

Abstract
The strongest susceptibility genes for the development of systemic lupus erythematosus (SLE) in humans are null mutants of classical pathway complement proteins. There is a hierarchy of disease susceptibility and severity according to the position of the missing protein in the activation pathway, with the severest disease associated with C1q deficiency. Here we demonstrate, using novel in vivo models of apoptotic cell clearance during sterile peritonitis, a similar hierarchical role for classical pathway complement proteins in vivo in the clearance of apoptotic cells by macrophages. Our results constitute the first demonstration of an impairment in the phagocytosis of apoptotic cells by macrophages in vivo in a mammalian system. Apoptotic cells are thought to be a major source of the autoantigens of SLE, and impairment of their removal by complement may explain the link between hereditary complement deficiency and the development of SLE.
Keywords: systemic lupus erythematosus, complement deficiency, C1q, transgenic mice, apoptosis
Introduction
Inherited deficiencies of the early components of the classical pathway of complement, particularly C1q and C4, are the strongest disease susceptibility genes for the development of systemic lupus erythematosus (SLE) that have been identified in humans 1. In addition, gene-targeted C1q-deficient (C1qa −/−) mice also develop a lupus-like disease characterized by antinuclear autoantibodies and proliferative glomerulonephritis 2. These observations show that there is an activity of the early components of the classical pathway that protects from the development of SLE. It is an important challenge to understand the mechanism of this protection.
C1q has been shown to bind specifically to the surface blebs of apoptotic keratinocytes 3. This finding, together with the observation that the common autoantigens targeted in SLE are localized in high concentrations on the surfaces of apoptotic cells 4, has led to the hypothesis that C1q deficiency may predispose to autoimmunity as a consequence of an impaired clearance of apoptotic cells 2 3. Immunization of mice with apoptotic thymocytes can induce the production of autoantibodies 5, supporting the hypothesis that apoptotic cells may provide a source of autoantigens. In mice deficient in C1q, we have observed an increased number of glomerular apoptotic bodies in the absence of histological evidence of glomerulonephritis and independently from C3 activation, supporting the hypothesis that C1q may be involved in the clearance of apoptotic cells 2 6.
Here we have used complement-deficient mice to determine the relative contribution of different complement proteins to the phagocytosis of apoptotic cells by both inflammatory and resident murine macrophages using novel in vivo peritoneal models of apoptotic cell clearance. We have also studied the uptake of apoptotic cells in vitro by macrophages from C1q-deficient humans. We report the first demonstration of an in vivo apoptotic cell clearance defect in a mammalian system that correlates closely with predisposition to the spontaneous development of SLE.
Materials and Methods
Mice.
C1qa −/− mice were generated as previously described 2 and were studied on the mixed (129/Sv × C57BL/6) and pure inbred 129/Sv and C57BL/6 (backcrossed for seven generations) genetic backgrounds as specified when appropriate. C4-deficient (C4 −/−) and C3-deficient (C3 −/−) mice used were on the mixed (129/Sv × C57BL/6) genetic background 7 8. All experiments described were conducted on mice matched for age (8–12 wk), strain, and sex. All animal work was conducted in accordance with institutional guidelines.
Apoptotic Cells.
Apoptosis was induced in the human Jurkat T cell line by exposure to UVB312nm (equivalent to 800 mJ/cm2), followed by 2-h culture in RPMI 1640/0.4% BSA. This resulted in a population of cells that was ∼70% apoptotic and >95% “viable.” Mouse thymocytes were obtained by mechanical dissociation of thymi from 3–5-wk-old mice and were induced to undergo apoptosis by 4-h culture in RPMI 1640/10% heat-inactivated FCS in the presence of 1μM dexamethasone (Sigma-Aldrich). This resulted in a population of cells that was ∼50% apoptotic and >95% viable. Apoptosis was confirmed by annexin V binding, TdT-mediated dUTP-biotin nick-end labeling (TUNEL) staining, and morphological changes including nuclear fragmentation and condensation, loss of cell volume, and membrane blebbing. Cells were considered viable when they excluded propidium iodide and trypan blue, and this indicated the presence of an “early” apoptotic cell population.
In Vivo Clearance of Apoptotic Cells by Peritoneal Macrophages.
Gene-targeted and control mice were injected intraperitoneally with 1 ml of sterile 4% thioglycollate to induce sterile peritonitis. 4 d later, the mice were injected intraperitoneally with 107 apoptotic Jurkat T cells or 1–3 × 107 apoptotic murine thymocytes in a volume of 200 μl. To study phagocytosis by resident peritoneal macrophages, mice were injected with apoptotic cells without prior treatment with thioglycollate. In all of the experiments, unless otherwise stated, the mice were killed after 30 min and the peritoneal cells were recovered by lavage with 5 ml of ice cold HBSS/5 mM EDTA. Guided by the results obtained with the initial kinetic experiments, 30 min was selected as the single time point for the majority of experiments where more than one group of complement-deficient mice was studied. Phagocytosis was scored on coded cytospins of peritoneal cells stained with Diff-Quik (Dade Behring AG) and was expressed as the percentage of macrophages ingesting apoptotic cells or as a phagocytic index (the number of ingested apoptotic cells per 100 macrophages). Apoptotic cells were considered phagocytosed when >50% of the cell volume was contained within the border of the macrophage, which is believed to underestimate the level of phagocytosis 9. In all of the experiments, the percentage of macrophages ingesting correlated closely with the phagocytic index (data not shown). Between 300 and 500 macrophages were scored in each sample. FACS® analysis of phagocytic uptake was performed by fluorescent labeling of the apoptotic cells before injection with 5- (and 6-) carboxytetramethylrhodamine, succinimidyl ester (5[6]-TAMRA, SE; Molecular Probes) as previously described 10 and by detecting macrophages after lavage with FITC-conjugated F4/80 (Caltag). Reconstitution experiments were performed by adding serum to the apoptotic cells immediately before injection to a final concentration of 20% vol/vol.
In Vitro Phagocytosis Assay.
PBMCs were isolated from C1q-deficient and normal humans and cultured as previously described 11 on coverslips in 24-well plates. Apoptotic Jurkat T cells (5 × 106 per well) were fed to the monocyte-derived macrophages between 7 and 10 d after isolation in the presence of 15% autologous serum for 1 h, and uningested cells were removed by repeated washing with PBS. The C1q-deficient serum was reconstituted with purified human C1q (Sigma-Aldrich) as indicated. Phagocytosis was scored on Diff-Quik–stained coded slides as described above.
Statistics.
Statistics were calculated using GraphPad Prism™ (version 2.0; GraphPad Software). Student's t test and one-way analysis of variance (ANOVA) with Bonferroni multiple comparison test was applied throughout.
Results
Phagocytosis of Apoptotic Jurkat T Cells during Sterile Peritonitis.
Complement-deficient and wild-type mice were injected intraperitoneally with apoptotic human Jurkat T cells 4 d after induction of sterile peritonitis with thioglycollate. Phagocytosis of apoptotic Jurkat T cells by elicited peritoneal macrophages was impaired in C1q-deficient animals (Fig. 1 A), resulting in a delay in the clearance of the apoptotic cells from the peritoneum (Fig. 1 B). The defect in phagocytosis was observed in C1q-deficient animals on both C57BL/6 and 129/Sv genetic backgrounds (Fig. 1 C). C1q deficiency did not alter the recruitment of macrophages into the peritoneum by thioglycollate. However, the genetic background of the mice used did have significant influence over the number of macrophages recruited in response to thioglycollate (C57BL/6 > 129/Sv × C57BL/6 > 129/Sv; Fig. 1 D). Comparison of the phagocytic uptake of an equivalent number of UV-treated and untreated Jurkat T cells confirmed that uptake was related to the level of apoptosis in the cell population (percentage of macrophages ingesting UV-treated cells = 49.1 ± 5.7% compared with 11.8 ± 1.8% with untreated cells [mean ± SEM], n = 4 in each group; P = 0.0008, Student's t test). FACS® analysis of the phagocytosis of 5(6)-TAMRA, SE fluorescently labeled apoptotic Jurkat T cells by F4/80+ macrophages corroborated the data obtained by manual counting of coded cytospin preparations (Fig. 1 E). Inflammatory macrophages of C4-deficient mice showed a defect in the phagocytic uptake of the apoptotic Jurkat T cells in vivo similar to that observed in C1qa −/− mice (Fig. 1 F), suggesting that the activation of the classical pathway and probably opsonization with C3 or C4 was the most likely mechanism for the clearance of apoptotic Jurkat T cells.
Figure 1.
Phagocytic clearance of apoptotic Jurkat T cells during sterile peritonitis. Mice were injected intraperitoneally with 107 apoptotic human Jurkat T cells 4 d after treatment with thioglycollate. (A) Graph showing the percentage of inflammatory macrophages from C57BL/6 mice phagocytosing apoptotic bodies at different time points after injection. The C1qa −/− mice (○) showed a significant delay in phagocytic uptake of the apoptotic cells compared with the wild-type mice (•) (P < 0.01 after 15 and 30 min, Student's t test; data shown corresponds to mean ± SEM of three to four mice in each group). (B) Graph showing the number of uningested apoptotic cells remaining free in the peritoneum of the mice shown in A. The C1qa −/− animals had a significant delay in the clearance of the apoptotic cells as a consequence of the impaired phagocytosis (P < 0.05 at the 15 and 60 min time points, Student's t test; data shown corresponds to mean ± SEM). (C) Scatter plots showing the percentage of elicited peritoneal macrophages containing phagocytosed apoptotic bodies 30 min after intraperitoneal injection of apoptotic Jurkat T cells. C1qa −/− mice (○) showed a significant reduction in the phagocytosis of apoptotic bodies compared with C1qa +/+ mice (•) irrespective of the genetic backgrounds of the experimental animals (horizontal bars denote means; P < 0.005 in each of the three experiments, Student's t test). (D) Peritoneal recruitment of macrophages in response to thioglycollate in wild-type (black bars), C1q-deficient (white bars), and C4-deficient (hatched bars) mice. Although neither C1q nor C4 deficiency affected macrophage recruitment, the number of macrophages elicited by thioglycollate was dependent on the genetic background of the animals used (C57BL/6 > 129/Sv × C57BL/6 > 129/Sv). The data shown represents the mean ± SEM of four to eight mice in each group. (E) FACS® analysis of the phagocytic uptake of 5(6)-TAMRA, SE–labeled apoptotic cells by F4/80+ peritoneal macrophages 30 min after the injection of apoptotic cells in C1q-deficient and wild-type mice (129/Sv × C57BL/6). The F4/80+, 5(6)-TAMRA, SE+ cells represent macrophages associated with apoptotic cells. Significantly fewer F4/80+ cells were associated with the 5(6)-TAMRA, SE+ apoptotic cells in the C1qa −/− animals. (F) Graph showing the percentage of phagocytosing macrophages 30 min after injection of 107 apoptotic Jurkat cells into C1qa −/−, C4 −/−, and wild-type (wt) 129/Sv × C57BL/6 mice during sterile peritonitis. The phagocytic uptake was significantly different between the three groups (P = 0.0002, one-way ANOVA). Both C1qa −/− (○) and C4 −/− (□) mice exhibited significantly impaired phagocytosis compared with the wild-type (•) mice (P < 0.001 and P < 0.01, respectively, Bonferroni multiple comparison test). Data shown is representative of three independent experiments. Horizontal bars denote means.






Phagocytosis of Apoptotic Murine Thymocytes during Sterile Peritonitis.
Although the observed phagocytic clearance of Jurkat T cells was specific for apoptotic cells, the possibility could not be ruled out that the dependence on complement in this system was due to the heterologous nature of the target apoptotic cells. Therefore, the experiments with the inflammatory macrophages were repeated using apoptotic murine thymocytes (Fig. 2 A). Irrespective of their genetic backgrounds, the C1qa −/− mice exhibited a defect in phagocytic uptake of apoptotic thymocytes by inflammatory macrophages compared with wild-type controls (129/Sv genetic background: 20.5 ± 1.4% in the C1qa −/− animals compared with 39.8 ± 6.9% [mean ± SEM] in the controls; P = 0.024, Student's t test; C57BL/6 genetic background: 14.7 ± 0.8% in C1qa −/− mice compared with 19.7 ± 0.9% in controls; P = 0.005, Student's t test). However, in contrast to the results obtained with the apoptotic human Jurkat T cells, phagocytic uptake of apoptotic thymocytes in the C4-deficient mice was only partially impaired compared with the C1qa −/− mice (Fig. 2 B). These findings suggest that, although activation of the classical pathway proteins after the binding of C1q plays a role in apoptotic cell clearance, C1q has a specific role in its own right in the clearance of apoptotic cells.
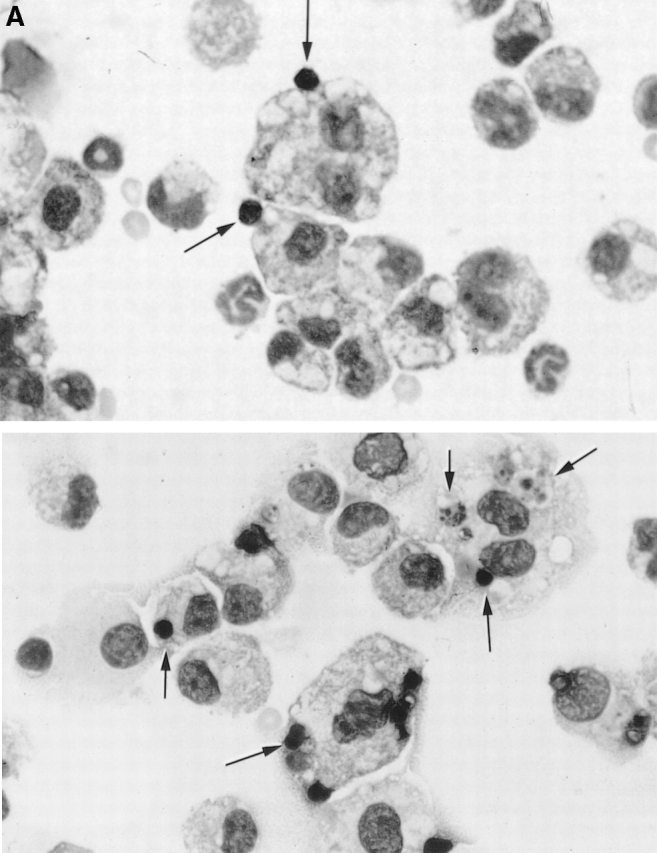
Figure 2.
Phagocytosis of apoptotic murine thymocytes by inflammatory macrophages. (A) Representative photomicrographs of cytospin preparations of peritoneal lavage cells from C1q-deficient (top) and wild-type (bottom) mice recovered 30 min after injection with 3 × 107 apoptotic thymocytes, 4 d after injection of thioglycollate. Arrows indicate apoptotic bodies. (B) Phagocytosis of apoptotic thymocytes by inflammatory macrophages of C1qa −/− (○), C4 −/− (□), and wild-type (wt; •) 129/Sv × C57BL/6 mice was significantly different (P < 0.0001, one-way ANOVA). Phagocytosis in the C1qa −/− mice was significantly lower than the uptake in the C4 −/− and the wild-type mice (P < 0.01 and P < 0.001, respectively, Bonferroni multiple comparison test); however, the C4 −/− mice also exhibited a defect in phagocytosis compared with control mice (P < 0.01, Bonferroni multiple comparison test). Horizontal bars denote means. (C) Reconstitution of C1q-deficient mice with wild-type serum. Apoptotic Jurkat T cells or murine thymocytes were suspended in a 20% vol/vol solution of C1q-deficient (open symbols) or -sufficient (closed symbols) mouse serum and injected into C1qa −/− (circles) and C1qa +/+ (squares) mice as indicated. The phagocytic uptake between the groups was significantly different 30 min after injection (P = 0.0001 in both separate experiments, one-way ANOVA). Phagocytosis in the reconstituted C1qa −/− mice injected with normal mouse serum was significantly higher than in the C1qa −/− mice injected with C1q-deficient serum when either Jurkat T cells or thymocytes were used (P < 0.01 in both experiments, Bonferroni multiple comparison tests). Horizontal bars denote means. Phagocytosis was scored on coded cytospin preparations. Mice used for the reconstitution experiments with apoptotic Jurkat cells and apoptotic thymocytes were on the C57BL/6 and 129/Sv genetic backgrounds, respectively.


We confirmed that deficiency of C1q was directly responsible for the impaired phagocytic uptake of apoptotic cells by performing reconstitution assays in vivo. C1qa −/− and C1qa +/+ mice were injected intraperitoneally with apoptotic cells in the presence of 20% normal or C1q-deficient mouse serum. Injection of apoptotic cells (both Jurkat T cells and murine thymocytes) in the presence of normal mouse serum significantly increased the phagocytic uptake in the C1q-deficient animals (Fig. 2 C) and showed that the presence of C1q at the time of injection of the apoptotic cells was a critical factor in their appropriate clearance.
Phagocytosis of Apoptotic Murine Thymocytes by Resident Peritoneal Macrophages In Vivo.
The role of C3 and the alternative pathway of complement in the clearance of apoptotic cells by inflammatory macrophages could not be assessed in vivo, as mice deficient in the alternative pathway exhibit an impaired inflammatory response to thioglycollate characterized by reduced neutrophil and macrophage recruitment (our unpublished data). To address the contributions of C3 and the alternative pathway of complement to the clearance of apoptotic murine thymocytes, resident peritoneal macrophages were studied in C1qa −/−, C4 −/−, C3 −/−, and wild-type mice in vivo. C1qa −/− mice were the only animals to exhibit a consistent defect in the phagocytosis of apoptotic cells by resident macrophages (Fig. 3), showing that C1q plays a significant role in the normal physiological clearance of apoptotic cells in vivo.
Figure 3.

Clearance of apoptotic mouse thymocytes by resident peritoneal macrophages. The phagocytic uptake by resident peritoneal macrophages 30 min after the intraperitoneal injection of 2 × 107 apoptotic cells was significantly reduced in C1qa −/− (○) mice compared with C4 −/− (□), C3 −/− (⋄), and wild-type (wt) 129/Sv × C57BL/6 (•) control mice. Data shown represents two pooled representative experiments (P < 0.01, P < 0.01, and P < 0.001, respectively, Bonferroni multiple comparison test; P = 0.0009, one-way ANOVA). Horizontal bars denote means. Phagocytosis was scored on coded cytospin preparations. Impaired phagocytic uptake by the resident macrophages in the C1qa −/− animals was observed in three separate experiments (data not shown).
Phagocytosis of Apoptotic Jurkat T Cells by Human Monocyte–derived Macrophages.
To establish the relevance of our observations in mice to humans, the role of C1q in the clearance of apoptotic cells was investigated by measuring the phagocytosis of apoptotic Jurkat T cells by monocyte-derived macrophages from three individuals with hereditary C1q deficiency. C1q-deficient macrophages, in the presence of autologous serum, showed a kinetic defect in the phagocytic uptake of apoptotic cells in vitro (Fig. 4 A) similar to that seen in the mice in vivo (Fig. 1 A) and consistent with other recent in vitro experiments using complement-depleted sera 12. Addition of purified human C1q to the C1q-deficient serum (final concentration of 75 μg/ml) rectified this defect (Fig. 4 A). Furthermore, phagocytosis of apoptotic cells by C1q-deficient macrophages from three different individuals (from two separate families) was similarly impaired compared with normal controls, and purified C1q protein corrected this in a dose-dependent manner (Fig. 4 B). Notably, serum taken from a C1q-deficient patient 30 min after treatment by infusion of 2 U of fresh frozen plasma (resulting in a serum C1q concentration of 27 μg/ml) when used in this phagocytosis assay also significantly increased the phagocytic uptake of apoptotic cells compared with autologous serum obtained before infusion (18.9 ± 3.6% before infusion and 74.5 ± 4.9% [mean ± SEM] after treatment with fresh frozen plasma; P = 0.0026, Student's t test).
Figure 4.
Phagocytosis of apoptotic Jurkat T cells by human monocyte–derived macrophages from three C1q-deficient patients. (A) The phagocytic uptake of apoptotic cells by C1q-deficient macrophages (○) in vitro showed a kinetic defect when compared with both of the normal controls (•, ▪) (at 60 min, P < 0.01 in both cases, Bonferroni multiple comparison test; P = 0.001, one-way ANOVA). When the C1q-deficient serum was reconstituted with 75 μg/ml of purified C1q (□), the phagocytic uptake was no longer significantly different from the controls. Data shown represents the mean ± SEM for triplicate samples. (B) C1q-deficient macrophages, from three individual patients, in autologous serum (○) showed a significant reduction in phagocytosis compared with normal controls (•) after 60-min incubation (P = 0.0038, Student's t test). Addition of purified human C1q protein to the autologous C1q-deficient serum (final concentration of 75 μg/ml in the serum) of one patient rectified the defect in the phagocytic uptake of apoptotic bodies in a dose-dependent manner. Data shown corresponds to the mean of three to four wells for each individual, and horizontal bars denote means.


Discussion
The phagocytosis of apoptotic cells is thought to be important in the resolution of inflammation 13, and multiple complementary ligand–receptor systems have been identified in vitro that may facilitate this process 14 15 16 17 18 19 20 21 22 23. Recently, complement has also been implicated in the phagocytosis of apoptotic cells by the observation that C1q binds specifically to the surface blebs of apoptotic cells 3 and that C3 depletion of human serum impairs uptake of apoptotic cells by human monocyte–derived macrophages in vitro 12. In this study, we have used complement-deficient mice to determine the relative contribution of different complement proteins to the phagocytosis of apoptotic cells in vivo. Our initial in vivo clearance experiments using apoptotic Jurkat human T cells indicated that complement did indeed play a significant role in the phagocytic clearance of apoptotic cells by inflammatory macrophages in vivo, consistent with previous observations in an in vitro human system 12. A significant, and equivalent, delay in the clearance of apoptotic cells was observed in both C1q- and C4-deficient mice, indicating that opsonization with C3 via classical pathway activation was the most likely mediator of clearance in this model. However, when apoptotic murine thymocytes were used instead of apoptotic human cells, differences were seen in the phagocytic uptake between inflammatory macrophages from C1q- and C4-deficient mice. Although the C4-deficient mice exhibited a defect in the phagocytic uptake of apoptotic murine cells by inflammatory macrophages, this defect was not as severe as that seen in the C1q-deficient animals. This result indicated that there is a hierarchy of importance of classical pathway complement proteins in the phagocytic clearance of apoptotic cells.
These observations indicated that the clearance of syngeneic apoptotic cells was dependent on C1q in two ways: an effect mediated directly by C1q, and a second effect mediated by C1q as an activator of the classical pathway. Clearance of apoptotic cells in vivo was more dependent on the second mechanism of classical pathway activation when a heterologous system was used (i.e., when human apoptotic cells were injected into mice), indicating the importance of using syngeneic cells in these assays to elucidate the independent role of C1q. This difference may be due to a reduction in the ability of human compared with mouse lymphocytes to regulate the activation of mouse complement or the presence of xenoreactive natural antibodies.
To address the role of C3 in the phagocytosis of apoptotic cells, resident peritoneal macrophages, as opposed to inflammatory macrophages induced by thioglycollate, were studied in vivo. C1q-deficient mice were the only complement-deficient animals to exhibit a defect in the phagocytosis of apoptotic cells by resident peritoneal macrophages in vivo, suggesting that C1q is the complement protein with the predominant role in the physiological phagocytosis of apoptotic cells in vivo in the absence of inflammation. However, there was a role for C4 in the clearance of apoptotic syngeneic thymocytes by inflammatory macrophages. These results imply that C1q and C4 may mediate the uptake of apoptotic cells by different mechanisms, either involving separate receptors or a single receptor that is only activated for uptake of C4/C3-coated cells on inflammatory macrophages. The lack of a role for C4 and C3 in the phagocytosis of apoptotic cells by resident peritoneal macrophages could be related to the activation state of the macrophages, as it is well recognized that murine resident peritoneal macrophages are unable to phagocytose C3-coated particles without additional stimuli, such as thioglycollate elicitation 24 25 26 27 28 29. However, these resident macrophages may be able to utilize C1q as an opsonin, presumably via direct interaction with one of the candidate C1q receptors 30 31 32 33. In the guinea pig, C1q is bound and degraded by both resident and thioglycollate-elicited peritoneal macrophages 34. This binding and degradation was increased by thioglycollate activation and found to be consistent with an increase in the number of receptors.
Apoptotic cells express on their surface blebs many of the autoantigens of SLE 4, and they also expose on their cellular membranes the negatively charged phospholipids that are the ligands of antiphospholipid autoantibodies 9 35. In addition, there is evidence that the enzymes specific to the cell death program may cleave and modify many autoantigens of SLE, which may reveal cryptic epitopes 36 37. These findings suggest that apoptotic cells are a major source of autoantigens in SLE and that an impairment of their physiological clearance may promote the development of autoimmunity. Indeed, macrophages from humans with SLE have been shown to exhibit a reduction in the phagocytic uptake of apoptotic cells in vitro 38. The data presented here demonstrate for the first time an in vivo deficiency in one of the clearance mechanisms of apoptotic cells in mammals. The proteins of the complement classical pathway showed a hierarchy in the clearance of apoptotic cells with the greatest dependence on the activity of C1q and with a lesser dependence on C4.
We previously showed the presence of glomerulonephritis and an excess of glomerular apoptotic bodies in C1q-deficient mice and in mice deficient in C1q, C2, and factor B 2 6. By contrast, there was no glomerulonephritis or increased number of glomerular apoptotic bodies in mice with C2 and factor B deficiency alone 6. The observations presented in this paper suggest that the increased presence of apoptotic cells in the glomeruli of mice deficient in C1q may be a direct result of impaired clearance of apoptotic cells in vivo. Furthermore, both sets of findings taken together indicate that deficiency of C1q, rather than the entire classical pathway, is the predominant cause of both the impaired clearance of apoptotic cells and the increased predisposition to spontaneous autoimmunity.
In support of these findings in murine models, C1q-deficient macrophages from three C1q-deficient humans with SLE showed a defect in the phagocytic uptake of apoptotic cells in vitro. This defect was corrected, in a dose-dependent manner, using purified C1q protein. Furthermore, experiments in vitro have shown that culture of human monocyte–derived macrophages in wells precoated with purified C1q partially inhibits the phagocytic uptake of apoptotic cells (Ogden, C., unpublished data). Collectively, these findings demonstrate an important role of the early proteins of the classical pathway in the clearance of apoptotic cells.
If macrophages fail to clear apoptotic cells normally, what might be the consequences? A second route for the uptake of apoptotic cells has been identified that may favor an autoimmune rather than an antiinflammatory response to apoptotic cells. Immature dendritic cells can phagocytose apoptotic cells and present apoptotic cell–derived antigens to MHC class I– and II-restricted T cells 39 40 41 in a dose-dependent manner 42. Furthermore, blockade of macrophage-mediated removal of a syngeneic apoptotic tumor cell line in vivo in mice led to an augmented CTL response when the mice were rechallenged with live tumor cells 43. These observations are consistent with the existence of a balance in vivo between macrophage- and dendritic cell–mediated clearance of apoptotic cells. Macrophages may maintain homeostasis by promoting the silent disposal of apoptotic cell debris, whereas clearance by dendritic cells favors an immune response. In C1q-deficient animals, a delay in the clearance of apoptotic cells by macrophages may tilt this balance toward an autoimmune response.
The establishment of an in vivo model for quantifying the phagocytosis of apoptotic cells by macrophages may provide a new approach for testing antiinflammatory therapies directed toward enhancing the resolution of inflammation by promoting macrophage-mediated removal of apoptotic cell debris. This model will also allow the mechanisms and kinetics of the clearance of apoptotic cells to be studied in mice with gene targeted mutations of other candidate molecules involved in apoptotic clearance pathways.
In conclusion, the findings of this study may help to resolve the association between hereditary complement deficiency and the development of SLE. The hierarchy of both the prevalence and severity of SLE according to the position of the missing protein in the classical pathway was paralleled by a similar hierarchy of the activity of classical pathway complement proteins in the clearance of apoptotic cells.
Acknowledgments
This work was supported by Wellcome Trust grant 054838. M. Andrews was funded by Wellcome Trust grant 047273.
Footnotes
Abbreviations used in this paper: ANOVA, analysis of variance; SLE, systemic lupus erythematosus.
References
- Morgan B.P., Walport M.J. Complement deficiency and disease. Immunol. Today. 1991;12:301–306. doi: 10.1016/0167-5699(91)90003-C. [DOI] [PubMed] [Google Scholar]
- Botto M., Dell'Agnola C., Bygrave A.E., Thompson E.M., Cook H.T., Petry F., Loos M., Pandolfi P.P., Walport M.J. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat. Genet. 1998;19:56–59. doi: 10.1038/ng0598-56. [DOI] [PubMed] [Google Scholar]
- Korb L.C., Ahearn J.M. C1q binds directly and specifically to surface blebs of apoptotic human keratinocytescomplement deficiency and systemic lupus erythematosus revisited. J. Immunol. 1997;158:4525–4528. [PubMed] [Google Scholar]
- Casciola-Rosen L.A., Anhalt G., Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J. Exp. Med. 1994;179:1317–1330. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mevorach D., Zhou J.L., Song X., Elkon K.B. Systemic exposure to irradiated apoptotic cells induces autoantibody production J. Exp. Med. 188 1998. 387 392b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell D.A., Taylor P.R., Cook H.T., Moss J., Bygrave A.E., Walport M.J., Botto M. C1q protects against the development of glomerulonephritis independently of C3 activation. J. Immunol. 1999;162:5676–5679. [PubMed] [Google Scholar]
- Fischer M.B., Ma M., Goerg S., Zhou X., Xia J., Finco O., Han S., Kelsoe G., Howard R.G., Rothstein T.L. Regulation of the B cell response to T-dependent antigens by classical pathway complement. J. Immunol. 1996;157:549–556. [PubMed] [Google Scholar]
- Wessels M.R., Butko P., Ma M., Warren H.B., Lage A.L., Carroll M.C. Studies of group B streptococcal infection in mice deficient in complement component C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity. Proc. Natl. Acad. Sci. USA. 1995;92:11490–11494. doi: 10.1073/pnas.92.25.11490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok V.A., Voelker D.R., Campbell P.A., Cohen J.J., Bratton D.L., Henson P.M. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 1992;148:2207–2216. [PubMed] [Google Scholar]
- Hess K.L., Babcock G.F., Askew D.S., Cook-Mills J.M. A novel flow cytometric method for quantifying phagocytosis of apoptotic cells. Cytometry. 1997;27:145–152. [PMC free article] [PubMed] [Google Scholar]
- Fadok V.A., Bratton D.L., Konowal A., Freed P.W., Westcott J.Y., Henson P.M. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mevorach D., Mascarenhas J.O., Gershov D., Elkon K.B. Complement-dependent clearance of apoptotic cells by human macrophages. J. Exp. Med. 1998;188:2313–2320. doi: 10.1084/jem.188.12.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savill J. Apoptosis in resolution of inflammation. J. Leukoc. Biol. 1997;61:375–380. doi: 10.1002/jlb.61.4.375. [DOI] [PubMed] [Google Scholar]
- Savill J., Dransfield I., Hogg N., Haslett C. Vitronectin receptor-mediated phagocytosis of cells undergoing apoptosis. Nature. 1990;343:170–173. doi: 10.1038/343170a0. [DOI] [PubMed] [Google Scholar]
- Savill J., Hogg N., Ren Y., Haslett C. Thrombospondin cooperates with CD36 and the vitronectin receptor in macrophage recognition of neutrophils undergoing apoptosis. J. Clin. Invest. 1992;90:1513–1522. doi: 10.1172/JCI116019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok V.A., Savill J.S., Haslett C., Bratton D.L., Doherty D.E., Campbell P.A., Henson P.M. Different populations of macrophages use either the vitronectin receptor or the phosphatidylserine receptor to recognize and remove apoptotic cells. J. Immunol. 1992;149:4029–4035. [PubMed] [Google Scholar]
- Platt N., Suzuki H., Kurihara Y., Kodama T., Gordon S. Role for the class A macrophage scavenger receptor in the phagocytosis of apoptotic thymocytes in vitro. Proc. Natl. Acad. Sci. USA. 1996;93:12456–12460. doi: 10.1073/pnas.93.22.12456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devitt A., Moffatt O.D., Raykundalia C., Capra J.D., Simmons D.L., Gregory C.D. Human CD14 mediates recognition and phagocytosis of apoptotic cells. Nature. 1998;392:505–509. doi: 10.1038/33169. [DOI] [PubMed] [Google Scholar]
- Moffatt O.D., Devitt A., Bell E.D., Simmons D.L., Gregory C.D. Macrophage recognition of ICAM-3 on apoptotic leukocytes. J. Immunol. 1999;162:6800–6810. [PubMed] [Google Scholar]
- Sambrano G.R., Steinberg D. Recognition of oxidatively damaged and apoptotic cells by an oxidized low density lipoprotein receptor on mouse peritoneal macrophagesrole of membrane phosphatidylserine. Proc. Natl. Acad. Sci. USA. 1995;92:1396–1400. doi: 10.1073/pnas.92.5.1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird D.A., Gillotte K.L., Horkko S., Friedman P., Dennis E.A., Witztum J.L., Steinberg D. Receptors for oxidized low-density lipoprotein on elicited mouse peritoneal macrophages can recognize both the modified lipid moieties and the modified protein moietiesimplications with respect to macrophage recognition of apoptotic cells. Proc. Natl. Acad. Sci. USA. 1999;96:6347–6352. doi: 10.1073/pnas.96.11.6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M.K., Bergmark C., Laurila A., Horkko S., Han K.H., Friedman P., Dennis E.A., Witztum J.L. Monoclonal antibodies against oxidized low-density lipoprotein bind to apoptotic cells and inhibit their phagocytosis by elicited macrophagesevidence that oxidation-specific epitopes mediate macrophage recognition. Proc. Natl. Acad. Sci. USA. 1999;96:6353–6358. doi: 10.1073/pnas.96.11.6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franc N.C., Heitzler P., Ezekowitz R.A., White K. Requirement for croquemort in phagocytosis of apoptotic cells in Drosophila . Science. 1999;284:1991–1994. doi: 10.1126/science.284.5422.1991. [DOI] [PubMed] [Google Scholar]
- Bianco C., Griffin F.M., Jr., Silverstein S.C. Studies of the macrophage complement receptor. Alteration of receptor function upon macrophage activation. J. Exp. Med. 1975;141:1278–1290. doi: 10.1084/jem.141.6.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin J.A., Griffin F.M., Jr. Augmentation of macrophage complement receptor function in vitro. I. Characterization of the cellular interactions required for the generation of a T-lymphocyte product that enhances macrophage complement receptor function. J. Exp. Med. 1979;150:653–675. doi: 10.1084/jem.150.3.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michl J., Pieczonka M.M., Unkeless J.C., Silverstein S.C. Effects of immobilized immune complexes on Fc- and complement-receptor function in resident and thioglycollate-elicited mouse peritoneal macrophages. J. Exp. Med. 1979;150:607–621. doi: 10.1084/jem.150.3.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin F.M., Jr., Griffin J.A. Augmentation of macrophage complement receptor function in vitro. II. Characterization of the effects of a unique lymphokine upon the phagocytic capabilities of macrophages. J. Immunol. 1980;125:844–849. [PubMed] [Google Scholar]
- Shaw D.R., Griffin F.M., Jr. Functional characteristics of the macrophage receptors for IgG-Fc and C3failure to detect C3 receptor-mediated extracellular cytolysis by mouse peritoneal macrophages. Cell. Immunol. 1984;84:317–323. doi: 10.1016/0008-8749(84)90103-5. [DOI] [PubMed] [Google Scholar]
- Aderem A.A., Wright S.D., Silverstein S.C., Cohn Z.A. Ligated complement receptors do not activate the arachidonic acid cascade in resident peritoneal macrophages. J. Exp. Med. 1985;161:617–622. doi: 10.1084/jem.161.3.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan E.N., Burgess W.H., Robinson S.L., Goodman E.B., McTigue K.J., Tenner A.J. Phagocytic cell molecules that bind the collagen-like region of C1q. Involvement in the C1q-mediated enhancement of phagocytosis. J. Biol. Chem. 1991;266:20345–20355. [PubMed] [Google Scholar]
- Peerschke E.I., Reid K.B., Ghebrehiwet B. Identification of a novel 33-kDa C1q-binding site on human blood platelets. J. Immunol. 1994;152:5896–5901. [PubMed] [Google Scholar]
- Ghebrehiwet B., Silvestri L., McDevitt C. Identification of the Raji cell membrane-derived C1q inhibitor as a receptor for human C1q. Purification and immunochemical characterization. J. Exp. Med. 1984;160:1375–1389. doi: 10.1084/jem.160.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klickstein L.B., Barbashov S.F., Liu T., Jack R.M., Nicholson-Weller A. Complement receptor type 1 (CR1, CD35) is a receptor for C1q. Immunity. 1997;7:345–355. doi: 10.1016/s1074-7613(00)80356-8. [DOI] [PubMed] [Google Scholar]
- Veerhuis R., Klar-Mohamad N., van Es L.A., Daha M.R. Enhanced binding and degradation of the C1q subcomponent of complement by thioglycollate-stimulated guinea pig peritoneal macrophages. Scand. J. Immunol. 1986;23:581–587. doi: 10.1111/j.1365-3083.1986.tb01991.x. [DOI] [PubMed] [Google Scholar]
- Casciola-Rosen L., Rosen A., Petri M., Schlissel M. Surface blebs on apoptotic cells are sites of enhanced procoagulant activityimplications for coagulation events and antigenic spread in systemic lupus erythematosus. Proc. Natl. Acad. Sci. USA. 1996;93:1624–1629. doi: 10.1073/pnas.93.4.1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen A., Casciola-Rosen L. Autoantigens as substrates for apoptotic proteasesimplications for the pathogenesis of systemic autoimmune disease. Cell Death Differ. 1999;6:6–12. doi: 10.1038/sj.cdd.4400460. [DOI] [PubMed] [Google Scholar]
- Casciola-Rosen L., Andrade F., Ulanet D., Wong W.B., Rosen A. Cleavage by granzyme B is strongly predictive of autoantigen statusimplications for initiation of autoimmunity. J. Exp. Med. 1999;190:815–826. doi: 10.1084/jem.190.6.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann M., Voll R.E., Zoller O.M., Hagenhofer M., Ponner B.B., Kalden J.R. Impaired phagocytosis of apoptotic cell material by monocyte-derived macrophages from patients with systemic lupus erythematosus. Arthritis Rheum. 1998;41:1241–1250. doi: 10.1002/1529-0131(199807)41:7<1241::AID-ART15>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Albert M.L., Sauter B., Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs Nature. 392 1998. 86 89b [DOI] [PubMed] [Google Scholar]
- Albert M.L., Pearce S.F., Francisco L.M., Sauter B., Roy P., Silverstein R.L., Bhardwaj N. Immature dendritic cells phagocytose apoptotic cells via alphavbeta5 and CD36, and cross-present antigens to cytotoxic T lymphocytes. J. Exp. Med. 1998;188:1359–1368. doi: 10.1084/jem.188.7.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba K., Turley S., Yamaide F., Iyoda T., Mahnke K., Inaba M., Pack M., Subklewe M., Sauter B., Sheff D. Efficient presentation of phagocytosed cellular fragments on the major histocompatibility complex class II products of dendritic cells. J. Exp. Med. 1998;188:2163–2173. doi: 10.1084/jem.188.11.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovere P., Vallinoto C., Bondanza A., Crosti M.C., Rescigno M., Ricciardi-Castagnoli P., Rugarli C., Manfredi A.A. Bystander apoptosis triggers dendritic cell maturation and antigen-presenting function. J. Immunol. 1998;161:4467–4471. [PubMed] [Google Scholar]
- Ronchetti A., Rovere P., Iezzi G., Galati G., Heltai S., Protti M.P., Garancini M.P., Manfredi A.A., Rugarli C., Bellone M. Immunogenicity of apoptotic cells in vivorole of antigen load, antigen-presenting cells, and cytokines. J. Immunol. 1999;163:130–136. [PubMed] [Google Scholar]