

Abstract
High systemic levels of osteoprotegerin (OPG) in OPG transgenic mice cause osteopetrosis with normal tooth eruption and bone elongation and inhibit the development and activity of endosteal, but not periosteal, osteoclasts. We demonstrate that both intravenous injection of recombinant OPG protein and transgenic overexpression of OPG in OPG−/2 mice effectively rescue the osteoporotic bone phenotype observed in OPG-deficient mice. However, intravenous injection of recombinant OPG over a 4-wk period could not reverse the arterial calcification observed in OPG−/− mice. In contrast, transgenic OPG delivered from mid-gestation through adulthood does prevent the formation of arterial calcification in OPG−/− mice. Although OPG is normally expressed in arteries, OPG ligand (OPGL) and receptor activator of NF-κB (RANK) are not detected in the arterial walls of wild-type adult mice. Interestingly, OPGL and RANK transcripts are detected in the calcified arteries of OPG−/− mice. Furthermore, RANK transcript expression coincides with the presence of multinuclear osteoclast-like cells. These findings indicate that the OPG/OPGL/RANK signaling pathway may play an important role in both pathological and physiological calcification processes. Such findings may also explain the observed high clinical incidence of vascular calcification in the osteoporotic patient population.
Keywords: osteoprotegerin, pathologic calcification, osteoporosis, vascular disease, atherosclerosis
Introduction
Osteoprotegerin (OPG; also known as osteoclastogenesis inhibitory factor [OCIF] or TNF receptor–like molecule [TR1]), a member of the TNF family of receptors, is a secreted protein that regulates bone mass by inhibiting osteoclast differentiation and activation 1 2 3. Overexpression of OPG in transgenic mice resulted in severe osteopetrosis associated with a decrease in osteoclasts in metaphyseal trabecular bone 1. In contrast, OPG-deficient mice (OPG−/−) exhibited an overall decrease in total bone density and a high incidence of fractures 4 5, reminiscent of postmenopausal osteoporosis in human populations 6. The early-onset osteoporosis observed in these mice is a result of increased bone resorption associated with increased numbers and activity of osteoclasts 4 6.
Surprisingly, OPG−/− mice also developed calcified lesions in the aorta and renal arteries 4. It is interesting to note that osteoporosis in humans is associated with a higher incidence of arterial calcification 7. Chemical composition studies indicate that these calcified vascular lesions include elements of bone matrix, such as hydroxyapatite, and proteins of bone, including growth factors and adhesion molecules (for review see reference 7). This dual phenomenon of osteoporotic loss of bone mass and accumulation of calcium minerals in arteries in the human population is an intriguing observation which is recapitulated in OPG−/− mice. Although OPG is expressed in arteries 1 4, the mechanism by which it regulates pathological calcification within the vasculature is unknown.
OPG exerts its inhibitory effects on osteoclastogenesis by binding to OPG ligand (OPGL; also known as TRANCE, RANKL, or osteoclast differentiation factor [ODF]), a TNF ligand family member 8 9 10 11. OPGL directly induces osteoclast differentiation from hematopoietic progenitor cells in the absence of vitamin D3, stromal cells, and glucocorticoids in a bone marrow coculture system. It also activates mature osteoclasts both in vivo and in vitro 8. Receptor activator of NF-κB (RANK), a TNF receptor family member 10, is expressed on the plasma membrane of osteoclasts and their precursors, and is the signaling receptor for OPGL 12 13 14. OPG functions as a secreted inhibitor of the RANK signaling pathway by binding to OPGL and competitively inhibiting the OPGL/RANK interaction on osteoclasts and their precursors.
The phenotype of OPG transgenic mice differs in significant ways from other murine models of osteopetrosis (for review see references 15 and 16). Genetic mutants such as op/op mice (CSF-1−/−), microphthalmic mice (mi/mi), and c-src −/− and c-fos −/− mice all exhibit osteopetrosis accompanied by impaired tooth eruption and/or retarded growth. The defects in these genetic mutants are generally associated with decreased bone resorption attributable to decreased osteoclast numbers or inactive osteoclasts. In general, the long bones of the characterized osteopetrotic mouse models are shortened in length and some mice also exhibit modest facial and cranial abnormalities. Osteopetrosis in OPG transgenic founder animals was severe in high expressors, yet occurred without shortening of the long bones or impaired tooth eruption 1. The unique features of the OPG osteopetrotic phenotype warrant further characterization of this mouse model.
In this study, we performed detailed analysis of three OPG transgenic lines that express different levels of OPG and determined the temporal onset of osteopetrosis. The analysis of these mice reveals that overexpression of OPG specifically inhibits development and activity of osteoclasts at the endosteal bone surface, whereas osteoclasts at the periosteal bone surface show signs of active resorption. We also demonstrate that systemic OPG, delivered either through injection of recombinant OPG, or through transgenic overexpression, rescues the osteoporosis phenotype of OPG knockout mice. However, transient introduction of OPG protein by injection failed to reverse the arterial calcification observed in OPG−/− mice, whereas transgenic expression of OPG prevented the formation of calcified lesions in arteries of adolescent mice. Interestingly, OPGL and RANK transcripts, which are expressed by osteoblastic stroma and osteoclast precursors 8 14, respectively, were also observed in cells associated with calcified arterial lesions of OPG knockout mice. Multinucleated cells which were cathepsin K+ and tartrate-resistant acid phosphatase (TRAP)+, but F4/80−, were also detected, indicating the presence of mature osteoclast-like cells in the calcified lesions. Taken together, these data indicate that arterial calcification may involve a process similar to osteogenesis and may in part be regulated by the OPG/OPGL/RANK signaling pathway.
Materials and Methods
Analysis of OPG Transgenic Mice.
RNA analysis, necropsy, histology, and bone density measurements were performed as previously described 1 4. Temporal onset of the osteopetrotic phenotype was analyzed as follows: transgenic embryos from the high expressing 22 line were obtained at embryonic day (E)15.5, E16.5, and E17.5. Mice were also obtained at days 1, 4, 7, 14, and 28 after birth. A small piece of tail tissue from each mouse was used to identify transgenic and normal mice by PCR analysis. Embryos and 1- and 4-d-old mice were processed whole for histological assessment. Femurs and tibias of mice at days 7, 14, and 28 were removed for histology and other analysis. All procedures involving mice were approved by the Amgen Laboratory Animal Research Committee.
Ultrastructural Analysis of Osteoclasts.
Mice selected for electron microscopic examination were perfused with 2.5% glutaraldehyde and 1.6% paraformaldehyde in 0.1 M sodium cacodylate buffer at pH 7.3. The femurs were removed and split lengthwise and immersed in 4°C fixative overnight. The specimens were then decalcified in 5% EDTA in 0.1 M sodium cacodylate buffer with the addition of 0.1% glutaraldehyde. After 2 wk at 4°C, the femurs were washed in buffer and postfixed in 1% aqueous osmium tetroxide before embedding in epoxy resin. 1-μm sections were stained with toluidine blue. Areas selected for electron microscopy were ultra thin sectioned and collected on to 200 mesh uncoated copper grids. The sections were stained with uranyl acetate and lead citrate before examination on a Philips CM120 transmission electron microscope.
Treatment of OPG−/− Mice with Recombinant OPG.
OPG−/− mice were generated as previously described 4 and aged to 8 wk. The severity of the phenotype in knockout mice was determined from serum alkaline phosphatase (ALP) levels obtained via retroorbital bleeds of lightly anesthetized mice, and treatment groups were stratified accordingly. OPG−/− mice were treated intravenously either with vehicle (PBS, n = 14) or a recombinant human–Fc fusion form of OPG (reference 17; 50 mg/kg three times per week, n = 13) for 4 wk. This high dose was selected to maintain circulating OPG activity even if there was a host immune response to the injected human protein. Age-matched wild-type control mice were treated (vehicle, n = 10 or recombinant OPG, n = 10) in a similar fashion. The effects of recombinant OPG treatment were monitored via serum ALP levels determined on day 0, before the initiation of treatment, and on days 1, 7, 14, 21, and 28. At the conclusion of the study all mice were radiographed with a Faxitron x-ray system (43855A), and the tibia and the thoracic aorta were harvested, fixed in zinc formalin, and processed for histological evaluation. An additional tibia was harvested and fixed in 70% EtOH for bone density analysis by peripheral quantitative computed tomography (pQCT).
Generation of OPG−/− Mice Bearing OPG Transgene.
Male OPG−/− mice were crossed with female mice from the 22 OPG transgenic line to generate OPG+/− mice with or without transgene. These F1 mice were mated to generate three groups used in the study: OPG−/− mice (n = 8); OPG−/− mice bearing OPG transgene (n = 9); and wild-type littermate controls (n = 10). These mice were aged to 8 wk for necropsy and further analysis.
Bone Densitometry.
Bone mineral density was determined from bones fixed in 70% EtOH at the proximal tibial metaphysis by pQCT (XCT-960M; Norland Medical Systems). Two 1-mm cross-sections of bone were analyzed (XMICE 5.2; Stratec Corporation) at 1.5 and 2 mm from the proximal end of the tibia to determine total (trabecular and cortical) bone mineral density, and an average value for both cross-sections is reported. A soft tissue separation threshold of 1,500 was used to define the boundary of the metaphyseal bone.
Histology, In Situ Hybridization, and Immunohistochemistry.
Sections of thoracic aorta were embedded in paraffin, sectioned on a microtome, stained with hematoxylin, and counter-stained with eosin. By this method, normal arterial smooth muscle stains light pink, and calcified tissue stains dark blue. Bones were decalcified with formic acid before paraffin embedding and sectioning. Tibial sections were reacted for TRAP, which stains osteoclasts red, and counter-stained with methyl green. Osteoclasts were counted in a 1-mm2 field adjacent to the metaphyseal growth plate and in a 0.5 mm × 1.5 mm area of the diaphysis, 3.0 mm distal to growth plate of the proximal tibia. Osteoblast surface area (mm) and trabecular bone area (mm2) were also determined in the same areas of the tibia. In situ hybridization and immunohistochemistry were performed as previously described 4 8.
Results
Overexpression of OPG in Transgenic Mice.
Three transgenic founder mice expressing varying levels of a rat OPG transgene predominately in the liver (Fig. 1 A) were bred to generate transgenic lines for analysis. High (line 22), intermediate (line 33), and low (line 45) expressing lines were established where F1 offspring derived from the three lines had different amounts of OPG protein in serum. An ELISA for OPG protein revealed that levels of circulating OPG spanned three orders of magnitude in the different lines (14 ± 4 ng/ml in line 45, 358 ± 79 ng/ml in line 33, and 1,112 ± 261 ng/ml in line 22, mean ± SD). The average serum OPG level in wild-type mice is <1 ng/ml. The rat protein existed as monomeric and dimeric forms in liver extracts and serum. On nonreducing SDS-PAGE, these forms comigrated with similar bands generated from Chinese hamster ovary cell–derived murine OPG protein (Fig. 1 B). A larger, presumably trimeric form, was also evident in serum.
Figure 1.

(A) Northern blot analysis of liver RNA from wild-type (line 30) and transgenic founder mice (lines 33, 22, and 45). (B) Western analysis of OPG from serum and liver extract of line 22 transgenic mice (+) or wild-type mice (−). Chinese hamster ovary cell–produced recombinant OPG (CHO-muOPG) is used as the positive control. (C) Hematoxylin and eosin–stained sections of femoral diaphysis from 6-wk-old wild-type (normal), low (HECR-45), moderate (HECR-33), and high (HECR-22) OPG-expressing F1 transgenic mice. Bone stains pink, and marrow cells stain dark blue. Green boxes represent the area used for histomorphometric quantitation of bone volume. (D) Bone volume as a percentage of tissue volume measurements were made in 6-wk-old femoral diaphysis 2–4 mm from the distal growth plate, and did not include cortical bone (see C–F, above). Values are mean ± SEM, n = 4–7 for F1 OPG transgenics and n = 16 for aged-matched littermates. (E) Osteoclast number per millimeter of growth plate length and active osteoclast surface as a percentage of growth plate length measurements were taken in the femoral metaphysis adjacent to the growth plate from 6-wk-old wild-type, low, moderate, and high OPG-expressing F1 mice. Osteoclasts considered for measurement were TRAP+ cells in contact with a bone surface and within three cell widths of the growth plate.
Phenotypic analysis of 6–10-wk-old OPG F1 transgenic mice revealed a profound increase in bone density in the long bones and vertebrae (Fig. 1 C and data not shown). Bone density correlated with levels of expression of the OPG transgene; trabecular bone volume as a percent of tissue volume in the femoral diaphysis increased from 2.64 ± 0.75% in normal littermates to 15.5 ± 2.1% (mean ± SEM) and 79.5 ± 2.2% with low (line 45) and high (line 22) expression of OPG respectively (Fig. 1 D). Quantitation of osteoclasts adjacent to the distal femoral growth plate in the primary spongiosa revealed an inverse correlation between expression levels of OPG and osteoclast numbers (Fig. 1 E). This is in agreement with previous data indicating that OPG is regulating osteoclast maturation and activation 1.
The dense appearance of the bone was similar to other osteopetrotic phenotypes but without endochondral clubbing or failure of tooth eruption. Elongation of the bones was normal in all OPG transgenic lines. In the high expressing 22 line, severe osteopetrosis was accompanied by splenomegaly and no apparent changes in other organ systems (data not shown). To address how normal bone elongation in OPG transgenic mice occurred even in the presence of severe osteopetrosis, the temporal onset of the osteopetrotic phenotype was examined in the high expressing 22 line. Expression of OPG mRNA was detected in embryonic liver at E12.5 (data not shown). Increased endochondral bone density was apparent by x-ray on E17.5 (data not shown) and by histology at birth (Fig. 2 A). Transgenic F1 progeny were clearly osteopetrotic on postnatal day 4, with progressive filling of the femoral medullary cavity with dense trabecular bone evident through postnatal day 28. Tooth eruption occurred normally in OPG transgenic mice. The mice exhibit normal growth rates and no signs of infertility past 1 yr of age. The late embryonic and postnatal increases in bone density were preceded and accompanied by reduced numbers of osteoclasts adjacent to hypertrophic chondrocytes below the metaphyseal growth plates of endochondral bones (Fig. 2 B). Clearly, in transgenic mice, OPG exerts its inhibitory effects on osteoclasts during the bone elongation and growth period. Thus, normal bone elongation in these mice occurs despite the early developmental onset of osteopetrosis.
Figure 2.

(A) TRAP staining of femoral metaphysis including the growth plate (black arrows) from E15.5, E17.5, and 1-d-old wild-type (WT) mice or high OPG expressing transgenic mice (OPG). Yellow arrowheads indicate TRAP+ osteoclasts. B, bone; M, marrow. (B) The number of osteoclasts per millimeter of growth plate length was quantitated in the metaphyseal region of both tibias at various ages (E15.5, E16.5, E17.5, day 1, day 4, week 1, week 2, and week 4). Osteoclasts considered for measurement were TRAP stain-positive, in contact with a bone surface, and within the margin of the hypertrophic cartilage zone.
Electron microscopy analysis of osteoclast structure provided insight into the osteopetrosis observed in OPG transgenic mice. Analysis of ultrastructural features of osteoclasts on the femoral endosteal bone surface indicates that trabecular osteoclasts in OPG transgenic mice show no evidence of active bone resorption or interaction with the bone surface (Fig. 3, compare A with B). Endosteal osteoclasts in OPG transgenic mice show no evidence of ruffled borders, Howship's lacunae, or vacuoles, all of which are evident in multinucleated osteoclasts actively resorbing bone in control mice. Mitochondria and lysosomes are abundant in the transgenic osteoclasts, and in this regard are similar to the activated osteoclasts seen in control mice. Interestingly, osteoclasts are abundant in OPG transgenic mice on periosteal bone surfaces adjacent to the growth plate, where active modeling or sculpting of bone occurs. In addition, osteoclasts on the periosteal surface of cortical bone show signs of active bone resorption (Fig. 3 C), indicating that OPG transgenic mice have a specific defect in endosteal osteoclast development. This finding is consistent with the previous observations that local levels of OPGL on the periosteal bone surface are relatively high compared with levels of OPGL on endosteal trabecular bone 8. The levels of OPG expression in these transgenic mice may be sufficient to block OPGL/RANK interactions in the endosteal envelope, yet may not be sufficient to block the high levels of OPGL present on periosteal bone surfaces, even in the highest expressing mice.
Figure 3.

(A) Activated osteoclast in the process of resorbing trabecular bone in the region of the diaphyseal–epiphyseal junction of the proximal femur of a wild-type mouse. The asterisks indicate lysosomal vacuoles, and the arrows indicate the formation of a ruffled border. (B) Resting or inactive osteoclast adjacent to trabecular bone from the region of the diaphyseal-epiphyseal junction of the proximal femur of an OPG transgenic mouse. (C) Activated osteoclast in a resorption lacuna on the periosteal femoral cortical bone of OPG transgenic mice. Note the formation of lysosomal vacuoles and a ruffled border. Bars: A and B, 2.5 μm; C, 5.0 μm.
Rescue of the Osteoporosis in OPG Knockout Mice.
In contrast to the phenotype of OPG transgenic mice, OPG−/− mice develop severe osteoporosis associated with increased number of osteoclasts 4. Osteoblast surfaces are also increased, indicating an increase in bone turnover rate that is presumably secondary and compensatory for the increased bone resorption. Interestingly, approximately two-thirds of OPG null mice also develop calcified lesions in the aorta and renal arteries 4. Since OPG mRNA expression is detected in bones as well as major arteries, we used two methods to investigate whether circulating OPG can rescue the bone and arterial phenotypes of OPG knockout mice. First, OPG transgenic mice were bred with OPG knockout mice to test the prophylactic capabilities of systemic OPG. We also tested the effects of OPG after the onset of osteoporosis and arterial calcification by injection of recombinant OPG protein into 8-wk-old OPG−/− mice.
High expressing OPG transgenic mice were bred into the OPG null background. Histological analysis of the metaphyseal region of the femur shows that 8-wk-old OPG−/− mice exhibit severe osteoporosis characterized by a significant decrease in trabecular bone density compared with wild-type mice (Fig. 4, compare A and B). In contrast, transgene-expressing OPG−/− mice exhibit significant increases in trabecular bone density (Fig. 4 C), close to the severity of osteopetrosis observed in high expressing OPG transgenic mice on the wild-type background (Fig. 1). The increase in bone density is also apparent on the whole body radiographs of mice from different groups (data not shown). The bone density in each group was quantified by pQCT measurements 18. Total bone density in OPG−/− mice is 353 ± 29 mg/cm3 versus 426 ± 39 mg/cm3 in wild-type mice (P < 0.005). OPG−/− mice bearing the transgene showed significantly increased total bone density of 637 ± 59 mg/cm3 (P < 0.005), which is comparable to bone density observed in wild-type mice bearing the transgene 4. These data indicate that circulating OPG can increase overall bone density independent of endogenous OPG expression.
Figure 4.

(A–C) Hematoxylin and eosin stained sections of the distal femur of (A) wild-type, (B) OPG−/− mice, and (C) OPG−/− mice bearing OPG transgene. OPG−/− mice (B) show osteoporotic bone structure and lack of secondary spongiosa compared to wild-type (A). Osteopetrosis occurs in OPG−/− mice bearing OPG transgene (C). (D–F) Polarized light microscopy of the cortical bone in mid-femoral diaphysis in wild-type mice (D), OPG−/− mice (E), and OPG−/− mice bearing OPG transgene (F). Note the woven bone in OPG−/− mice (E), a characteristic of high bone turnover rate, compared to highly lamellar bone of OPG−/− mice bearing OPG transgene (F). (G–I) TRAP-stained sections of wild-type mice(G), OPG−/− mice (H), and OPG−/− mice bearing OPG transgene (I). Note the significant increase in osteoclasts (black arrows) in OPG−/− mice (H) compared with OPG−/− mice bearing OPG transgene (I). The visible increase in osteoblasts (yellow arrowheads) indicates a compensatory increase in bone formation in OPG−/− mice.
Increased bone density in OPG−/− mice bearing the transgene can be attributable to decreased bone resorption. Polarization microscopy of the femoral cortical bones of these mice showed a highly lamellar collagen deposition pattern, consistent with a slow remodeling rate (Fig. 4 F). This is in contrast to the woven bone observed in OPG−/− mice. The rate of bone turnover is also reflected in osteoclast and osteoblast numbers (data not shown). Histological staining reveals decreased levels of osteoclasts and osteoblasts in OPG−/− mice bearing the transgene compared with OPG−/− mice (Fig. 4H and Fig. I). Thus, systemic OPG expression starting from E12 prevents the onset of the osteoporotic phenotype of OPG−/− mice by decreasing the rate of bone resorption, and also results in bone of more ordered lamellar structure by reducing bone remodeling.
To test effects of systemic OPG after onset of the osteoporotic phenotype, recombinant human OPG–Fc fusion protein was injected intravenously into 8-wk-old OPG−/− mice. These mice were treated at a dose of 50 mg/kg per day, three times per week, for 4 wk. The OPG-injected knockout mice show a significant increase in bone density compared with mock-treated OPG−/− mice as indicated by radiographic analysis (Fig. 5A–D). As controls, wild-type mice were also injected either with recombinant OPG or with PBS solution. The increased bone density was most prominent in the metaphysis of long bones, sites of growth-related remodeling, but was also evident in the shafts of long bones. pQCT analysis at the proximal tibial metaphysis showed that trabecular bone density in the OPG injected OPG−/− mice is 641 ± 42 mg/cm3 versus 238 ± 16 mg/cm3 in the mock-treated OPG−/− mice (Fig. 5E, Fig. F, and Fig. I). The wild-type control groups showed trabecular densities of 394 ± 39 mg/cm3 in mock-treated versus 639 ± 51 mg/cm3 in OPG-treated mice (Fig. 5G–I). The increased bone density in OPG-injected mice can be correlated with decreased bone resorption as osteoclast numbers are markedly decreased. The rate of bone formation also is very likely reduced, as osteoblast numbers are significantly lower in OPG-injected groups than in mock-treated groups (Fig. 5 J). The serum ALP level in OPG−/− mice is elevated to ∼400 IU/liter compared with 100–150 IU/liter in wild-type mice, probably due to increased osteoblast activity in OPG−/− mice (Fig. 5 K). After commencement of treatment with recombinant OPG in OPG−/− mice, serum ALP levels decreased to 158 IU/liter within 3 d, and further injections caused an additional decrease in ALP to wild-type levels. Clearly, systemic OPG can decrease bone resorption and reverse the osteoporotic phenotype of OPG−/− mice, and this is associated with a decrease in the rate of bone turnover.
Figure 5.
4-wk treatment of 8-wk-old OPG−/− and wild-type mice with human OPG–Fc fusion protein (50 mg/kg, three times per week) increases bone density. (A–D) Radiographs of the hind limbs of OPG−/− (A and B) and wild-type (C and D) mice treated with either vehicle (A and C) or OPG–Fc protein (B and D). Yellow arrows indicate area where increase in bone density is apparent after OPG treatment. (E–H) Computed tomographic images of OPG −/− (E and F) and wild-type (G and H) mice generated from a 1-mm cross-section of bone 2 mm distal to the proximal articular surface of the tibia. White indicates the areas of greatest bone mineral content. Note the increase in the bone mineral content in OPG–Fc–treated mice (F and H) compared with vehicle-treated mice (E and G). (I) Bone density, as determined by pQCT of OPG−/− and wild-type mice, is increased in the proximal tibia after 4 wk of OPG treatment. (J) Osteoclast number per mm2 bone (NOc/BA) and osteoblast surface relative to bone surface (ObS%BS) are increased in OPG−/− mice compared with wild-type controls. Treatment of these mice with OPG–Fc for 4 wk suppresses these markers of bone metabolism to subphysiologic levels. (K) Serum ALP levels in OPG−/− mice are elevated, indicating the systemic nature of the increased bone turnover. 3 d after beginning treatment with OPG–Fc protein, serum ALP levels in OPG−/− mice fall to within the normal range and remain decreased for the duration of the treatment period. (Mean ± SEM, asterisk indicates P < 0.01 compared with OPG−/−; No. symbol indicates P < 0.01 compared with wild-type.)


Effects of OPG on the Calcified Lesions of OPG Knockout Mice.
Previously, we showed that approximately two-thirds of OPG−/− mice develop calcified lesions in large arteries, including the aorta and renal arteries 4. In recombinant OPG injection experiments, 9 out of 14 OPG−/− mice injected with recombinant protein still showed incidence of aortic calcification (Fig. 6 C). 9 out of 13 OPG−/− mice in the PBS-treated control group exhibited calcified lesions in the aorta (Fig. 6 B). Thus, 4 wk of continuous treatment of OPG did not reduce the incidence of aortic calcification. In contrast, among eight OPG−/− mice expressing the ApoE–OPG transgene, none showed any calcification in major arteries (Fig. 6 F). Five out of eight OPG−/− mice from the same litters showed calcified lesions as expected (Fig. 6 E). Taken together, these results indicate that circulating OPG has preventative, but not reversing effects on the formation of arterial calcified lesions. Timing of OPG administration could influence the severity of arterial calcification.
Figure 6.

(A–C) Effects of recombinant OPG–Fc protein on calcified aorta of OPG−/− mice. Hematoxylin and eosin–stained sections of the descending thoracic aorta of (A) age-matched wild-type mice, (B) OPG−/− mice treated with PBS, and (C) OPG−/− mice treated with OPG–Fc (50 mg/kg three times per week) are shown. Calcified lesions are indicated by black arrowheads and dark purple staining. Note that protein therapy with OPG–Fc does not have any effect on arterial calcification. (D–F) Effects of transgenic OPG expression on aorta of OPG−/− mice. Aortic sections of (D) wild-type mice, (E) OPG−/− mice, and (F) OPG−/− mice bearing OPG transgene. Note the absence of the calcified lesions in F.
Closer examination of OPG knockout mice revealed the presence of large multinucleated cells associated with calcified arteries (Fig. 7 D). Interestingly, in situ analysis showed that RANK is expressed in these cells, whereas little or no RANK expression was observed in uncalcified regions of arteries of OPG−/− mice and in arteries of wild-type mice (Fig. 7, compare F with C). OPGL expression is also detected above background in this region, possibly on cells with close proximity to the large multinucleated cells (Fig. 7, compare E with B). Since OPGL and RANK have been shown to be expressed by cells of the osteoblast and osteoclast lineages, respectively, we were curious about the nature of cells in calcified areas of the OPG−/− arterial wall. Calcified lesions have been shown to contain cells expressing many osteoblast markers, including hydroxyapatite, osteocalcin, collagen type I, and osteopontin 19 20 21 22 23 24. However, the presence of osteoclasts or their equivalent in calcified lesions of the arterial wall has not been well characterized to date. Many multinucleated cells associated with RANK expression within the calcified lesions of OPG−/− arteries were TRAP+ and Cathepsin K+ (Fig. 8A–C). These TRAP+ and Cathepsin K+ multinuclear cells were negative for F4/80, a macrophage marker (data not shown). Thus, these multinucleated cells behave very similar to mature osteoclasts. The calcification-associated cell mass contained other cell types as well. Some multinucleated cells were negative for TRAP, Cathepsin K, and F4/80 stains and thus could not be identified (Fig. 8 D). However, CD3+ T cells and F4/80+ macrophages were present, indicating an inflammatory response (Fig. 8E–H).
Figure 7.
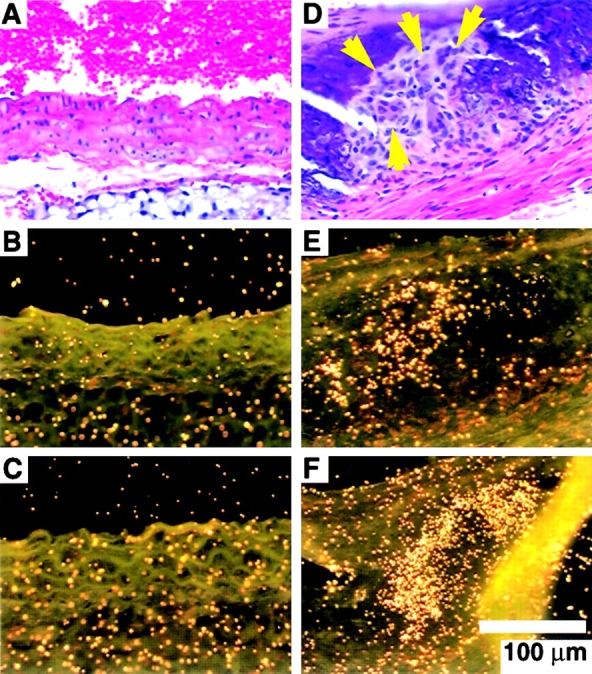
The expression of OPGL and RANK mRNAs near calcified lesions by in situ hybridization. (A–C) Normal appearance of and OPGL/RANK expression in murine aortic tissue. (A) Hematoxylin and eosin–stained section of smooth muscle in the aortic wall of wild-type mice. (B) OPGL and (C) RANK are not normally expressed in adult murine aortas. (D–F) Calcified lesion in OPG −/− mice associated with large multinuclear cells (yellow arrows) and OPGL/RANK mRNA expression. (D) Hematoxylin and eosin–stained section of calcified lesion in the aortic wall of an OPG −/− mouse. (E) OPGL and (F) RANK mRNAs are detectable by in situ hybridization in the calcified regions where multinucleated cells are present (D).
Figure 8.

Immunohistochemistry showing the presence of osteoclast-like cells (Cathepsin K+ and TRAP+) and T cell infiltrates (CD3+) in calcified lesions of OPG −/− mice. Large multinuclear cells (A–D, yellow arrows associated with calcified lesions are apparent with hematoxylin and eosin staining (A). Some of these cells stain positive with a Cathepsin K antibody (B) and TRAP stain (C). (D) Other large multinuclear cells are not positive for Cathepsin K, TRAP, or F4/80 macrophage marker (Cathepsin K immunohistochemistry is shown), thus the identity of these cells could not be determined. (E–H) Calcified regions of OPG−/− aortas are also associated with infiltrates of inflammatory cells that are apparent with (E) hematoxylin and eosin staining. These infiltrates primarily consist of CD3+ T cells (F) with a few F4/80+ macrophages (G, black arrow). These cells did not overlap with Cathepsin K+ cells (H).
Discussion
Transgenic mice expressing OPG under the control of the human apolipoprotein E gene promoter and its liver-specific enhancer exhibit severe osteopetrosis 1. Circulating OPG in these mice functions by inhibiting osteoclast differentiation from hematopoietic monocyte/macrophage precursor cells and by inhibiting activation of existing osteoclasts. Other mouse osteopetrotic models characterized to date contain loss-of-function mutations that affect osteoclast development or function (for review see reference 16). A major phenotypic difference between OPG transgenic mice and other osteopetrotic models is the absence of impaired tooth eruption and “clubbing” of long bones, which occur in all the mutant models.
Analysis of three transgenic lines that overexpress OPG reveal that the severity of osteopetrosis correlates directly with serum OPG levels. High expressing lines exhibited the most drastic phenotype with no discernible open marrow space in the midshaft region of long bones. In the two highest expressing lines, the histological appearance of endochondral growth plates paralleled those in classical osteopetrosis, being characterized by the presence of cartilage remnants from the primary spongiosa seen encased in bone in the diaphysis of the femur. It is not likely that the phenotypic differences that are observed between OPG transgenic mice and other osteopetrotic models are due to a difference in severity of osteopetrosis. Onset of osteopetrosis in OPG transgenic mice is evident at E17.5 and progresses during the postnatal bone growth and elongation period, indicating that temporal exertion of OPG's osteoclast inhibitory effect also is not responsible for the phenotypic differences. Electron microscopy analysis revealed an important difference in the osteoclast populations of OPG transgenic mice. The few trabecular osteoclasts present in the OPG transgenic mouse bones appear morphologically inactive, lacking vacuolated spaces and failing to form resorptive pits on the bone surface. In contrast, osteoclasts at the periosteal bone surface were numerous and exhibited active signs of bone resorption.
The most likely explanation for the differences between endosteal trabecular osteoclasts and periosteal osteoclasts is that transgenic delivery of OPG to the periosteal envelope is insufficient to completely block OPGL on periosteal bone, where high levels of OPGL interact in a juxtacrine fashion with RANK on osteoclast precursors. In contrast to OPG transgenic mice, other mouse osteopetrosis models, such as op/op mice (CSF-1−/−), microphthalmic mice (mi/mi), and c-src −/− and c-fos −/− mice, contain loss-of-function mutations, and all osteoclasts in both the endosteal and periosteal envelopes are defective in different stages of differentiation and activation (for review see references 15 and 16). Alternatively, the periosteal osteoclasts in OPG transgenic mice may be functional because OPG is specific only for a subpopulation of osteoclasts resident in the endosteal envelope. In this scenario, OPG could control the specific regulatory pathway in trabecular endosteal osteoclasts and thus bypass the stimulation of bone resorption for tooth eruption and periosteal remodeling of the long bones. However, OPGL-deficient mice have been generated recently and exhibit a classic osteopetrosis phenotype with defective tooth eruption and shortened and club-shaped long bones due to complete lack of mature osteoclasts 25. These results suggest that the pathway regulated by OPG affects the differentiation of all, rather than a subpopulation of, osteoclasts. The unique features of the osteopetrosis observed in the OPG transgenic mice may result from differences in the ability of systemic OPG to effectively inhibit local concentrations of OPGL on different bone surfaces.
Consistent with osteopetrosis observed in OPG transgenic mice, the OPG−/− mice exhibited severe osteoporosis characterized by trabecular and cortical bone porosity, thinning of parietal bones, and a high incidence of fractures 4. Unexpectedly, OPG−/− mice also exhibited an increase in vascular calcification in the aorta and renal arteries. Since OPG is expressed in bone as well as major arteries, it was not clear what effect circulating OPG would have on the osteoporosis and calcified lesions observed in OPG−/− mice. We show that high levels of circulating OPG expressed as a transgene on the OPG null background resulted in osteopetrosis. Injection of recombinant OPG into osteoporotic OPG−/− mice resulted in an increase in trabecular bone associated with a decreased bone turnover rate. These results suggest that circulating OPG can be used as a therapeutic agent to treat osteoporosis before and after the onset of osteolytic disease. Recently, OPG was shown to protect against bone loss caused by various resorptive hormones and cytokines, such as IL-1β, TNF-α, parathyroid hormone (PTH), parathyroid hormone related peptide (PTHrp), and 1α,25-dihydroxyvitamin D3 17. All of these factors are implicated in human bone disease associated with increased osteoclast activity, such as postmenopausal osteoporosis, localized bone loss in rheumatoid arthritis, osteoitis fibrosa associated with renal failure, and hypercalcemia in hyperparathyroidism, sarcoid, and cancer. Hence, OPG could have utility in multiple clinical indications as a therapeutic agent against bone loss.
The aorta and renal arteries are sites of endogenous OPG expression, and it is these arteries that develop calcified lesions in OPG-deficient mice 4. These arteries are also the sites most frequently exhibiting calcification in human patients with atherosclerosis. However, the calcification in OPG knockout mouse arteries occurs in the absence of fat deposition and other vascular lesions commonly seen in atherosclerosis patients. Furthermore, although pathological calcification of arteries occurs late in the atherosclerotic process, the onset of arterial calcification is early in the OPG knockout mice. The rescue experiments show that expression of OPG as a transgene on an OPG null background prevented the onset of arterial calcification, indicating that systemic OPG plays a protective role in major arteries. However, injection of a high dose of recombinant OPG into adult OPG−/− mice did not affect the incidence of arterial calcification, indicating that OPG cannot reverse the calcification process. It is not clear from our data whether the size of calcified lesions stopped increasing in the OPG-injected group. Delivery of OPG to younger animals, or treatment for >4 wk, will be necessary to determine whether OPG can stop further progression of vascular calcification.
Areas of arterial calcification have been reported to show similar morphological characteristics to lamellar bones, such as trabeculae, lacunae, and islands of marrow 26 27. Calcified lesions have also been shown to contain many of the bone matrix proteins, such as collagen type I, osteocalcin, osteonectin, osteopontin, matrix γ-carboxyglutamic acid (GLA) protein, and bone morphogenic protein 2 20 21 22. Watson et al. have previously defined a population of aortic cells that spontaneously calcify in vitro, which they termed calcifying vascular cells 28. These cells produce osteoblast-specific markers, including osteocalcin and hydroxyapatite. Consistent with these observations, cells in the calcified lesions of OPG−/− arteries express OPGL, a molecule expressed on osteoblastic stroma 8. Although these cells could represent calcifying vascular cells, we cannot rule out the possibility that the OPGL+ cells detected in the arteries of OPG−/− mice are T cells or some other cell type. The presence of functional osteoclasts in calcified arteries has been questionable thus far. At least some multinucleated cells in the calcified arteries of OPG−/− mice are positive for TRAP stain as well as for Cathepsin K, and negative for F4/80, which is typical of osteoclasts. Furthermore, RANK, which is expressed in osteoclasts and their precursors 14, was also detected by in situ in the cell mass associated with calcified lesions. These findings indicate the presence of osteoclasts or osteoclast-like cells in the calcified lesions, suggesting that vascular calcification may result in a process involving cellular mechanisms similar to those exhibited during physiological calcification processes.
Genetic studies using inbred mice have suggested that multiple genes are involved in formation of aortic calcification 29. Several loss-of-function mutations in both human and mouse extracellular matrix (ECM) protein genes suggest that ECM calcification is a passive process and “active inhibition” is necessary to avoid pathological calcification (for review see reference 30). The most prominent example is the matrix GLA protein–deficient mice. Unlike the OPG−/− mice, these mice show extensive calcifications not only in major arteries but also in muscular arteries and cartilage 31. Although OPG is not an extracellular matrix protein, it does seems to be involved in active inhibition of calcified lesion formation in arteries. Not only endogenous OPG but also systemic OPG in the OPG−/− background can actively inhibit calcification in arteries. OPG therapy in the osteoporotic population may potentially lower the incidence of arterial calcification.
Footnotes
Abbreviations used in this paper: ALP, alkaline phosphatase; E, embryonic day; OPG, osteoprotegerin; OPGL, osteoprotegerin ligand; pQCT, peripheral quantitative computed tomography; RANK, receptor activator of NF-κB; TRAP, tartrate-resistant acid phosphatase.
References
- Simonet W.S., Lacey D.L., Dunstan C.R., Kelley M., Chang M.S., Luethy R., Nguyen H.Q., Wooden S., Bennett L., Boone T. Osteoprotegerina novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–319. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- Yasuda H., Shima N., Nakagawa N., Mochizuki S., Yano K., Fujise N., Sato Y., Goto M., Yamaguchi K., Kuriyama M. Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG)a mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology. 1998;139:1329–1337. doi: 10.1210/endo.139.3.5837. [DOI] [PubMed] [Google Scholar]
- Tan K.B., Harrop J., Reddy M., Young P., Terrett J., Emery J., Moore G., Truneh A. Characterization of a novel TNF-like ligand and recently described TNF ligand and TNF receptor superfamily genes and their constitutive and inducible expression in hematopoietic and non-hematopoietic cells. Gene. 1997;204:35–46. doi: 10.1016/s0378-1119(97)00509-x. [DOI] [PubMed] [Google Scholar]
- Bucay N., Sarosi I., Dunstan C.R., Morony S., Tarpley J., Capparelli C., Scully S., Tan H.L., Xu W.L., Lacey D.L. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998;12:1260–1268. doi: 10.1101/gad.12.9.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno A., Amizuka N., Irie K., Murakami A., Fujise N., Kanno T., Sato Y., Nakagawa N., Yasuda H., Mochizuki S. Severe osteoporosis in mice lacking osteoclastogenesis inhibitory factor/osteoprotegerin. Biochem. Biophys. Res. Commun. 1998;247:610–615. doi: 10.1006/bbrc.1998.8697. [DOI] [PubMed] [Google Scholar]
- Pacifici R. Estrogen, cytokines, and pathogenesis of post-menopausal osteoporosis. J. Bone Miner. Res. 1996;11:1043–1051. doi: 10.1002/jbmr.5650110802. [DOI] [PubMed] [Google Scholar]
- Parhami F., Demer L.L. Arterial calcification in face of osteoporosis in ageingcan we blame oxidized lipids? Curr. Opin. Lipidol. 1997;8:312–314. doi: 10.1097/00041433-199710000-00010. [DOI] [PubMed] [Google Scholar]
- Lacey D.L., Timms E., Tan H.L., Kelley M.J., Dunstan C.R., Burgess T., Elliott R., Colombero A., Elliott G., Scully S. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- Wong B.R., Rho J., Arron J., Robinson E., Orlinick J., Chao M., Kalachikov S., Cayani E., Bartlett F., Sr., Frankel W.N. TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J. Biol. Chem. 1997;272:25190–25194. doi: 10.1074/jbc.272.40.25190. [DOI] [PubMed] [Google Scholar]
- Anderson D.M., Maraskovsky E., Billingsley W.L., Dougall W.C., Tometsko M.E., Roux E.R., Teepe M.C., DuBose R.F., Cosman D., Galibert L. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. 1997;390:175–179. doi: 10.1038/36593. [DOI] [PubMed] [Google Scholar]
- Yasuda H., Shima N., Nakagawa N., Yamaguchi K., Kinosaki M., Mochizuki S., Tomoyasu A., Yano K., Goto M., Murakami A. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. USA. 1998;95:3597–3602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimi E., Akiyama S., Tsurukai T., Okahashi N., Kobayashi K., Udagawa N., Nishihara T., Takahashi N., Suda T. Osteoclast differentiation factor acts as a multifunctional regulator in murine osteoclast differentiation and function. J. Immunol. 1999;163:434–442. [PubMed] [Google Scholar]
- Nakagawa N., Kinosaki M., Yamaguchi K., Shima N., Yasuda H., Yano K., Morinaga T., Higashio K. RANK is the essential signaling receptor for osteoclast differentiation factor in osteoclastogenesis. Biochem. Biophys. Res. Commun. 1998;253:395–400. doi: 10.1006/bbrc.1998.9788. [DOI] [PubMed] [Google Scholar]
- Hsu H., Lacey D.L., Dunstan C.R., Solovyev I., Colombero A., Timms E., Tan H.L., Elliott G., Kelley M.J., Sarosi I. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc. Natl. Acad. Sci. USA. 1999;96:3540–3545. doi: 10.1073/pnas.96.7.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filvaroff E., Derynck R. Bone remodellinga signalling system for osteoclast regulation. Curr. Biol. 1998;8:679–682. doi: 10.1016/s0960-9822(98)70434-8. [DOI] [PubMed] [Google Scholar]
- Felix R., Hofstetter W., Cecchini M.G. Recent developments in the understanding of the pathophysiology of osteopetrosis. Eur. J. Endocrinol. 1996;134:143–156. doi: 10.1530/eje.0.1340143. [DOI] [PubMed] [Google Scholar]
- Morony S., Capparelli C., Lee R., Shimamoto G., Boone T., Lacey D.L., Dunstan C.R. A chimeric form of osteoprotegerin inhibits hypercalcemia and bone resorption induced by IL-1beta, TNF-alpha, PTH, PTHrP, and 1, 25(OH)2D3. J. Bone Miner. Res. 1999;14:1478–1485. doi: 10.1359/jbmr.1999.14.9.1478. [DOI] [PubMed] [Google Scholar]
- Beamer W.G., Donahue L.R., Rosen C.J., Baylink D.J. Genetic variability in adult bone density among inbred strains of mice. Bone. 1996;18:397–403. doi: 10.1016/8756-3282(96)00047-6. [DOI] [PubMed] [Google Scholar]
- Schmid K., McSharry W.O., Pameijer C.H., Binette J.P. Chemical and physicochemical studies on the mineral deposits of the human atherosclerotic aorta. Atherosclerosis. 1980;37:199–210. doi: 10.1016/0021-9150(80)90005-2. [DOI] [PubMed] [Google Scholar]
- Bostrom K., Watson K.E., Horn S., Wortham C., Herman I.M., Demer L.L. Bone morphogenetic protein expression in human atherosclerotic lesions. J. Clin. Invest. 1993;91:1800–1809. doi: 10.1172/JCI116391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giachelli C.M., Bae N., Almeida M., Denhardt D.T., Alpers C.E., Schwartz S.M. Osteopontin is elevated during neointima formation in rat arteries and is a novel component of human atherosclerotic plaques. J. Clin. Invest. 1993;92:1686–1696. doi: 10.1172/JCI116755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien K.D., Kuusisto J., Reichenbach D.D., Ferguson M., Giachelli C C., Alpers E., Otto C.M. Osteopontin is expressed in human aortic valvular lesions. Circulation. 1995;92:2163–2168. doi: 10.1161/01.cir.92.8.2163. [DOI] [PubMed] [Google Scholar]
- Ginsberg B., van Haarlem L.J.M., Soute B.A.M., Ebberink R.H.M., Vermeer C. Characterization of a GLA-containing protein from calcified human atherosclerotic plaques. Arteriosclerosis. 1990;10:991–995. doi: 10.1161/01.atv.10.6.991. [DOI] [PubMed] [Google Scholar]
- Katsuda S., Okada Y., Minamoto T., Oda Y., Matsui Y., Nakanishi I. Collagens in human atherosclerosis. Immunohistochemical analysis using collagen type-specific antibodies. Arterioscler. Thromb. 1992;12:494–502. doi: 10.1161/01.atv.12.4.494. [DOI] [PubMed] [Google Scholar]
- Kong Y.Y., Yoshida H., Sarosi I., Tan H.L., Timms E., Capparelli C., Morony S., Oliveira-dos-Santos A.J., Van G., Itie A. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–323. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- Bunting C.H. The formation of true bone with cellular (red) marrow in a sclerotic aorta. J. Exp. Med. 1906;8:365–376. doi: 10.1084/jem.8.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haust M.D., Geer J.C. Mechanism of calcification in spontaneous aortic arteriosclerotic lesions of the rabbit. An electron microscopic study. Am. J. Pathol. 1970;60:329–346. [PMC free article] [PubMed] [Google Scholar]
- Watson K.E., Bostroem K., Ravindranath R., Lam T., Norton B., Demer L.L. TGF-beta 1 and 25-hydroxycholesterol stimulate osteoblast-like vascular cells to calcify. J. Clin. Invest. 1994;93:2106–2113. doi: 10.1172/JCI117205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao J.H., Xie P.Z., Fishbein M.C., Kreuzer J., Drake T.A., Demer L.L., Lusis A.J. Pathology of atheromatous lesions in inbred and genetically engineered mice. Genetic determination of arterial calcification. Arterioscler. Thromb. 1994;14:1480–1497. doi: 10.1161/01.atv.14.9.1480. [DOI] [PubMed] [Google Scholar]
- Schinke T., McKee M.D., Karsenty G. Extracellular matrix calcificationwhere is the action? Nat. Genet. 1999;21:150–151. doi: 10.1038/5928. [DOI] [PubMed] [Google Scholar]
- Luo G., Ducy P., McKee M.D., Pinero G.J., Loyer E., Behringer R.R., Karsenty G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78–81. doi: 10.1038/386078a0. [DOI] [PubMed] [Google Scholar]