

Abstract
Lymphocytes deficient in the T cell costimulatory molecule CD28 exhibit defects in cell survival, clonal expansion, and differentiation into effector cells. It is known that CD28-mediated signaling results in the upregulation of the Bcl family member Bcl-XL. To investigate the role that Bcl-XL plays in the various functions of CD28, we expressed Bcl-XL in CD28-deficient primary T lymphocytes using retrovirus-mediated gene transfer. T cells were activated in vitro and infected with Bcl-XL or control retroviruses; this method allows gene expression in activated, cycling cells. Expression of Bcl-XL in naive T cells was achieved by reconstitution of the immune system of lethally irradiated recipient mice with retrovirus-infected purified bone marrow stem cells from CD28−/− or wild-type donor mice. Our studies demonstrate that Bcl-XL prolongs the survival of CD28−/− T cells but does not restore normal proliferation or effector cell development. These results indicate that the various functions of CD28 can be dissociated, and provide an experimental approach for testing the roles of downstream signals in the functions of cellular receptors such as CD28.
Keywords: CD4+ T lymphocyte, retrovirus-mediated gene transfer, bone marrow reconstitution, costimulation, Th2 differentiation
Introduction
The functional responses of T lymphocytes are initiated by the recognition of antigens and costimulators expressed on professional APCs 1 2. The best-described costimulatory pathway involves the B7 molecules on APCs and their receptor, CD28, on T cells 3 4. Ligation of CD28 by B7 promotes the survival of T cells by stimulating expression of the antiapoptotic protein Bcl-XL 5 6 7 and enhances IL-2 production and sustained IL-2–driven T cell proliferation 8. In addition, CD28 is required for the development of Th2 effectors from naive CD4 T cells. Mice lacking CD28 show reduced T cell clonal expansion in response to antigen, and a marked defect in Th2-dependent responses 9 10 11.
We have initiated a gene complementation approach for identifying the key intracellular signals triggered by CD28, and for determining how these signals relate to functional responses. The principle of this approach is to express, by retrovirus-mediated gene transfer, genes encoding various signaling or antiapoptotic proteins in normal T cells lacking CD28. By assaying the functional responses of these reconstituted T cells to various stimuli, we can determine if the expressed genes restore any or all of the defects of CD28−/− T cells. The first gene we have chosen to establish this approach is Bcl-XL, for several reasons. First, Bcl-XL is rapidly induced in T cells upon antigen recognition and costimulation via CD28. Second, defective survival is a major abnormality in CD28−/− T cells, and it is possible that this alone accounts for the multiple functional defects associated with the absence of CD28. The following studies demonstrate that expression of Bcl-XL in CD28−/− T cells both in vitro and in vivo prolongs the survival of these cells but does not correct the observed defects in proliferation and Th2 differentiation. Thus, CD28 is involved in multiple biological responses of T cells, and these responses are likely to be mediated by different intracellular biochemical pathways.
Materials and Methods
Mice.
DO.11 transgenic mice expressing a TCR recognizing the hen egg white ovalbumin peptide, OVA 323–339, in the context of the MHC class II molecule I-Ad 12 were obtained from Dr. Dennis Loh (Hoffman-La Roche, Inc., Nutley, NJ). Crosses of DO.11 mice with CD28-deficient mice were obtained from Dr. Jeff Bluestone (University of Chicago, Chicago, IL). BALB/c mice, 6–8 wk old, were purchased from The Jackson Laboratory. All mice were bred and maintained in accordance with the guidelines of the Committee on Animals of the Harvard Medical School, and those prepared by the Committee of Care and Use of Laboratory Animal Resources, National Research Council.
Generation of Recombinant Retrovirus.
The murine stem cell virus (MSCV) 2.2 vector 13 modified to contain an internal ribosomal binding site, IRES (Novagen), and the cDNA of green fluorescent protein (GFP) (hGFP S65T; CLONTECH Laboratories, Inc.) were gifts of Dr. Bill Sha (University of California at Berkeley, Berkeley, CA; 14). In this vector, a foreign cDNA is inserted in front of the IRES element and the GFP cDNA so that one long transcript encompassing the foreign cDNA and the GPF cDNA is generated under control of the LTR from the pCMV mutant myeloproliferative sarcoma virus. The human Bcl-XL cDNA 15 was a gift of Dr. Gabriel Nunez (University of Michigan, Ann Arbor, MI). Human Bcl-XL has been demonstrated to be functional in mouse cells 16. The Bcl-XL cDNA was cloned into MSCV2.2 IRES GFP as a XhoI-XhoI fragment to generate MSCV2.2 Bcl-XL IRES GFP (MS-Bcl-XL).
Virus used for infection of primary T cells was produced by transfecting the ecotropic packaging cell line Phoenix E (obtained from Dr. G. Nolan, Stanford University, Stanford, CA) with MS or MS-Bcl-XL plasmid described above. Supernatants were collected at days 2 and 3 after transfection and either used directly for infection of target cells or snap frozen in liquid nitrogen and stored at −80°C. The titer of different virus stocks was determined by infecting a fixed number of NIH3T3 cells and measuring GFP expression in infected cells at 48 h after infection. Virus used for infection of bone marrow stem cells was produced by transfecting the vesicular stomatitis virus G protein producing packaging cell line, 293 GPG 17, with control or Bcl-XL plasmid. The pseudotyped virus in the cell supernatant was harvested over 7 consecutive days and concentrated by ultracentrifugation to titers of 108–109 infectious particles per milliliter.
T Cell Culture and Infection with Recombinant Retrovirus In Vitro.
Spleen and lymph node cells were collected from DO.11 wild-type (WT) and CD28−/− mice. The cells were depleted of red blood cells, purified by centrifugation on a Ficoll Paque gradient (Amersham Pharmacia Biotech), and activated at 3 × 106 cells/ml with 1 μg/ml OVA 323–339 peptide in 24-well plates in a total volume of 1 ml. The culture medium consisted of RPMI 1640 supplemented with 2 mM glutamine, penicillin, streptomycin, nonessential amino acids, sodium pyruvate, 10 mM Hepes (all from GIBCO BRL), 5 × 10−5 M 2-ME (Bio-Rad), and 10% heat-inactivated FCS (Sigma-Aldrich). After 24 h, the cells were collected, counted, and replated in a 24-well plate at 1.25 × 106 cells/ml in 2 ml retroviral supernatant containing 8 μg/ml lipofectamine (GIBCO BRL). The culture plates were then centrifuged at 1,800 rpm for 45 min at room temperature. The following day, 1.5 ml of the viral inoculum was replaced with fresh culture medium. The cells were split 1:2 or 1:3 on that day or the day after, depending on cell density, and were collected, Ficolled, and analyzed by flow cytometry at day 3 after infection. The cells were then either used directly in restimulation cultures or, if indicated, stained with TCR (KJ1-26) 18 and CD4-specific antibodies and sorted on a MoFlo high-speed cell sorter (see below). For restimulation, 2.5 × 105 cells were cultured with 2.5 × 106 mitomycin C–treated BALB/c splenocytes (APCs) and 0.1 or 1 μg/ml OVA peptide in a volume of 1 ml/well in a 24-well plate.
Transplantation of Retrovirus-infected Purified Bone Marrow Stem Cells.
Sca-1+Lin− bone marrow stem cells were isolated from femoral bone marrow of WT DO.11 or CD28−/− DO.11 mice and cultured in 50 ng/ml murine stem cell factor (mSCF), 50 ng/ml human flt3 ligand, 200 U/ml murine IL-6, and 200 U/ml murine IL-3, all from R&D Systems. After 2–3 d, the cells were infected with pseudotyped virus as follows. Concentrated virus was added to precooled cells in the presence of polybrene (8 μg/ml; Sigma-Aldrich), and after an initial incubation at 4°C, the cells were transferred to a 37°C incubator and left for an additional period of 2–3 h. Then, the cells were washed and injected into lethally (1,200 rads) irradiated 10–12-wk-old male BALB/c mice by tail vein injection.
Assays of Proliferation and Cytokine Production.
Proliferation of T cells was assayed by incubating 2.5 × 104 T cells with 2.5 × 105 mitomycin C–treated BALB/c splenocytes and a range of different peptide concentration for 1–3 d. During the final 8–10 h of culture, the cells were pulsed with 1 μCi of 3[H]thymidine and then harvested for liquid scintillation counting. The concentrations of cytokines in culture supernatants were measured on days 1–3 by ELISA using anticytokine antibodies from BD PharMingen. Cytokine production by individual cells was assessed by intracellular cytokine staining (see below).
Flow Cytometry.
T cell cultures were analyzed for the presence of the TCR and CD4 by incubation with biotinylated KJ1-26 followed by incubation with PE-conjugated streptavidin (BD PharMingen) and CyC-conjugated anti–murine CD4 antibody (BD PharMingen). All surface stains were done in PBS, 1% BSA, 0.01% sodium azide and the cells were analyzed immediately after staining without fixation. When cell sorting was required, a MoFlo high-speed cell sorter (Cytomation) was used.
For intracellular cytokine stains, 5 × 105 cells in a volume of 200 μl medium were activated in the presence of 10 ng/ml PMA (Sigma-Aldrich) and 1 μM calcium ionophore (A23187; Sigma-Aldrich) in 96-well plates for 4–6 h. During the last 2 h of activation, brefeldin A was included to give a final concentration of 10 μg/ml. The cells were washed once in PBS, fixed for 10 min with 200 μl 2% (wt/vol) formaldehyde (Polysciences) in PBS, and washed twice with PBS and once with 0.5% (wt/vol) saponin (Sigma-Aldrich), 1% (wt/vol) BSA (Sigma-Aldrich), and 0.01% (wt/vol) sodium azide (PBS/saponin). The cells were stained with PE-conjugated antibodies against murine IL-2, IL-4, IFN-γ, or a PE-conjugated rat IgG1 isotype control. All antibodies were diluted to 10 μg/ml in PBS/saponin. The cells were subsequently washed twice with PBS/saponin and once with PBS, and then stained for surface CD4 as described above. Immediately after the surface stain, the cells were washed twice with PBS, resuspended in PBS/1% BSA, and analyzed by FACS®.
Western Blots.
T cells infected with control virus or Bcl-XL virus were lysed in RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% deoxycholic acid, 0.5% SDS, and 50 mM Tris-HCl, pH 8.0) to give a concentration of 108 cells/ml. Proteins in cell lysates from 2 × 106 cells were separated by SDS-PAGE in a 15% gel, blotted onto a polyvinylidene difluoride (PVDF) membrane, and probed with 1 μg/ml of a rabbit polyclonal Bcl-XL/S antiserum (Santa Cruz Biotechnology) followed by incubation with a horseradish peroxidase–conjugated anti–rabbit antibody (ICN Biomedicals), and developed using an enhanced chemiluminescence kit (Amersham Pharmacia Biotech).
Results
Retrovirus-mediated Expression of Bcl-XL in T Cells Activated In Vitro.
To establish the system for introducing genes into CD28−/− T cells, we activated DO.11 WT and DO.11 CD28−/− T cells for 24 h with OVA peptide and then incubated the cells with virus expressing GFP only (control virus) or virus expressing human Bcl-XL (Bcl-XL virus). Analysis of GFP expression at 72 h after infection showed that >30% of the DO.11 T cells were GFP+ (Fig. 1 A). This was true even for the CD28−/− T cells, despite lower levels of proliferation and expansion compared with WT cells. Western blot analysis of the infected cells demonstrated that Bcl-XL protein was present in WT cells infected with the control virus, was virtually undetectable in the CD28−/− cells, and was readily detectable in CD28−/− cells infected with the Bcl-XL virus (Fig. 1 B). Thus, the retrovirus infection method leads to the expression of the introduced cDNA.
Figure 1.
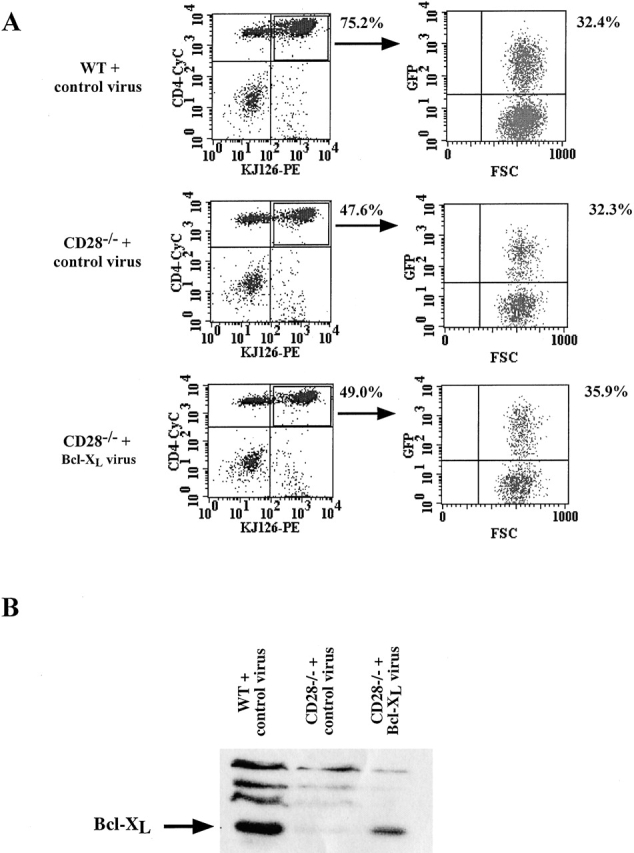
Retrovirus-mediated expression of Bcl-XL in DO.11 WT and DO.11 CD28−/− primary T lymphocytes. T cells were activated in vitro with OVA peptide and APCs and infected on day 1 with retrovirus encoding either GFP alone (control virus) or Bcl-XL and GFP (Bcl-XL virus). (A) Cells were collected and stained at day 3 after viral infection with antibodies to CD4 and the DO.11 TCR (KJ1-26) and analyzed for GFP expression in CD4+KJ1-26+ cells by flow cytometry. The number beside the FACS® plot represents the percentage of CD4+KJ1-26+ T cells. FSC, forward scatter. (B) Cells infected with retrovirus containing GFP alone or GFP/Bcl-XL as in A were cultured for an additional 4 d in the presence of APCs and OVA peptide, collected, lysed, and analyzed by Western blotting with a polyclonal antiserum specific for Bcl-XL/S protein.
Bcl-XL Expression Does Not Enhance Proliferation or Th2 Differentiation of In Vitro–infected CD28−/− T Cells.
After confirming that the retrovirus-introduced Bcl-XL protein was expressed in T cells lacking CD28, we asked whether Bcl-XL expression in CD28−/− T cells could correct the previously described defects in survival, proliferation, and Th2 differentiation. CD28−/− T cells were activated in vitro with OVA peptide and APCs, and the cells were infected with either control virus or Bcl-XL virus. The viable cells were then collected, cultured with medium alone (no stimulus) or IL-2, and assayed at various time points for total cell numbers as well as the percentage of CD4+KJ1-26+ cells that were GFP+ (Fig. 2). In the absence of IL-2, the number of recovered viable cells remained constant for up to 8 d. However, the proportion of GFP+ cells in the Bcl-XL virus–infected population increased progressively (Fig. 2). This result suggests that Bcl expression promotes cell survival but not expansion. Consistent with this result, when the cells were cultured in IL-2, the total numbers of recovered cells increased in both the control virus– and Bcl-XL virus–infected populations but the proportion of GFP+ cells remained constant (Fig. 2). Therefore, expression of Bcl-XL does not provide a selective growth advantage to T cells induced to proliferate with IL-2.
Figure 2.

Expression of Bcl-XL promotes the survival of CD28−/− T cells. T cells were activated with OVA peptide and APCs and infected with control virus or Bcl-XL virus as in the legend to Fig. 1. 3 d after infection, the cells were collected, Ficolled (to remove dead cells, antigen, and APCs), and put into culture with medium alone (no stimulus) or IL-2 at 50 U/ml. Cells were then harvested from the wells and counted 2, 4, 6, and 8 d after reculture (left). Aliquots of the cells were stained with anti-CD4 and anti–KJ1-26 and analyzed by flow cytometry. The percentage of CD4+KJ1-26+ GFP+ cells was then determined (right).
To assess the role of Bcl-XL in T cell proliferation induced by antigen and APCs, the virally infected cells (WT with control virus, CD28−/− with control virus, and CD28−/− with Bcl-XL virus) were sorted into GFPhigh KJ1-26+CD4+ populations. The sorted cells were cultured for an additional 3 d with APCs and OVA peptide (to expand cell numbers for staining) and re-analyzed for GFP expression on day 3. At this time, >90% of the CD4 T cells infected with Bcl-XL virus and sorted on the basis of high GFP expression remained GFPhigh (Fig. 3 A). The sorted populations were stimulated with antigen and APCs, and proliferation was assayed. These experiments showed that the CD28−/− T cells proliferated much less than the WT T cells, and that Bcl-XL expression did not restore normal proliferation (Fig. 3 B). In some experiments, a small increase in the proliferative response was observed on day 3 in the CD28−/− lymphocytes expressing Bcl-XL. This was most likely due to the fact that Bcl-XL expression promoted the survival of greater numbers of CD28−/− T cells, thus leading to a gradual increase in total cell numbers in each culture well and therefore an increase in total DNA synthesis per well.
Figure 3.
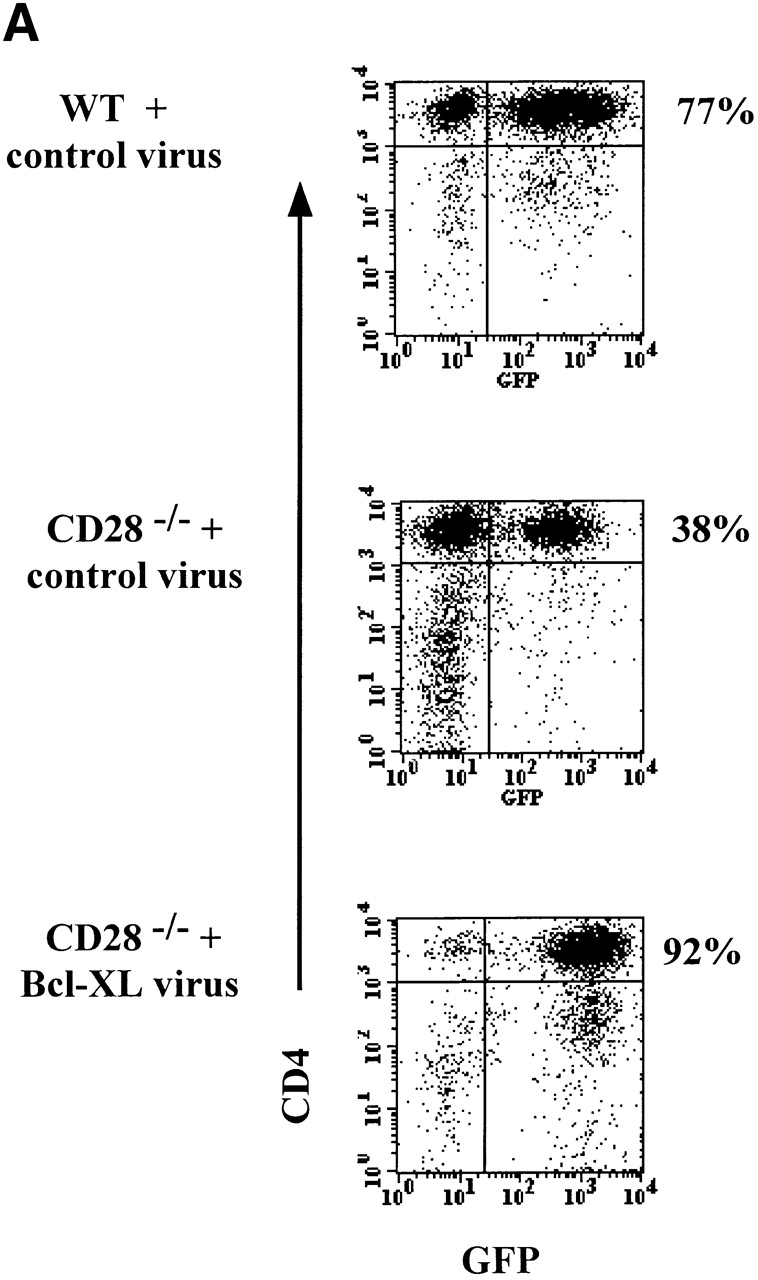

Expression of Bcl-XL in CD28−/− T cells does not restore normal proliferation. DO.11 WT or DO.11 CD28−/− T cells activated in vitro were infected with control or Bcl-XL virus on day 1 (as described in the legend to Fig. 1) and harvested 3 d later. Using a Cytomation MoFlo high speed sorter, the CD4+KJ1-26+ cells expressing high levels of GFP were collected. The cells were then plated in 24-well plates at 2.5 × 105 T cells/well with 2.5 × 106 APCs and 1 μg/ml peptide or in 96-well plates at 2.5 × 104 T cells/well with 2.5 × 105 APCs and a range of OVA peptide concentrations. (A) After 4 d of culture in 24-well plates with APCs and OVA peptide, the cells were collected and stained with antibodies to CD4. The cells were then analyzed by flow cytometry for GFP expression in the CD4+ population. (B) The microcultures of DO.11 WT or DO.11 CD28−/− GFPhigh T cells fractions stimulated with APCs and OVA peptide were pulsed with [3H]thymidine for 6 h on days 1 and 3, and the incorporated radioactivity was measured.
To evaluate the effect of Bcl-XL on T cell differentiation, the WT or CD28−/− DO.11 lymphocytes infected with control or Bcl-XL virus were sorted into GFPhighKJ1-26+CD4+ populations as described above. The GFPhigh populations were then restimulated with OVA peptide and APCs, and culture supernatants were analyzed for IL-2, IL-4, and IL-5 by ELISA. As demonstrated in Fig. 4, CD28−/− T cells produced much less of these cytokines than did WT T cells, and Bcl-XL expression did not restore cytokine production. Therefore, although expression of Bcl-XL clearly enhances the survival of CD28−/− lymphocytes, it does not correct the defects in effector cell development that are seen in these costimulator-deficient cells.
Figure 4.

Expression of Bcl-XL does not restore normal cytokine production in CD28−/− T cells. Activated DO.11 WT (black bars) or DO.11 CD28−/− GFPhigh T cells (expressing control virus [white bars] or Bcl-XL virus [hatched bars]) were generated as described in the legend to Fig. 3. These cells were then cultured with OVA peptide (1 μg/ml) and mitomycin C–treated APCs in 24-well plates. After 12 h of restimulation, supernatants were harvested and analyzed for IL-2, IL-4, and IL-5 by ELISA.
Expression of Bcl-XL in Naive CD28−/− T Cells by Reconstitution of Lethally Irradiated Mice with Retrovirus-infected Bone Marrow Stem Cells.
One of the limitations of the in vitro infection method is that only cells that are cycling can be infected, thus restricting this technique to activated T cells. To express genes in naive T cells, we infected Sca-1+Lin− purified bone marrow stem cells from WT or CD28−/− DO.11 TCR transgenic mice, and used the transduced bone marrow to reconstitute lethally irradiated BALB/c recipients. CD4+KJ1-26+ cells were observed in the peripheral blood from 5–6 wk after transfer (Fig. 5 A). The numbers of CD4+KJ1-26+ cells in the spleen and lymph nodes were similar in mice reconstituted with DO.11 WT bone marrow stem cells or with CD28−/− DO.11 bone marrow stem cells (Fig. 5 A), and 15–50% of the mature DO.11 T cells that developed also expressed GFP (Fig. 5 B). Thus, expression of Bcl-XL does not alter the maturation of the T cells. However, Bcl-XL expression did give the cells a survival advantage, because in mice reconstituted with the Bcl-XL virus, the percentage of GFP+ cells among mature lymphocytes was significantly greater than in mice reconstituted with control virus–infected precursors.
Figure 5.


Reconstitution of lethally irradiated mice with purified bone marrow stem cells infected with recombinant Bcl-XL retrovirus. Lethally irradiated BALB/c mice were reconstituted with stem cells purified from bone marrow of DO.11 WT or DO.11 CD28−/− mice infected with control retrovirus or with retrovirus expressing Bcl-XL. (A) Spleen and lymph nodes were harvested at 6–7 wk after transfer and analyzed for KJ1-26 and CD4 expression by flow cytometry. The BALB/c control was a nonirradiated age- and sex-matched mouse. Irradiated BALB/c mice that did not receive any bone marrow died within 10 d of treatment. (B) FACS® analysis of GFP expression in CD4+KJ1-26+ cells from peripheral blood, spleen, and lymph nodes of BALB/c mice reconstituted with purified DO.11 CD28−/− bone marrow stem cells infected with Bcl-XL virus.
To evaluate the function of Bcl-XL in the bone marrow–reconstituted CD28−/− T cells, CD4+ T cells were collected from the spleen and lymph nodes of reconstituted mice and cultured with APCs and OVA peptide. Proliferative responses of the naive T cells were measured on days 1 and 2 of culture. As was found in the previously described in vitro retroviral infection experiments, expression of Bcl-XL in the naive CD28−/− lymphocytes via bone marrow reconstitution did not restore their proliferative capacity (Fig. 6).
Figure 6.

Proliferation of naive T cells from mice reconstituted with stem cells infected with Bcl-XL virus. Naive CD4+ T cells isolated from the lethally irradiated mice reconstituted with bone marrow stem cells infected with control or Bcl-XL virus (see Fig. 5) were cultured with mitomycin C–treated APCs and a range of OVA peptide in 96-well plates. Plates were pulsed with [3H]thymidine for 6 h on days 1 and 2 of activation, and the incorporated radioactivity was measured.
The previous experiments investigating the role of Bcl-XL in the effector differentiation of CD28−/− T cells relied on infecting the cells after T cell activation by antigen had taken place. We were therefore interested to determine if expression of Bcl-XL in naive CD28−/− T cells could promote the production of IL-4 by these cells. Lymph node T cells from bone marrow–reconstituted mice were cultured with antigen and APCs, and IL-4 production was measured by intracellular staining, gating on GFP+ cells. As was observed with the introduction of Bcl-XL into in vitro–activated CD28−/− T cells, the presence of Bcl-XL at the initiation of activation did not restore differentiation into IL-4–producing effector cells (Fig. 7).
Figure 7.
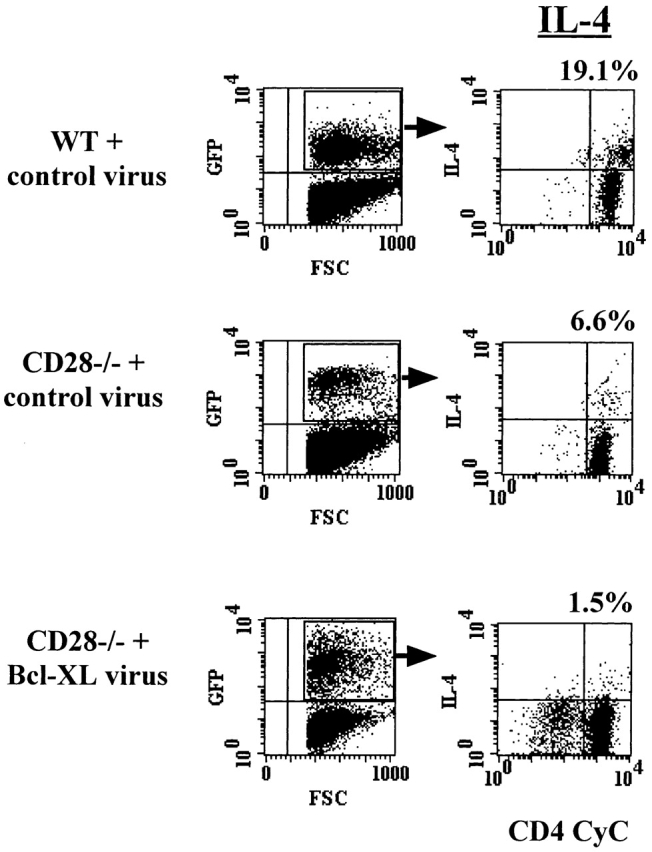
Expression of Bcl-XL in naive T cells does not restore normal cytokine production in DO.11 CD28−/− T cells. Naive spleen and lymph node CD4+ lymphocytes from the retrovirus-infected stem cell–reconstituted mice described in the legend to Fig. 5 were activated for 4 d in vitro with 1 μg/ml of OVA peptide and APCs and analyzed for IL-4 production by intracellular cytokine staining. FSC, forward scatter.
Discussion
The studies described in this paper were designed to establish a method for gene complementation in lymphocytes lacking selected proteins and to use this method to begin to define the specific functions of various molecules associated with CD28-mediated signals. It was essential to express genes in primary lymphocytes so that we could assay a variety of responses as well as target lymphocytes from various knockout mouse strains. In these initial experiments, we have asked if the antiapoptotic protein Bcl-XL can correct the functional defects of T cells lacking CD28.
We chose to focus on Bcl-XL, as this antiapoptotic protein is directly induced by CD28 costimulation. Furthermore, the failure of sustained responses of CD28−/− T cells has been postulated to result primarily from an absence of Bcl-XL and decreased cell survival. Using a retroviral construct that gives efficient expression from a bicistronic message, we have achieved high expression of the marker gene (GFP) as well as the gene of interest (Bcl-XL) in primary naive and activated T cells. Western blot analysis of bulk cultures infected with Bcl-XL GFP virus showed that these cell populations express the Bcl-XL protein. We also found that cells infected with the Bcl-XL virus had the expected survival advantage over cells infected with control virus, thereby demonstrating that the introduced Bcl-XL gene was functional. The ability to use the GFP-containing vector for gene expression both in in vitro–activated lymphocytes and in bone marrow–derived cells greatly increases the potential utility of this retroviral method. Infection of in vitro–activated lymphocytes allows for rapid screening of candidate genes for effector function at early and late stages of lymphocyte activation, whereas stable gene expression in bone marrow–derived cells allows us to examine the consequences of expressing selected genes in developing lymphocytes and in naive (unstimulated) lymphocytes.
The analyses of CD28−/− T cells that do or do not express Bcl-XL demonstrate that Bcl-XL promotes the survival of CD28−/− T cells but does not correct the defects in proliferation and effector differentiation. The ability of Bcl-XL to promote the survival of CD28−/− T cells is consistent with the hypothesis that reduced expression of this antiapoptotic protein is most likely responsible for the defective survival of these cells 5 6 7. Some studies have suggested that CD28 may be important for sustained T cell proliferation primarily through the ability of CD28-mediated signals to induce antiapoptotic proteins 6, rather than by promoting the cycling of T cells. Our data indicate that CD28 plays an additional role in T cell proliferation unrelated to its ability to promote survival, because restoring survival by expressing Bcl-XL is not sufficient to restore T cell proliferation.
CD28−/− T cells also show markedly deficient Th2 differentiation, and Bcl-XL does not correct this defect. One explanation for this result may be that Th2 differentiation requires cell cycling 19, CD28−/− T cells do not cycle normally, and Bcl-XL expression does not correct this defect. Alternatively, CD28 may stimulate Th2 differentiation by promoting the activation of transcription factors that enhance Th2 gene expression, or by making the promoters of Th2-specific genes accessible to transcription factors 20 21 22. It is likely that Bcl-XL does not directly enhance transcription factor activity or chromatin accessibility, and therefore does not correct the Th2 differentiation defect.
In summary, this study demonstrates that CD28-induced Bcl-XL–mediated survival function can be separated from the roles of CD28 in T cell proliferation and differentiation. Therefore, other CD28-induced signals must be necessary to stimulate normal cell expansion and differentiation. The experimental system described in this paper is currently being used to express a variety of constitutively active signaling molecules in T cells lacking CD28. Such a gene complementation approach provides a powerful method for identifying various downstream mediators important in the function of cellular receptors such as CD28.
Acknowledgments
We thank Bill Sha and Ken Murphy for help and generous gifts of retroviral constructs, Zoher Ghogawala for advice on retroviral techniques, Shu Wei Jiang for cell sorting, and Ted Mau and Peggy Russell for technical assistance.
This work was supported by National Institutes of Health grants AI35297 and AI25022 (A.K. Abbas), a grant from Deutsche Forschungsgemeinschaft (C. Klein), and a Basic Research award from the Glaxo Wellcome Institute for Digestive Health (P.G. Andres).
Footnotes
Abbreviations used in this paper: GFP, green fluorescent protein; WT, wild-type.
P.G. Andres and A.K. Abbas's present address is Department of Pathology, School of Medicine, University of California at San Francisco, San Francisco, CA 94143.
C.A. London's present address is Department of Veterinary Surgery and Radiology, School of Veterinary Medicine, University of California at Davis, Davis, CA 95616.
References
- Lanzavecchia A., Lezzi G., Viola A. From TCR engagement to T cell activationa kinetic view of T cell behavior. Immunity. 1998;96:1–4. doi: 10.1016/s0092-8674(00)80952-6. [DOI] [PubMed] [Google Scholar]
- Abbas A.K., Murphy K.M., Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383:787–793. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- Gause W.C., Halvorson M.J., Lu P., Greenwald R., Linsley P., Urban J.F., Finkelman F.D. The function of costimulatory molecules and the development of IL-4 producing T cells. Immunol. Today. 1997;18:115–120. doi: 10.1016/s0167-5699(97)01005-0. [DOI] [PubMed] [Google Scholar]
- Lenschow D.J., Walunas T.L., Bluestone J.A. CD28/B7 system of costimulation. Annu. Rev. Immunol. 1996;14:233–258. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
- Boise L.H., Minn A.J., Noel P.J., June C.H., Accavitti M.A., Lindsten T., Thompson C.B. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL . Immunity. 1995;3:87–98. doi: 10.1016/1074-7613(95)90161-2. [DOI] [PubMed] [Google Scholar]
- Noel P.J., Boise L.H., Green J.M., Thompson C.B. CD28 costimulation prevents cell death during primary T cell activation. J. Immunol. 1996;157:636–642. [PubMed] [Google Scholar]
- Sperling A.I., Auger J.A., Ehst B.D., Rulifson I.C., Thompson C.B., Bluestone J.A. CD28/B7 interactions deliver a unique signal to naive T cells that regulates cell survival but not early proliferation. J. Immunol. 1996;157:3909–3917. [PubMed] [Google Scholar]
- Umlauf S.W., Beverly B., Lantz O., Schwartz R.D. Regulation of interleukin 2 gene expression by CD28 costimulation in mouse T-cell clonesboth nuclear and cytoplasmic RNAs are regulated with complex kinetics. Mol. Cell. Biol. 1995;15:3197–3205. doi: 10.1128/mcb.15.6.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahinian A., Pfeffer K., Lee K.P., Kundig T.M., Kishihara K., Wakeham A., Kawai K., Ohashi P.S., Thompson C.B., Mak T.W. Differential T cell costimulatory requirements in CD28-deficient mice. Science. 1993;261:609–612. doi: 10.1126/science.7688139. [DOI] [PubMed] [Google Scholar]
- Lenschow D.J., Herold K.C., Rhee L., Patel B., Koons A., Qin H.-Y., Fuchs E., Singh B., Thompson C.B., Bluestone J.A. CD28/B7 regulation of Th1 and Th2 subsets in the development of autoimmune diabetes. Immunity. 1996;5:285–293. doi: 10.1016/s1074-7613(00)80323-4. [DOI] [PubMed] [Google Scholar]
- Rulifson I.C., Sperling A.I., Fields P.E., Fitch F.W., Bluestone J.A. CD28 costimulation promotes the production of Th2 cytokines. J. Immunol. 1997;158:658–665. [PubMed] [Google Scholar]
- Murphy K.M., Heimberger A.B., Loh D.Y. Induction by antigen of intrathymic apoptosis of CD4+CD8+ TCRlo thymocytes in vivo. Science. 1990;250:1720–1723. doi: 10.1126/science.2125367. [DOI] [PubMed] [Google Scholar]
- Hawley R.G., Fong A.Z.C., Burns B.F., Hawley T.S. Transplantable myeloproliferative disease induced in mice by an interleukin 6 retrovirus. J. Exp. Med. 1992;176:1149–1163. doi: 10.1084/jem.176.4.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang W., Ranganath S.H., Weindel K., Bhattacharaya D., Murphy T.L., Sha W.C., Murphy K.M. Inhibition of Th1 development mediated by GATA-3 through an IL-4 independent mechanism. Immunity. 1998;9:745–755. doi: 10.1016/s1074-7613(00)80671-8. [DOI] [PubMed] [Google Scholar]
- Boise L.H., Gonzalez-Garcia M., Postema C.E., Ding L., Lindsten T., Turka L.A., Mao X., Nunez G., Thompson C.B. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- Chao D.T., Linette G.P., Boise L.H., White L.S., Thompson C.B., Korsmeyer S.J. Bcl-XL and Bcl-2 repress a common pathway of cell death. J. Exp. Med. 1995;182:821–828. doi: 10.1084/jem.182.3.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ory D.S., Neugeboren B.A., Mulligan R.C. A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus pseudotypes. Proc. Natl. Acad. Sci. USA. 1996;93:11400–11406. doi: 10.1073/pnas.93.21.11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrack P., Shimonkevitz R., Hannum C., Haskins K., Kappler J.W. The major histocompatibility complex–restricted antigen receptor on T cells. IV. An antiidiotypic antibody predicts both antigen and I-specificity. J. Exp. Med. 1983;158:1635–1646. doi: 10.1084/jem.158.5.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird J.J., Brown D.R., Mullen A.C., Moskowitz N.H., Mahowald M.A., Sider J.R., Gajewsky T.F., Wang C.-R., Reiner S.L. Helper T cell differentiation is controlled by the cell cycle. Immunity. 1998;9:229–237. doi: 10.1016/s1074-7613(00)80605-6. [DOI] [PubMed] [Google Scholar]
- Brown M.A., Hural J. Functions of IL-4 and control of its expression. Crit. Rev. Immunol. 1997;17:1–32. doi: 10.1615/critrevimmunol.v17.i1.10. [DOI] [PubMed] [Google Scholar]
- Agarwal S., Rao A. Modulation of chromatin structure regulates cytokine gene expression during T cell differentiation. Immunity. 1998;9:765–775. doi: 10.1016/s1074-7613(00)80642-1. [DOI] [PubMed] [Google Scholar]
- Glimcher L.H., Singh H. Transcription factors in lymphocyte development—T and B cells get together. Cell. 1999;96:13–23. doi: 10.1016/s0092-8674(00)80955-1. [DOI] [PubMed] [Google Scholar]