

Abstract
CD1b and CD1c are antigen-presenting molecules that mediate recognition of bacterial lipids by T cells, but it is currently not known whether these two molecules are redundant or are specialized to perform different immunological functions. Here, we show that the distribution of CD1c in human dendritic cells was characterized by a high ratio of cell surface to intracellular molecules, whereas CD1b showed a reciprocal pattern of distribution. In contrast to the accumulation of CD1b in lysosomal major histocompatibility complex class II compartments, intracellular CD1c molecules accumulated in other endocytic compartments, most likely early and late endosomes. Deletion of the cytoplasmic tail of CD1c, containing a tyrosine-based internalization motif, abolished most of its intracellular localization. Functional studies using T cells specific for defined lipid antigens revealed that in contrast to CD1b-mediated antigen presentation, antigen presentation by CD1c was resistant to drugs inhibiting endosomal acidification and was independent of endosomal localization of CD1c. Taken together, these results support the hypothesis that CD1b and CD1c are specialized to survey the lipid content of different intracellular compartments.
Keywords: CD1, Mycobacterium tuberculosis, antigen presentation, intracellular trafficking, lipids
Introduction
The human CD1 locus encodes five nonpolymorphic CD1 genes, four of which are known to encode expressed proteins related in structure and evolution to MHC class I molecules 1. These proteins, designated CD1a, CD1b, CD1c, and CD1d, are expressed on the surfaces of dendritic cells (DCs), and all have been implicated in the presentation of specific lipid and glycolipid ligands to T cells 2 3 4 5. Several mycobacterial lipids presented by CD1b have been identified 6 7, and recently the first lipid presented by CD1c has also been characterized as a mycobacterial glycolipid 8. T cell responses to a semisynthetic analogue of this CD1c-presented lipid were elevated in the blood of human subjects previously infected with Mycobacterium tuberculosis compared with naive control subjects, underscoring the potential importance of the CD1 system in the immune response to pathogens 8. The genomes of a variety of mammals, including humans, sheep, rabbits, guinea pigs, and rhesus macaques, contain multiple copies of CD1 genes that encode diversified forms of CD1 genes 6. The forces driving and maintaining this diversification of CD1 genes in various mammals are not known, but we have previously speculated that this may be due at least in part to the specialization of different CD1 proteins to perform subtly but significantly different roles in the immune response to pathogens encountered by these different species 9.
To evaluate the hypothesis of functional specialization of CD1 isoforms, we have studied the intracellular distribution and antigen-presenting properties of the human CD1c protein compared with the previously documented features of CD1b. Studies on the intracellular localization of CD1b demonstrate that this protein is prominently found in late endosomal and lysosomal compartments 10. Localization of CD1b to these intracellular compartments is dependent on its nine–amino acid cytoplasmic tail, which contains a tetrapeptide that conforms to a tyrosine-based targeting motif denoted as YxxZ (where Y is tyrosine, x represents any amino acid, and Z is a bulky hydrophobic amino acid) 11. The localization of CD1b to late endosomes and lysosomes is consistent with the finding that presentation of lipid antigens by CD1b is in most cases highly sensitive to agents that interfere with normal endosomal acidification 12. In the current study, we demonstrate significantly different intracellular localization and antigen presentation properties for CD1b and CD1c. Our results provide support for the hypothesis that the diversification of CD1 proteins into multiple isoforms reflects their specialization for presentation of different subsets of antigens accumulating in distinct intracellular compartments. This may help to explain the extensive duplication and diversification of CD1 proteins observed in humans and other mammalian species.
Materials and Methods
Cell Lines, Antigens, and Antibodies.
T cell lines DN1 12, CD8.1 2 8, and SP-F14 13 have been described. An extract of total lipids of M. tuberculosis strain H37Ra was prepared as described previously 2 4. Tetanus toxoid was obtained from the Massachusetts Department of Public Health Laboratory (Jamaica Plain, MA). The 15-mer tetanus toxoid peptide used in this study corresponded to residues 947–961 of tetanus toxin (FNNFTVSFWLRVPKV). HeLa cells were obtained from the American Type Culture Collection. C1R cells were obtained from P. Cresswell (Yale University, New Haven, CT). Immature monocyte-derived DCs were obtained by culturing the adherent fraction of normal human PBMCs in the presence of GM-CSF and IL-4 for 3 d 5. Antibodies used in this study that have been described previously 11 14 were BCD1b3 (anti-CD1b, IgG1), F10/21A3 (anti-CD1c, IgG1), anti-CD1b rabbit antiserum R3, P3 (IgG1 control), and the rabbit anti–human lysosome-associated membrane protein 1 (Lamp-1) antiserum. Other antibodies used were L161 (anti-CD1c, IgG1; Immunotech), W6/32 (anti–HLA-A,-B,-C; American Type Culture Collection), L243 (anti–HLA-DR; American Type Culture Collection), HC-10 (anti–HLA-B,-C; gift of Dr. H. Ploegh, Harvard Medical School, Boston, MA), and H68.4 (anti-transferrin receptor [TfR]; gift of Dr. S. Amigorena, Institut Curie, Paris, France). Commercially available antibodies and secondary labeling reagents used were: CY3- and FITC-conjugated goat F(ab′)2 anti–mouse Ig, CY3- or FITC-conjugated goat F(ab′)2 anti–rabbit IgG (Jackson Immunoresearch Laboratories), horseradish peroxidase (HRP)-conjugated protein A or goat anti–rabbit IgG antibody (Zymed Laboratories), rabbit anti–mouse IgG (Dako), and 10-nm gold–conjugated protein A (from G. Posthuma, University of Utrecht, Utrecht, The Netherlands).
Construction of CD1c Mutants and Transfections.
Plasmids for expression of the wild-type CD1c protein (CD1c.WT) and CD1b have been described 15. A cDNA encoding the tail-deleted form (CD1c.TD) was generated by PCR using CD1c.WT as template DNA. Primers used were 5′-CTCAACTCTACGTCTTTGTTTCG-3′ (bases ∼570–590 of the human T cell lymphotropic virus [HTLV] LTR region from the vector pSRα-NEO) and 5′-TCGGGATCCTACTTCTTAAACCATAACACAAGGAC-3′ (minus strand 3′ end of CD1c). The amplified fragment was cloned and sequenced, and the Xho1 to BamH1 region was inserted into pSRα-NEO. Stable transfectants were produced by electroporation (C1R cells) and the CaPO4 method (HeLa cells) as described 11. For transient transfection, HeLa cells were transfected with 1.5 μg of each plasmid DNA using Lipofectamine™ lipid reagent (Life Technologies).
Flow Cytometry and Microscopy Studies.
Flow cytometry and standard immunofluorescence or confocal microscopy were done as described previously 11 16. Samples were mounted for fluorescence microscopy using the ProLong™ Antifade kit (Molecular Probes).
For immunoelectron microscopy, ultrathin cryosections of immature DCs were stained with either BCD1b3 (anti-CD1b) or L161 (anti-CD1c) followed by a rabbit anti–mouse Ig antibody revealed by staphylococcal protein A conjugated to 10-nm gold particles. Procedures used for preparation of electron microscopy (EM) samples and for quantitative analysis of immunogold staining 11 have been described previously.
Subcellular Fractionation.
Immature DCs were radiolabeled with a mixture of [35S]methionine and [35S]cysteine (NEN Life Science Products) at 100 μCi/ml for 22 h. Subsequent Percoll fractionation, enzymatic assays, and immunoblotting were performed as described 17. Membranes of the Percoll fractions were treated with 200 U of PNGase F (New England Biolabs, Inc.) for 12 h to deglycosylate CD1 molecules before immunoblotting. Dilutions for antibodies were: TfR (H68.4; 1:10 dilution of hybridoma culture supernatant), MHC class I (HC-10; 1:200 ascites), Lamp-1 (93B1; 1:1,500 rabbit serum), or CD1b (R3; 1:1,000 rabbit serum). Primary antibodies were revealed by HRP-coupled protein A or goat anti–rabbit IgG antibodies (1:40,000 dilution) and incubation with the HRP substrate to induce chemiluminescence (Renaissance™; NEN Life Science Products), which was detected by autoradiography (Biomax light films; Eastman Kodak Co.). To determine the total protein distribution, 10 μl of each fraction was counted in a β-scintillation counter. For immunoprecipitations, 500 μl of the fractions was added to 1 ml of lysis buffer (0.5% Triton X-100) for 30 min, followed by a 30-min centrifugation at 14,000 rpm in a microfuge. Proteins were sequentially immunoprecipitated using the anti-CD1c (F10/21A3), anti-CD1b (BCD1b3), anti–MHC class II (L243), and anti–MHC class I (W6/32) antibodies (5 μg bound to protein G–Sepharose beads; Amersham Pharmacia Biotech). The CD1b and CD1c immunoprecipitates were treated with PNGase F (50 U) for 12 h at 37°C. Samples were boiled, reduced, and separated by SDS-PAGE. Dried gels were incubated with an MR film (Eastman Kodak Co.) and a Dupont intensifying screen at −80°C for 6 h to 3 d.
Antigen Presentation Assays.
Cytolytic T cell assays were done as described 12 18. Determination of the kinetics of antigen presentation by CD1c compared with MHC class II was done using measurement of intracellular Ca2+ mobilization as the readout for T cell activation. Immature DCs were incubated for various times with either 10 μg/ml of total lipid extract from M. tuberculosis, 0.1 mg/ml of intact tetanus toxoid protein, or 75 ng/ml of an antigenic 15-mer peptide from tetanus toxoid, and then immediately placed in an ice water bath. T cell lines (CD8.1 to assess antigen presentation by CD1c, and SP-F14 to assess presentation by MHC class II) were incubated with 1.5 μg/ml Indo-1 for 40 min at 37°C, washed, aliquoted, and applied to a flow cytometer (MO-FLO cytometer; Cytomation) for 30 s to determine the baseline fluorescence at 405 vs. 485 nm of unstimulated Indo-1–loaded T cells after excitation with an argon laser at 364 nm. Subsequently, the Indo-1–loaded T cells were quickly pelleted together in a microfuge with equal numbers of immature DCs that had been incubated at 37°C for various periods of time with antigen. After incubation of the pelleted cells together for 1.5 min at 37°C, the samples were resuspended in 500 μl of RPMI 1640 medium and applied to the flow cytometer for quantification of the increase in free [Ca2+]i as monitored by the change in the ratio of fluorescence emissions at 405 vs. 485 nm (Indo-1 index).
Results and Discussion
Different Distribution of CD1b and CD1c in Immature DCs.
Initially, the distribution of CD1b and CD1c in immature DCs derived from PBMCs was analyzed by dual immunofluorescence staining and confocal microscopy. This showed strong cell surface and weaker, scattered punctate intracellular staining for CD1c (Fig. 1 A, left). This staining pattern was the inverse of that observed for CD1b, which had strong intracellular staining and relatively weaker cell surface staining (Fig. 1 A, middle). The overlay of CD1c and CD1b staining revealed vesicles containing only CD1c (red) or CD1b (green), and also vesicles containing both CD1 proteins (yellow) (Fig. 1 A, right; arrowheads). A detailed immunofluorescence analysis was also performed on CD1b- and CD1c-transfected HeLa cells and showed that whereas CD1b mainly colocalizes with Lamp-1–positive compartments, CD1c was mainly found in compartments positive for the TfR (data not shown). To assess the subcellular distribution of CD1b and CD1c more precisely, immunogold labeling of ultrathin cryosections of immature DCs and EM were performed. CD1b was found on the plasma membrane (PM), in endosomes, and in lysosomal MHC class II compartments (MIICs; Fig. 1 B, top). Interestingly, although very well expressed at the PM, fewer endosomal structures that labeled for CD1c could be found and CD1c labeling was totally absent from MIICs (Fig. 1 B, bottom). A quantitative EM analysis revealed that the mean cell surface expression was not significantly different for CD1b and CD1c (Fig. 1 C, left), which was consistent with FACS® analysis showing equal cell surface expression of CD1b and CD1c (not shown). In contrast, the mean value for the amount of endosomal labeling (including MIIC) was about four times higher for CD1b compared with CD1c (Fig. 1 C, middle), and the mean for the ratio of endosomal to PM staining determined for each individual section revealed a similar fourfold increased level of intracellular CD1b staining relative to CD1c (Fig. 1 C, right). Altogether, these results demonstrated that the relative distributions of CD1b and CD1c in immature DCs were significantly different, suggesting different pathways of intracellular trafficking for these proteins.
Figure 1.
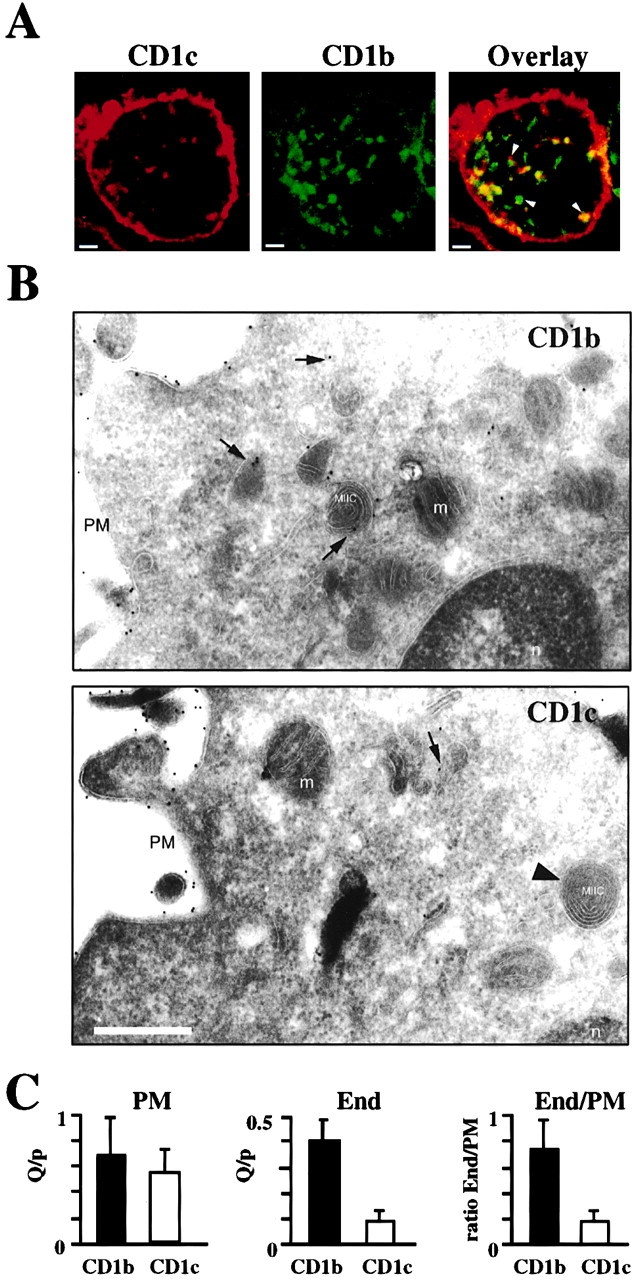
Intracellular distribution of CD1b versus CD1c in immature DCs. (A) Confocal microscopy of immature DCs that were fixed, permeabilized, and double labeled with CY3-conjugated anti-CD1c mAb (F10/21A3) and CY2-conjugated anti-CD1b mAb (BCD1b3). Single channel analysis is shown for CD1c (left) and for CD1b (middle). The overlay (right) revealed vesicles containing both CD1b and CD1c (yellow), and vesicles containing only CD1c (red) or CD1b (green) (arrowheads). Bars, 2 μm. (B) Electron micrographs of cryosections of immature DCs labeled with CD1b-specific mAb BCD1b3 (top) or CD1c-specific mAb L161 (bottom). CD1b labeling was found at the PM, in endosomes (arrows) and MIICs, but not in mitochondria (m) or nuclei (n). Expression of CD1c was restricted to PM and endosomes (arrows) and was not found in MIICs (arrowhead). Bar, 0.5 μm. (C) Quantitation of the CD1b or CD1c staining in immunoelectron micrographs. The mean PM expression (the number of gold particles [Q] over a unit length of membrane [p]; left) for CD1b and CD1c was not significantly different (P = 0.48; for CD1b, n = 11, and for CD1c, n = 10). Quantitation of endosomal (End) distribution (including lysosomes and MIICs) of the CD1 proteins in the same samples revealed fourfold more CD1b labeling compared with CD1c (P = 0.0001; middle). In addition, for each individual section the ratio of endosomal to PM staining (End/PM) was determined (right), and again the mean of these ratios was three- to fourfold higher for CD1b compared with CD1c (P = 0.0002). The plasma membrane gold particle count was 404 for CD1b and 310 for CD1c labeling on surface areas of 615 and 544 U, respectively. Similarly, endosomal gold particles counted were 231 for CD1b and 27 for CD1c on surface areas of 535 and 262 U, respectively.
To analyze this difference in distribution on a larger population of cells, we used a biochemical approach based on subcellular fractionation by Percoll gradients to isolate MIICs. Since no antibody for detection of CD1c by immunoblotting was available, the distribution of CD1c in these Percoll fractions was determined by specific immunoprecipitation. Therefore, immature DCs were labeled for 22 h with radioactive methionine/cysteine to achieve steady state labeling of cellular proteins. Initial characterization of the Percoll gradients was carried out using enzymatic assays or Western immunoblotting to localize specific markers of various subcellular compartments, which demonstrated that lysosomal compartments were efficiently separated from the PM, early and late endosomes, and the endoplasmic reticulum (Fig. 2 A). Next, the distribution of CD1b and CD1c in the Percoll gradient fractions was analyzed and compared with MHC class I and MHC class II by immunoprecipitation. This revealed that the distribution of CD1b was similar to that of MHC class II molecules, with one peak in the lysosomal fractions 3 and 4 and a second peak in fractions 8 and 9 containing PM and endosomes (Fig. 2 B). In fact, CD1b accumulated even more prominently in lysosomes than MHC class II molecules. In contrast, CD1c molecules were not found in the lysosomal fractions and accumulated in fractions 8 and 9, very similar to the distribution of MHC class I molecules.
Figure 2.

Accumulation of CD1b but not CD1c in lysosomal compartments and MIICs. Immature DCs were labeled for 22 h with radioactive [35S]methionine/cysteine, homogenized, and subsequently fractionated by Percoll gradient centrifugation. (A) The relative distribution of the enzymes alkaline phosphodiesterase (APDE, ▵) and β-hexosaminidase (B-HEX, •) was determined to monitor the presence of plasma membrane or lysosomes in the Percoll gradient fractions, respectively. The distribution of total proteins (▪) is also shown. Membranes of each fraction were isolated and deglycosylated with PNGase F before immunoblotting with antibodies specific for TfR, Lamp-1, MHC class I, and CD1b and detection by chemiluminescence and autoradiography. (B) The distribution of MHC class II, MHC class I, CD1b, and CD1c molecules in Percoll fractions was compared by immunoprecipitation. Proteins from the postnuclear supernatant (PNS) were immunoprecipitated with either the specific antibody (Sp) or an isotype control antibody (Co). Only the CD1b and CD1c molecules were deglycosylated with PNGase F before SDS-PAGE.
Cytoplasmic Tail Dependence of Endosomal Localization of CD1c.
The endosomal localization of CD1b is dependent on a tyrosine-based motif (YQ NI) contained within its cytoplasmic tail 10 11. The cytoplasmic tail of CD1c contains a very similar four–amino acid tyrosine-based motif with only a single amino acid difference (N to D at position Y+2) compared with the CD1b motif. To analyze the importance of the cytoplasmic tail of CD1c in its endosomal localization, a tail-deleted molecule (CD1c.TD) was stably expressed by transfection in HeLa cells. A comparison of the staining pattern of the wild-type CD1c protein (CD1c.WT) and CD1c.TD by immunofluorescence revealed striking differences between these molecules. Whereas a clear punctate intracellular staining pattern was observed for CD1c.WT (Fig. 3 A, top) consistent with an endosomal localization of a substantial fraction of these molecules, CD1c.TD showed prominent cell surface staining and almost no punctate intracellular staining (Fig. 3 A, bottom). Vesicular staining with an antiserum specific for Lamp-1 was equivalent in both transfectants (data not shown).
Figure 3.


Cytoplasmic tail dependence of the endosomal distribution of CD1c. (A) Transfected HeLa cells stably expressing CD1c.WT (top) and CD1c.TD (bottom) were fixed, permeabilized, and labeled with the specific anti-CD1c mAb F10/21A3. Bars, 10 μm. (B) HeLa cells were transiently transfected with equimolar quantities of plasmid encoding CD1c.WT, CD1c.TD, or with vector alone. After 48 h, intact cells were analyzed by flow cytometry for surface expression of CD1c. Results shown are mean values of the mean fluorescence intensity (MFI) for three independent samples of both transfections.
As a second approach to assess the role of the cytoplasmic tail of CD1c in its intracellular localization, we examined the effect of cytoplasmic tail deletion on cell surface expression of CD1c by FACS®. HeLa cells were transiently transfected with equimolar quantities of plasmid encoding CD1c.WT or CD1c.TD or with vector alone, and 2 d later the cell surface expression of CD1 molecules was analyzed by FACS®. This showed that the tail-deleted form of CD1c was expressed at threefold higher levels at the cell surface compared with CD1c.WT (Fig. 3 B), consistent with its failure to accumulate in intracellular compartments and subsequent redistribution to the cell surface. Thus, as previously shown for CD1b, the cytoplasmic tail was necessary for the endosomal localization of CD1c.
Characterization of the CD1c-mediated Lipid Antigen Presentation.
To determine if the different intracellular distribution of CD1b and CD1c molecules was linked to differences in antigen presentation function, we analyzed the dependence on endosomal acidification of presentation of mycobacterial lipids by these molecules. Two different lysosomotropic agents, chloroquine and concanamycin B, were used at increasing doses to determine the requirement for endosomal acidification for exogenous lipid antigen presentation by CD1b- or CD1c-transfected B lymphoblastoid cells (CIR). Both agents produced a complete inhibition of CD1b-mediated antigen presentation, whereas CD1c-mediated presentation was only slightly affected by concanamycin B at high doses and not at all by chloroquine (Fig. 4 A). The differential effect of chloroquine on antigen presentation by CD1b or CD1c could also be reproduced using immature DCs as APCs in this in vitro system, excluding the possibility that this effect was cell line specific (data not shown). This difference in dependence on endosomal acidification could be interpreted as evidence that the association of lipids with CD1c either occurred directly at the PM or in early endosomes, which are known to have a neutral pH. The classic experiment to distinguish between the association of an antigen and its presenting molecule at the PM as opposed to an intracellular compartment is to fix the APC and subsequently analyze its capacity to present an exogenously added antigen. However, this approach was not successful for this study for technical reasons, most likely due to the CD1c–lipid complex at the cell surface being extremely sensitive to fixation by a variety of different agents (data not shown).
Figure 4.

Characterization of CD1c-mediated lipid antigen presentation. (A) C1R B lymphoblastoid cells stably transfected with CD1b (circles) or CD1c (squares) were labeled with 51Cr and coincubated overnight at a constant antigen concentration (CD1b: 60 μg/ml mycolic acid; CD1c: 13 μg/ml of M. tuberculosis total lipid extract) and various concentrations of either chloroquine (0.02–62.5 μM; filled symbols) or concanamycin B (0.02–62.5 nM; open symbols). These target cells were then analyzed by a standard 4-h chromium release cytolysis assay as described in Materials and Methods using the CD1c-specific T cell line CD8.1 and the CD1b-specific T cell line DN1. (B) HeLa cells transfected with vector alone (mock; ▴) or with CD1c.WT (▪) or CD1c.TD (•) plasmid for stable expression were labeled with 51Cr. These cells were incubated overnight with increasing doses of antigen (M. tuberculosis total lipid extract), then harvested, washed once with PBS, and combined with the T cell line CD8.1 at an E/T ratio of 15:1 for 4 h. (C) Immature DCs were incubated with antigens presented by CD1c (10 μg/ml of total lipid extract from M. tuberculosis containing the Hex-1–PIP antigen) or MHC class II (0.1 mg/ml tetanus toxoid protein [TTprot] or 75 ng/ml of an antigenic 15-mer peptide from tetanus toxoid [TTpep]) for various times. The presentation of antigens was measured by the increase in [Ca2+]i in the T cells specific for the CD1c-presented lipid antigen Hex-1–PIP (▪) or MHC class II–presented TTpep (▴) or TTprot (•), and was expressed as a percentage of maximal Indo-1 index after the subtraction of background values (T cells plus APCs without antigen). Specific T cell lines used were CD8.1 for CD1c and SP-14 for MHC class II. Results shown are representative of three independent experiments.
To circumvent this technical problem, we took advantage of the tail deletion effect on the intracellular distribution of CD1c in HeLa cells (Fig. 3 B) and compared the capacity of the wild-type and tail-deleted forms of CD1c to present lipid antigens. Surprisingly, this revealed that CD1c.TD had no detectable deficiency in the presentation of the mycobacterial glycolipid antigen hexose 1–phosphoisoprenoid (Hex-1–PIP) to CD1c-restricted T cells (Fig. 4 B). This result was confirmed using CD1c.WT- or CD1c.TD-expressing CIR cells as APCs (data not shown). These results, along with the absence of an effect of lysosomotropic agents on antigen presentation by CD1c, suggested that association of this glycolipid antigen with CD1c occurred at the cell surface rather than in an endosomal compartment.
A second approach to examine this question was based on the premise that the kinetics of presentation for an antigen that associates with its presenting molecule at the cell surface (like MHC class II with antigenic peptides) should be more rapid than antigen presentation requiring internalization and processing by the APC (like presentation of intact protein antigens by MHC class II). Therefore, the kinetics of presentation of a mycobacterial glycolipid antigen (Hex-1–PIP) by CD1c was compared with the kinetics of MHC class II–restricted antigen presentation after incubation of the APCs with either intact tetanus toxoid protein antigen (TTprot) or an antigenic 15-mer peptide derived from this protein (TTpep). The time required to achieve 50% of the maximal response (t max1/2) to lipid antigen presentation by CD1c was ∼8 min, which was similar to that of the TTpep presentation by MHC class II, with a t max1/2 of 5 min. In contrast, the t max1/2 of the TTprot presentation by MHC class II was substantially longer, at ∼39 min (Fig. 4 C). These results were consistent with the idea that the association of lipid antigen with CD1c occurred at or near the cell surface, since the kinetics of presentation was much more similar to that of MHC class II–mediated presentation of exogenous peptides than to the kinetics of presentation of an intact protein antigen. In contrast, a separate series of experiments showed no difference in the kinetics of presentation of the lipid antigen mycolic acid by CD1b compared with that of intact tetanus toxoid presentation by MHC class II (data not shown), consistent with the proposed association of lipid antigens with CD1b in a late endocytic compartment.
In summary, the differences in subcellular distribution of CD1b and CD1c correlated with marked differences in the functional properties of these molecules in antigen presentation. One major difference observed between the CD1b- and CD1c-mediated antigen presentation in this study was related to their requirement for endosomal acidification for normal expression of antigen-presenting function (Fig. 4 A). Consistently, only CD1b localizes to the most acidic endocytic compartments, whereas CD1c accumulates on the cell surface and in early endosomes (Fig. 1 and Fig. 2). Although the Hex-1–PIP lipid seemed to associate with CD1c at the cell surface in our in vitro assay, the intracellular localization of CD1c may very well be important for the presentation of other CD1c-restricted lipid antigens that have yet to be defined or during the course of a live mycobacterial infection. In fact, preliminary results show that CD1c was found to colocalize with mycobacteria in phagolysosomes (data not shown), and studies currently in progress are assessing the presentation of CD1c-restricted lipids in the context of intracellular infection of APCs.
The recently identified CD1c-presented glycolipid Hex-1–PIP is unique among the known CD1-presented antigens in that its hydrophobic moiety contains only a single alkyl chain, in contrast to the dual or branched alkyl chains found in all of the previously described CD1b-presented lipid antigens 6 8. Although data on the spectrum of lipids presented by CD1c are still extremely limited, it is intriguing to speculate that CD1b and CD1c molecules may predominantly present two structurally different classes of lipids, in that they contain either a dual or a single alkyl chain motif, respectively. In this context, the finding that lipids can be sorted to various sites within the cell according to the specific structure and composition of their alkyl tails is of great interest 19. Although a recent study observed a similar resistance to inhibitors of endosomal acidification for presentation of a putative lipid antigen by another human CD1 molecule, CD1a 20, the CD1a ligand has not yet been identified and therefore no conclusions about a structural analogy to the CD1c ligand are possible.
Taken together, the data currently available on the intracellular localization of three human CD1 proteins (CD1a, CD1b, and CD1c) are consistent with each of these being focused predominantly at a different level within the endocytic system, supporting the hypothesis that the different isoforms of CD1 expressed by a single individual have evolved complementary functions that are to survey the lipid content of different subcellular compartments 9. An important goal for future studies will be to determine whether the specific antigens presented by each of these different CD1 proteins are also distributed within APCs in a pattern that overlaps the localization and trafficking patterns of the particular CD1 protein that presents them.
Acknowledgments
We give special thanks to Maris Handley for expert assistance in the use of the Cytomation MO-FLO cytometer and to Gene Lai for assistance with confocal microscopy. We would also like to thank Drs. H. Ploegh, M. Fukuda, S. Amigorena, H. Spits, and P. Cresswell for providing valuable reagents used in this study.
This work was supported by grants from the National Institutes of Health (AI40135 and AI45889) and the American Cancer Society to S.A. Porcelli. V. Briken was supported by a grant from the Human Frontiers Science Program Organization (HFSPO), and R.M. Jackman by a predoctoral fellowship from the National Science Foundation.
References
- Porcelli S.A. The CD1 familya third lineage of antigen-presenting molecules. Adv. Immunol. 1995;59:1–98. doi: 10.1016/s0065-2776(08)60629-x. [DOI] [PubMed] [Google Scholar]
- Rosat J.P., Grant E.P., Beckman E.M., Dascher C.C., Sieling P.A., Frederique D., Modlin R.L., Porcelli S.A., Furlong S.T., Brenner M.B. CD1-restricted microbial lipid antigen-specific recognition found in the CD8+ alpha beta T cell pool. J. Immunol. 1999;162:366–371. [PubMed] [Google Scholar]
- Joyce S., Woods A.S., Yewdell J.W., Bennink J.R., De S.A., Boesteanu A., Balk S.P., Cotter R.J., Brutkiewicz R.R. Natural ligand of mouse CD1d1cellular glycosylphosphatidylinositol. Science. 1998;279:1541–1544. doi: 10.1126/science.279.5356.1541. [DOI] [PubMed] [Google Scholar]
- Beckman E.M., Porcelli S.A., Morita C.T., Behar S.M., Furlong S.T., Brenner M.B. Recognition of a lipid antigen by CD1-restricted αβ+ T cells. Nature. 1994;372:691–694. doi: 10.1038/372691a0. [DOI] [PubMed] [Google Scholar]
- Beckman E.M., Melian A., Behar S.M., Sieling P.A., Chatterjee D., Furlong S.T., Matsumoto R., Rosat J.P., Modlin R.L., Porcelli S.A. CD1c restricts responses of mycobacteria-specific T cells. Evidence for antigen presentation by a second member of the human CD1 family. J. Immunol. 1996;157:2795–2803. [PubMed] [Google Scholar]
- Porcelli S.A., Modlin R.L. The CD1 systemantigen-presenting molecules for T cell recognition of lipids and glycolipids. Annu. Rev. Immunol. 1999;17:297–329. doi: 10.1146/annurev.immunol.17.1.297. [DOI] [PubMed] [Google Scholar]
- Porcelli S.A., Segelke B.W., Sugita M., Wilson I.A., Brenner M.B. The CD1 family of lipid antigen-presenting molecules. Immunol. Today. 1998;19:362–368. doi: 10.1016/s0167-5699(98)01289-4. [DOI] [PubMed] [Google Scholar]
- Moody D.B., Ulrichs T., Muehlecker W., Young D.C., Gurcha S.S., Grant E.P., Rosat J.P., Brenner M.B., Costello C.E., Besra G.S., Porcelli S.A. CD1c-mediated T cell recognition of isoprenoid glycolipids in M. tuberculosis infection. Nature. 2000;404:848–888. doi: 10.1038/35009119. [DOI] [PubMed] [Google Scholar]
- Jackman R.M., Moody D.B., Porcelli S.A. Mechanisms of lipid antigen presentation by CD1. Crit. Rev. Immunol. 1999;19:49–63. [PubMed] [Google Scholar]
- Sugita M., Jackman R.M., van Donselaar E., Behar S.M., Rogers R.A., Peters P.J., Brenner M.B., Porcelli S.A. Cytoplasmic tail-dependent localization of CD1b antigen-presenting molecules to MIICs. Science. 1996;273:349–352. doi: 10.1126/science.273.5273.349. [DOI] [PubMed] [Google Scholar]
- Jackman R.M., Stenger S., Lee A., Moody D.B., Rogers R.A., Niazi K.R., Sugita M., Modlin R.L., Peters P.J., Porcelli S.A. The tyrosine-containing cytoplasmic tail of CD1b is essential for its efficient presentation of bacterial lipid antigens. Immunity. 1998;8:341–351. doi: 10.1016/s1074-7613(00)80539-7. [DOI] [PubMed] [Google Scholar]
- Porcelli S., Morita C.T., Brenner M.B. CD1b restricts the response of human CD4−8− T lymphocytes to a microbial antigen. Nature. 1992;360:593–597. doi: 10.1038/360593a0. [DOI] [PubMed] [Google Scholar]
- Roncarolo M.G., Yssel H., Touraine J.L., Bacchetta R., Gebuhrer L., de Vries J.E., Spits H. Antigen recognition by MHC-incompatible cells of a human mismatched chimera. J. Exp. Med. 1988;168:2139–2152. doi: 10.1084/jem.168.6.2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant E.P., Degano M., Rosat J.P., Stenger S., Modlin R.L., Wilson I.A., Porcelli S.A., Brenner M.B. Molecular recognition of lipid antigens by T cell receptors. J. Exp. Med. 1999;189:195–205. doi: 10.1084/jem.189.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porcelli S., Brenner M.B., Greenstein J.L., Balk S.P., Terhorst C., Bleicher P.A. Recognition of cluster of differentiation 1 antigens by human CD4−CD8− cytolytic T lymphocytes. Nature. 1989;341:447–450. doi: 10.1038/341447a0. [DOI] [PubMed] [Google Scholar]
- Briken V., Lankar D., Bonnerot C. New evidence for two MHC class II-restricted antigen presentation pathways by overexpression of a small G protein. J. Immunol. 1997;159:4653–4658. [PubMed] [Google Scholar]
- Bonnerot C., Briken V., Brachet V., Lankar D., Cassard S., Jabri B., Amigorena S. syk protein tyrosine kinase regulates Fc receptor gamma-chain-mediated transport to lysosomes. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:4606–4616. doi: 10.1093/emboj/17.16.4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner M.B., McLean J., Yang S.Y., van der Poel J.J., Pious D., Strominger J.L. Clonal T lymphocyte recognition of the fine structure of the HLA-A2 molecule. J. Immunol. 1985;135:384–390. [PubMed] [Google Scholar]
- Mukherjee S., Soe T.T., Maxfield F.R. Endocytic sorting of lipid analogues differing solely in the chemistry of their hydrophobic tails. J. Cell Biol. 1999;144:1271–1284. doi: 10.1083/jcb.144.6.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita M., Grant E.P., van Donselaar E., Hsu V.W., Rogers R.A., Peters P.J., Brenner M.B. Separate pathways for antigen presentation by CD1 molecules. Immunity. 1999;11:743–752. doi: 10.1016/s1074-7613(00)80148-x. [DOI] [PubMed] [Google Scholar]