagged human rBid in the pET-15b vector was expressed in competent BL21 Escherichia coli and purified as described 17.
Immunodepletion of Bid from Jurkat Cytosol.
Anti–human Bid antibodies (C-20; Santa Cruz Biotechnology, Inc.; or PBS alone for the mock control) were incubated in 325 μl PBS containing 4.5% protein A– and protein G–agarose (Amersham Pharmacia Biotech) for 3 h at 4°C with rocking. The antibody-bound protein A/G beads were washed in buffer A and then incubated with 170 μg of Jurkat cytosol at 4°C for 18 h with rocking. The agarose beads were then pelleted and the resulting supernatants were labeled as C (−Bid) or C (mock) for Bid-depleted or mock-depleted cytosol, respectively. Immunodepletion of Bid was verified by Western blotting.
In Vitro Assays.
Purified mitochondria (10–20 μg) were mixed either with an equivalent amount of cytosol (10–20 μg) or an equivalent volume of buffer A alone as indicated. GrB (0.5 μg) was added for 30 min at room temperature in the presence or absence of 100 μM zVAD-fmk. The mixtures were then spun for 5 min at 16,000 g (14,000 rpm in an Eppendorf tabletop microfuge). The supernatants were transferred to fresh tubes and the pellets (mitochondria) were resuspended in a volume of buffer A equivalent to the initial sample volume. Pellets and supernatants were then mixed with 6× SDS loading buffer, boiled for 10 min, and loaded onto 15% SDS–polyacrylamide gels. Proteins were resolved at 200 V for ∼50 min and subsequently transferred to nitrocellulose (Micron Separations Inc.) at 150 mA for 1.25 h in a semidry blotting apparatus (Tyler Instruments Inc.). Membranes were blocked overnight in 5% milk proteins (Carnation) in PBST (PBS plus 0.1% Tween 20 [Fisher Scientific]). Proteins were visualized with a monoclonal anti–human cytochrome c antibody (1:2,000), followed by a goat anti–mouse HRP-conjugated secondary antibody (1:3,000), followed by enzyme-linked chemiluminescence (Amersham Pharmacia Biotech). Immunoblotting for Bid and Bax was performed as for cytochrome c with the following modifications. Rabbit anti–mouse Bid (which cross-reacts with human) was used at 1:4,000 to 1:8,000. The goat anti–rabbit HRP-conjugated secondary was used at 1:20,000. Rabbit anti–human Bax was used at 1:400 to 1:1,000.
Alkaline Extraction of Mitochondria.
Mitochondria were incubated under the conditions indicated. After incubation, mitochondria were centrifuged at 16,000 g (14,000 rpm in an Eppendorf tabletop microfuge) for 10 min at 4°C. The supernatants (preextraction supernatants) were removed, and to them was added 6× SDS loading buffer followed by boiling for 10 min. The mitochondria were resuspended in 0.1 M Na2CO3 for 30 min on ice. After this incubation, the extracted mitochondria were centrifuged at 100,000 g (39,000 rpm in a Beckman Ti100.2 rotor) for 10 min at 4°C. The supernatants were discarded and the pellets were resuspended in a volume of buffer A equivalent to the volume of the starting samples as well as 6× SDS loading buffer. These samples were boiled for 10 min and the extracted pellets and preextraction supernatants were resolved on 15% polyacrylamide gels followed by transfer to nitrocellulose. The blots were probed for Bax as outlined above.
Whole Cell Assays.
Jurkat cells were treated with grB and Ad in the presence or absence of 100 μM zVAD-fmk and the mitochondrial and cytosolic fractions were separated as described previously 39. Immunoblotting was performed as outlined above.
Alkaline Extraction of Whole Cells.
Jurkat cells (8 × 106/sample) were treated as indicated. After treatment, cells were washed once with PBS and then resuspended in 400 μl digitonin lysis buffer (75 mM NaCl, 1 mM NaH2PO4, 8 mM Na2HPO4, 250 mM sucrose, and 191 μg/ml digitonin). Cells were incubated on ice for 15 min, followed by centrifugation at 300 g (2,000 rpm in an Eppendorf microfuge) at 4°C. The pellet, containing nuclei and unbroken cells, was discarded. The supernatant was then centrifuged at 16,000 g (14,000 rpm in an Eppendorf microfuge) for 15 min at 4°C. The supernatant contained the cytosol. The pellet, containing mitochondria, was subjected to alkaline extraction with 400 μl of 0.1 M Na2CO3 for 30 min on ice followed by centrifugation at 100,000 g (39,000 rpm in a Beckman 100.2 rotor) for 10 min at 4°C. The supernatant was discarded and the membrane pellets were resuspended with 6× SDS loading buffer followed by boiling. The preextraction supernatant (cytosol) and the extracted mitochondrial proteins were resolved by SDS-PAGE, followed by transfer to nitrocellulose and immunoblotting for Bax.
Densitometric Analysis.
Blots were scanned at a resolution of 400 pixels per inch using an Agfa Arcus II scanner with Agfa FotoLook Software (v3.0) and imported into Adobe Photoshop® (v5.5). The scanned images were saved as grayscale PICT files and imported into Fuji Photo Film Co. Image Gauge software (v3.0). An area of 84 square pixels was then placed about each band and the pixel density was determined by the software. Values were plotted as pixel density per unit area and are displayed as arbitrary units.
Results
A Cytosolic Factor Is Required for GrB-mediated Cytochrome c Release.
We have previously shown that grB can induce the loss of inner mitochondrial membrane potential and cytochrome c efflux from mitochondria through a caspase-independent mechanism 39. To investigate whether grB acted alone or in concert with additional factors, we developed an in vitro system that used purified mitochondria from Jurkat cells. Jurkat mitochondria were incubated either with grB alone or Jurkat cytosol and grB in various combinations for 30 min at room temperature either in the presence or absence of the broad-spectrum caspase inhibitor, zVAD-fmk. After this incubation, the mixture was centrifuged and the mitochondrial pellets were separated from the supernatants. Proteins in the separated samples were resolved by electrophoresis and examined for the presence of cytochrome c by immunoblotting (Fig. 1).
Figure 1.
GrB-mediated cytochrome c release from isolated mitochondria requires Bid. Purified Jurkat mitochondria were treated with grB and/or Jurkat cytosol for 30 min at room temperature in the presence or absence of 100 μM zVAD-fmk. The mitochondria/cytosol mixture was then centrifuged and the pellets (mitochondria) and supernatants (cytosol) were resolved on 15% polyacrylamide gels followed by transfer to nitrocellulose and immunoblotting for cytochrome c. M, mitochondria; C, cytosol; C (mock), mock immunodepleted cytosol; C (−Bid), cytosol immunodepleted for Bid. These data are representative of three independent experiments.


In the absence of treatment, cytochrome c was detectable only in the mitochondrial pellet and not in the supernatant (Fig. 1 A, lane 1, and compare lanes 12 and 2). When cytosol (Fig. 1 A, lanes 13 and 3) or grB (Fig. 1 A, lanes 14 and 4) was added separately to mitochondria, cytochrome c release was not detected. However, when grB was added to mitochondria in conjunction with cytosol, cytochrome c efflux from mitochondria was observed (Fig. 1 A, compare lanes 15 and 5). Importantly, the same results were observed in the presence of 100 μM zVAD-fmk (Fig. 1 A, compare lanes 33 and 24). Therefore, cytochrome c release did not occur through a direct interaction of grB with mitochondria. Rather, a zVAD-fmk–insensitive cytosolic factor (or factors) was required.
Bid Is Required for GrB-mediated Cytochrome c Release.
One possible candidate for the cytosolic factor required by grB to elicit cytochrome c release was the proapoptotic Bcl-2 family member Bid. As outlined in the introduction, Bid has been shown to play a role in apoptosis induced after ligation of the Fas and TNF receptors 17 18 19. In addition to the caspase cleavage site at Asp59, Bid contains an excellent cleavage site for grB (IEAD75). We, and others, have recently shown that in the presence of grB, in vitro–transcribed/translated Bid is cut into a 14-kD product 18 35. Moreover, Bid cleavage occurs in whole cells after treatment with grB and Ad 35.
To determine if grB-mediated cytochrome c efflux was dependent on Bid, we immunodepleted Bid from the cytosolic extract before its addition to mitochondria plus grB. The efficiency of Bid immunodepletion was assessed by Western blotting (Fig. 1 B). Mock-immunodepleted cytosol generated the same cytochrome c efflux pattern as nonimmunodepleted cytosol (Fig. 1 A, compare lanes 6 and 16 with lanes 15 and 5). In contrast, when the Bid-depleted cytosol was added to mitochondria in the presence of grB, no release of cytochrome c was observed (Fig. 1 A, compare lanes 7 and 17). When rBid was added back to the immunodepleted cytosol, cytochrome c efflux was restored but only upon the addition of grB (Fig. 1 A, lanes 8 and 18). These data suggested that the loss of cytochrome c from mitochondria in response to grB treatment was dependent on the presence of cytosolic Bid.
To confirm the dependence of cytochrome c release on Bid, mitochondria were treated with rBid in the absence of cytosol and in the presence or absence of grB. Without grB, rBid was unable to induce cytochrome c release from mitochondria (Fig. 1 A, lanes 9 and 19). Importantly, in contrast to rBid alone, treatment of mitochondria with rBid in conjunction with grB led to the complete loss of cytochrome c (Fig. 1 A, lanes 10 and 20). These results imply that grB-dependent proteolytic cleavage of Bid leads to the generation of a fragment that is active in mediating effects on mitochondria.
These experiments were repeated in the presence of zVAD-fmk with identical results (Fig. 1 A, compare lanes 21–29 to 30–38). Thus, the grB-mediated cytochrome c efflux in this system occurred independently of caspases, consistent with the caspase-independent cytochrome c loss in whole cells seen after treatment with grB and Ad 39. Collectively, these data show that Bid is necessary for grB-mediated cytochrome c release from mitochondria, after direct activation by this proteinase.
GrB-mediated Target Cell Death Induces Translocation of gtBid from the Cytosol to Mitochondria.
In response to ligation of Fas or TNF receptors, Bid is cleaved by caspase-8 to generate 15-kD tBid. Translocation of tBid from the cytosol to mitochondria has been shown to be critical for cytochrome c release to occur 17 18 19. We wished to know if the smaller, granzyme-cleaved gtBid fragment also translocated to mitochondria in whole cells in response to grB and Ad.
Jurkats cells were treated either with soluble anti-Fas antibodies or with grB and Ad for the times indicated in the presence or absence of zVAD-fmk. After treatment, cells were rendered permeable with digitonin and the mitochondrial fraction was separated from the cytosol as described previously 39. The proteins from these fractions were examined for Bid by immunoblotting (Fig. 2 A). We first confirmed the translocation of tBid in response to anti-Fas treatment (Fig. 2 A, lanes 1–12). In the absence of zVAD-fmk, cytosolic Bid was cleaved into 15-kD tBid, which was detectable in the mitochondrial fraction within 2–4 h after anti-Fas addition (Fig. 2 A, lanes 5 and 6). After longer incubation times, a greater quantity of tBid was seen in the mitochondrial fraction (Fig. 2 A, compare lanes 5 and 6). In the presence of zVAD-fmk, the generation of tBid in response to Fas receptor ligation and its accumulation in mitochondria was not observed. These data are consistent with those published previously 17 18.
Figure 2.

Cytosolic Bid is cleaved and translocates to mitochondria in response to grB and Ad. (A) Jurkat targets were treated with either anti-Fas antibodies (lanes 1–12) or grB and Ad (lanes 13–20) in the presence or absence of 100 μM zVAD-fmk. After incubation at 37°C for the times indicated, the plasma membranes of cells were rendered permeable with digitonin. The membrane fraction (containing mitochondria) was then separated from the cytosol by centrifugation. The pellets (mitochondrial fraction) were resolved on 15% polyacrylamide gels followed by transfer to nitrocellulose and immunoblotting for Bid using a rabbit polyclonal anti-Bid antiserum. (B) The supernatant blots from A were stripped and reprobed for cytochrome c using a monoclonal anti–cytochrome c antibody. These data are representative of three independent experiments.
In response to treatment with grB and Ad, 14-kD gtBid was generated that was detectable in the mitochondrial fraction within 1 h after treatment (Fig. 2 A, lanes 15 and 16). In addition to 14-kD gtBid, 15-kD tBid was also generated which translocated to mitochondria (Fig. 2 A, lane 16). However, in the presence of zVAD-fmk only 14-kD gtBid was seen in the mitochondria (Fig. 2 A, lane 20), indicating that the 15-kD band was likely caspase-8–generated tBid.
The blots shown in Fig. 2 A were stripped and reprobed for cytochrome c as a control for mitochondrial dysfunction (Fig. 2 B). After anti-Fas treatment, cytochrome c release was observed (Fig. 2 B, lanes 1–6), detectable cytochrome c release corresponding to the appearance of detectable tBid at the mitochondria (compare lanes 4–6, Fig. 2 A with lanes 4–6, Fig. 2 B). In the presence of zVAD-fmk, no cytochrome c release was observed after anti-Fas treatment (Fig. 2 B, compare lanes 1–6 and 7–12), consistent with the caspase dependence of Fas-mediated cytochrome c release in Jurkats. Similarly, grB-dependent loss of cytochrome c from mitochondria was observed (Fig. 2 B, lane 16) at the same time as gtBid was detected associated with mitochondria (compare lanes 13–20, Fig. 2 A, with lanes 13–20, Fig. 2 B). However, unlike anti-Fas treatment, cytochrome c efflux occurred with similar kinetics in the presence of zVAD-fmk after treatment with grB and Ad (Fig. 2 B, compare lanes 15 and 16 with 19 and 20). Thus, in whole cells, Bid cleavage by grB generated 14-kD gtBid, which translocated to mitochondria coincident with the efflux of cytochrome c.
Bax Is Integrated into the Mitochondrial Membrane after GrB and Adenovirus Treatment.
Like Bid, another proapoptotic Bcl-2 family member, Bax, has been implicated in cytochrome c release 44 45 46 47. In some cases, Bid activation leads to a translocation or conformational change in Bax and its subsequent insertion into the outer mitochondrial membrane 48 49 50 51 52. To investigate whether Bax might be involved in grB-mediated cytochrome c release, we used our whole cell killing system as indicated. Mitochondria were isolated from Jurkat targets after treatment with grB and Ad and then subjected to alkaline extraction in 0.1 M Na2CO3, which strips away proteins peripherally associated with membranes while leaving integral membrane proteins intact 52. Bax was found in the cytosolic fraction and peripherally associated with mitochondria (data not shown) but not integrated into the mitochondrial membrane in control cells (Fig. 3, compare lanes 1 and 6) or cells treated with either grB alone (Fig. 3, lanes 2 and 7) or Ad alone (Fig. 3, lanes 3 and 8). However, after incubation with both grB and Ad together, the levels of cytosolic Bax decreased (Fig. 3, compare lanes 1 and 4) and Bax was found integrated into mitochondria (Fig. 3, lane 9). Loss of Bax from the cytosol and its appearance in the mitochondrial membrane were also observed in the presence of zVAD-fmk (Fig. 3, lane 10). These data suggest that in response to treatment with grB and Ad, Bax becomes integrated into the mitochondrial membranes of targets through a caspase-independent process.
Figure 3.

Bax is integrated into the mitochondrial membrane in response to grB and Ad. Jurkat targets were treated either with nothing (control [ctrl]), grB alone, Ad alone, or grB and Ad (grB/Ad) in the presence or absence of 100 μM zVAD-fmk for 3 h at 37°C. After incubation, the plasma membranes of cells were rendered permeable with digitonin. The permeable cells were then centrifuged to separate the pellets (containing mitochondria) from the supernatants (cytosol). Pellets were subjected to an alkaline extraction with 0.1 M Na2CO3 for 30 min on ice followed by centrifugation. The pellets and the prealkaline extraction supernatants were resolved on 15% polyacrylamide gels. Proteins were transferred to nitrocellulose and the blots were probed with polyclonal anti-Bax antibodies. These data are representative of three independent experiments.
GrB-mediated Integration of Bax into the Mitochondrial Membrane Is Dependent on Bid.
As both gtBid translocation to mitochondria and Bax integration into mitochondrial membranes occurred in response to grB and Ad in whole cells (Fig. 2 A and 3), we sought to elucidate the relationship between Bax and Bid in grB-mediated cytochrome c release. To this end, we treated isolated Jurkat mitochondria under various conditions followed by alkaline extraction (Fig. 4). Bax was not found integrated with either mitochondria alone or mitochondria treated with Jurkat cytosol (Fig. 4, lanes 1 and 2). However, when mitochondria were exposed to cytosol in conjunction with grB, Bax was integrated into the membrane as indicated by the increase in the intensity of Bax immunolabeling in alkali-treated mitochondria (Fig. 4, lane 3). When mock-immunodepleted cytosol was used in place of regular Jurkat cytosol, no difference in Bax insertion was observed (Fig. 4, lane 4). In contrast, when Bid was immunodepleted from Jurkat cytosol and the cytosol subsequently added to mitochondria in combination with grB, Bax insertion was abrogated (Fig. 4, lane 5). Indeed, in the absence of any other cytosolic components, rBid in combination with grB was able to induce Bax integration into the mitochondrial membrane (Fig. 4, lane 7). Detectable levels of Bax are found peripherally associated with Jurkat mitochondria (data not shown), which provided a source of Bax in the absence of cytosol. Presumably, the requirement for Bax insertion was gtBid, as Bax insertion did not occur with rBid in the absence of grB (Fig. 4, lane 6). We observed the same results in the presence of zVAD-fmk (Fig. 4, lanes 8–12), which indicated that in the granzyme system, caspases are not required for Bax insertion. GrB-mediated Bax integration into rat heart mitochondria was also dependent on Bid when HeLa cytosolic extracts were used (data not shown)
Figure 4.

GrB-mediated Bax integration into mitochondria requires Bid. Purified Jurkat mitochondria (M) were treated with grB and cytosol (C) for 30 min at room temperature in the presence or absence of 100 μM zVAD-fmk. Mitochondria were also treated with grB in the presence of mock-immunodepleted cytosol (C [mock]) and cytosol immunodepleted for Bid (C [−Bid]) as well as rBid. After a 30-min incubation at room temperature, the mitochondria were subjected to an alkaline extraction with 0.1 M Na2CO3 for 30 min on ice followed by centrifugation. The pellets were resuspended in buffer A and resolved on 15% polyacrylamide gels, followed by transfer to nitrocellulose and immunoblotting for Bax. These data are representative of three independent experiments.
Bcl-2 Does Not Block GrB-mediated Bid Cleavage and Translocation but Does Prevent Bax Insertion and Cytochrome c Release.
Both Bid and Bax have been implicated in cytochrome c release 17 18 19 44 45 46 47. In our system, Bax insertion into mitochondria did not occur unless Bid was also present and processed by grB (Fig. 4). We wanted to determine if both Bid and Bax were required for grB-mediated cytochrome c release and if so, to determine the order of recruitment of these proteins. The easiest method to investigate this would have been to immunodeplete Bax from cytosolic extracts and examine this effect on mitochondria in the presence of grB. However, Jurkat mitochondria have significant levels of peripherally associated Bax after isolation (data not shown), precluding such an experimental approach. Nevertheless, Bcl-2 overexpression has been shown to block Bax translocation to mitochondria and subsequent cytochrome c release 53 54 55 56. We therefore examined the effect of grB on Bid and Bax in Jurkat cells overexpressing Bcl-2 and in vector-only transfectants (Fig. 5).
Figure 5.
Bcl-2 blocks grB-mediated Bax insertion and cytochrome c release but not Bid processing and translocation. (A) Vector-only transfected controls (neo) and Bcl-2–overexpressing Jurkats were treated with grB and Ad for the times indicated. After incubation, cells were fractionated and the proteins were resolved by SDS-PAGE on 15% gels and transferred to nitrocellulose. Cytosols were probed with a monoclonal anti–cytochrome c antibody. (B) The mitochondrial fraction from A was probed for Bid using a polyclonal anti-Bid antiserum. (C) Jurkat and Bcl-2–overexpressing Jurkat cells were treated with grB and Ad as indicated followed by subfractionation as described in Materials and Methods. The mitochondria were subjected to alkaline extraction followed by SDS-PAGE on 15% gels and transferred to nitrocellulose. Blots were probed with polyclonal anti-Bax antibodies. (D) Mitochondrial fractions from C were overexposed, scanned, and then analyzed with Image Gauge software to determine relative band intensities.

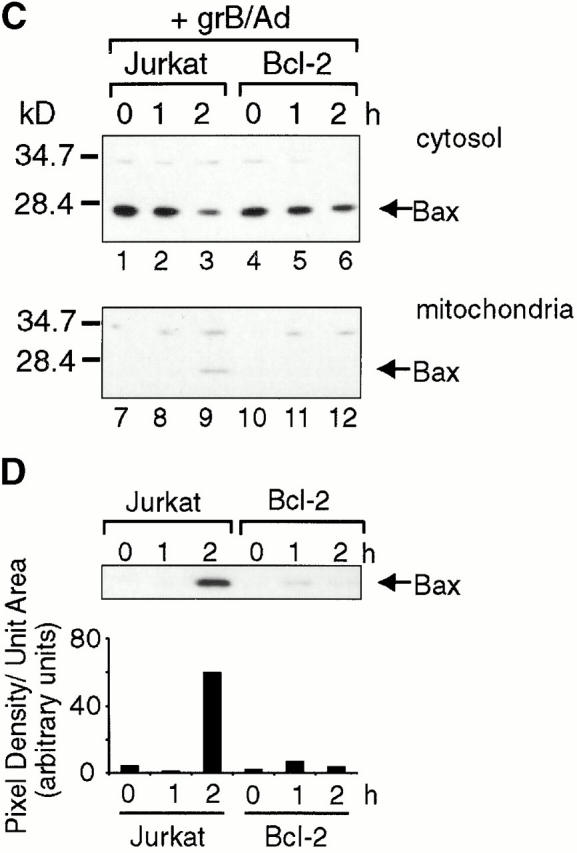
Cells were manipulated as described for Fig. 2. As with parental Jurkat cells (Fig. 2 B), cytochrome c release occurred in the vector-only transfectants (neo), detectable within 1 h after treatment with grB and Ad (Fig. 5 A, lanes 1–4). However, cytochrome c was not detected in supernatants of Bcl-2 overexpressers under identical conditions (Fig. 5 A, lanes 5–8). Therefore, as has been previously published for a variety of apoptotic stimuli 40 57 58, Bcl-2 blocked cytochrome c release in our system.
We next examined Bid cleavage and translocation. Similar to parental Jurkat cells (Fig. 2), Bid was processed to gtBid in the neo clones and detectable gtBid was found associated with mitochondria within 1 h after grB and Ad treatment (Fig. 5 B, lanes 1–4). Importantly, gtBid was also found to be associated with mitochondria in the Bcl-2 overexpressers with identical kinetics to the vector-only controls (Fig. 5 B, lanes 5–8).
Finally, we investigated the effect of Bcl-2 overexpression on Bax integration into the mitochondrial membrane (Fig. 5 C). For these experiments, cells were manipulated as described for Fig. 3. The ideal control for these experiments would have been to compare the vector-only (neo) transfectants with the Bcl-2 overexpressers as for Bid and cytochrome c above (Fig. 5A and Fig. B). However, our neo clones displayed unusually low levels of Bax relative to the parental Jurkat line (data not shown). Therefore, we compared the effects on Bax integration in the Bcl-2 overexpressers to the parental Jurkat line. Bax integration in Jurkat cells was detectable within 2 h after grB and Ad treatment (Fig. 5 C, lanes 1–3). However, no Bax incorporation was observed in the Bcl-2–overexpressing cells (Fig. 5 C, lanes 4–6). A slight difference in the quantity of cytosolic Bax between parental Jurkat and Bcl-2 overexpressers was seen when the blots were scanned and the intensity of the bands measured. At time zero, the intensity of the Bax band of the Bcl-2 overexpressers was ∼13% lower than that of the parental Jurkat line (data not shown). The fact that there was slightly less cytosolic Bax in the Bcl-2 transfectants might suggest that the absence of Bax in the mitochondrial fraction simply resulted from less starting material. However, when the blots containing the mitochondrial fractions were overexposed overnight (Fig. 4 D) and the intensity of the bands corresponding to Bax was measured (Fig. 4 D), a nearly 15-fold increase in intensity of Bax was seen in the parental Jurkat mitochondria after 2 h. In contrast, no more than a threefold increase was seen with the Bcl-2 transfectants. If Bax integration into mitochondria occurred despite the presence of Bcl-2, an ∼13% decrease in the starting levels of Bax would still allow it to be detected in mitochondria, particularly with longer exposures. However, virtually no Bax was detected at longer exposures. Collectively, these data indicated that the overexpression of Bcl-2 blocked grB-mediated Bax integration into mitochondria and cytochrome c release, but did not affect Bid processing and translocation.
Discussion
The induction of apoptosis in a pathogenic cell under attack by a CTL is known to involve grB activation of caspases, notably caspase-3. However, this paradigm does not explain the caspase-independent arm of CTL-mediated death. Our previous experiments demonstrated that mitochondrial changes might be an important component of the death machinery activated by CTLs 39. As cytochrome c efflux in response to grB occurred in a caspase-independent fashion, grB was either acting directly on mitochondria to induce cytochrome c release, or indirectly through a cellular substrate that was not a caspase. Data presented here indicate that grB alone is unable to elicit the release of cytochrome c from purified mitochondria (Fig. 1 and Fig. 2). Rather, the proapoptotic Bcl-2 family members Bid and Bax are required in conjunction with grB.
We examined whether Bid was required for cytochrome c efflux in an in vitro system using purified Jurkat mitochondria and purified Jurkat cytosol. Immunodepletion of Bid from the cytosol abolished, whereas reintroduction of recombinant Bid (rBid) to the Bid-depleted cytosol restored the ability of grB to induce cytochrome c efflux. These data are consistent with those already published, indicating that the immunodepletion of Bid from TNF/cycloheximide-treated cytosolic extracts prevented cytochrome c release 19. However, in our system, grB had to be present in conjunction with cytosol, suggesting that Bid is processed either directly or through other granzyme-activated proteinases.
In the absence of cytosol, the addition of rBid to mitochondria was an insufficient stimulus for cytochrome c release. However, cytochrome c efflux was accomplished by adding grB and rBid to mitochondria, indicating that caspases were clearly not required to mediate this effect. These data contrast with the process of Bid cleavage characterized for the Fas pathway that depends on caspase-8 to process Bid 17 18.
In response to ligation of the Fas receptor, caspase-8 cleavage of Bid at Asp59 generates a 15-kD tBid COOH-terminal fragment. Removal of the NH2 terminus of Bid exposes the otherwise inaccessible BH3 domain found in the COOH terminus of full-length Bid 59 60. Once formed, tBid rapidly translocates to mitochondria to induce cytochrome c release through an ill-defined mechanism 17 18 19. Processing of Bid by grB at Asp75 generates a 14-kD gtBid product 18 that, like tBid, contains the BH3 domain. Despite the different site of cleavage and the smaller size of the cleavage product, gtBid was found localized to mitochondria in whole cells in response to treatment with grB and Ad. As the BH1, BH2, and BH3 domains of Bcl-2/Bcl-XL can act as a binding pocket for the BH3 domains of other Bcl-2 family members 20 61, freeing the BH3 domain of Bid may allow it to heterodimerize with Bcl-2/Bcl-XL on the mitochondrial membrane. In support of this notion, one study reported that recombinant tBid had a 10-fold higher affinity for glutathione S-transferase (GST)-Bcl-XL than did full-length Bid 18. In addition, mutations in Bid's BH3 domain that prevented its binding to Bcl-2/Bcl-XL also blocked cytochrome c release and apoptosis 17 19 20. However, these BH3 mutations did not block translocation of Bid to mitochondria. In addition to exposing the BH3 domain, the removal of the NH2 terminus of Bid increases the surface hydrophobicity of tBid and alters its surface charge 59 60. It is these changes, rather than exposure of the BH3 domain, which may allow tBid to target membranes. Nevertheless, by whatever mechanism tBid associates with mitochondria, the regions of tBid that are required for this process are clearly also present in gtBid.
In the absence of zVAD-fmk, tBid was also generated in response to grB and Ad treatment 35. Although not required for apoptosis, grB can cleave and activate caspase-8 35. As tBid is generated by caspase-8, it remained possible that despite the presence of gtBid, cytochrome c release resulted from tBid translocation alone. However, we showed here that in the presence of zVAD-fmk, cytochrome c release occurred but only gtBid was detected. Importantly, detectable cytochrome c release paralleled detectable gtBid translocation either in the absence or presence of zVAD-fmk. This indicated that the smaller gtBid was equivalent to the larger caspase-8–generated tBid in its ability to induce cytochrome c efflux from mitochondria. Therefore, our in vitro and in vivo data demonstrate for the first time that caspase-independent cytochrome c release is dependent on grB-mediated cleavage of Bid and the subsequent translocation to mitochondria of gtBid.
The mechanism by which Bid induces cytochrome c release remains elusive. Nevertheless, mounting evidence suggests that, at least in some cases, the relocalization of tBid functions to recruit another proapoptotic Bcl-2 family member, Bax, to mitochondria 56. Bax recruitment may occur through a tBid-induced conformational change in Bax that allows it to insert into the mitochondrial outer membrane 55. In Jurkat cells after treatment with grB and Ad, Bax was found to integrate into mitochondrial membranes in both the presence and absence of zVAD-fmk. These results are consistent with data showing that in the presence of recombinant tBid, Bax translocation to mitochondria and its integration into the outer membrane do not rely on caspases 53 55 56. Here we show that Bax integration in response to grB also occurred with purified mitochondria in a caspase-independent fashion, but only when Bid was present in cytosolic extracts. Thus, as described previously for tBid, gtBid was able to recruit Bax to mitochondria.
What role Bax may play in gtBid-induced cytochrome c release is unclear. Bax itself has been shown to induce cytochrome c release in isolated mitochondria 44 45 46 47 and when overexpressed in whole cells 62. Recently, Bax was shown to interact with the voltage-dependent anion channel (VDAC) in the outer mitochondrial membrane to form a cytochrome c conducting channel 45 63 64. Bax may also interact with the adenine nucleotide transporter (ANT) on the inner mitochondrial membrane to cause a permeability transition leading to an indirect release of cytochrome c 65. Cyclosporine, an inhibitor of the permeability transition, blocked this Bax-mediated cytochrome c release 65. However, other evidence indicates that Bax-mediated cytochrome c release is cyclosporine insensitive and therefore independent of the permeability transition 55. This argues that the manner in which Bax affects cytochrome c release may be differentially regulated in different cells and/or in response to different apoptotic stimuli. In the granzyme system, both inner mitochondrial membrane potential loss and cell death seen in whole cells are cyclosporine independent 39 40, arguing that the recruitment of Bax to mitochondria and its role in cytochrome c release are independent of the permeability transition.
Bcl-2 has been reported to block cytochrome c release in response to a variety of apoptotic stimuli 57 58. Further, Bcl-2 blocks both translocation of Bax from the cytosol to mitochondria and its subsequent insertion into the outer membrane 53 54 55 56. In response to grB and Ad treatment, no cytochrome c release was seen in Jurkat cells overexpressing Bcl-2. However, even with Bcl-2 overexpression, grB-dependent Bid cleavage still occurred and gtBid was found relocalized to mitochondria. Despite gtBid translocation to mitochondria in the presence of Bcl-2, no Bax integration into the mitochondrial membrane was observed. The Bcl-2–antagonized integration of Bax correlated with the absence of cytochrome c release in response to grB and Ad. Thus, whereas gtBid was localized to mitochondria in the Bcl-2–overexpressing cells, no cytochrome c release was observed in the absence of Bax integration. We cannot rule out the possibility that the levels or functions of other Bcl-2 family members may be altered in the Bcl-2 transfectants and that the effects of these proteins may influence the movement of Bax to mitochondria. Nevertheless, there is now growing evidence in the literature to link Bid with Bax recruitment to mitochondria 55 56 66. In addition, the literature supports a model whereby Bax recruitment can be antagonized by Bcl-2 53 54 67. Finally, evidence suggests that Bax is able to induce the efflux of cytochrome c from mitochondria 44 45 47 48 56 68. We have not shown that cytochrome c release absolutely depends on Bax integration into the mitochondrial outer membrane in the granzyme system. However, the fact that cytochrome c release does not occur in the absence of Bax integration coupled with the available literature renders this hypothesis plausible.
Collectively, our data support a model in which grB proteolytically cleaves Bid to generate a 14-kD gtBid fragment that translocates to mitochondria through a caspase-independent mechanism (Fig. 6). gtBid localized to mitochondria then recruits Bax to the mitochondrial outer membrane through a mechanism antagonized by Bcl-2. Once integrated, Bax initiates cytochrome c release from mitochondria through a cyclosporine-independent mechanism.
Figure 6.
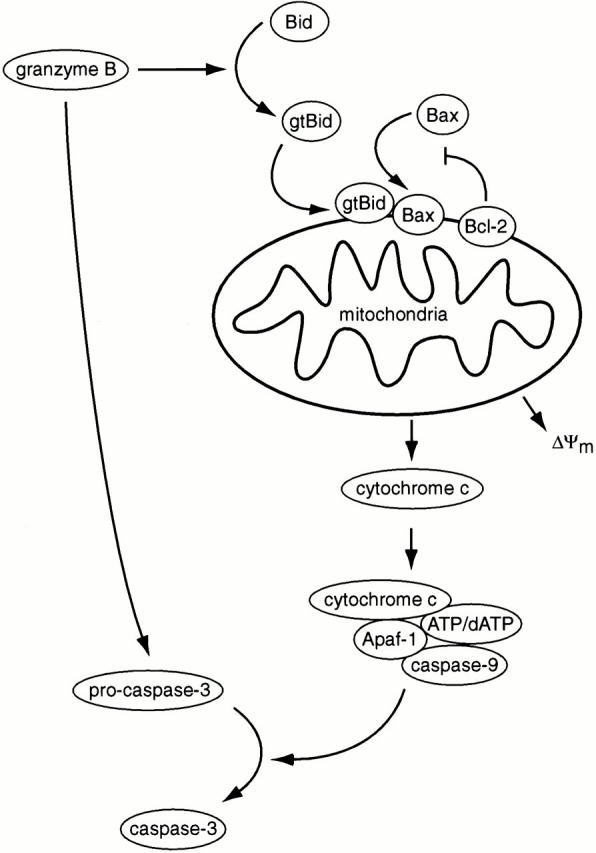
Model for regulation by Bcl-2 family members of grB-mediated cytochrome c release. GrB may activate caspase-3 directly, or indirectly through the proteolytic activation of Bid. Bid is cleaved to generate gtBid that translocates to mitochondria where it recruits Bax through a mechanism antagonized by Bcl-2. Bax becomes integrated into the mitochondrial membrane and induces cytochrome c release. Cytochrome c acts as a cofactor with Apaf-1, caspase-9, and ATP/dATP to foster the proteolytic activation of caspase-3. ΔΨm, inner mitochondrial membrane potential.
It has been suggested that caspase inhibitors may be effective in the treatment of autoimmune disorders and suppression of transplant rejection. Our results imply that these alone would be ineffective. Rather, therapeutic interventions that block both caspase and mitochondrial pathways would be required. The relative importance of these mechanisms in cell death in vivo is currently under investigation.
Acknowledgments
We wish to thank Stuart Farrow for the Bcl-2 cDNA, Jack Gauldie for the replication-deficient Ad, and Xiaodong Wang for the Bid cDNA and anti-Bid antiserum. We also thank Tracy Sawchuk and Irene Shostak for their invaluable assistance with cell culture.
This work was supported by grants from the Medical Research Council of Canada (J.A. Heibein, M.J. Pinkoski, and M. Barry), the Alberta Heritage Foundation for Medical Research (I.S. Goping and R.C. Bleackley), the National Cancer Institute of Canada (R.C. Bleackley), and The Howard Hughes Foundation for Medical Research (R.C. Bleackley).
Footnotes
Abbreviations used in this paper: Ad, adenovirus; DISC, death-inducing signaling complex; grB, granzyme B; HRP, horseradish peroxidase; t, truncated.
References
- Atkinson E., Bleackley R.C. Mechanisms of lysis by cytotoxic T cells. Crit. Rev. Immunol. 1995;15:359–384. doi: 10.1615/critrevimmunol.v15.i3-4.90. [DOI] [PubMed] [Google Scholar]
- Darmon A.J., Pinkoski M.J., Bleackley R.C. Granule-mediated cytotoxicity. In: Kumar S., editor. ApoptosisBiology and Mechanisms. Springer-Verlag; Berlin/Heidelberg: 1999. pp. 103–125. [DOI] [PubMed] [Google Scholar]
- Kägi D., Ledermann B., Bürki K., Zinkernagel R.M., Hengartner H. Molecular mechanisms of lymphocyte-mediated cytotoxicity and their role in immunological protection and pathogenesis in vivo . Annu. Rev. Immunol. 1996;14:207–232. doi: 10.1146/annurev.immunol.14.1.207. [DOI] [PubMed] [Google Scholar]
- Shresta S., Pham C.T., Thomas D.A., Graubert T.A., Ley T.J. How do cytotoxic lymphocytes kill their targets? Curr. Opin. Immunol. 1998;10:581–587. doi: 10.1016/s0952-7915(98)80227-6. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan A.M., O'Rourke K., Tewari M., Dixit V.M. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81:505–512. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- Muzio M., Chinnaiyan A.M., Kischkel F.C., O'Rourke K., Shevchenko A., Ni J., Scaffidi C., Bretz J.D., Zhang M., Gentz R. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signalling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- Boldin M.P., Goncharov T.M., Goltsev Y.V., Wallach D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell. 1996;85:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- Nicholson D.W., Thornberry N.A. Caspaseskiller proteases. Trends Biochem. Sci. 1997;22:299–306. doi: 10.1016/s0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- Thornberry N.A., Lazebnik Y. Caspases, enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- Earnshaw W.C., Martins L.M., Kaufmann S.H. Mammalian caspasesstructure, activation, substrates, and functions during apoptosis. Annu. Rev. Biochem. 1999;68:383–424. doi: 10.1146/annurev.biochem.68.1.383. [DOI] [PubMed] [Google Scholar]
- Scaffidi C., Fulda S., Srinivasan A., Friesen C., Li F., Tomaselli K.J., Debatin K.-M., Krammer P.H., Peter M.E. Two CD95 (APO-1/Fas) signaling pathways. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:1675–1687. doi: 10.1093/emboj/17.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe B.B., Schuler M., Echeverri F., Green D.R. Caspase-3 is the primary activator of apoptotic DNA fragmentation via DNA fragmentation factor-45/inhibitor of caspase-activated DNase inactivation. J. Biol. Chem. 1999;274:30651–30656. doi: 10.1074/jbc.274.43.30651. [DOI] [PubMed] [Google Scholar]
- Stroh C., Schulze-Osthoff K. Death by a thousand cutsan ever increasing list of caspase substrates. Cell Death Differ. 1998;5:997–1000. doi: 10.1038/sj.cdd.4400451. [DOI] [PubMed] [Google Scholar]
- Porter A.G., Jänicke R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:88–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- Enari M., Skahira H., Yokoyama H., Okawa K., Iwamatsu A., Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;393:43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- Kothakota S., Azuma T., Reinhard C., Klippel A., Tang J., Chu K., McGarry T., Kirschner M., Koths K., Kwiatkowski D., Williams L. Caspase-3-generated fragment of gelsolineffector of morphological change in apoptosis. Science. 1997;278:294–298. doi: 10.1126/science.278.5336.294. [DOI] [PubMed] [Google Scholar]
- Luo X., Budihardjo I., Zou H., Slaughter C., Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- Li H., Zhu H., Xu C.-J., Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- Gross A., Yin X.-M., Wang K., Wei M.C., Jockel J., Milliman C., Erdjument-Bromage H., Tempst P., Korsmeyer S.J. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumour necrosis factor-R1/Fas death. J. Biol. Chem. 1999;274:1156–1163. doi: 10.1074/jbc.274.2.1156. [DOI] [PubMed] [Google Scholar]
- Wang K., Yin X.-M., Chao D.T., Milliman C.L., Korsmeyer S.J. BIDa novel BH3 domain-only death agonist. Genes Dev. 1996;10:2859–2869. doi: 10.1101/gad.10.22.2859. [DOI] [PubMed] [Google Scholar]
- Zou H., Henzel W.J., Liu X., Lutschg A., Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- Li P., Nijhawan D., Budihardjo I., Srinivasula S.M., Ahmad M., Alnemri E.S., Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Poe M., Blake J.T., Boulton D.A., Gammon M., Sigal N.H., Wu J.K., Zweerink H.J. Human cytotoxic granzyme B. Its purification from granules and the characterization of substrate and inhibitor specificity. J. Biol. Chem. 1991;266:98–103. [PubMed] [Google Scholar]
- Harris J.L., Peterson E.P., Hudig D., Thornberry N.A., Craik C.S. Definition and redesign of the extended substrate specificity of granzyme B. J. Biol. Chem. 1998;273:27364–37373. doi: 10.1074/jbc.273.42.27364. [DOI] [PubMed] [Google Scholar]
- Vande Craen M., Vandenbrande I., Declercq W., Irmler M., Beyaert R., Tschopp J., Fiers W., Vandenabeele P. Cleavage of caspase family members by granzyme Ba comparative study in vitro . Eur. J. Immunol. 1997;27:1296–1299. doi: 10.1002/eji.1830270535. [DOI] [PubMed] [Google Scholar]
- Darmon A.J., Nicholson D.W., Bleackley R.C. Activation of the apoptotic protease CPP32 by cytotoxic T-cell-derived granzyme B. Nature. 1995;377:446–448. doi: 10.1038/377446a0. [DOI] [PubMed] [Google Scholar]
- Martin S.J., Amarante-Mendes G.P., Shi L., Chuang T.H., Casiano C.A., O'Brien G.A., Fitzgerald P., Tan E.M., Bokoch G.M., Greenberg A.H., Green D.R. The cytotoxic cell protease granzyme B initiates apoptosis in a cell-free system by proteolytic processing and activation of the ICE/CED-3 family protease, CPP32, via a novel two-step mechanism. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:2407–2416. [PMC free article] [PubMed] [Google Scholar]
- Quan L.T., Tewari M., O'Rourke K., Dixit V., Snipas S.J., Poirier G.G., Ray C., Pickup D.J., Salvesen G.S. Proteolytic activation of the cell death protease Yama/CPP32 by granzyme B. Proc. Natl. Acad. Sci. USA. 1996;93:1972–1976. doi: 10.1073/pnas.93.5.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnaiyan A.R., Orth K., Hanna W.L., Duan H.J., Poirier G.G., Froelich C.J., Dixit V.M. Cytotoxic T cell-derived granzyme B activates the apoptotic protease ICE-LAP3. Curr. Biol. 1996;6:897–899. doi: 10.1016/s0960-9822(02)00614-0. [DOI] [PubMed] [Google Scholar]
- Gu Y., Sarnecki C., Fleming M.A., Lippke J.A., Bleackley R.C., Su M.S. Processing and activation of CMH-1 by granzyme B. J. Biol. Chem. 1996;271:10816–10820. doi: 10.1074/jbc.271.18.10816. [DOI] [PubMed] [Google Scholar]
- Srinivasula S., Fernandes-Alnemri T., Zangirlli J., Robertson N., Armstrong R., Wang L., Trapani J., Tomaselli K., Litwack G., Alnemri E. The Ced-3/interleukin 1β converting enzyme-like homolog Mch6 and the lamin-cleaving enzyme Mch2α are substrates for the apoptotic mediator CPP32. J. Biol. Chem. 1996;271:27099–27106. doi: 10.1074/jbc.271.43.27099. [DOI] [PubMed] [Google Scholar]
- Orth K., Chinnaiyan A., Garg M., Froelich C., Dixit V. The CED-3/ICE-like protease Mch2 is activated during apoptosis and cleaves the death substrate lamin A. J. Biol. Chem. 1996;271:16443–16446. [PubMed] [Google Scholar]
- Duan H., Orth K., Chinnaiyan A.M., Poirier G.G., Froelich C.J., He W.-W., Dixit V.M. ICE-LAP6, a novel member of the ICE/Ced-3 gene family, is activated by the cytotoxic T cell protease granzyme B. J. Biol. Chem. 1996;271:16720–16724. doi: 10.1074/jbc.271.28.16720. [DOI] [PubMed] [Google Scholar]
- Fernandes-Alnemri T., Armstrong R.C., Krebs J., Srinivasula S.M., Wang L., Bullrich F., Fritz L.C., Trapani J.A., Tomaselli K.J., Litwack G., Alnemri E.S. In vitro activation of CPP32 and Mch3 by Mch4, a novel human apoptotic cysteine protease containing two FADD-like domains. Proc. Natl. Acad. Sci. USA. 1996;93:7464–7469. doi: 10.1073/pnas.93.15.7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry M., Heibein J.A., Pinkoski M.J., Lee S.-F., Moyer R.W., Green D.R., Bleackley R.C. Granzyme B short circuits the need for caspase 8 activity during granule-mediated CTL killing by directly cleaving Bid. Mol. Cell. Biol. 2000;20:3781–3794. doi: 10.1128/mcb.20.11.3781-3794.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson E.A., Barry M., Darmon A.J., Shostak I., Turner P.C., Moyer R.W., Bleackley R.C. Cytotoxic T lymphocyte-assisted suicide. Caspase 3 activation is primarily the result of the direct action of granzyme B. J. Biol. Chem. 1998;273:21261–21266. doi: 10.1074/jbc.273.33.21261. [DOI] [PubMed] [Google Scholar]
- Darmon A.J., Ley T.J., Nicholson D.W., Bleackley R.C. Cleavage of CPP32 by granzyme B represents a critical role for granzyme B in the induction of target cell DNA fragmentation. J. Biol. Chem. 1996;271:21709–21712. doi: 10.1074/jbc.271.36.21709. [DOI] [PubMed] [Google Scholar]
- Shi L., Chen G., MacDonald G., Bergeron L., Li H., Miura M., Rotello R.J., Miller D.K., Li P., Sheshardri T. Activation of an interleukin-1 converting enzyme-dependent apoptosis pathway by granzyme B. Proc. Natl. Acad. Sci. USA. 1996;93:11002–11007. doi: 10.1073/pnas.93.20.11002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heibein J.A., Barry M., Motyka B., Bleackley R.C. Granzyme B-mediated loss of mitochondrial inner membrane potential (ΔΨm) and cytochrome c release are caspase-independent. J. Immunol. 1999;163:4683–4693. [PubMed] [Google Scholar]
- MacDonald G., Shi L., VandeVelde C., Lieberman J., Greenberg A.H. Mitochondria-dependent and -independent regulation of granzyme B–induced apoptosis. J. Exp. Med. 1999;189:131–143. doi: 10.1084/jem.189.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputo A., Parrish J.C., James M.N., Powers J.C., Bleackley R.C. Electrostatic reversal of serine proteinase substrate specificity. Proteins. 1999;35:415–424. [PubMed] [Google Scholar]
- Karasuyama H., Melchers F. Establishment of mouse cell lines which constitutively secrete large quantities of interleukin 2, 3, 4 or 5, using modified cDNA expression vectors. Eur. J. Immunol. 1988;18:97–104. doi: 10.1002/eji.1830180115. [DOI] [PubMed] [Google Scholar]
- Samali A., Cai J., Zhivotovsky B., Jones D.P., Orrenius S. Presence of a pre-apoptotic complex of pro-caspase-3, Hsp60 and Hsp10 in the mitochondrial fraction of Jurkat cells. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:2040–2048. doi: 10.1093/emboj/18.8.2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priault M., Chaudhuri B., Clow A., Camougrand N., Manon S. Investigation of bax-induced release of cytochrome c from yeast mitochondria. Eur. J. Biochem. 1999;260:684–691. doi: 10.1046/j.1432-1327.1999.00198.x. [DOI] [PubMed] [Google Scholar]
- Narita M., Shimizu S., Ito T., Chittenden T., Lutz R.J., Matsuda H., Tsujimoto Y. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc. Natl. Acad. Sci. USA. 1998;95:14681–14686. doi: 10.1073/pnas.95.25.14681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskes R., Antonsson B., Osen-Sand A., Montessuit S., Richter C., Sadoul R., Mazzei G., Nichols A., Martinou J.-C. Bax-induced cytochrome c release from mitochondria is independent of the permeability transition pore but highly dependent on Mg2+ ions. J. Cell Biol. 1998;143:217–224. doi: 10.1083/jcb.143.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jürgensmeier J.M., Xie Z., Deveraux Q., Ellerby L., Bredesen D., Reed J.C. Bax directly induces release of cytochrome c from isolated mitochondria. Proc. Natl. Acad. Sci. USA. 1998;95:4997–5002. doi: 10.1073/pnas.95.9.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonsson B., Montessuit S., Lauper S., Eskes R., Martinou J.-C. Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem. J. 2000;345:271–278. [PMC free article] [PubMed] [Google Scholar]
- Gross A., Jockel J., Wei M.C., Korsmeyer S.J. Enforced dimerization of BAX results in its translocation, mitochondrial dysfunction and apoptosis. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:3878–3885. doi: 10.1093/emboj/17.14.3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolter K.G., Hsu U.-T., Smith C.L., Nechushtan A., Xi X.-G., Youle R.J. Movement of Bax from the cytosol to mitochondria during apoptosis. J. Cell Biol. 1997;139:1281–1292. doi: 10.1083/jcb.139.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechushtan A., Smith C.L., Hsu Y.-T., Youle R.J. Conformation of the Bax C-terminus regulates subcellular location and cell death. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:2330–2341. doi: 10.1093/emboj/18.9.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goping I.S., Gross A., Lavoie J.N., Nguyen M., Jemmerson R., Roth K., Korsmeyer S.J., Shore G.C. Regulated targeting of BAX to mitochondria. J. Cell Biol. 1998;143:1–9. doi: 10.1083/jcb.143.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy K.M., Ranganathan V., Farnsworth M.L., Kavallaris M., Lock R.B. Bcl-2 inhibits Bax translocation from cytosol to mitochondria during drug-induced apoptosis of human tumor cells. Cell Death Differ. 2000;7:102–111. doi: 10.1038/sj.cdd.4400597. [DOI] [PubMed] [Google Scholar]
- Murphy K.M., Streips U.N., Lock R.B. Bcl-2 inhibits a Fas-induced conformational change in the Bax N-terminus and Bax mitochondrial translocation. J. Biol. Chem. 2000;275:17225–17228. doi: 10.1074/jbc.C900590199. [DOI] [PubMed] [Google Scholar]
- Eskes R., Desagher S., Antonsson B., Martinou J.-C. Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol. Cell. Biol. 2000;20:929–935. doi: 10.1128/mcb.20.3.929-935.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desagher S., Osen-Sand A., Nichols A., Eskes R., Montessuit S., Lauper S., Maundrell K., Antonsson B., Martinou J.-C. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J. Cell Biol. 1999;144:891–901. doi: 10.1083/jcb.144.5.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluck R.M., Bossy-Wetzel E., Green D.R., Newmeyer D.D. The release of cytochrome c from mitochondriaa primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- Yang J., Liu X., Bhalla K., Kim C.N., Ibrado A.M., Cai J., Pen T.-I., Jones D.P., Wang X. Prevention of apoptosis by Bcl-2release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- McDonnell J.M., Fushman D., Milliman C.L., Korsmeyer S.J., Cowburn D. Solution structure of the proapoptotic molecule BIDa structural basis for apoptotic agonists and antagonists. Cell. 1999;96:625–634. doi: 10.1016/s0092-8674(00)80573-5. [DOI] [PubMed] [Google Scholar]
- Chou J.J., Li H., Salvasen G.S., Yuan J., Wagner G. Solution structure of BID, an intracellular amplifier of apoptotic signaling. Cell. 1999;96:615–624. doi: 10.1016/s0092-8674(00)80572-3. [DOI] [PubMed] [Google Scholar]
- Sattler M., Liang H., Nettesheim D., Meadows R.P., Harlan J.E., Eberstadt M., Yoon H.S., Shuker S.B., Chang B.S., Minn A.J. Structure of Bcl-xL-Bak peptide complexrecognition between regulators of apoptosis. Science. 1997;275:983–986. doi: 10.1126/science.275.5302.983. [DOI] [PubMed] [Google Scholar]
- Pastorino J.G., Chen S.-T., Tafani M., Snyder J.W., Farber J.J. The overexpression of Bax produces cell death upon induction of the mitochondrial permeability transition. J. Biol. Chem. 1998;273:7770–7775. doi: 10.1074/jbc.273.13.7770. [DOI] [PubMed] [Google Scholar]
- Shimizu S., Narita M., Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- Shimizu S., Tsujimoto Y. Proapoptotic BH3-only Bcl-2 family members induce cytochrome c release, but not mitochondrial membrane potential loss, and do not directly modulate voltage-dependent anion channel activity. Proc. Natl. Acad. Sci. USA. 2000;97:577–582. doi: 10.1073/pnas.97.2.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzo I., Brenner C., Zamzami N., Jürgensmeier J.M., Susin S.A., Vieira H.L., Prevost M.C., Xie Z., Matsuyama S., Reed J.C., Kroemer G. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–2031. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- Ruffolo S.C., Breckenridge D.G., Nguyen M., Goping I.S., Gross A., Korsmeyer S.J., Li H., Yuan J., Shore G.C. BID-dependent and BID-independent pathways for BAX insertion into mitochondria. Cell Death Differ. 2000;In press. doi: 10.1038/sj.cdd.4400739. [DOI] [PubMed] [Google Scholar]
- Murphy K.M., Streips U.N., Lock R.B. Bax membrane insertion during Fas(CD95)-induced apoptosis precedes cytochrome c release and is inhibited by Bcl-2. Oncogene. 1999;18:5991–5999. doi: 10.1038/sj.onc.1203001. [DOI] [PubMed] [Google Scholar]
- Finucane D.M., Bossy-Wetzel E., Waterhouse N.J., Cotter T.G., Green D.R. Bax-induced caspase activation and apoptosis via cytochrome c release from mitochondria is inhibitable by Bcl-xL. J. Biol. Chem. 1999;274:2225–2233. doi: 10.1074/jbc.274.4.2225. [DOI] [PubMed] [Google Scholar]