

Abstract
Somatic hypermutation and isotype switch recombination occur in germinal center B cells, are linked to transcription, and are similarly affected by deficiency in MutS homologue (MSH)2. Class-switch recombination is abrogated by disruption of genes encoding components of the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs)/Ku complex and likely involves nonhomologous end joining (NHEJ). That somatic hypermutation might also be associated with end joining is suggested by its association with the creation of deletions, duplications, and sites accessible to terminal transferase. However, a requirement for NHEJ in the mutation process has not been demonstrated. Here we show that somatic mutation in mice deficient in NHEJ can be tested by introduction of rearranged immunoglobulin and T cell receptor transgenes: the transgene combination not only permits reconstitution of peripheral lymphoid compartments but also allows formation of germinal centers, despite the wholly monoclonal nature of the lymphocyte antigen receptors in these animals. Using this strategy, we confirm that somatic hypermutation like class-switching can occur in the absence of recombination-activating gene (RAG)1 but show that the two processes differ in that hypermutation can proceed essentially unaffected by deficiency in DNA-PKcs activity.
Keywords: immunoglobulin gene, B lymphocyte, switch recombination, end-joining, mutation
Introduction
Three types of genetic transaction occur at the Ig gene loci that play a role in antigen receptor diversification: V(D)J joining, IgH class-switching, and somatic hypermutation. The regulation of all three processes is in some way linked to transcription. Although switching and hypermutation are clearly distinct (they target different, albeit linked, regions and each can take place independently of the other), they show some striking parallels. Both are specific to the secondary antibody repertoire and take place in germinal center (GC) B cells; they preferentially act at similar consensus sequences, and the efficiency and local target specificity of both processes is affected by deficiency in MutS homologue (MSH)2 1 2 3 4 5.
Here we focus on the role of nonhomologous end joining (NHEJ). The absence of class-switching in B cells that lack components of the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs)/Ku complex make it highly likely that NHEJ is involved in switching 6 7 8. The situation with regard to hypermutation is unknown. The finding that hypermutation is accompanied by the creation of sites accessible to terminal deoxynucleotidyl transferase (TdT) in the V region 9 and that observations that hypermutation can lead to deletions and duplications (in addition to the physiological single nucleotide substitutions) 9 10 11 could be interpreted as indicating a role for NHEJ in the hypermutation process. Indeed, the ability of TdT to interact with components of the DNA-PKcs/Ku complex 12 13 could provide a mechanism for its recruitment to the sites of DNA breaks. However, it is equally possible that the breaks, duplications, and TdT-accessible sites are generated as byproducts of aberrant hypermutation events without NHEJ constituting a necessary physiological intermediate.
Testing whether NHEJ is required for the somatic hypermutation of Ig genes cannot simply be accomplished by analyzing hypermutation in mice defective in NHEJ, as NHEJ is required to achieve successful antigen receptor assembly and lymphocyte production. The same difficulty similarly exists in testing whether the RAG proteins, needed for the initiation of V(D)J joining, are also required for hypermutation. To address this problem, we have developed a model in which the requirement for V(D)J recombination is bypassed through the provision of rearranged Ig and TCR transgenes. We find that despite the entirely monoclonal nature of both B and T cell compartments, such mice can develop GCs in which hypermutation takes place. The results reveal that hypermutation can proceed both in RAG1-deficient mice and SCID mice, which lack a functional DNA-PKcs 14 15.
Materials and Methods
Mice.
MD4 mice (on a C57BL/6 background) 16 were obtained from The Jackson Laboratory, and DO-TCR mice (on a BALB/c background) 17 were from D. Loh (Washington University School of Medicine, St. Louis, MO). After rederivation into our barriered facility, both lines were crossed with Scid/Scid 14 or Rag1 −/− mice 18 on a C57BL/6 background to yield animals of the required genotype. Mice were genotyped by PCR of tail DNA (MD4: primers 5′-GCGACTCCATCACCAGCGAT and 5′-ACCACAGACCAGCAGGCAGA amplify across the Vκ/Jκ rearrangement [430-bp product] with inclusion of 5′-CTGGAGCCCTAGCCAAGGAT, allowing generation of a control PCR product [264-bp product] from the germline Jκ locus; DO-TCR: primers 5′-TGGCTCTACAGTGAGTTTGGTGCCA and 5′-TGCAGCTGGATGGGATGAGCCAAGG amplify the transgenic Vα/Jα rearrangement [380-bp product]; RAG1: primers 5′-GGACATAGCGTTGGCTACCC [within the inserted Neo cassette] and 5′-GAAAGGACTTCACTGGGCTCT [within RAG1] generate an ∼650-bp product from the targeted allele with the inclusion of AGATGTCTCAAAGTCATGGGC-3′ [within RAG1] that generates an 87-bp product from the wild-type allele; SCID: primers 5′-GTCAGTCTCATGTTGCCAATG and 5′-GTTGGCCCCTGCTAACTTTC generate a 1,300-bp product, which is cut by AluI to yield 208- and 18-bp fragments from the SCID allele and a 236-bp product from the wild-type allele). A 214-bp BglII–HaeIII fragment of the SCID PCR product from sorted GC B cells was cloned into pBluescript for sequence analysis to confirm lack of reversion of the SCID mutation.
FACS® Analysis of Lymphocytes.
Splenic lymphocytes purified on Lympholyte-M (Cedarlane Laboratories Ltd.) were stained with FITC-conjugated rabbit anti–mouse IgM and PE-conjugated rat anti-CD45R(B220) (clone RA3-6B2; PharMingen and Life Technologies) or PE-conjugated anti-CD4 and FITC-conjugated anti-CD8 (PharMingen). The HyHEL10 idiotope was detected using mAb 7.6 (reference 19; obtained from J.R. Drake, Trudeau Institute, Saranac Lake, NY) and FITC-conjugated anti–mouse γ2a (PharMingen).
Immunohistology.
Peyer's patches were quick-frozen in Tissue-Tek OCT (Sakura) using liquid N2, and sections (6–8 μM) were prepared using a microtome cryostat. After fixing in cold acetone (10 min) and air drying, sections were stained with rat anti-CD45R(B220), FITC-conjugated peanut agglutinin (PNA; Vector Laboratories), or rabbit anti-Ki67 (Dianova). Sections were developed with biotinylated anti–rabbit or anti–rat Ig (Jackson ImmunoResearch Laboratories) and alkaline phosphatase conjugated to streptavidin or anti-FITC–Ig (Roche).
Analysis of Somatic Mutation.
DNA was prepared using a QIAGEN minikit from GC B cells sorted from Peyer's patches on the basis of their PNAhiCD45R(B220)+ phenotype. The transgenic Vκ was amplified in a 30-cycle PCR using PfuTurbo polymerase (Stratagene) and primers 5′-CAGCTCGAGATTGTGCTAACTCAGTCTCC and 5′-CAGAGATCTACACCTGATCTGAGAATGGA (452-nucleotide product) and cloned into Bluescript for sequencing; primers 5′-CAGGGTACCCGAAGATGGTTTTCACACCT and 5′-CAGGAGCTCGTCCCATCACTGAATGTGAT yielded an 897-bp product that included the transgenic Vκ and additional intronic Jκ 3′ flank. Sequence databases were corrected for germline differences between Vκ transgene copies as well as any effects of clonality before computation of mutations as previously described 9. The genotype of the mouse was confirmed in all studies by PCR analysis of DNA prepared from the PNAlo population performed as described for the tail DNA.
Results
Our strategy for analyzing hypermutation in mice carrying genetic deficiencies rendering them incapable of productive antigen receptor assembly was to reconstitute the B and T cell compartments in these mice using Ig and TCR transgenes, hoping that such genetically reconstituted mice expressing monoclonal B and T cell antigen receptors could nevertheless form GCs.
Multiple Transgene Copies in MD4 Mice All Act as Hypermutation Targets.
MD4 mice, which carry an IgM–IgD,κ transgene specific for hen egg lysozyme 16, were used to provide the Ig transgene. As not all Ig transgenes are capable of acting as hypermutation targets (possibly reflecting a sensitivity to integration site), we first checked that the transgenic Vκ, PCR-amplified from the Peyer's patch GC B cells, had indeed been subjected to hypermutation. The MD4 Vκ sequences revealed an abundance of somatic mutations (Fig. 1). However, in addition to the scattered mutations characteristic of antibody hypermutation, some recurrent mutations were identified that were common to samples obtained from different individual MD4 animals. These recurrent mutations could be most readily explained by postulating that the MD4 mice carry multiple κ transgene copies that differ in the germline by virtue of mutations in the Vκ domain. This explanation was confirmed by sequencing the transgenic Vκ from tail DNA. Thus, MD4 mice carry four distinct HyHEL10 Vκ transgenes (differing by 0, 1, 2, or 3 mutations from the canonical sequence); all four copies act as efficient hypermutation targets.
Figure 1.
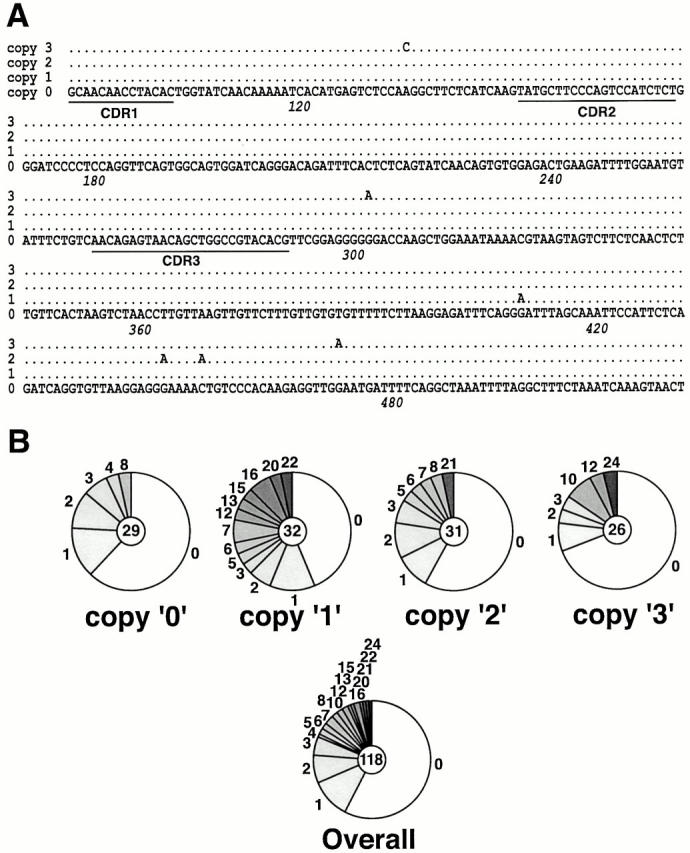
Somatic mutation of the Vκ transgene in MD4 mice. (A) Sequences of the four distinct transgenic rearranged Vκ domains that are deduced to exist in the germline of MD4 mice. The transgenic rearranged Vκ was PCR amplified from tail DNA and cloned into Bluescript. Of 33 clones sequenced, 5 derived from copy 0, 7 from copy 1, and 11 each from copies 2 and 3. The copies are named according to the number of nucleotide substitutions by which each diverges from the consensus sequence (copy 0). Nucleotide 1 corresponds to the first nucleotide of Kabat codon 1. (B) Prevalence of somatic mutations in the four transgenic Vκ genes PCR amplified from sorted GC B cells from Peyer's patches of MD4 mice. The number in the center of the pie chart indicates the number of sequences contributing to each database, with the segments of the pie indicating the number of sequences carrying 0, 1, 2, etc. mutations.
The differences between the germline Igκ transgene copies could reflect mutations that were artificially introduced during the in vitro manipulations of the transgene DNA construct before creation of the MD4 mice. Alternatively, they could have arisen during replication/recombination in vivo, possibly before transgene integration into the chromosome.
GC Formation in Reconstituted Rag1−/− and SCID Mice Monoclonal for B and T Cell Antigen Receptors.
Crossing of the MD4 transgene into a RAG1-deficient or SCID background allowed reconstitution of a peripheral B cell compartment as well as the development of Peyer's patches (Fig. 2A–C). Additional provision of the DO11.10 α/β TCR transgene (specific for OVA323–329 in the context of IAd; reference 17) allowed T cell reconstitution, triggering the development of both CD4+ and CD8+ subsets. We wondered whether a GC reaction could be induced in the reconstituted mice despite the fact that both B and T cells expressed a solitary antigen receptor specificity with, presumably, a consequently limited range of antigen responsiveness. Histological examination of Peyer's patches revealed that GCs were present in essentially all of the sections analyzed from 5 mo normal or MD4/DO-TCR(Rag1 +/+) transgenic mice with an average of 0.8 GCs per B cell cluster and individual animals exhibiting a range of 0.5–1.0 GCs/clusters. In the MD4/DO-TCR-reconstituted Rag1 −/− and SCID mice, GCs could also develop, although at lower frequency: on average there were 0.3 GCs per B cell cluster but with a range extending from 0 to 1.0 GCs per cluster (Fig. 2 D). These clusters of PNAhi cells detected in the Peyer's patches of the reconstituted mice were usually smaller than those in control animals. No GCs were detected in the B cell clusters of Rag1 −/− mice that had been reconstituted with the MD4 transgene alone (Fig. 2 D, right).
Figure 2.

Reconstitution of peripheral lymphocyte compartments in MD4/DO-TCR(Rag1 −/−) and MD4/DO-TCR(Scid/Scid) mice. (A and B) Flow cytometric analyses of splenic lymphocytes double stained for CD45R(B220) and IgM, CD4 and CD8, and CD45R(B220) and the HyHEL10 idiotype as indicated. (C) Reconstitution of Peyer's patches. Peyer's patches were classified as very small, small, or normal (light gray, dark gray, and black bars, respectively), and the histogram depicts the average number of each size (based on four to six mice per group). (D) Immunohistological examination of Peyer's patches from an MD4/DO-TCR(Rag1 −/−) mouse (left) and an MD4(Rag1 −/−) mouse (right). Sequential sections have been stained for CD45R(B220), PNA, and Ki67 (a marker of proliferating cells). GCs (which were also found to stain for FDC markers; not shown) can be seen as small PNA+Ki67+ clusters within the B cell follicles (marked B) of MD4/DO-TCR(Rag1 −/−) but not MD4(Rag1 −/−) mice, as indicated by arrowheads.
Somatic Hypermutation in the Absence of RAG1 or Active DNA-PKcs.
GC B cells from the Peyer's patches of MD4/DO-TCR(Rag1 −/−) mice were sorted by virtue of their CD45R(B220)+PNAhi phenotype. Sequence analysis of the PCR-amplified transgenic Vκ segments revealed that the Vκ transgenes had accumulated multiple somatic mutations, and there was no clear difference in the extent of transgene mutation in the B cells from RAG1-deficient and RAG1-proficient mice, as judged by the average number of mutations per mutated sequence, the distribution of mutations across the mutation domain, the transition bias, or the G/C to A/T ratio (Fig. 3 and Table ).
Figure 3.
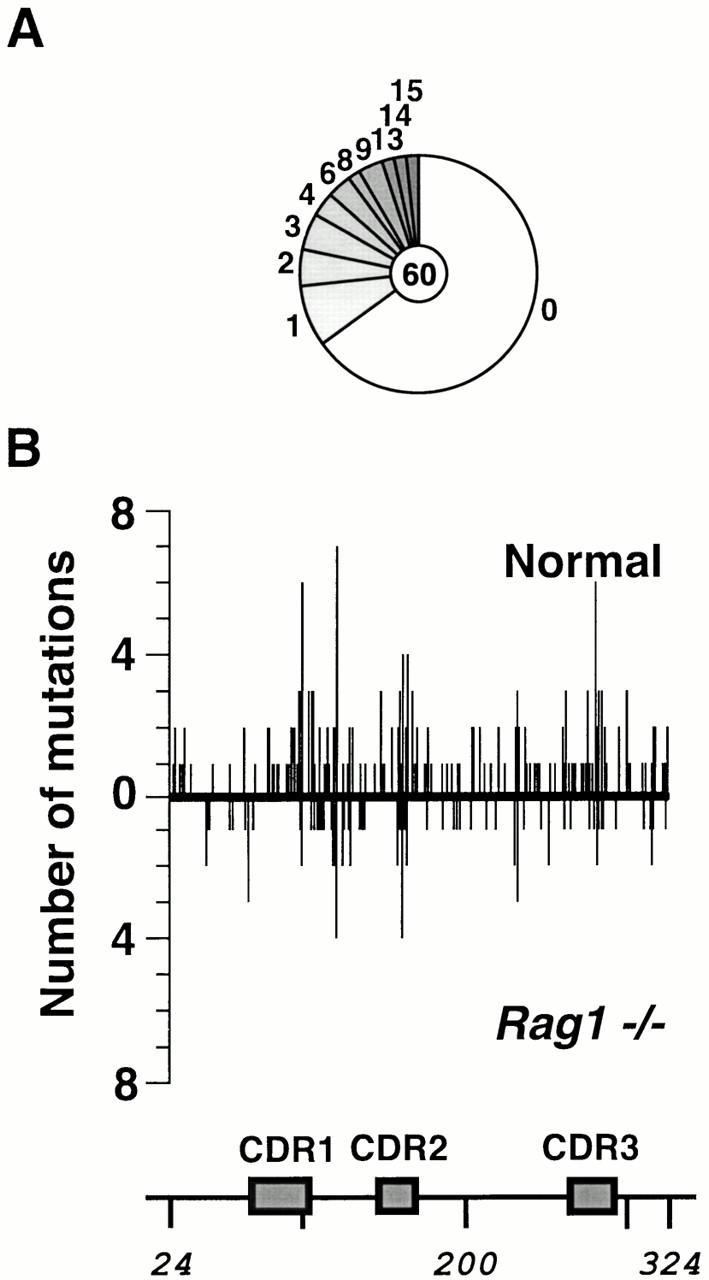
Somatic mutation of the Vκ transgene in MD4/DO-TCR(Rag1 −/−) mice. (A) Sequence diversity due to somatic mutation of the transgenic Vκ PCR amplified from sorted GC B cells from Peyer's patches of MD4/DO-TCR(Rag1 −/−) mice. The mutations identified in samples obtained from five mice (analyzed in two pools) have been combined. (B) Comparison of the distribution of mutations across the Vκ domain in MD4 (above the line) and MD4/DO-TCR(Rag1 −/−) mice (below the line).
Table 1.
Nucleotide Substitution Preferences
| To | ||||||
|---|---|---|---|---|---|---|
| From | Background | T | C | G | A | Total |
| T | Normal | – | 8.3 | 4.0 | 4.0 | 16 |
| Rag1 −/− | – | 4.3 | 0.9 | 6.0 | 11 | |
| SCID | – | 5.2 | 2.8 | 3.2 | 11 | |
| C | Normal | 15 | – | 4.6 | 3.3 | 22 |
| Rag1 −/− | 14 | – | 2.5 | 5.0 | 21 | |
| SCID | 2.3 | – | 4.0 | 16 | 23 | |
| G | Normal | 2.5 | 6.0 | – | 13 | 22 |
| Rag1 −/− | 5.3 | 2.1 | – | 16 | 23 | |
| SCID | 2.9 | 7.9 | – | 23 | 34 | |
| A | Normal | 12 | 7.0 | 19 | – | 38 |
| Rag1 −/− | 8.7 | 9.5 | 25 | – | 43 | |
| SCID | 12 | 6.5 | 14 | – | 33 | |
Single nucleotide substitutions on the Vκ sense strand are expressed as a percentage of the total number of mutations identified and have been corrected for base composition of the target. Database statistics are as follows: Normal, 303 mutations (none clonally related) in 118 sequences with 50 mutated sequences giving 6.1 mutations per mutated sequence; Rag1 −/−, 108 mutations (6 clonally related) in 60 sequences with 21 mutated sequences giving 5.1 mutations per mutated sequence; SCID, 267 mutations (48 clonally related) in 95 sequences with 55 mutated sequences giving 4.9 mutations per mutated sequence.
A similar approach to the analysis of mutations in SCID mice also revealed abundant somatic mutations. Again, analysis of the distribution of mutations over the Vκ domain and its 3′ flank as well as of the nucleotide substitution preferences does not reveal any major divergence from the pattern of somatic mutation in normal mice, although the proportion of mutations at GC base pairs is increased from 44% in normal (or RAG1-deficient) mice to 57% in MD4/DO-TCR(SCID) mice (Fig. 4A and Fig. C; Table ). It is interesting that deletions, which have previously been proposed to be generated by somatic hypermutation in human B cells 9 10 11, also occasionally accompany somatic hypermutation in mice even in the absence of DNA-PKcs (Fig. 4 B).
Figure 4.
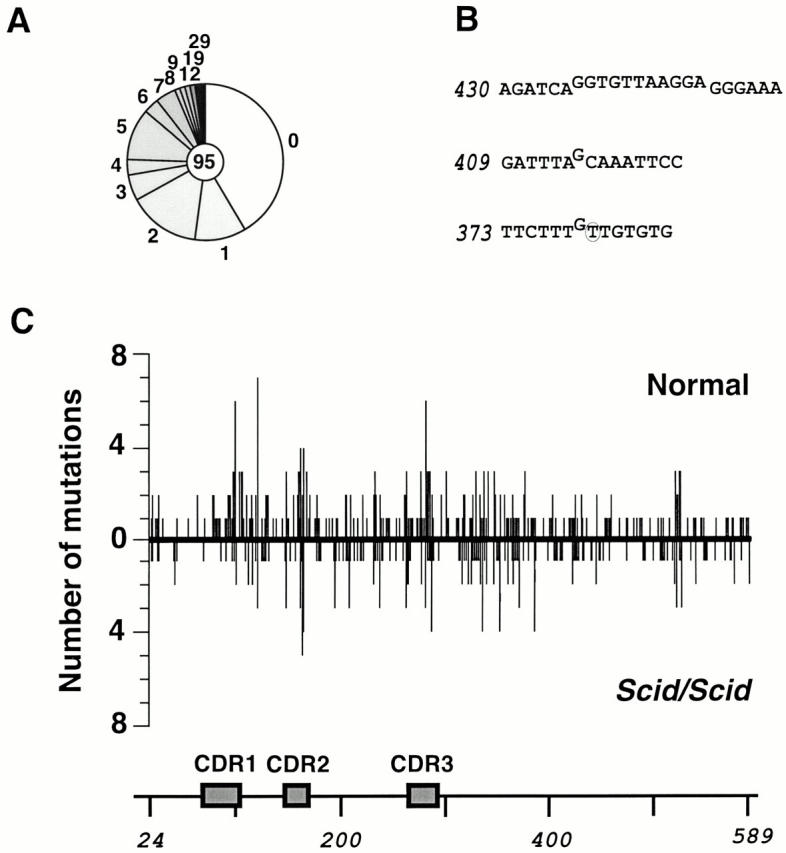
Somatic mutation of the Vκ transgene in MD4/DO-TCR(Scid/Scid) mice. (A) Sequence diversity due to somatic mutation of the transgenic Vκ PCR amplified from sorted GC B cells from Peyer's patches of MD4/DO-TCR(Scid/Scid) mice. The mutations identified in samples obtained from five mice (analyzed in pools of two mice, two mice, and one mouse) have been combined. (B) Mutations other than nucleotide substitutions identified in the transgenic Vκ in MD4/DO-TCR(Scid/Scid) mice. Numbers indicate the first nucleotide in the sequence string (see Fig. 1 A). Deleted nucleotides are shown above the line, and single nucleotide substitutions are circled, with the novel base being specified. The deletion in sequence string 409 forms part of a mutational dynasty, indicating that it did not arise from a PCR artefact. Deletions were also identified among transgenic Vκ sequences from GC B cells of normal MD4 mice. (C) Comparison of the distribution of mutations across the Vκ domain in MD4 (above the line) and MD4/DO-TCR(Scid/Scid) mice (below the line).
We were careful to exclude the possibility that the GCs in MD4/DO-TCR(SCID) mice were not simply populated by DNA-PKcs + revertant founder B cells, which were then able to expand and undergo hypermutation owing to their DNA-PK+ phenotype. We therefore amplified the relevant portion of the DNA-PKcs gene from DNA extracted from two distinct pools of sorted CD45R (B220)+PNAhi B cells from MD4/DO-TCR(SCID) mice that had also been used for Vκ mutation analysis and had revealed abundant Vκ somatic mutation. Sequence analysis revealed no case of reversion of the TAA-406 stop codon (or indeed any other mutation) out of 16 templates sequenced. Thus, somatic hypermutation can clearly occur in SCID B cells.
Discussion
GCs are found within the Peyer's patches of MD4/DO-TCR(Rag1 −/−) and MD4/DO-TCR(SCID) mice. This presumably means that, despite the monoclonal nature of both B and T cell receptors in these animals, there are environmental antigens in the gut that yield both B and T cell epitopes of sufficient affinity to allow activation and productive interaction of the MD4-BCR–expressing B cells with DO-TCR T cells. The same strategy used here could therefore be extrapolated to monitor somatic hypermutation in other mutant mice incapable of productive antigen receptor assembly (e.g., Ku70- and Ku80-deficient mice).
Deficiency in RAG1 has no apparent effect on the extent or pattern of mutation. That hypermutation can proceed without RAG1 is not unanticipated in view of previous observations referred to by Zheng et al. 20 using lymphocyte-repopulated animals. However, that mutation can proceed in genetically reconstituted SCID mice is, to our knowledge, the first identification of a gene product involved in DNA metabolism that is differentially required for class-switching and somatic hypermutation.
The ability of hypermutation to proceed essentially unaffected as regards both extent and pattern in the absence of an active DNA-PKcs makes it improbable that NHEJ is required for hypermutation. Rather, it is likely that double-strand DNA breaks are either not necessary intermediates in the hypermutation process, or, if they are, that such breaks are resolved (as in homologous recombination) by templating on the sister chromatid during the G2/S phase of the cell cycle with such template-dependent DNA synthesis possibly being error prone.
Acknowledgments
We thank Theresa Langford for help with animal handling and Andrew Johnson for flow cytometry.
M. Bemark was supported by a grant from the Swedish Cancer Society.
Footnotes
M. Bemark and J.E. Sale contributed equally to this work.
References
- Phung Q.H., Winter D.B., Cranston A., Tarone R.E., Bohr V.A., Fishel R., Gearhart P.J. Increased hypermutation at G and C nucleotides in immunoglobulin variable genes from mice deficient in the MSH2 mismatch repair protein. J. Exp. Med. 1998;187:1745–1751. doi: 10.1084/jem.187.11.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey S., Bertocci B., Delbos F., Quint L., Weill J.-C., Reynaud C.-A. Mismatch repair deficiency interferes with the accumulation of mutations in chronically stimulated B cells and not with the hypermutation process. Immunity. 1998;9:127–134. doi: 10.1016/s1074-7613(00)80594-4. [DOI] [PubMed] [Google Scholar]
- Rada C.A., Ehrenstein M.R., Neuberger M.S., Milstein C. Somatic hypermutation in MSH2 deficient mice is more focussed on intrinsic hotspots suggesting targeting to be a two stage process. Immunity. 1998;9:135–141. doi: 10.1016/s1074-7613(00)80595-6. [DOI] [PubMed] [Google Scholar]
- Ehrenstein M.R., Neuberger M.S. Deficiency in Msh2 affects the efficiency and local sequence specificity of immunoglobulin class-switch recombinationparallels with somatic hypermutation. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:3484–3490. doi: 10.1093/emboj/18.12.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader C.E., Edelmann W., Kucherlapati R., Stavnezer J. Reduced isotype switching in splenic B cells from mice deficient in mismatch repair enzymes. J. Exp. Med. 1999;190:323–330. doi: 10.1084/jem.190.3.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolink A., Melchers F., Andersson J. The SCID but not the RAG-2 gene product is required for S mu-S epsilon heavy chain class switching. Immunity. 1996;5:319–330. doi: 10.1016/s1074-7613(00)80258-7. [DOI] [PubMed] [Google Scholar]
- Casellas R., Nussenzweig A., Wuerffel R., Pelanda R., Reichlin A., Suh H., Qin X.F., Besmer E., Kenter A., Rajewsky K. Ku80 is required for immunoglobulin isotype switching. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:2404–2411. doi: 10.1093/emboj/17.8.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manis J.P., Gu Y., Lansford R., Sonoda E., Ferrini R., Davidson L., Rajewsky K., Alt F.W. Ku70 is required for late B cell development and immunoglobulin heavy chain class switching. J. Exp. Med. 1998;187:2081–2089. doi: 10.1084/jem.187.12.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sale J.E., Neuberger M.S. TdT accessible breaks are scattered over the V domain in a constitutively hypermutating cell line. Immunity. 1998;9:859–869. doi: 10.1016/s1074-7613(00)80651-2. [DOI] [PubMed] [Google Scholar]
- Goossens T., Klein U., Küppers R. Frequent occurrence of deletions and duplications during somatic hypermutationimplications for oncogenic translocations and heavy chain disease. Proc. Natl. Acad. Sci. USA. 1998;95:2463–2468. doi: 10.1073/pnas.95.5.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson P.C., de Boutellier O., Liu Y.-J., Potter K., Banchereau J., Capra J.D., Pascual V. Somatic hypermutation introduces insertions and deletions into immunoglobulin V genes. J. Exp. Med. 1998;187:59–70. doi: 10.1084/jem.187.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan K.N., Gangi-Peterson L., Sorscher D.H., Wang J., Gathy K.N., Mahajan N.P., Reeves W.H., Mitchell B.S. Association of terminal deoxynucleotidyl transferase with Ku. Proc. Natl. Acad. Sci. USA. 1999;96:13926–13931. doi: 10.1073/pnas.96.24.13926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mickelsen S., Snyder C., Trujillo K., Bogue M., Roth D.B., Meek K. Modulation of terminal deoxynucleotidyltransferase activity by the DNA-dependent protein kinase. J. Immunol. 1999;163:834–843. [PubMed] [Google Scholar]
- Bosma G.C., Custer R.P., Bosma M.J. A severe combined immunodeficiency mutation in the mouse. Nature. 1983;301:527–530. doi: 10.1038/301527a0. [DOI] [PubMed] [Google Scholar]
- Beamish H.J., Jessberger R., Priestley R.A., Blunt T., Kysela B., Jeggo P.A. The C-terminal conserved domain of DNA-PKcs, missing in the SCID mouse, is required for kinase activity. Nucleic Acids Res. 2000;28:1506–1513. doi: 10.1093/nar/28.7.1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodnow C.C., Crosbie J., Adelstein S., Lavoie T.B., Smith-Gill S.J., Brink R.A., Pritchard-Briscoe H., Wotherspoon J.S., Loblay R.H., Raphael K. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature. 1988;334:676–682. doi: 10.1038/334676a0. [DOI] [PubMed] [Google Scholar]
- Murphy K.M., Heimberger A.B., Loh D.Y. Induction by antigen of intrathymic apoptosis of CD4+CD8+TCRlo thymocytes in vivo. Science. 1990;250:1720–1723. doi: 10.1126/science.2125367. [DOI] [PubMed] [Google Scholar]
- Spanopoulou E., Roman C.A.J., Corcoran L.M., Schlissel M.S., Silver D.P., Nemazee D., Nussenzweig M., Shinton S.A., Hardy R.R., Baltimore D. Functional immunoglobulin transgenes guide ordered B-cell differentiation in Rag-1 deficient mice. Genes Dev. 1994;8:1030–1042. doi: 10.1101/gad.8.9.1030. [DOI] [PubMed] [Google Scholar]
- Grivel J.C., Ferrier P., Renard N., Jolly G., Jarry T., Leserman L. Rapid induction of anti-idiotypic responses to unmodified monoclonal antibodies from syngeneic mice following primary immunization. J. Immunol. Methods. 1993;158:173–182. doi: 10.1016/0022-1759(93)90211-o. [DOI] [PubMed] [Google Scholar]
- Zheng B., Han S., Spanopoulou E., Kelsoe G. Immunoglobulin gene hypermutation in germinal centres is independent of the RAG-1 V(D)J recombinase. Immunol. Rev. 1998;162:133–141. doi: 10.1111/j.1600-065x.1998.tb01436.x. [DOI] [PubMed] [Google Scholar]