

Abstract
Using intravital microscopy, we examined the role played by B1 receptors in leukocyte trafficking across mouse mesenteric postcapillary venules in vivo. B1 receptor blockade attenuated interleukin (IL)-1β–induced (5 ng intraperitoneally, 2 h) leukocyte–endothelial cell interactions and leukocyte emigration (∼50% reduction). The B1 receptor agonist des-Arg9bradykinin (DABK), although inactive in saline- or IL-8–treated mice, caused marked neutrophil rolling, adhesion, and emigration 24 h after challenge with IL-1β (when the cellular response to IL-1β had subsided). Reverse transcriptase polymerase chain reaction and Western blot revealed a temporal association between the DABK-induced response and upregulation of mesenteric B1 receptor mRNA and de novo protein expression after IL-1β treatment. DABK-induced leukocyte trafficking was antagonized by the B1 receptor antagonist des-arg10HOE 140 but not by the B2 receptor antagonist HOE 140. Similarly, DABK effects were maintained in B2 receptor knockout mice. The DABK-induced responses involved the release of neuropeptides from C fibers, as capsaicin treatment inhibited the responses. Treatment with the neurokinin (NK)1 and NK3 receptor antagonists attenuated the responses, whereas NK2, calcitonin gene-related peptide, or platelet-activating factor receptor antagonists had no effect. Substance P caused leukocyte recruitment that, similar to DABK, was inhibited by NK1 and NK3 receptor blockade. Mast cell depletion using compound 48/80 reduced DABK-induced leukocyte trafficking, and DABK treatment was shown histologically to induce mast cell degranulation. DABK-induced trafficking was inhibited by histamine H1 receptor blockade. Our findings provide clear evidence that B1 receptors play an important role in the mediation of leukocyte–endothelial cell interactions in postcapillary venules, leading to leukocyte recruitment during an inflammatory response. This involves activation of C fibers and mast cells, release of substance P and histamine, and stimulation of NK1, NK3, and H1 receptors.
Keywords: kinin B1 receptor, leukocyte–endothelial cell interaction, intravital microscopy, C fiber, mast cell
Introduction
The kinins, specifically bradykinin (BK) and its biologically active metabolite des-Arg9bradykinin (DABK), are produced at sites of inflammation and are important inflammatory mediators 1. The effects of these kinins are brought about by interaction with specific G protein–coupled receptors, of which there are two clearly defined and cloned types: B1 and B2 2. BK produces its acute proinflammatory effects via the activation of constitutively expressed B2 receptors. The action of BK is characterized by increased blood flow and vascular permeablitity 2 3. The effects of DABK, in contrast, are mediated by B1 receptors. B1 receptors are not normally expressed, but are induced selectively during the host inflammatory response. Activation of B1 receptor by its endogenous ligand (DABK in rodents and Lys-DABK in humans) produces a proinflammatory profile similar to that of B2 receptor activation, but is also characterized by a stimulation of inflammatory cell accumulation 4. B1 receptors, similar to other proinflammatory proteins induced during inflammation (such as cyclooxygenase [COX]-2 and inducible nitric oxide synthase [iNOS]), are induced by certain immunostimulants, including IL-1β 4. For this reason, the kinin B1 receptor may be an innovative target in the development of novel antiinflammatory strategies.
Knowledge of the contribution of B1 receptors to the regulation of the inflammatory process may be important, as B2 receptors undergo rapid agonist-induced desensitization 5, a process possibly linked to B1 receptor induction 6, and are unlikely to be involved in subacute and chronic inflammatory conditions 1. In contrast, B1 receptors, once induced, do not undergo agonist-induced desensitization to any appreciable extent 5 and are better equipped to mediate the development and progression of a chronic inflammatory response. It has therefore been proposed that an integrated system develops from B2 receptor–mediated acute inflammation to a sustained B1 receptor–mediated chronic inflammation, the latter mainly characterized by cellular infiltration 1.
Previous studies using models of experimental inflammation have shown that B1 receptor activation plays a role in the process of neutrophil accumulation 7 8 9. However, the step(s) activated by B1 receptors in the promotion of neutrophil extravasation have not been investigated, and it is not known whether B1 receptor activation can trigger leukocyte interactions with the endothelium of postcapillary venules.
Intravital microscopy techniques have been used over the past 10 years to elucidate the mechanisms involved in leukocyte accumulation 10. Upon application of an inflammatory stimulus, neutrophils roll on the endothelium of inflamed postcapillary venules, an action mediated by selectins CD62P, CD62E, or CD62L 11. During this phase, the neutrophil can be activated by various inflammatory mediators, such as substance P, and become firmly adherent 12. This phenomenon is mediated by integrin activation and increased avidity for their counterligands 12. After adhesion, leukocytes undergo cytoskeletal reorganization leading to the actual emigration process known as diapedesis.
This study was designed to investigate the cellular and molecular mechanism(s) by which B1 receptor activation produces neutrophil accumulation in vivo. To achieve this we have used (a) a method of observation that accurately quantifies the interaction between the circulating leukocyte and the endothelium of inflamed postcapillary venules, and (b) correlated this with kinin B1 receptor mRNA and protein expression. We have also investigated the mechanisms involved with appropriate histological techniques and via the use of selective agents to inhibit possible intermediary pathways.
Materials and Methods
Animals.
Male Swiss albino mice (10–15 g) were purchased from Banton and Kingsman and maintained on a standard chow pellet diet with tap water ad libitum. Animals were housed in groups of eight per cage in a room with controlled lighting (lights on 8:00–20:00) in which the temperature was maintained at 21–23°C, and the animals were used 3–4 d after their arrival. Animal work was performed according to Home Office regulations. Guidance on the operation on animals was from the Scientific Procedures Act (1986).
Intravital Microscopic Studies.
Mice were anesthetized with diazepam (60 mg/kg subcutaneously) and Hypnorm® (0.7 mg/kg fentanyl citrate and 20 mg/kg fluanisone intramuscularly). Cautery incisions were made along the abdominal region, and the mesenteric vascular bed was exteriorized and placed on a viewing Plexiglas stage. The preparation was mounted on a Zeiss Axioskop FS with a water immersion objective lens (magnification: ×40; Zeiss), and an eyepiece (magnification: ×10; Zeiss) was used to observe the microcirculation. The preparation was transilluminated with a 12-V, 100-W halogen light source. A Hitachi charge-coupled device color camera (model KPC571) acquired images that were displayed onto a Sony Trinitron color video monitor (model PVM 1440QM) and recorded on a Sony super-VHS video cassette recorder (model SVO-9500 MDP) for subsequent offline analysis. A video time-date generator (FOR.A video timer; model VTG-33) projected the time, date, and stopwatch function onto the monitor. Mesenteries were superfused with bicarbonate-buffered solution at 37°C (g/liter: NaCl, 7.71; KCl, 0.25; MgSO4, 0.14; NaHCO3, 1.51; and CaCl2, 0.22 [pH 7.4], gassed with 5% CO2/95% N2) at a rate of 2 ml/min. The temperature of the stage was maintained at 37°C. This procedure has no effect on rectal temperature or blood pressure (our unpublished observations). RBC velocity was measured in venules by using an optical doppler velocimeter (Microcirculation Research Institute, Texas A&M University). Venular blood flow was calculated from the product of mean RBC velocity (V mean = centerline velocity/1.6) and microvascular cross-sectional area, assuming a cylindrical geometry. Wall shear rate was calculated by the Newtonian definition: shear rate = 8,000× (V mean/diameter) 13. One to three randomly selected postcapillary venules (diameter between 20 and 40 μm; length of at least 100 μm) were observed for each mouse
Analysis of the Inflammatory Response Elicited by IL-1β.
Murine recombinant IL-1β (gift from Dr. R.C. Newton, Dupont Life Sciences Enterprise, Wilmington, DE) was used to induce neutrophil rolling, adhesion, and emigration. Adapting a protocol successfully used for the rat mesentery 14 15, mice received either IL-1β (5 ng intraperitoneally in 0.25 ml of sterile saline) or vehicle alone, and the mesenteric vascular bed was prepared for microscopic observation 2 or 24 h later, as described previously 16. The extent of the inflammatory response elicited by IL-1β was analyzed by measuring white blood cell velocity (V WBC) in mm/s. Cell adhesion was quantified by counting, for each vessel, the number of adherent neutrophils in a 100-μm length (see Fig. 1). Leukocyte emigration from the microcirculation into the tissue was quantified by counting the number of cells that had emigrated up to 50 μm away from the wall of the 100-μm vessel segments (see Fig. 1).
Figure 1.
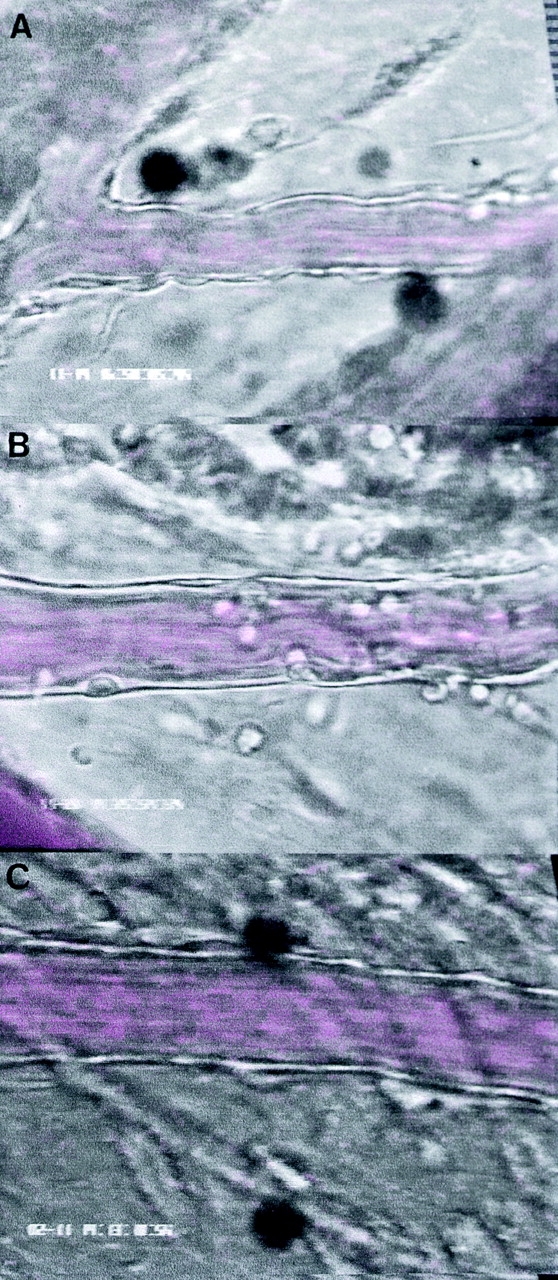
Videomicroscopy images of leukocyte trafficking responses in mouse mesenteric postcapillary venules in vivo in response to (A) saline (0.25 ml intraperitoneally, 2 h), (B) IL-1β (5 ng intraperitoneally, 2 h), and (C) IL-1β (5 ng intraperitoneally, 24 h).
Assessment of the Involvement of B1 Receptors.
The involvement of B1 receptors in the cellular response to IL-1β was evaluated using the selective B1 receptor antagonist des-arg10HOE 140 (d-Arg-[Hyp3,Thi5,d-Tic7,Oic8]-BK, 25 nmol intraperitoneally; gift from Dr. Gillian Burgess, Novartis Institute for Medical Sciences, London, UK), which was coinjected with IL-1β. Neutrophil accumulation was quantified at the 2-h time point.
Analysis of the Inflammatory Response Elicited by DABK.
To determine whether activation of B1 receptors by itself led to neutrophil accumulation, the effect of DABK (Bachem) given 24 h after IL-1β (5 ng) was also tested. In this set of experiments, mice received DABK (10–30 nmol; reference 7), and the cellular response assessed 2 or 4 h later by videomicroscopy. Two types of controls were used for each experiment: one group of animals was pretreated with sterile saline (0.25 ml) in place of IL-1β 24 h before DABK, and another group received sterile saline (0.25 ml) instead of DABK 24 h after IL-1β injection. The specificity of IL-1β pretreatment was controlled for by administering the neutrophil activator human rIL-8 (0.5 μg; from the National Institute of Biological Standards and Controls, South Mimms, UK).
Histology.
Histological studies were carried out to identify general histological structures, to enable clear identification of polymorphonuclear and mononuclear cells, and to visualize mesenteric mast cells. After saline perfusion through the heart, whole mount sections of mesentery were prepared on microscope slides and fixed in 4% paraformaldehyde overnight. General structures were identified by staining with hematoxylin and eosin to enable identification of polymorphonuclear and mononuclear cells. For mast cell identification, sections were fixed in 2% paraformaldehyde/2% glutaraldehyde overnight and stained with toluidine blue (which elicits a metachromatic reaction only in the granules of mast cells and basophils) as described previously 17. The number of mast cells were counted for six different fields per sample with a high powered objective (magnification: ×40). Cells in a 100-μm2 area were counted. The proportion of mast cells that were degranulated was expressed as a percentage of the total number of mast cells.
B1 Receptor Gene Expression in Inflamed Mesenteries.
polyA+ mRNA was extracted (Micro-FastTrack Kit; Invitrogen) from homogenates of mesentery, stored at −70°C, from IL-1β– (5 ng, 24 h) and saline-treated mice. polyA+ mRNA was resuspended in 20 μl of Tris (pH 7.5), and 2-μl aliquots were reverse transcribed in a 20-μl reaction mixture containing 150 ng of random hexanucleotides, 2 mM dNTPs, 50 mM Tris-HCl (pH 8.3), 75 mM KCl, 3 mM MgCl2, 10 mM dithiothreitol, and 200 U SuperScript II RNAse H− reverse transcriptase (RT; Life Technologies). PCR experiments were performed with 2 μl of the first-strand DNA in a 25-μl reaction mixture containing 200 μM dNTPs, 1.5 mM MgCl2, 25 pmol of specific forward and reverse primers, and 1 U of recombinant Taq DNA polymerase (Life Technologies), and were amplified for 30 cycles with a Biometra-TRIO thermocycler at 95°C for 1 min, 60°C for 2 min, and 72°C for 2 min, followed by a final extension step at 72°C for 10 min.
Mouse B1 receptor cDNA was amplified with the sense primer (5′-GCGGAAATCTACCTGGCTAACTTG-3′) at base position 392–415 and antisense primer (5′-CAGTCACGGGGAGGAGGAAACC-3′) at base position 806–827 of rat B1 receptor cDNA 18, which are conserved sequences in the mouse B1 receptor cDNA 19. The quality of RNA samples was evaluated by RT-PCR using glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-specific primers (sense primer, 5′-AAGGTGAAGGTCGGAGTCAACG-3′ [position 8–29] and antisense primer 5′-GGCAGAGATGATGACCCTTTTGGC-3′ [position 362–339]). A plasmid containing the mouse B1 receptor cDNA 19 was used as the template for the positive PCR control reaction. PCR products were detected by electrophoresis on 2% agarose gels stained with ethidium bromide.
In some experiments, 4 μl of first-strand cDNA was transferred to a separate tube, and six serial 1:2 dilutions were made before PCR amplification using the above conditions. Product intensity was determined using densitometry, and log plots of starting amount of cDNA versus product intensity were constructed.
B1 Receptor Protein Expression in Inflamed Mesenteries.
Mice received either IL-1β (5 ng intraperitoneally) or saline alone, and 24 h later, the mesenteric vascular bed was rapidly excised and frozen in liquid nitrogen. Samples were crushed on dry ice and lysed in ice-cold lysis buffer (20 mM Tris-HCl [pH 7.4], 50 mM NaCl, 50 mM NaF, 5 mM EDTA, and 20 mM Na4P2O7·10H2O). Lysates were vortexed and centrifuged for 15 min at 25,000 rpm and 4°C. The protein concentration of the supernatant was determined using Coomassie brilliant blue with BSA as the standard. Western blot analysis was performed as follows: after addition of an equal volume of SDS-PAGE sample buffer (20 mM Tris-HCl [pH 6.8], 2% SDS [wt/vol], 20% glycerol [vol/vol], 0.025% bromophenol blue [wt/vol], and 10% 2-ME [vol/vol]), the lysates were heated for 3 min at 100°C. Lysates containing equal amounts of protein (20 μg) were separated on a 12% polyacrylamide gel (wt/vol) followed by electrotransfer to enhanced chemiluminescence (ECL) nitrocellulose membrane (Immobilon-P; Millipore). The membrane was blocked by incubation for 60 min in 5% nonfat dry milk and 0.1% Tween-20 in PBS (PBS-T), washed twice with PBS-T, and incubated with B1 receptor antipeptide antibody (termed A15C [20]; dilution 1:20,000) overnight at 4°C. After six washes with PBS-T, blots were incubated 2 h with a 1:5,000 dilution of a peroxidase-coupled donkey anti–rabbit antibody, followed by six washes with PBS-T. Visualization of the antibody–protein complex was achieved using an ECL detection reagent (ECL kit; Amersham Pharmacia Biotech) followed by exposure to scientific imaging film (Kodak) for 1–5 min. Full-range molecular weight markers were from Amersham Pharmacia Biotech. In preadsorption experiments, the antibody was incubated with its corresponding peptide at a concentration of 10 μg/ml at 4°C overnight.
Mechanisms of the Cellular Response to DABK.
To confirm that the effect of DABK was via the activation of B1 receptors, 24 h after sensitization of the mice with IL-1β (5 ng), DABK (30 nmol) was coinjected with either the B1 receptor antagonist des-arg10HOE 140 (25 nmol intraperitoneally) or the chemically related selective B2 receptor antagonist HOE 140 (d-Arg-[Hyp3,Thi5,d-Tic7,Oic8]-BK (25 nmol intraperitoneally; gift from Hoechst, Frankfurt, Germany). These doses were chosen from preliminary observations (not shown). The mesenteric vascular bed was visualized, and the neutrophil response was assessed 2 h later.
To determine whether the effects of the B1 receptor agonist involved activation of sensory C fibers, mice were pretreated with a single subcutaneous injection of capsaicin (50 mg/kg; Sigma-Aldrich) 4 d before the administration of IL-1β. This capsaicin treatment produces >90% depletion of sensory neuropeptide content in mice 21.
To ascertain the involvement of specific neuropeptide receptors, histamine H1 receptors, platelet activating factor (PAF), and/or mast cells, either the calcitonin gene-related peptide (CGRP) antagonist, CGRP-(8–37) (300 nmol/kg intravenously, 15 min 22; Bachem); the neurokinin (NK)1 receptor antagonist SR 140333, (S)1-(2-[3-(3,4-dichlorophenyl)-1-(3-isopropoxyphenylacetyl)pip eridin-3- yl]ethyl)-4-phenyl-1-azoniabicyclo[2.2.2] octane chloride (0.1 μmol/kg intravenously, 30 min [23]); the NK2 receptor antagonist MEN 11420, (cyclo [Asn(β-d-GlcNAc)-Asp-Trp-Phe-Dap-Leu]cyclo [2β-5β]) (0.1 μmol/kg intravenous 15-min pretreatment 24); the NK3 receptor antagonist SR 142801, (S)-(N)-(1-(3-(1-benzoyl-3-(3,4-dichlorophenyl)piperidin-3-yl)-propyl)-4-phenylepipiperidin-4-y)-N-methylacetamide (4 μmol/kg intravenously for 15 min 25) (all gifts from Dr. C.A. Maggi, synthesized in the Chemistry Department of Menarini Research, Florence, Italy); the H1 receptor antagonist triprolidine (1 mg/kg intraperitoneally, 20 min [26]; Sigma-Aldrich), the PAF antagonist WEB 2086 26, 3-[4-(2-chlorphenyl)-9-methyl-6H-thienol[3,2-f][1,2,4]tria-zolo-[4,3-a] [1,4]-diazepine-2-yl]-1-(morpholinyl)-1-propanon (5 mg/kg intravenously, 20 min [27]; Boheringer); or the mast cell degranulating agent compound 48/80 (1.2 mg/kg intraperitoneally, 72 h [28, 29]; Sigma-Aldrich) were administered before DABK, and the neutrophil response was assessed at the 2-h time point.
In other experiments, substance P (7.5 nmol intraperitoneally), compound 48/80 (1.2 mg/kg intraperitoneally), or PAF (1 μg, intraperitoneally) were administered, and the neutrophil response assessed at the 1- or 2-h time point. In control experiments, substance P was tested against SR 140333, MEN 11420, or SR 142801, with compound 48/80 against triprolidine and PAF against WEB 2086 in the doses specified above.
Kinin B2 Receptor Knockout Mice.
B2 receptor knockout mice (BK2r−/−) were produced by homologous recombination in embryonic stem cells and generated on a mixed background of J129 × C57 as described previously (30; gift from Merck Research Laboratories, Terlings Park, UK). Wild-type mice derived from the same background were used as controls. Animals were bred and housed in specific pathogen-free facilities and used when their weight was between 11 and 16 g. The effect of DABK (30 nmol intraperitoneally) and saline was tested in BK2r−/− mice 24 h after IL-1β treatment (5 ng intraperitoneally), and the cellular response compared with that observed in the wild-type BK2r+/+ animals, following the protocols detailed above.
Data Analysis.
All values in the figures and text are expressed as mean ± SEM of n observations, where n represents the number of animals studied. Data sets were examined by one- and two-way analysis of variance, and individual group means were compared with Student's unpaired t test. A P value <0.05 was considered significant.
Results
Leukocyte–Endothelial Cell Interactions in Postcapillary Venules of the Mouse Mesentery.
In control animals injected with saline, leukocytes rolled in vivo on the endothelium at high speed (V WBC = 54.9 ± 6.9 mm/sec, n = 11). In contrast, cells moved significantly (P < 0.05) more slowly (V WBC = 9.1 ± 2.1 mm/sec, n = 7) 2 h after intraperitoneal administration of IL-1β (5 ng). This reduction in V WBC was accompanied by a significant increase in cell adhesion and emigration (Table and Fig. 1). Treatment with the B1 receptor antagonist des-arg10HOE 140 (25 nmol/mouse intraperitoneally) significantly (P < 0.05) reduced IL-1β–induced leukocyte rolling, adhesion, and emigration at 2 h (Table ). By 24 h, the inflammatory response to IL-1β, as assessed by the cellular response, had subsided (Table and Fig. 1).
Table 1.
Effect of IL-1β on Leukocyte–Endothelial Interactions In Vivo: Assessment of the Role of B1 Receptors
| Treatment (time) | V WBC | Adherent PMNs | Emigrated PMNs |
|---|---|---|---|
| (mm/s) | |||
| Saline (2 h) | 54.9 ± 6.9 (n =10) | 1.3 ± 0.4 (n =10) | 0.2 ± 0.1 (n =10) |
| IL-1β (2 h) | 9.1 ± 2.1 (n = 6) | 11.4 ± 1.4 (n = 6) | 6.7 ± 0.9 (n = 6) |
| IL-1β/DAH (2 h) | 31.7 ± 2.8 (n = 6) | 5.5 ± 0.7 (n = 6) | 3.6 ± 0.5 (n = 6) |
| IL-1β (24 h) | 49.5 ± 4.7 (n = 6) | 2.0 ± 0.6 (n = 6) | 1.4 ± 0.3 (n = 6) |
Mice received either saline (0.25 ml intraperitoneally). IL-1β (5 ng intraperitoneally), or IL-1β/des-arg10HOE 140 (DAH, 2.5 nmol intraperitoneally). Data are mean ± SEM for animals.
Characterization of DABK-induced Leukocyte Recruitment.
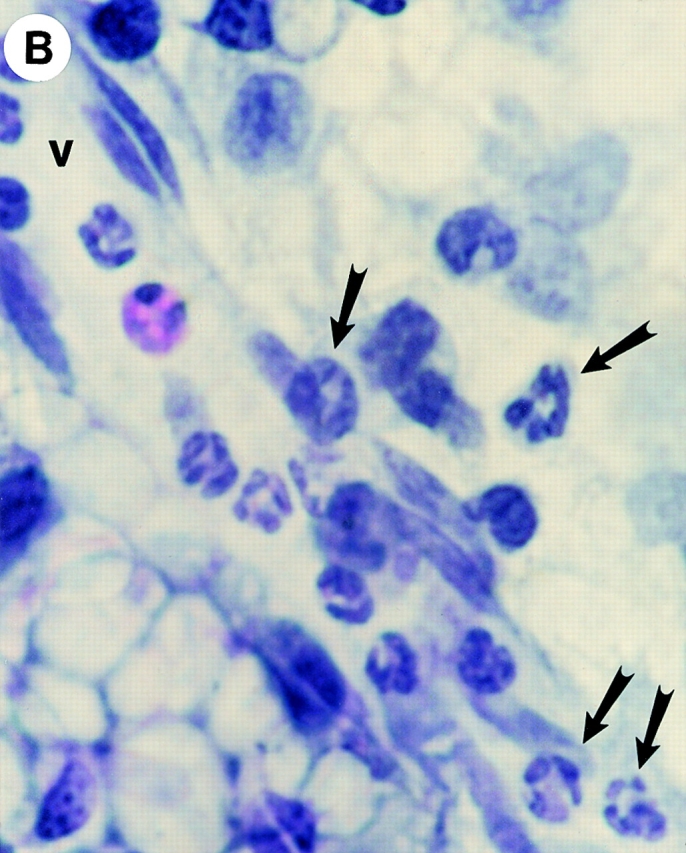
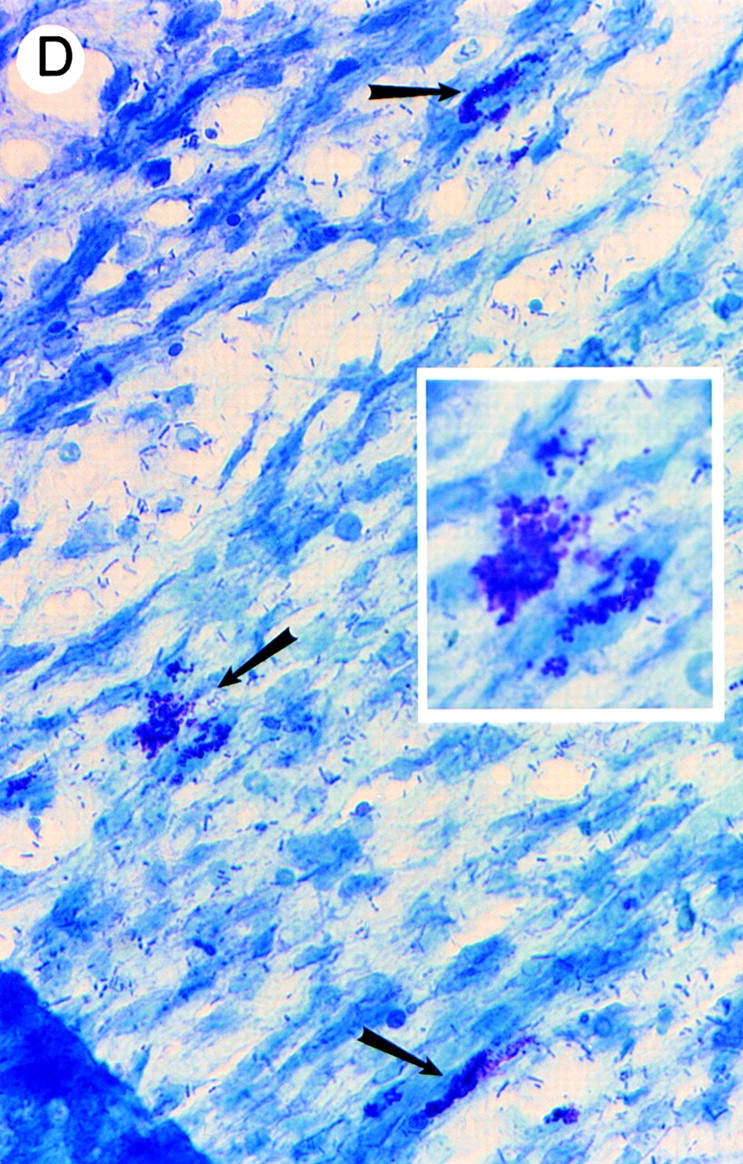
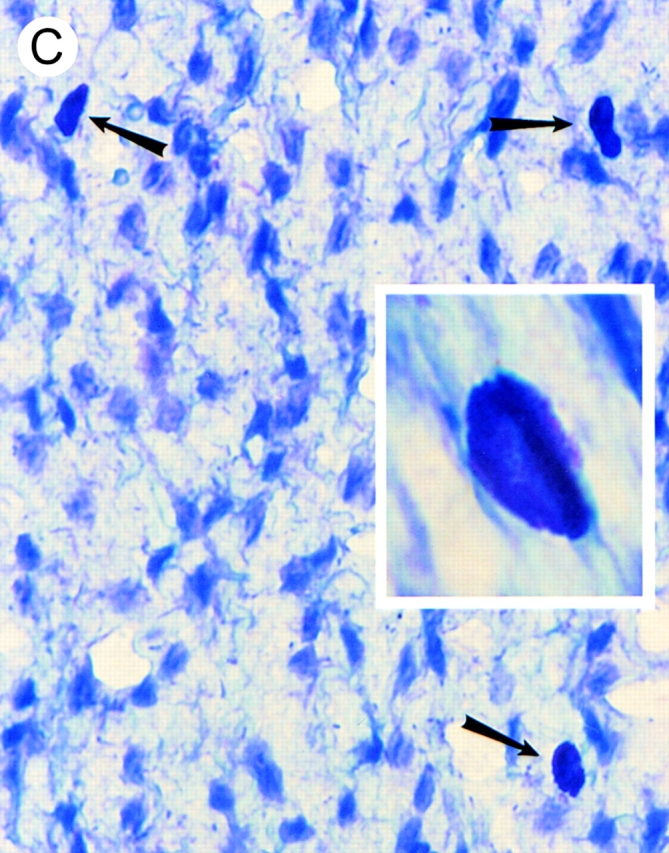
To investigate the effects of B1 receptor activation on leukocyte–endothelial cell interactions in vivo, microscopic visualization of postcapillary venules was performed after the administration of the B1 receptor agonist DABK. 24 h after challenge with IL-1β, DABK but not saline triggered intense leukocyte rolling, adhesion, and emigration at 2 and 4 h (Fig. 2). These effects of DABK were only seen in animals pretreated with IL-1β, and not in those pretreated with saline or the chemokine IL-8 (Fig. 2; see Fig. 4). The vast majority of cells infiltrating the subendothelial matrix were neutrophils, as demonstrated by histological analysis (Fig. 3). Further analysis revealed that in mesenteries from animals treated with DABK (2 h), 71.0 ± 6.7% of mast cells had degranulated (Fig. 3; n = 3). In contrast, only 4.3 ± 1.5% (P < 0.05) were degranulated in control (saline-treated, 2 h) animals (n = 3). No significant change in total mast cell number was observed between groups (control, 2.03 ± 0.03 × 103 cells/mm2; DABK treated, 1.77 ± 0.12 × 103 cells/mm2; P > 0.05).
Figure 2.
Leukocyte–endothelial cell interactions in mouse mesenteric postcapillary venules in vivo in response to B1 receptor activation with DABK (30 nmol intraperitoneally). (A) Leukocyte rolling velocity, (B) leukocyte adhesion, and (C) leukocyte emigration were examined 2 and 4 h after DABK or saline administration. Mice received either DABK (•) or saline (▪) 24 h after IL-1β (time = 0). The effect of DABK in saline- (○, 24 h) or IL-8–treated (□, 24 h) mice is also shown. Data are mean ± SEM for n = 5–8 animals per group. *P < 0.05 versus saline-treated values.
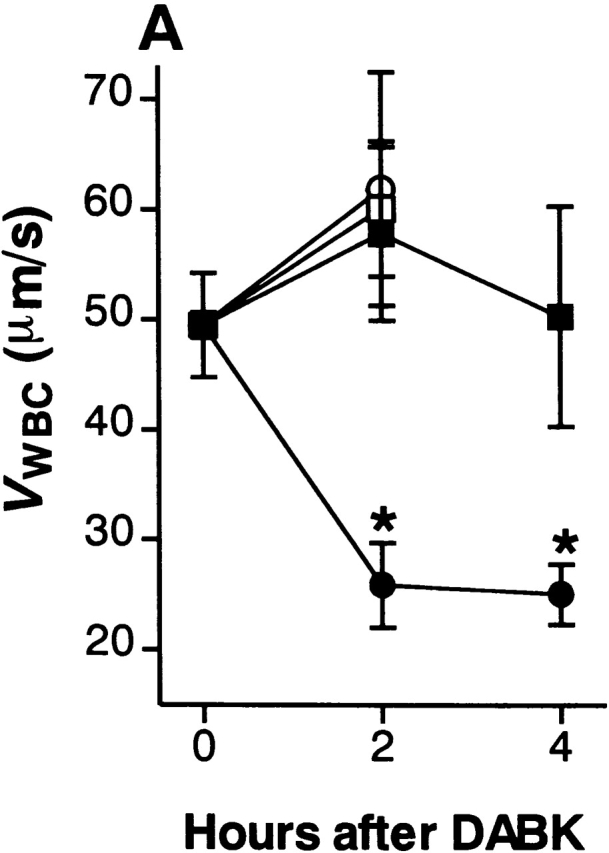
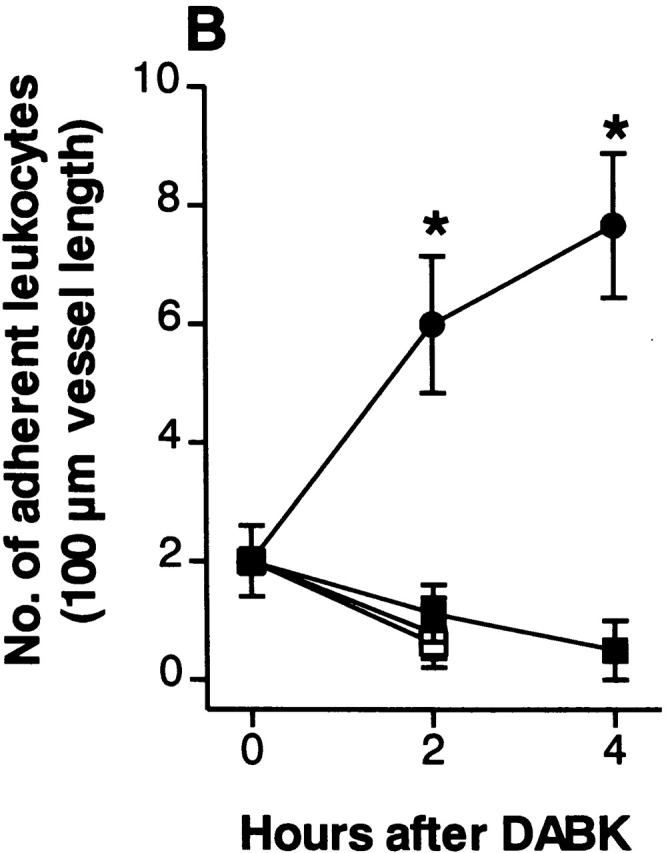
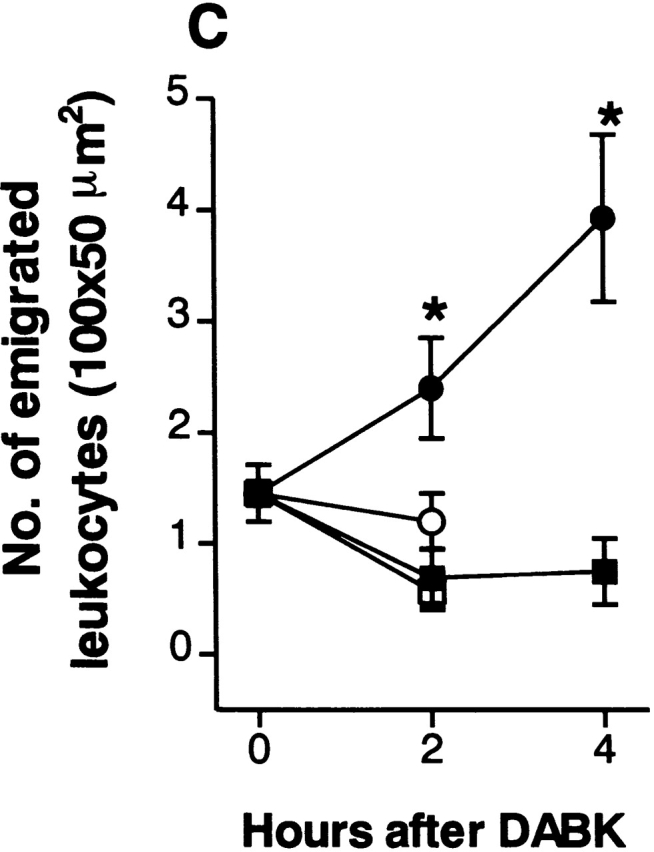
Figure 4.
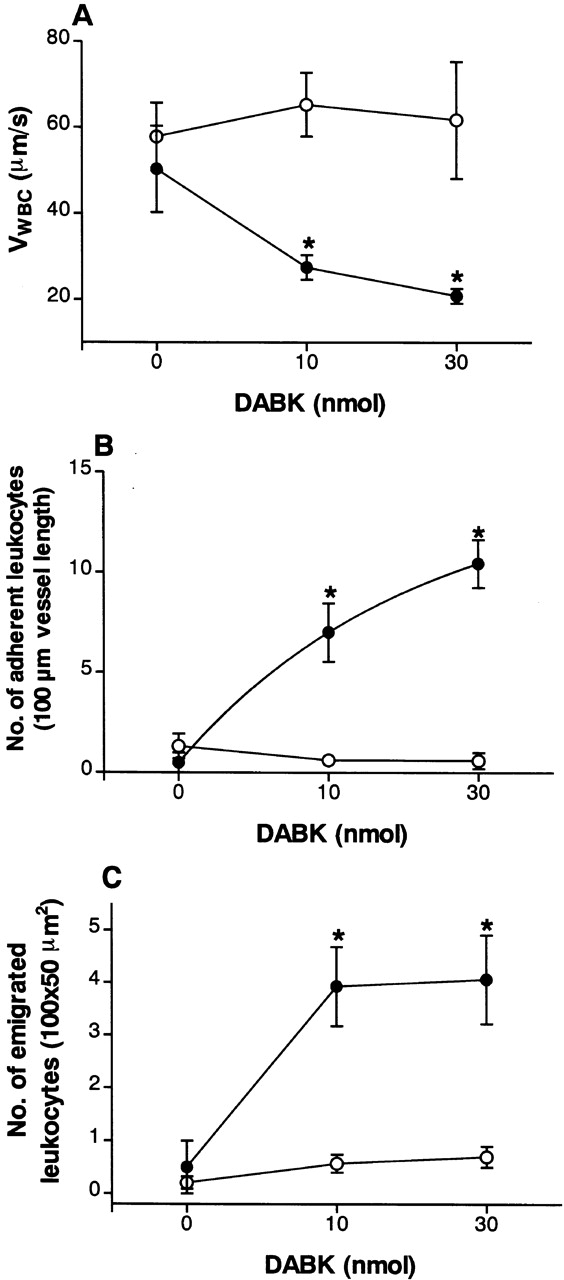
DABK causes dose-related changes in (A) leukocyte rolling velocity, (B) adhesion, and (C) emigration in mouse mesenteric postcapillary venules. Mice received DABK (10–30 nmol in 0.25 ml saline, intraperitoneally) 24 h after IL-1β (•; 5 ng in 0.25 ml saline intraperitoneally) or saline alone (○; 0.25 ml intraperitoneally). Leukocyte responses were examined 2 h after DABK treatment. Data are mean ± SEM for n = 5–8 animals per group. *P < 0.05 versus saline-treated values.
Figure 3.
Identification of leukocytes by (A and B) hematoxylin and eosin staining and (C and D) mast cells by toluidine blue staining of mesenteries from mice treated with either (A, B, and D) DABK (2 h) 24 h after IL-β or (C) saline (2 h). (A) Mesenteric section showing a postcapillary venule (v) and arteriole (a); original magnification: ×20. (B) Postcapillary venule (v) with a predominance of polymorphonuclear cells (arrowheads) either attached within or emigrated from the venule (original magnification: ×100). (C) Nondegranulated mast cells (arrowheads) in saline-treated mice (original magnification: ×40; inset original magnification: ×100). (D) Degranulated mast cells (arrowheads) in DABK/IL-1–treated mice (original magnification: ×40; inset original magnification: ×100).

DABK-induced leukocyte recruitment was a time- (Fig. 2) and dose-dependent (Fig. 4) process. A maximal decrease in V WBC (48% reduction from basal) was observed 2 h after DABK treatment. This led to a 200% increase in cell adhesion at 2 h, which increased further to 280% at 4 h. The number of emigrated leukocytes increased by 65% at 2 h and 171% at 4 h after DABK (Fig. 2).
There was no difference in RBC (centerline) velocity, venule diameter, and shear rate for any of the experimental groups. An apparent reduction in shear rate was seen in the IL-1β (24 h)/DABK (4 h) group; however this did not reach statistical significance compared with the IL-1β (24 h)/saline (4 h) group (Table ).
Table 2.
Hemodynamic Parameters of Venules Studied
| Treatment (time) | No. of venules | Venule diameter | Centerline velocity | Calculated wall shear rate |
|---|---|---|---|---|
| μm | mm/s | s−1 | ||
| Saline (2 h) | 18 | 27.9 ± 2.1 | 1.5 ± 0.1 | 287 ± 22 |
| IL-1β (2 h) | 10 | 29.2 ± 2.2 | 1.3 ± 0.1 | 235 ± 24 |
| IL-1β/DAH (2 h) | 18 | 26.8 ± 0.9 | 1.4 ± 0.1 | 269 ± 17 |
| IL-1β (24 h)/saline (2 h) | 11 | 29.6 ± 1.7 | 1.5 ± 0.1 | 265 ± 21 |
| IL-1β (24 h)/saline (4 h) | 6 | 25.6 ± 1.3 | 1.4 ± 0.1 | 270 ± 32 |
| IL-1β (24 h)/DABK (2 h) | 9 | 33.5 ± 2.9 | 1.7 ± 0.3 | 253 ± 32 |
| IL-1β (24 h)/DABK (4 h) | 9 | 24.0 ± 1.4 | 1.5 ± 0.1 | 208 ± 13 |
Mice received either saline (0.25 ml), IL-1β (5 ng intraperitoneally), IL-1β (5 ng)/des-arg10HOE 140 (DAH; 25 nmol intraperitoneally), or IL-1β (5 ng intraperitoneally, 24 h) followed by DABK (2 or 4 h). Data are mean ± SEM.
Molecular Expression of B1 Receptor mRNA After IL-1β Treatment.
RT-PCR of mesenteric mRNA from saline-treated mice revealed minimal expression of B1 receptor mRNA. In contrast, a marked increase in expression was consistently detected in mesenteries taken from mice 24 h after IL-1β treatment (Fig. 5 A; n = 3). A similar pattern of B1 receptor mRNA induction was observed in mesenteries from BK2r−/− mice (Fig. 5 A; n = 3). GAPDH mRNA was detected in all mesenteries tested, the level of which was consistent within strains. Fig. 5 (inset) shows that the PCR analyses of both the B1 receptor and GAPDH genes were amplified over several cycles that gave rise to a linear relationship between starting amount of cDNA and product intensity.
Figure 5.
Upregulation of the B1 receptor by IL-1β in mouse mesenteric vascular bed. (A) Detection of B1 receptor mRNA by RT-PCR. B1 receptor (435 bp) and GAPDH (363 bp) PCR products amplified from polyA+ RNA extracted from mesenteric vascular beds from either saline- (0.25 ml, intraperitoneally) or IL-1β–treated (5 ng in 0.25 ml saline, intraperitoneally) mice. Positive PCR control is a mouse B1 receptor cDNA plasmid. Results are representative of tissue from at least three animals per group. Inset: PCR product yield as a function of initial amount of cDNA. Serial dilutions of cDNA before PCR results in a log plot in which a linear relationship between starting amount of cDNA and product intensity is observed. B1 receptor and GAPDH curves are parallel (P > 0.05). (B) Western blot analysis of the expression of mouse B1 receptor using the IgG purified from A15C (reference 27; dilution 1:20,000) on mesenteric tissues (20 μg protein per lane). Mesenteric tissue were taken from either saline- (0.25 ml intraperitoneally) or IL-1β–treated (5 ng in 0.25 ml saline intraperitoneally) mice. Results are representative of tissue from four experiments. + anti B1 represents inhibition of detection of the 39-kD band when the antibody is preincubated (overnight at 4°C) with the A15C peptide at a concentration of 10 μg/ml. Molecular mass marker proteins were run simultaneously. Their molecular masses (kD) are shown on the left side of the figure.


Induction of B1 Receptor Protein by IL-1β Treatment.
Western blot analysis of mesenteric tissue protein collected from IL-1β–pretreated mice showed a band with an apparent molecular mass of ∼39 kD (Fig. 5 B; n = 4) corresponding to the mouse B1 receptor 19. This band was not observed when tissue from saline-treated mice was used (n = 4). The specificity of the response of the A15C antibody was further demonstrated by the ability of the free A15C peptide 20 to inhibit the presence of this band (Fig. 5 B).
Receptor Specificity of DABK-induced Leukocyte Trafficking. The effects of DABK on leukocyte recruitment were significantly attenuated by the concomitant administration of the B1 receptor selective antagonist des-arg10HOE 140 (25 nmol, n = 6; Fig. 6), whereas HOE 140 (25 nmol, n = 6; Fig. 6) had no effect.
Figure 6.
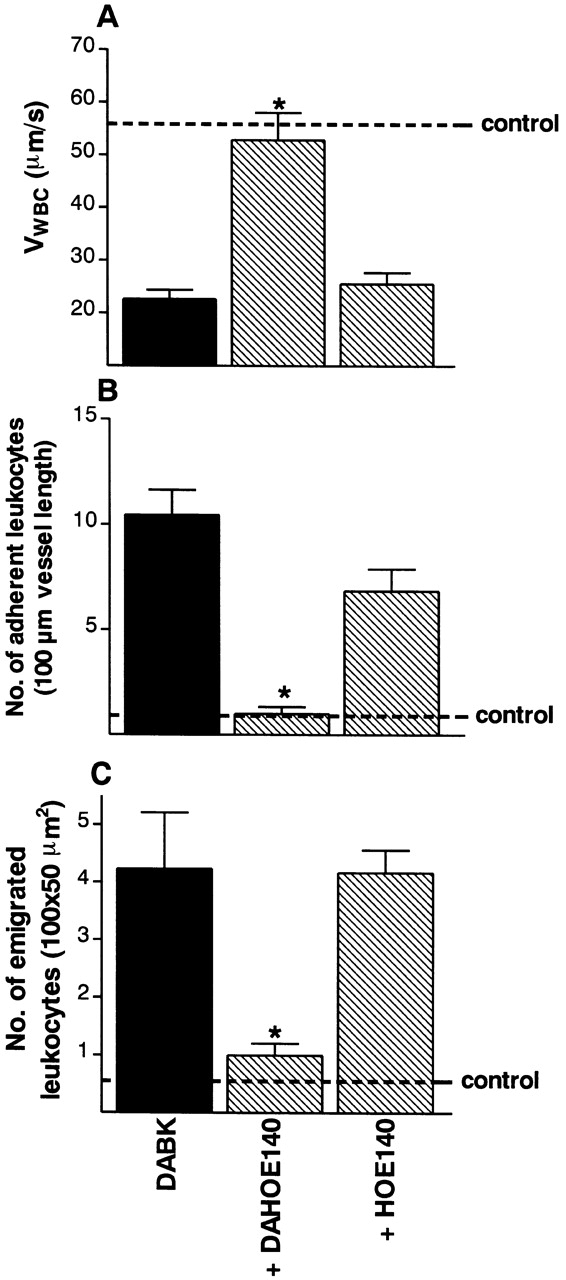
The effect of B1 or B2 receptor blockade on the changes in leukocyte kinetics induced by DABK (30 nmol). Mice were injected with IL-1β (5 ng) 24 h before DABK together with saline, des-arg10HOE 140 (DAHOE 140; B1 receptor antagonist, 25 nmol) or HOE 140 (B2 receptor antagonist, 25 nmol). Leukocyte kinetics (A) rolling velocity, (B) adhesion, and (C) emigration were examined 2 h after DABK treatment. Data are mean ± SEM for n = 5–8 animals per group. *P < 0.05 versus saline-treated values.
Mechanisms of the Enhanced Leukocyte–Endothelial Cell Interactions Induced by B1 Receptor Activation.
Systemic treatment with capsaicin (n = 6) significantly reduced DABK-induced effects on leukocyte accumulation, producing a similar level of inhibition on rolling, adhesion, and emigration (Fig. 7). Treatment with SR 140333 (n = 6) produced a similar extent of inhibition to that observed with capsaicin (Fig. 7). Neither MEN 11420 (n = 5), CGRP-(8–37) (n = 5), nor WEB 2086 (n = 4) had any significant effect on the response to DABK (Fig. 7). SR 142801 (n = 4), compound 48/80 (n = 6), and triprolidine (n = 4) significantly (P < 0.05) inhibited leukocyte rolling and cell adhesion (Fig. 7), but to a lesser extent than was observed with SR 140333 or capsaicin (Fig. 7).
Figure 7.
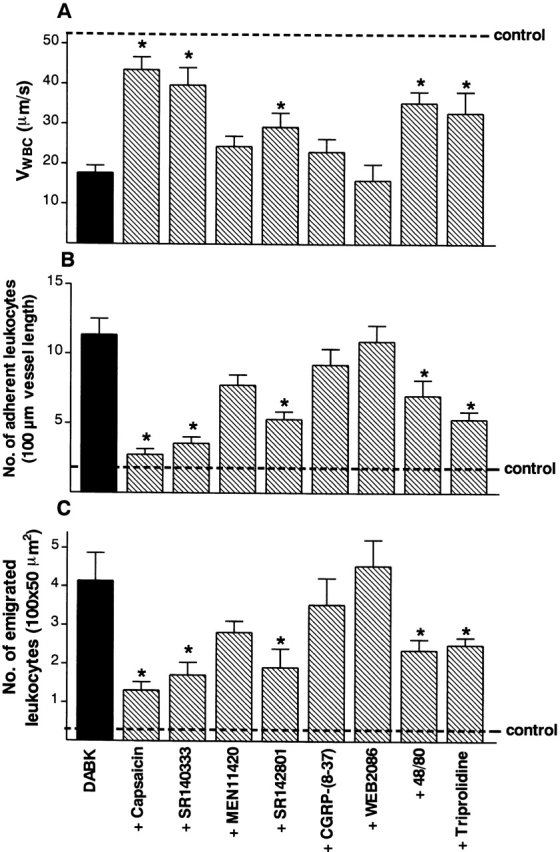
Effect of capsaicin, compound 48/80, and a range of selective receptor antagonists on DABK-induced (30 nmol intraperitoneally) changes in (A) leukocyte rolling velocity, (B) leukocyte adhesion, and (C) leukocyte emigration at the 2-h time point. Values from saline-treated control animals were V WBC = 52.8 ± 6.6 μm/s; adherent leukocytes = 1.9 ± 0.5 cells/100 μm; emigrated leukocytes = 0.2 ± 0.1 cells/100 × 50 μm2. The following specific treatments were used: capsaicin (50 mg/kg subcutaneously, 4 d); CGRP-(8–37) (300 nmol/kg intravenously, 15 min); SR 140333 (0.1 μmol/kg intravenously, 30 min); MEN 11420 (0.1 μmol/kg intravenously, 15 min); SR 142801 (4 μmol/kg intravenously, 15 min); WEB 2086 (5 mg/kg intravenously, 20 min); compound 48/80 (1.2 mg/kg intraperitoneally, 72 h [reference 23]); and triprolidine (1 mg/kg intraperitoneally, 20 min [reference 26]). Data are mean ± SEM for n = 6 animals per group. *P < 0.05 versus DABK-treated values.
Leukocyte–Endothelial Cell Interactions in Response to Substance P.
24 h after challenge with IL-1β, substance P (7.5 nmol) promoted leukocyte recruitment from mesenteric postcapillary venules, with V WBC, cell adhesion, and emigration all being affected (Fig. 8). This response to substance P was significantly reduced by treatment with SR 140333 (n = 4; Fig. 8). In contrast, MEN 11420 (0.1 μmol/kg, n = 4) had no significant effect on the response to substance P (Fig. 8). SR 142801 (4 μmol/kg, n = 4) also inhibited substance P–induced effects; however, this inhibition was not as marked as that achieved with SR 140333 (Fig. 8).
Figure 8.
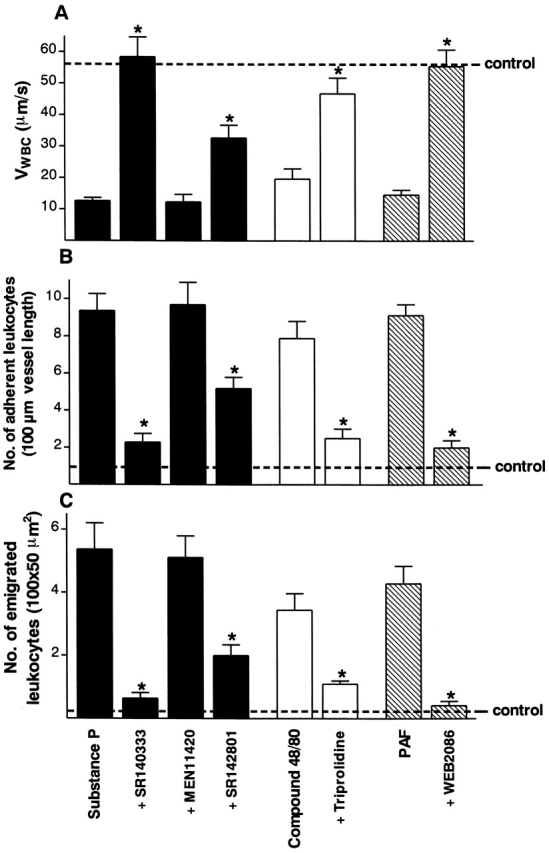
Effect of a range of selective receptor antagonists on substance P– (7.5 nmol intraperitoneally), compound 48/80– (1.2 mg/kg intraperitoneally), and PAF-induced (1 μg intraperitoneally) changes in (A) leukocyte rolling velocity, (B) leukocyte adhesion, and (C) leukocyte emigration at the 2-h time point. Values from saline-treated control animals are V WBC = 58.7 ± 9.2 μm/s; adherent leukocytes = 1.0 ± 0.5cells/100 μm; emigrated leukocytes = 0.7 ± 0.1 cells/100 × 50 μm2). The following specific treatments were used: SR 140333 (0.1 μmol/kg intravenously, 30 min); MEN 11420 (0.1 μmol/kg intravenously, 15 min); SR 142801 (4 μmol/kg intravenously, 15 min); triprolidine (1 mg/kg intraperitoneally, 20 min); and WEB 2086 (5 mg/kg intravenously, 20 min). Data are mean ± SEM for n = 6 animals per group. *P < 0.05 versus substance P–, compound 48/80–, or PAF-treated values.
Leukocyte–Endothelial Cell Interactions in Response to Compound 48/80 and PAF.
Acute administration of compound 48/80 (1.2 mg/kg intraperitoneally, 1 h) produced an inflammatory response characterized by slow leukocyte rolling, adhesion, and emigration (Fig. 8). These effects were significantly reduced by pretreatment with the H1 receptor antagonist triprolidine (n = 4; Fig. 8). Histological assessment showed that 93 ± 7% (n = 3) of mesenteric mast cells had degranulated after compound 48/80 treatment.
PAF (1 μg) promoted leukocyte recruitment from mesenteric postcapillary venules, with V WBC, cell adhesion, and emigration all being affected (Fig. 8). This response to PAF was abolished by treatment with WEB 2086 (n = 4; Fig. 8).
Leukocyte–Endothelial Cell Interactions in BK2r−/− Mice.
Neither DABK (30 nmol intraperitoneally) nor saline treatment 24 h after IL-1β treatment (5 ng intraperitoneally) revealed any significant difference in leukocyte–endothelial cell interactions between the wild-type (BK2r+/+) and knockout (BK2r−/−) mice (Fig. 9). An apparent 23% reduction of DABK-induced cell emigration was observed in BK2r−/− relative to wild-type mice; however, this did not reach statistical significance (P = 0.13, n = 6). Neither DABK nor saline treatment 24 h after IL-1β treatment produced any significant effect on RBC (centerline) velocity, venule diameter, or shear rate in either genotype (Table ).
Figure 9.
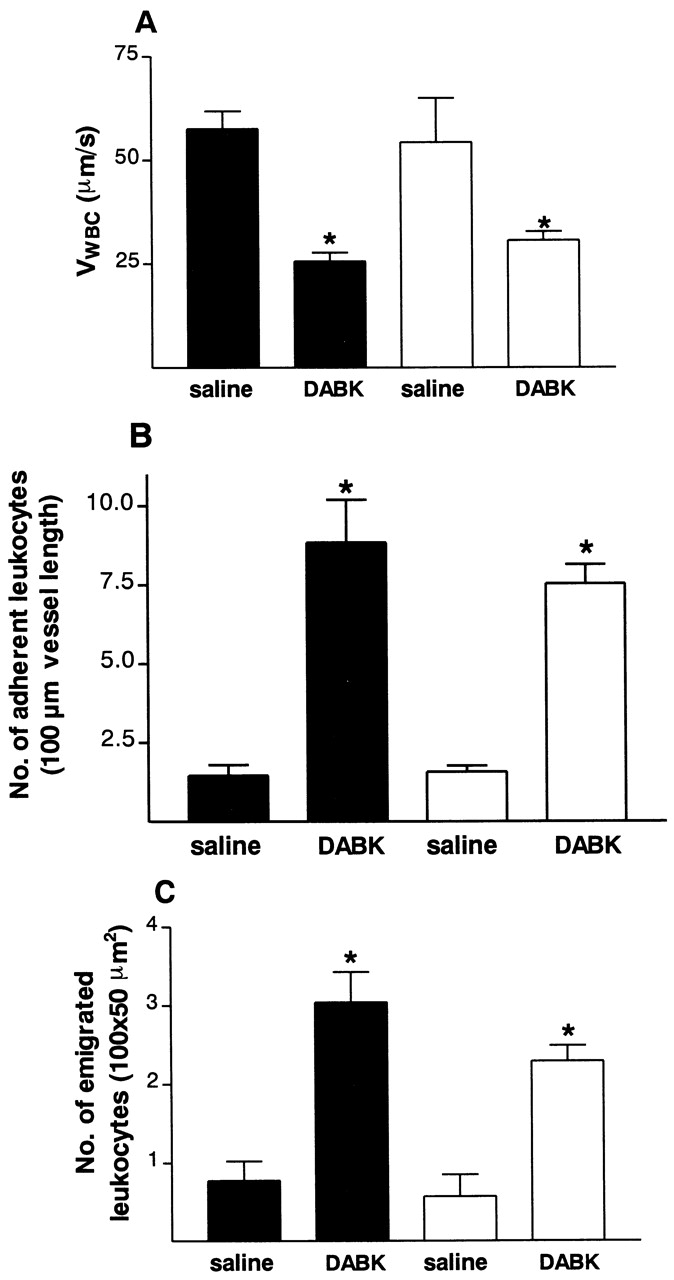
Leukocyte kinetics. (A) rolling velocity, (B) adhesion, and (C) emigration in the mesenteric vascular bed of BK2r−/− (white bars) and BK2r+/+ (black bars) mice. Mice were injected with IL-1β (5 ng intraperitoneally) 24 h before DABK (30 nmol intraperitoneally). No significant difference (P > 0.05) in any of the parameters measured was observed in BK2r−/− compared with BK2r+/+ mice 2 h after DABK treatment. Data are mean ± SEM for n = 5–8 animals per group. *P < 0.05 versus saline-treated values within the same genetic background.
Table 3.
Hemodynamic Parameters of Venules Studied: BK2r−/− Mice
| Mouse genotype (treatment) | No. of venules | Venule diameter | Centerline velocity | Calculated wall shear rate |
|---|---|---|---|---|
| μm | mm/s | s−1 | ||
| Wild-type (saline) | 11 | 28.0 ± 2.1 | 1.7 ± 0.1 | 318.0 ± 47.1 |
| BK2r−/− (saline) | 7 | 28.7 ± 1.9 | 1.5 ± 0.1 | 257.0 ± 19.1 |
| Wild-type (IL-1β/DABK) | 13 | 26.9 ± 1.6 | 1.4 ± 0.1 | 273.7 ± 25.2 |
| BK2r−/− (IL-1β/DABK) | 25 | 30.0 ± 0.8 | 1.4 ± 0.1 | 239.1 ± 9.5 |
Mice received either saline (0.25 ml intraperitoneally) or IL-1β (5 ng intraperitoneally, 24 h) followed by DABK (2 h). Data are mean ± SEM.
Discussion
This study describes, for the first time, a functional role for the B1 receptor in the processes regulating the initial steps of leukocyte extravasation in experimental inflammation. We have demonstrated that B1 receptor activation with DABK, 24 h after treatment with the cytokine IL-1β but not after treatment with saline or IL-8, triggers leukocyte rolling on and adhesion to the endothelium and emigration from mouse mesenteric postcapillary venules. Additionally, these responses were temporally associated with de novo B1 receptor mRNA and protein expression. Furthermore, we have shown that the induction of the receptor and reactivity to DABK are not affected in BK2r−/− mice. We have also characterized the mechanisms involved, identifying substance P (probably released from sensory C fibers) and mast cell degranulation as mechanisms involved in mediating the DABK-induced cellular response.
IL-1β is a proinflammatory cytokine that plays an integral role in the initiation of leukocyte–endothelial cell interactions during an inflammatory response 14 31. Application of IL-1β causes leukocyte recruitment across mouse mesenteric postcapillary venules similar to that seen within the rat mesenteric microcirculation 14 15. This response to IL-1β involves induction and activation of B1 receptors, as coadministration of the selective B1 receptor antagonist des-arg10HOE 140 reduced IL-1β–induced leukocyte rolling, adhesion, and emigration from postcapillary venules. The extent of inhibition obtained (∼50%) is in line with that observed in gross models of neutrophil extravasation 7.
The association between B1 receptor activation and leukocyte trafficking was studied using the B1 receptor selective agonist DABK. The 24-h time point after IL-1β was chosen because the cellular response to the cytokine itself had subsided. DABK produced a dose- and time-dependent sequential response characterized by leukocyte rolling followed by adhesion and then emigration. Histochemical studies confirmed that >90% of the leukocytes involved in this response were neutrophils. The time dependency of these phenomena suggests a sequential process in which each step is a prerequisite for the next, as observed for other inflammatory stimuli in the same vascular bed 16. Importantly, DABK had no effect in animals treated with saline or previously inflamed with the chemokine IL-8. Together, these data show that in the mouse mesenteric microcirculation, functional B1 receptors are not constitutively expressed but are induced selectively by the cytokine IL-1β.
The functional response to the B1 receptor agonist correlated with induction of the receptor. IL-1β has been shown to be an optimal inducer of B1 receptor expression on several cell types in vitro 32 33. This includes human embryo lung fibroblasts where IL-1β upregulates B1 receptor mRNA expression at both a transcriptional and posttranscriptional level 20 34. However, no study to date has shown an ability of this cytokine to induce B1 receptor mRNA and protein expression in vivo. In this study, IL-1β treatment caused an upregulation of B1 receptor mRNA expression, leading to de novo protein synthesis. A modest basal expression of the B1 receptor mRNA was seen in saline-injected animals. However, this was not associated with detectable expression of the protein. These results suggest that although low-level expression of B1 receptor mRNA is constitutively expressed in the mesenteric microvasculature, an appropriate inflammatory stimulus is required to achieve optimal transcription and translation.
The molecular studies were supported by the functional data. DABK-induced leukocyte rolling, adhesion, and emigration was mediated by B1 receptors, as the selective B1 receptor antagonist des-arg10HOE 140 almost abolished the effects of the agonist (90–95% reduction), whereas the chemically related B2 receptor selective antagonist HOE 140 was without effect. This cellular response was not due to a nonspecific alteration in local hemodynamics, as no significant change in blood flow or shear rate was observed. The lack of involvement of B2 receptors in DABK responses was confirmed by the experiments performed with the BK2r−/− mice. Quantitatively equal responses to DABK were achieved in BK2r+/+ mice and BK2r−/− mice. Taken together, these data show that IL-1β induces expression of functional B1 receptors in a manner independent of B2 receptor expression.
We next studied the mechanism(s) by which B1 receptor activation causes leukocyte recruitment. Other studies have proposed that DABK-induced neutrophil accumulation is mediated indirectly, as DABK does not directly modify neutrophil function in vitro 7, whereas neuropeptide receptor antagonists 7 block neutrophil accumulation in gross models of experimental inflammation in vivo 8. We hypothesized that the DABK-induced rolling, adhesion, and emigration may be mediated by the release of neuropeptides from sensory C fibers. Therefore we investigated the effects of C fiber desensitization with capsaicin using a validated protocol providing >90% depletion of sensory neuropeptide content 21. Using this protocol, DABK-induced leukocyte rolling, adhesion, and emigration were attenuated by 73, 91, and 71%, respectively. Therefore, activation of C fibers is pivotal in mediation of B1 receptor–induced leukocyte recruitment.
A substantial body of evidence suggests an important role of proinflammatory neuropeptides, such as substance P and CGRP, in C fiber–mediated neurogenic inflammation 35 36. Neuropeptides produce their effects via the selective activation of G protein–coupled receptors. Three neurokinin receptors—NK1, NK2, and NK3—have been characterized based on the relative order of potency of their corresponding endogenous agonists: substance P, neurokinin-A, and neurokinin-B, respectively 37. To identify the neuropeptide(s) involved in the B1 receptor–mediated leukocyte–endothelial cell interaction, we used a range of selective antagonists with established selectivity for neuropeptide receptors 38. Blockade of NK1 receptors with the selective antagonist SR 140333 produced an inhibition (rolling, adhesion, and emigration reduced by 60, 82, and 60%, respectively) similar to that observed with capsaicin. The dose of SR 140333 used in this study was previously validated for selectivity at NK1 receptors 23. The order of potency for neurokinins at their receptors 39 would suggest that the B1 receptor–mediated enhancement of leukocyte–endothelial cell interactions is mediated predominantly by substance P. This possibility is further confirmed by the lack of activity of the selective NK2 receptor antagonist MEN 11420. A partial role for NK3 receptors in the response may be possible, as blockade with the NK3 receptor antagonist, SR 142801, reduced DABK-induced rolling, adhesion, and emigration by 33, 64, and 50%, respectively. NK-B, the preferred endogenous agonist for the NK3 receptor, is not expressed to any appreciable extent in the periphery 37. However, it is of interest to note that functional NK3 receptors have been shown to be present in mesenteric vascular venous beds 40. It is therefore possible that during inflammation, these receptors might be activated by substance P. This was confirmed by the observation that substance P–induced leukocyte rolling, adhesion, and emigration were also reduced by SR 140333 and SR 142801. The lack of activity of the selective CGRP receptor antagonist, CGRP-(8–37), precludes an involvement of this neuropeptide in the observed response.
A recent study has illustrated the ability of a relatively high dose of BK to cause B2 receptor–mediated leukocyte recruitment in rat mesentery postcapillary venules 41. This response was shown to be mediated by PAF release, possibly from the endothelium or macrophages 41. However, this high-dose BK treatment was accompanied by a significant reduction in shear rate, which may have been a contributory factor in the leukocyte response observed. In contrast, in this study, no change in shear rate was observed after DABK treatment. Furthermore, the PAF antagonist WEB 2086 had no effect on the response to DABK, whereas it abolished the response to PAF itself. These data imply that distinct mechanisms exist for kinin receptor–mediated leukocyte recruitment, with B1 receptors acting predominantly via the release of neuropeptides, and B2 receptors activating PAF release 41.
Mast cells play an important role in the initiation and progression of the inflammatory response 42. Of particular relevance is their role in cellular accumulation 42, and also their role in neurogenic inflammation via a close relationship and interaction with sensory C fibers 43. Indeed, it has been demonstrated that C fibers and mast cells are often in intimate proximity 44 and share biochemical and functional cooperativity 45 46 47 48 49. Therefore, we investigated the involvement of mast cells in the DABK-induced leukocyte recruitment response by depleting (>80%) peritoneal mast cells with the selective serosal mast cell degranulator, compound 48/80, using a well-established protocol 28 29. The efficacy of this protocol was confirmed histologically and by the demonstration that acute administration of compound 48/80 itself induces leukocyte recruitment. This effect of compound 48/80 is most likely due to degranulation of mast cells, as pretreatment of mice with the H1 receptor triprolidine attenuated the response by 70–80%. Compound 48/80 is known to be inactive on neutrophil and/or endothelial cells, but it can promote leukocyte–endothelial cell interactions in vitro only when cocultured with mast cells 42. Mast cell depletion with compound 48/80 significantly attenuated the response to DABK, suggesting that mast cells play an important role in mediation of this response. Further support for an involvement of mast cells in the DABK-induced response was provided by histological studies demonstrating intense mast cell degranulation after DABK treatment. Additionally, as with the acute response to compound 48/80, the DABK-induced response was suppressed by the H1 receptor antagonist supporting a role for histamine. PAF is unlikely to be involved, as the response was not affected by the PAF antagonist WEB 2086 27. Thus it is clear that both C fibers and mast cells are involved in mediating the B1 receptor–induced leukocyte trafficking. The exact relationship and sequence of events between the C fiber– and mast cell–dependent pathways is less clear. It is possible that DABK causes substance P release, which in turn activates mast cells, as this neuropeptide is known to do 46 47 50 51, thereby stimulating the process of neutrophil extravasation 49. Conversely, DABK may also activate mast cells, resulting in the release of mediators such as histamine, serotonin, or mast cell tryptase, which are known to activate the release of neuropeptides from C fibers 45 52 47 53. Further studies are required to determine the precise cellular location of the kinin B1 receptor. However the evidence presented in this study suggests that the receptor may be expressed on C fibers and/or mast cells.
The molecular mechanisms of substance P– and histamine-induced leukocyte trafficking are known to involve the specific upregulation and rapid mobilization of certain adhesion molecules for PMNs; e.g., substance P causes the upregulation and mobilization of P-selectin, intercellular adhesion molecule (ICAM)-1, vascular cell adhesion molecule (VCAM)-1, and endothelial leukocyte adhesion molecule (ELAM)-1 54 55 56 57, and histamine-induced leukocyte recruitment is known to be mediated in part by P-selectin 55 56 57 58. Given our findings implicating substance P and histamine in the response to DABK, it is likely that DABK-induced leukocyte trafficking involves similar mechanisms. However, confirmation of this will be the subject of further study.
Conclusion.
To summarize, we have shown that IL-1β treatment induces the de novo expression of functional B1 receptors in the mouse mesentery. Activation of these receptors induces interactions between circulating leukocytes and the venular endothelium, leading to leukocyte emigration from postcapillary venules. The B1 receptor–mediated cellular response is dependent on activation of both C fibers and mast cells, the effects of which are mediated by tachykinin NK1, NK3, and histamine H1 receptor activation. In conclusion, we propose that inhibition of early leukocyte–endothelial cell interactions leading to reduced neutrophil recruitment represents an important additional mechanism for the antiinflammatory effects provided by B1 receptor antagonism. B1 receptors may be involved in certain inflammatory pathologies such as inflammatory hyperalgesia 4 59 and endotoxic shock 60, and have been implicated in atheromatous disease 61 and myocardial ischemia 62. Thus, the B1 receptor is clearly a target for the development of novel antiinflammatory drugs with a potent action on the events that lead to leukocyte recruitment.
Acknowledgments
The authors thank Dr. J.L. Bascands (Institut National de la Santé et de la Recherche Médicale, Toulouse, France) for the supply of the B1 receptor antibody and Dr. J.B. Pesquero (Escola Paulista de Medicine, Sao Paulo, Brazil) for the supply of the B1 receptor plasmid. We also thank Professor S.M. Oliani and Mr. A.S. Damazo (Instituto de Biociencias, Letras e Ciencias Exatas-Universidade Estadual Paulista Julio Mesquita Filho, Sao José do Rio Preto, Brazil) for helping with the histological analyses.
This study and P.G. McLean were supported by the British Heart Foundation (BHF; PG 97013). Dr. Ahluwalia is funded by a BHF Intermediate Fellowship, and Dr. Perretti is a Research Fellow of the Arthritis Research Campaign.
Footnotes
Abbreviations used in this paper: BK2r−/−, B2 receptor knockout mice; BK, bradykinin; CGRP, calcitonin gene-related peptide; DABK, des-Arg9bradykinin; ECL, enhanced chemiluminescence; NK, neurokinin; PAF, platelet-activating factor; PBS-T, 0.1% Tween-20 in PBS; RT, reverse transcriptase; V WBC, white blood cell velocity.
References
- Ahluwalia A., Perretti M. B1 receptors as a new inflammatory target. Trends Pharmacol. Sci. 1999;20:100–104. doi: 10.1016/s0165-6147(99)01321-8. [DOI] [PubMed] [Google Scholar]
- Regoli D., Barabe J. Pharmacology of bradykinin and related kinins. Pharmacol. Rev. 1980;32:1–46. [PubMed] [Google Scholar]
- Bhoola K.D., Figueroa C.D., Worthy K. Bioregulation of kininskallikreins, kininogens, and kininases. Pharmacol. Rev. 1992;44:1–80. [PubMed] [Google Scholar]
- Marceau F., Hess J.F., Bachvarov D.R. The B1 receptors for kinins. Pharmacol. Rev. 1998;50:357–386. [PubMed] [Google Scholar]
- Faussner A., Proud D., Towns M., Bathon J.M. Influence of the cytosolic carboxyl termini of human B1 and B2 kinin receptors on receptor sequestration, ligand internalization, and signal transduction. J. Biol. Chem. 1998;273:2617–2623. doi: 10.1074/jbc.273.5.2617. [DOI] [PubMed] [Google Scholar]
- Phagoo S.B., Poole S., Leeb-Lundberg L.M. Autoregulation of bradykinin receptorsagonists in the presence of interleukin-1beta shift the repertoire of receptor subtypes from B2 to B1 in human lung fibroblasts. Mol. Pharmacol. 1999;56:325–333. doi: 10.1124/mol.56.2.325. [DOI] [PubMed] [Google Scholar]
- Ahluwalia A., Perretti M. Involvement of bradykinin B1 receptors in the polymorphonuclear leukocyte accumulation induced by IL-1 beta in vivo in the mouse. J. Immunol. 1996;156:269–274. [PubMed] [Google Scholar]
- Vianna R.M., Calixto J.B. Characterization of the receptor and the mechanisms underlying the inflammatory response induced by des-Arg9-BK in mouse pleurisy. Br. J. Pharmacol. 1998;123:281–291. doi: 10.1038/sj.bjp.0701590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perron M.S., Gobeil F.J., Pelletier S., Regoli D., Sirois P. Involvement of bradykinin B1 and B2 receptors in pulmonary leukocyte accumulation induced by Sephadex beads in guinea pigs. Eur. J. Pharmacol. 1999;376:83–89. doi: 10.1016/s0014-2999(99)00348-9. [DOI] [PubMed] [Google Scholar]
- Panes J., Granger D.N. Leukocyte-endothelial cell interactionsmolecular mechanisms and implications in gastrointestinal disease. Gastroenterology. 1998;114:1066–1090. doi: 10.1016/s0016-5085(98)70328-2. [DOI] [PubMed] [Google Scholar]
- Kansas G.S. Selectins and their ligandscurrent concepts and controversies. Blood. 1996;88:3259–3287. [PubMed] [Google Scholar]
- Newton R.A., Thiel M., Hogg N. Signaling mechanisms and the activation of leukocyte integrins. J. Leukoc. Biol. 1997;61:422–426. doi: 10.1002/jlb.61.4.422. [DOI] [PubMed] [Google Scholar]
- Bienvenu K., Granger D.N. Molecular determinants of shear rate-dependent leukocyte adhesion in postcapillary venules. Am. J. Physiol. 1993;264:H1504–H1508. doi: 10.1152/ajpheart.1993.264.5.H1504. [DOI] [PubMed] [Google Scholar]
- Wakelin M.W., Sanz M.J., Dewar A., Albelda S.M., Larkin S.W., Boughton-Smith N., Williams T.J., Nourshargh S. An anti-platelet–endothelial cell adhesion molecule-1 antibody inhibits leukocyte extravasation from mesenteric microvessels in vivo by blocking the passage through the basement membrane. J. Exp. Med. 1996;184:229–239. doi: 10.1084/jem.184.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tailor A., Flower R.J., Perretti M. Dexamethasone inhibits leukocyte emigration in rat mesenteric post-capillary venulesan intravital microscopy study. J. Leukoc. Biol. 1997;62:301–308. doi: 10.1002/jlb.62.3.301. [DOI] [PubMed] [Google Scholar]
- Lim L.H., Solito E., Russo-Marie F., Flower R.J., Perretti M. Promoting detachment of neutrophils adherent to murine postcapillary venules to control inflammationeffect of lipocortin 1. Proc. Natl. Acad. Sci. USA. 1998;95:14535–14539. doi: 10.1073/pnas.95.24.14535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochi H., Hirani W.M., Yuan Q., Friend D.S., Austen K.F., Boyce J.A. T helper cell type 2 cytokine–mediated comitogenic responses and CCR3 expression during differentiation of human mast cells in vitro. J. Exp. Med. 1999;190:267–280. doi: 10.1084/jem.190.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni A., Chai K.X., Chao L., Chao J. Molecular cloning and expression of rat bradykinin B1 receptor. Biochim. Biophys. Acta. 1998;1442:177–185. doi: 10.1016/s0167-4781(98)00163-8. [DOI] [PubMed] [Google Scholar]
- Pesquero J.B., Pesquero J.L., Oliveira S.M., Roscher A.A., Metzger R., Ganten D., Bader M. Molecular cloning and functional characterization of a mouse bradykinin B1 receptor gene. Biochem. Biophys. Res. Commun. 1996;220:219–225. doi: 10.1006/bbrc.1996.0384. [DOI] [PubMed] [Google Scholar]
- Schanstra J.P., Bataille E., Marin C.M., Barascud Y., Hirtz C., Pesquero J.B., Pecher C., Gauthier F., Girolami J.P., Bascands J.L. The B1-agonist [des-Arg10]-kallidin activates transcription factor NF- kappaB and induces homologous upregulation of the bradykinin B1- receptor in cultured human lung fibroblasts. J. Clin. Invest. 1998;101:2080–2091. doi: 10.1172/JCI1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggi C.A., Geppetti P., Santicioli P., Frilli S., Giuliani S., Furio M., Theodorsson E., Fusco B., Meli A. Tachykinin-like immunoreactivity in the mammalian urinary bladdercorrelation with the functions of the capsaicin-sensitive sensory nerves. Neuroscience. 1988;26:233–242. doi: 10.1016/0306-4522(88)90140-6. [DOI] [PubMed] [Google Scholar]
- Hall J.M., Brain S.D. Interaction of amylin with calcitonin gene-related peptide receptors in the microvasculature of the hamster cheek pouch in vivo. Br. J. Pharmacol. 1999;126:280–284. doi: 10.1038/sj.bjp.0702272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emonds-Alt X., Doutremepuich J.D., Heaulme M., Neliat G., Santucci V., Steinberg R., Vilain P., Bichon D., Ducoux J.P., Proietto V. In vitro and in vivo biological activities of SR140333, a novel potent non-peptide tachykinin NK1 receptor antagonist. Eur. J. Pharmacol. 1993;250:403–413. doi: 10.1016/0014-2999(93)90027-f. [DOI] [PubMed] [Google Scholar]
- Lecci A., Tramontana M., Giuliani S., Maggi C.A. Role of tachykinin NK1 and NK2 receptors on colonic motility in anesthetized ratseffect of agonists. Can. J. Physiol. Pharmacol. 1997;75:582–586. [PubMed] [Google Scholar]
- Inoue H., Nagata N., Koshihara Y. Involvement of tachykinin receptors in oedema formation and plasma extravasation induced by substance P, neurokinin A, and neurokinin B in mouse ear. Inflamm. Res. 1996;45:316–323. doi: 10.1007/BF02252943. [DOI] [PubMed] [Google Scholar]
- Harris J.G., Flower R.J., Watanabe K., Tsurufuji S., Wolitzky B.A., Perretti M. Relative contribution of the selectins in the neutrophil recruitment caused by the chemokine cytokine-induced neutrophil chemoattractant (CINC) Biochem. Biophys. Res. Commun. 1996;221:692–696. doi: 10.1006/bbrc.1996.0658. [DOI] [PubMed] [Google Scholar]
- Perretti M., Flower R.J. Modulation of IL-1-induced neutrophil migration by dexamethasone and lipocortin 1. J. Immunol. 1993;150:992–999. [PubMed] [Google Scholar]
- Getting S.J., Flower R.J., Parente L., de Medicis R., Lussier A., Woliztky B.A., Martins M.A., Perretti M. Molecular determinants of monosodium urate crystal-induced murine peritonitisa role for endogenous mast cells and a distinct requirement for endothelial-derived selectins. J. Pharmacol. Exp. Ther. 1997;283:123–130. [PubMed] [Google Scholar]
- Das A.M., Flower R.J., Perretti M. Eotaxin-induced eosinophil migration in the peritoneal cavity of ovalbumin-sensitized micemechanism of action. J. Immunol. 1997;159:1466–1473. [PubMed] [Google Scholar]
- Borkowski J.A., Ransom R.W., Seabrook G.R., Trumbauer M., Chen H., Hill R.G., Strader C.D., Hess J.F. Targeted disruption of a B2 bradykinin receptor gene in mice eliminates bradykinin action in smooth muscle and neurons. J. Biol. Chem. 1995;270:13706–13710. doi: 10.1074/jbc.270.23.13706. [DOI] [PubMed] [Google Scholar]
- Nourshargh S., Larkin S.W., Das A., Williams T.J. Interleukin-1-induced leukocyte extravasation across rat mesenteric microvessels is mediated by platelet-activating factor. Blood. 1995;85:2553–2558. [PubMed] [Google Scholar]
- McLean P.G., Perretti M., Ahluwalia A. Inducible expression of the kinin B1 receptor in the endotoxemic heartmechanisms of des-Arg9bradykinin-induced coronary vasodilation. Br. J. Pharmacol. 1999;128:275–282. doi: 10.1038/sj.bjp.0702743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukagoshi H., Shimizu Y., Horie T., Fukabori Y., Iwamae S., Hisada T., Ishizuka T., Iizuka K., Dobashi K., Mori M. Regulation by interleukin-1beta of gene expression of bradykinin B1 receptor in MH-S murine alveolar macrophage cell line. Biochem. Biophys. Res. Commun. 1999;259:476–482. doi: 10.1006/bbrc.1999.0798. [DOI] [PubMed] [Google Scholar]
- Zhou X., Polgar P., Taylor L. Roles for interleukin-1β, phorbol ester and a post-transcriptional regulator in the control of bradykinin B1 receptor gene expression. Biochem. J. 1998;330:361–366. doi: 10.1042/bj3300361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer P. Capsaicincellular targets, mechanisms of action, and selectivity for thin sensory neurons. Pharmacol. Rev. 1991;43:143–201. [PubMed] [Google Scholar]
- Schaffer M., Beiter T., Becker H.D., Hunt T.K. Neuropeptidesmediators of inflammation and tissue repair? Arch. Surg. 1998;133:1107–1116. doi: 10.1001/archsurg.133.10.1107. [DOI] [PubMed] [Google Scholar]
- Maggi C.A., Patacchini R., Rovero P., Giachetti A. Tachykinin receptors and tachykinin receptor antagonists. J. Auton. Pharmacol. 1993;13:23–93. doi: 10.1111/j.1474-8673.1993.tb00396.x. [DOI] [PubMed] [Google Scholar]
- Betancur C., Azzi M., Rostene W. Nonpeptide antagonists of neuropeptide receptorstools for research and therapy. Trends Pharmacol. Sci. 1997;18:372–386. doi: 10.1016/s0165-6147(97)01109-7. [DOI] [PubMed] [Google Scholar]
- Regoli D., Boudon A., Fauchere J.L. Receptors and antagonists for substance P and related peptides. Pharmacol. Rev. 1994;46:551–599. [PubMed] [Google Scholar]
- D'Orleans-Juste P., Claing A., Telemaque S., Warner T.D., Regoli D. Neurokinins produce selective venoconstriction via NK-3 receptors in the rat mesenteric vascular bed. Eur. J. Pharmacol. 1991;204:329–334. doi: 10.1016/0014-2999(91)90860-s. [DOI] [PubMed] [Google Scholar]
- Shigematsu S., Ishida S., Gute D.C., Korthuis R.J. Concentration-dependent effects of bradykinin on leukocyte recruitment and venular hemodynamics in rat mesentery. Am. J. Physiol. 1999;277:H152–H160. doi: 10.1152/ajpheart.1999.277.1.H152. [DOI] [PubMed] [Google Scholar]
- Kubes P., Granger D.N. Leukocyte-endothelial cell interactions evoked by mast cells. Cardiovasc. Res. 1996;32:699–708. [PubMed] [Google Scholar]
- Geppetti P., Holzer P. Neurogenic Inflammation 1999. CRC Press; New York: pp. 324 [Google Scholar]
- Stead R.H., Tomioka M., Quinonez G., Simon G.T., Felten S.Y., Bienenstock J. Intestinal mucosal mast cells in normal and nematode-infected rat intestines are in intimate contact with peptidergic nerves. Proc. Natl. Acad. Sci. USA. 1987;84:2975–2979. doi: 10.1073/pnas.84.9.2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhoff M., Vergnolle N., Young S.H., Tognetto M., Amadesi S., Ennes H.S., Trevisani M., Hollenberg M.D., Wallace J.L., Caughey G.H. Agonists of proteinase-activated receptor 2 induce inflammation by a neurogenic mechanism. Nat. Med. 2000;6:151–158. doi: 10.1038/72247. [DOI] [PubMed] [Google Scholar]
- Yano H., Wershil B.K., Arizono N., Galli S.J. Substance P-induced augmentation of cutaneous vascular permeability and granulocyte infiltration in mice is mast cell dependent. J. Clin. Invest. 1989;84:1276–1286. doi: 10.1172/JCI114295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanahan F., Denburg J.A., Fox J., Bienenstock J., Befus D. Mast cell heterogeneityeffects of neuroenteric peptides on histamine release. J. Immunol. 1985;135:1331–1337. [PubMed] [Google Scholar]
- Tomoe S., Iwamoto I., Tomioka H., Yoshida S. Comparison of substance P-induced and compound 48/80-induced neutrophil infiltrations in mouse skin. Int. Arch. Allergy Immunol. 1992;97:237–242. doi: 10.1159/000236126. [DOI] [PubMed] [Google Scholar]
- Walsh D.T., Weg V.B., Williams T.J., Nourshargh S. Substance P-induced inflammatory responses in guinea-pig skinthe effect of specific NK1 receptor antagonists and the role of endogenous mediators. Br. J. Pharmacol. 1995;114:1343–1350. doi: 10.1111/j.1476-5381.1995.tb13354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foreman J.C., Jordan C.C., Oehme P., Renner H. Structure-activity relationships for some substance P-related peptides that cause wheal and flare reactions in human skin. J. Physiol. (Lond.). 1983;335:449–465. doi: 10.1113/jphysiol.1983.sp014543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lembeck F., Holzer P. Substance P as neurogenic mediator of antidromic vasodilation and neurogenic plasma extravasation. Naunyn Schmiedebergs Arch. Pharmacol. 1979;310:175–183. doi: 10.1007/BF00500282. [DOI] [PubMed] [Google Scholar]
- Griesbacher T., Rainer I. 5-hydroxytryptamine release from skin mast cells in vivo induced by peptide but not by nonpeptide ligands for bradykinin receptors. Immunopharmacology. 1999;43:195–201. doi: 10.1016/s0162-3109(99)00103-4. [DOI] [PubMed] [Google Scholar]
- Saria A., Hua X., Skofitsch G., Lundberg J.M. Inhibition of compound 48/80–induced vascular protein leakage by pretreatment with capsaicin and a substance P antagonist. Naunyn Schmiedebergs Arch. Pharmacol. 1984;328:9–15. doi: 10.1007/BF00496097. [DOI] [PubMed] [Google Scholar]
- Smith C.H., Barker J.N., Morris R.W., MacDonald D.M., Lee T.H. Neuropeptides induce rapid expression of endothelial cell adhesion molecules and elicit granulocytic infiltration in human skin. J. Immunol. 1993;151:3274–3282. [PubMed] [Google Scholar]
- Baluk P., Bertrand C., Geppetti P., McDonald D.M., Nadel J.A. NK1 receptors mediate leukocyte adhesion in neurogenic inflammation in the rat trachea. Am. J. Physiol. 1995;268:L263–L269. doi: 10.1152/ajplung.1995.268.2.L263. [DOI] [PubMed] [Google Scholar]
- Quinlan K.L., Song I.S., Naik S.M., Letran E.L., Olerud J.E., Bunnett N.W., Armstrong C.A., Caughman S.W., Ansel J.C. VCAM-1 expression on human dermal microvascular endothelial cells is directly and specifically up-regulated by substance P. J. Immunol. 1999;162:1656–1661. [PubMed] [Google Scholar]
- Matis W.L., Lavker R.M., Murphy G.F. Substance P induces the expression of an endothelial-leukocyte adhesion molecule by microvascular endothelium. J. Invest. Dermatol. 1990;94:492–495. doi: 10.1111/1523-1747.ep12874665. [DOI] [PubMed] [Google Scholar]
- Asako H., Kurose I., Wolf R., DeFrees S., Zheng Z.L., Phillips M.L., Paulson J.C., Granger D.N. Role of H1 receptors and P-selectin in histamine-induced leukocyte rolling and adhesion in postcapillary venules. J. Clin. Invest. 1994;93:1508–1515. doi: 10.1172/JCI117129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins M.N., Campbell E., Dray A. Antinociceptive activity of the bradykinin B1 and B2 receptor antagonists, des-Arg9, [Leu8]-BK and HOE 140, in two models of persistent hyperalgesia in the rat. Pain. 1993;53:191–197. doi: 10.1016/0304-3959(93)90080-9. [DOI] [PubMed] [Google Scholar]
- Whalley E.T., Solomon J.A., Modafferi D.M., Bonham K.A., Cheronis J.C. CP-0127, a novel potent bradykinin antagonist, increases survival in rat and rabbit models of endotoxin shock. Agents Actions Suppl. 1992;38:413–420. [PubMed] [Google Scholar]
- Raidoo D.M., Ramsaroop R., Naidoo S., Muller-Esterl W., Bhoola K.D. Kinin receptors in human vascular tissuetheir role in atheromatous disease. Immunopharmacology. 1997;36:153–160. doi: 10.1016/s0162-3109(97)00015-5. [DOI] [PubMed] [Google Scholar]
- Bouchard J.F., Chouinard J., Lamontagne D. Role of kinins in the endothelial protective effect of ischaemic preconditioning. Br. J. Pharmacol. 1998;123:413–420. doi: 10.1038/sj.bjp.0701619. [DOI] [PMC free article] [PubMed] [Google Scholar]