

Abstract
Adenosine deaminase (ADA) is a purine catabolic enzyme that manages levels of the biologically active purines adenosine and 2′-deoxyadenosine in tissues and cells. ADA-deficient mice die at 3 wk of age from severe respiratory distress. This phenotype is progressive and is linked to perturbations in pulmonary purine metabolism. The inflammatory changes found in the lungs of ADA-deficient mice included an accumulation of activated alveolar macrophages and eosinophils. These changes were accompanied by a pronounced enlargement of alveolar spaces and increases in mucus production in the bronchial airways. The alveolar enlargement was found to be due in part to abnormal alveogenesis. Lowering adenosine and 2′-deoxyadenosine levels using ADA enzyme therapy decreased the pulmonary eosinophilia and resolved many of the lung histopathologies. In addition, genetically restoring ADA to the forestomach of otherwise ADA-deficient mice prevented adenine metabolic disturbances as well as lung inflammation and damage. These data suggest that disturbances in purinergic signaling mediate the lung inflammation and damage seen in ADA-deficient mice.
Keywords: eosinophil, asthma, emphysema, alveolar macrophage, adenosine deaminase
Introduction
Adenosine deaminase (ADA) is an essential enzyme of purine catabolism that is responsible for the hydrolytic deamination of adenosine and 2′-deoxyadenosine to inosine and 2′-deoxyinosine 1. These biochemical pathways are essential for maintaining homeostasis, as both ADA substrates have substantial signaling properties. Adenosine engages G protein–coupled receptors on the surface of target cells to evoke a variety of cellular responses 2, whereas 2′-deoxyadenosine is cytotoxic via mechanisms that interfere with cellular growth and differentiation 3 4 or the promotion of apoptosis 5 6. ADA deficiency in humans results in marked accumulations of both of these signaling molecules, and these accumulations are associated with a variety of cellular phenotypes. The most thoroughly studied phenotype has been the effect of this enzyme deficiency on the immune system. ADA deficiency results in a combined immunodeficiency characterized by a severe T, B, and NK cell lymphopenia 7 8. Most studies identify the accumulation of 2′-deoxyadenosine as the metabolic basis for this lymphopenia 3 9, although evidence exists to suggest that engagement of adenosine receptors may be involved 10 11. Additional phenotypes associated with ADA deficiency are not as well characterized and include bony and renal abnormalities 12, hepatocellular damage 13, neurological disorders 14, and pulmonary insufficiency 7. The metabolic basis for these phenotypes and the mechanisms involved are unknown, in part due to the lack of adequate models with which to study the effects of ADA deficiency on these systems.
We have recently used a two-stage genetic engineering strategy to generate ADA-deficient mice 15. The initial knockout of the murine Ada gene resulted in a prenatal lethality that prevented the analysis of postnatal consequences of ADA deficiency 16 17. This prenatal lethality was overcome with an ADA minigene under the control of a trophoblast-specific promoter to restore ADA specifically to the placenta of otherwise ADA-deficient fetuses 15 18. This was sufficient to rescue ADA-deficient fetuses and resulted in postnatal ADA-deficient mice amenable to the analysis of the phenotypic and metabolic consequences of ADA deficiency. ADA-deficient mice developed a combined immunodeficiency that was linked with profound disturbances in purine metabolism 15 19.
In addition to immunodeficiency, ADA-deficient mice developed other phenotypes noted in ADA-deficient humans 12, including bony and renal abnormalities and pulmonary insufficiency 15. The most severe of these phenotypes was the pulmonary insufficiency. ADA-deficient mice began to show signs of respiratory distress as early as postpartum day 12. This distress increased in severity, and the mice died between postpartum days 19 and 25. Initial examination of this phenotype revealed severe lung inflammation in association with severe purine metabolic disturbances including the accumulation of adenosine and to a lesser extent 2′-deoxyadenosine 15.
Purinergic signaling has been implicated to play a role in lung inflammation. Most notable are the well-recognized effects of adenosine in asthma 20. Clinical evidence linking adenosine to this disease state includes the detection of elevated adenosine levels in bronchial alveolar lavage fluid (BALF) collected from asthmatics 21; the observation that inhaled adenosine elicits bronchoconstriction in individuals suffering from asthma 22; the expression of adenosine receptors is altered in patients with airway inflammation 23; and theophylline, an adenosine receptor antagonist, has a well-recognized therapeutic benefit in this disease 24. In addition, there are many in vitro studies that implicate adenosine as a modulator of inflammatory processes that are central to asthma. These include adenosine's ability to enhance 25 or directly evoke 26 mediator release from mast cells, and to influence eosinophil function 27 28 29. Adenosine signaling has also been implicated in regulating the function of other inflammatory cells such as macrophages 30 31 32 and neutrophils 33 34. Despite these lines of evidence, a causative link between adenosine signaling and lung inflammation, as well as the cell types and mechanisms involved, are unclear. In the current study, we characterized the lung inflammation and damage occurring in ADA-deficient mice. Moreover, we used ADA enzyme therapy to demonstrate a relationship between adenosine and 2′-deoxyadenosine levels and the inflammation that results in ADA-deficient animals. The ADA-deficient mice described exhibited many features of lung disease, including defects in alveogenesis, activation of alveolar macrophages, lung eosinophilia, and mucus hypersecretion. These pulmonary features were closely associated with disturbances in the concentrations of ADA substrates, suggesting that perturbations in signaling pathways accessed by these substrates are involved. This model will provide a unique approach to examining the specific roles of adenosine signaling in vivo.
Materials and Methods
Transgenic Mice.
ADA-deficient mice were generated and genotyped as described previously 15 16. All mice used in these studies were on a mixed background of 129/Sv and FVB/N strains 18. Control mice were either wild-type animals or mice heterozygous for the null Ada allele 15. Animal care was in accordance with institutional and National Institutes of Health guidelines. All mice were housed in cages equipped with microisolator lids and maintained under strict containment protocols. No evidence of bacterial, parasitic, or fungal infection was found. In addition, serologies on cage littermates were negative for 12 of the most common murine viruses.
Histological Analysis and Immunofluorescence.
Aged-matched control and experimental animals were killed, and the lungs were infused with 0.1–0.5 ml of fixative (4% paraformaldehyde in PBS), depending on age, before fixation overnight at 4°C. Fixed lung samples were rinsed in PBS, dehydrated, and embedded in paraffin. Sections (5 μm) were collected on microscope slides and stained with hematoxylin and eosin (H&E; Shandon-Lipshaw) or periodic acid-Schiff (PAS; EM Science), according to manufacturer's instructions. Immunofluorescence of lungs for the expression of murine eosinophil granule major basic protein 1 (mMBP-1) was performed according to established procedures 35. Sections were reacted with antiserum from a rabbit immunized with purified mMBP-1, followed by detection using FITC-conjugated anti–rabbit IgG.
Quantification of Lung Histopathology.
The size of alveolar airways was determined by measuring mean chord lengths 36 on H&E-stained lungs. Representative images were digitized, and a grid consisting of 53 black lines at 10.5-μm intervals was overlaid on the image. This line grid was subtracted from the lung images using Image-Pro® Plus (Media Cybernetics) image analysis software, and the resultant lines were measured and averaged to give the mean chord length of the alveolar airways. The final mean chord lengths represent averages from 12 nonoverlapping images of each lung specimen and are given in micrometers.
The extent of mucus production in bronchial airways was determined by quantitating the amount of PAS-stained material in the bronchial airways using Image-Pro® Plus analysis software. PAS-stained material was identified on digitized images, and the pixel intensities of each color channel (red, blue, and green) were averaged. This was repeated for each image, and the values were averaged and used to determine the area (M) and intensity (I) of PAS-stained material in bronchial airways. In addition, the area (A) of the total epithelium (including PAS-stained material) was determined. The mucus index was determined using the following equation: M × I/A. Final indices were results of an average of eight images per lung encompassing large and small bronchial airways.
Peripheral Blood Cell Counts.
Mice were anesthetized, and a heparinized syringe was used to collect 200–500 μl of blood from the subaxilary artery. Samples were immediately analyzed for complete blood cell counts using an H1 Analyzer (Technicon Instruments).
Bronchial Alveolar Lavages.
Mice were anesthetized with avertin, and a blunted 21-gauge needle was secured into the trachea. Lungs were lavaged five times with 0.25 ml PBS, and 0.95–1 ml of pooled lavage fluid was recovered. Samples were centrifuged at 2,500 rpm for 5 min to recover cells, and supernatant from these spins was collected and stored at −70°C for the analysis of cytokines. BALF cells were resuspended in 200 μl PBS; total cell counts were determined from an aliquot counted using a hemocytometer, and another aliquot cytospun onto microscope slides was stained with Diff-Quik (Dade) for cellular differentials. 400 cells per sample were identified and counted under oil immersion.
ELISA Assays.
IFN-γ, IL-4, and IL-5 levels in BALF were determined using specific murine OptEIA™ ELISA kits from BD PharMingen. For the analysis of IgE levels, blood was collected from the heart of anesthetized mice and the serum was separated by centrifugation at 3,000 rpm for 10 min at 4°C. A murine OptiEA ELISA kit from BD PharMingen was used to quantitate total serum IgE levels.
ADA Enzyme Therapy and Analysis of ADA Enzyme Activity.
Polyethylene glycol–modified ADA (PEG-ADA), also known as ADAGEN®, was obtained through collaboration with Enzon, Inc. Control or ADA-deficient mice were anesthetized and injected intramuscularly with 10 μl of PEG-ADA corresponding to ∼2.5 U of ADA enzymatic activity. Injections were given either chronically every 4 d starting at postpartum day 4, or acutely, as one injection on postpartum day 18. Levels of ADA enzyme activity in tissues were measured according to established procedures 15.
Quantification of Adenosine and 2′-Deoxyadenosine.
Mice were anesthetized, the thoracic cavity was exposed, and the lungs were removed and frozen rapidly in liquid nitrogen. Adenine nucleosides were extracted from frozen lungs using 0.4 N perchloric acid as described 37, and adenosine and 2′-deoxyadenosine were separated and quantitated using reversed phase HPLC 37.
Results
Severe Lung Inflammation and Damage Are Found in the Lungs of ADA-deficient Mice.
ADA-deficient mice begin to show signs of respiratory distress as early as postpartum day 12. This distress was characterized by rapid and labored breathing that became increasingly severe. These mice became cyanotic and died between postpartum days 19 and 25 15. Lung inflammation was examined on postpartum day 18 to assess the nature of the respiratory distress in ADA-deficient mice (Fig. 1). An increase in inflammatory cells was seen throughout the lungs, with a specific increase of enlarged and foamy macrophages (Fig. 1 b). Closer examination of cells collected from BALF showed alveolar macrophages engulfing eosinophils (Fig. 1 f). In addition, multinucleated giant cells were prominent in the BALF (Fig. 1 g). Histological analysis also suggested a pronounced infiltration of eosinophils throughout the lungs (Fig. 1b and Fig. c, arrows). Lung sections were reacted with a polyclonal antibody against mMBP-1 35 to confirm that the cells were eosinophils. Results from these studies (Fig. 1 d) confirm the accumulation of eosinophils in ADA-deficient lungs. Eosinophils were found in interstitiary and lumenal spaces throughout the lung, with high concentrations accumulating around bronchioles and pulmonary blood vessels (Fig. 1b–d, arrows). Intense mMBP-1 immunoreactivity was noted in alveolar macrophages surrounding focal points of eosinophil accumulation (Fig. 1 d, yellow arrows), confirming the engulfment of eosinophils by activated macrophages. A large increase in eosinophils was also found in BALF (Fig. 1 e), and there was a twofold increase in circulating eosinophils (Table ). Inflammation was not seen in other tissues that were examined, including the gastrointestinal tract, thymus, spleen, liver, and kidney (data not shown). These results demonstrated that ADA-deficient mice developed pronounced pulmonary inflammation characterized by the accumulation of activated alveolar macrophages and eosinophils.
Figure 1.

Morphological and cytological changes in the lungs of ADA-deficient mice. (a) H&E-stained control lung at postpartum day 18. (b) H&E-stained ADA-deficient lung at postpartum day 18. Arrows indicate areas of inflammation. Notice the pronounced enlargement of alveolar spaces (AS) and the thickening of pulmonary blood vessels (BV). (c) High magnification of an H&E-stained ADA-deficient lung demonstrating eosinophil infiltration (arrows) around bronchioles (B) and pulmonary blood vessels (BV). (d) Immunolocalization of eosinophils in an 18-d-old ADA-deficient lung, using a rabbit polyclonal antibody raised against mMBP-1 followed by detection with FITC immunofluorescence. Notice intense staining of inflammatory cells with anti–MBP-1 (arrows). mMBP-1 immunoreactivity was also detected in alveolar macrophages (AM). Cytological analysis of cells collected from BALF from the lung of an 18-d-old ADA-deficient mouse showing (e) eosinophils (arrows), (f) alveolar macrophages engulfing eosinophils (arrows), and (g) multinucleated giant cells. Bars, (a–d) 100 μm; (e–g) 10 μm.
Table 1.
Peripheral Blood Cell Counts
| n | RBC | WBC | Lymphocyte | Monocyte | Eosinophil | Neutrophil | |
|---|---|---|---|---|---|---|---|
| Control | 5 | 6.95 ± 0.2 | 1,564 ± 179 | 1,138 ± 134 | 170 ± 42 | 22 ± 7 | 238 ± 66 |
| ADA-deficient | 4 | 6.38 ± 0.2 | 1,263 ± 156 | 563 ± 48 | 225 ± 100 | 48 ± 8 | 385 ± 111 |
| ADA-deficient +PEG-ADA | 3 | 6.17 ± 0.2 | 1,187 ± 396 | 623 ± 107 | 207 ± 162 | 23 ± 12 | 303 ± 151 |
Peripheral blood cell counts are from18-d-old control and ADA-deficient mice or ADA-deficient mice 72 h after a single dose of PEG-ADA on day 18.
Evidence of tissue damage and lung remodeling accompanied the inflammatory changes in ADA-deficient lungs. Examination of the alveolar airways demonstrated a prominent increase in the size of alveolar spaces as well as increases in the thickness of the smooth muscle of pulmonary blood vessels (Fig. 1 b). Hypertrophy of the bronchial epithelium was common, as was a progressive increase in mucus production and an accumulation of mucus and cellular debris in the bronchial airways (Fig. 2). Collectively, these results demonstrated that the pulmonary inflammation seen in ADA-deficient mice was associated with histopathological changes in the lung.
Figure 2.

Mucus hypersecretion in the bronchial airways of ADA-deficient mice. Lung sections were stained with PAS for the detection of neutral mucins. (a) Control lung at postpartum day 18. (b) ADA-deficient lung at postpartum day 18. (c) ADA-deficient lung at postnatal day 21. Arrows in b and c denote PAS-positive material indicative of increased mucus production. B, bronchiole. Bars, 100 μm.
Developmental Defects in Alveogenesis Precede Lung Inflammation in ADA-deficient Mice.
The severe inflammation and damage seen on day 18 prompted us to examine the development of this phenotype. At birth, control and ADA-deficient lungs were histologically similar (Fig. 3, a and b). At postpartum day 5, the overall morphology (Fig. 3c and Fig. d) suggested that there was an increase in alveolar airway size. Secondary septation of the alveoli occurred between postpartum days 5 and 10 in control mice (Fig. 3 e); however, alveolar size remained enlarged in ADA-deficient lungs at postpartum day 10 (Fig. 3 f). Quantitation of alveolar size (Fig. 3 g) verified that there was a significant difference in alveolar size at day 5, suggesting that alveolar formation in ADA-deficient mice was disturbed early in life and worsened by day 10. Lung inflammation was not seen in ADA-deficient lungs at day 0 and day 5 as determined by H&E staining (Fig. 3) and mMBP-1 immunostaining (data not shown). Slight inflammation was seen at day 10 (Fig. 3 f), and increased numbers of alveolar macrophages and eosinophils were consistently seen at day 15 (data not shown; 15). These results demonstrated that there was a defect in alveogenesis in ADA-deficient mice and that these defects preceded the appearance of lung inflammation.
Figure 3.


Defects in alveogenesis in ADA-deficient mice. Lungs from age-matched control and ADA-deficient mice were collected and processed for H&E staining. (a) Control lung at postpartum day 0. (b) ADA-deficient lung at day 0. (c) Control lung at day 5. (d) ADA-deficient lung at day 5. (e) Control lung at day 10 demonstrating the septation of presumptive alveoli into mature alveolar sacs. (f) ADA-deficient lung at day 10 demonstrating enlarged alveolar spaces. Panels a–f are at the same magnification; bars, 250 μm. (g) The size of alveolar airways was determined in control (white bars, n = 4) and ADA-deficient (black bars, n = 4) lungs by measuring mean chord lengths (in μm) of alveolar airways in H&E-stained lungs. Values are given as mean μm ± SE from four separate age-matched control and ADA-deficient lung pairs at each developmental stage; *P ≤ 0.05.
Serum IgE and BALF IL-5 Levels Are Elevated in ADA-deficient Mice.
Elevated IgE and eosinophilia are features noted in some subsets of ADA-deficient patients 8 38 39. Total serum IgE levels were assessed to determine whether elevations in IgE were associated with the lung eosinophilia seen in ADA-deficient mice. Serum IgE levels were slightly increased on postpartum day 18 (Fig. 4 a); however, when the ratios of IgE to total Ig were compared in control and ADA-deficient mice (Fig. 4 b), a prominent bias towards IgE production was seen in ADA-deficient animals.
Figure 4.
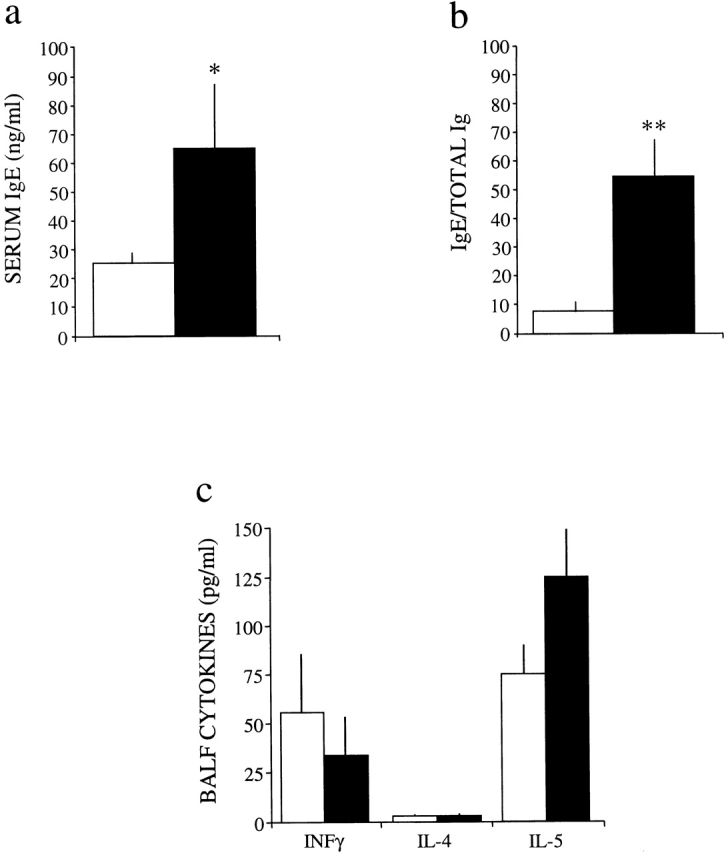
Levels of serum IgE and BALF cytokines in control (white bars) and ADA-deficient (black bars) mice. (a) Total IgE levels were measured in the serum of 18-d-old control (n = 11) and ADA-deficient (n = 8) mice. Mean total serum IgE values are given as ng/ml ± SE. Total serum Ig [IgG + IgM] levels are decreased 67% in ADA-deficient mice (reference 15). (b) Ratios of IgE to Ig in control and ADA-deficient mice, demonstrating a significant bias towards hyper IgE in ADA-deficient mice. Statistical significance was determined using Student's t test analysis; *P ≤ 0.100, **P ≤ 0.05. (c) The levels of IFN-γ, IL-4, and IL-5 were measured in BALF collected from 18-d-old control (n = 10) and ADA-deficient (n = 7) mice. Mean values are given as pg/ml ± SE.
Cytokine levels in BALF of 18-d-old ADA-deficient mice were examined to investigate whether the lung inflammation seen was associated with a Th2 cytokine profile. Though there was a trend toward a reduction in the levels of IFN-γ in ADA-deficient BALF, the differences were not significant (Fig. 4 c). There was no difference in the levels of IL-4 in ADA-deficient BALF; however, slight increases in IL-5 were noted (Fig. 4 c). These findings suggested that IL-5 signaling may be involved in the eosinophilia seen in ADA-deficient mice.
ADA Enzyme Therapy Results in the Reduction of BALF and Circulating Eosinophils and a Decrease in Mucus Production by Bronchial Airways.
PEG-ADA enzyme therapy is a lifesaving strategy used to treat ADA-deficient patients 40. ADA-deficient mice maintained on weekly injections of PEG-ADA from birth did not show signs of abnormal alveogenesis, lung eosinophilia, or respiratory distress, and survived as long as they were maintained on PEG-ADA (data not shown). To determine if PEG-ADA treatments could reverse the pulmonary phenotype after its onset, ADA-deficient mice were allowed to develop severe lung inflammation (day 18) and were subsequently treated with PEG-ADA. Within 24–48 h, their respiratory status was noticeably improved, and these animals recovered and survived as long as they were maintained on PEG-ADA (data not shown). Cell populations were monitored in BALF collected 72 h after treatment with PEG-ADA to determine what effect PEG-ADA treatment had on lung inflammation (Fig. 5). The most striking observation was a substantial decrease in the number of eosinophils in BALF of ADA-deficient mice treated with PEG-ADA. The numbers of alveolar macrophages were not reduced at 72 h after PEG-ADA treatment, but were reduced after 2 wk of enzyme therapy (Fig. 5). These findings suggested that PEG-ADA enzyme therapy led to a rapid reduction in lung eosinophilia in ADA-deficient mice.
Figure 5.

Reversible lung eosinophilia in ADA-deficient mice treated with PEG-ADA. Total cellular differentials were determined on cells collected from BALF of 18-d-old control mice (n = 19, white bars), ADA-deficient mice (n = 12, black bars), and ADA-deficient mice treated with PEG-ADA and examined 3 d later (n = 5, hatched bars) or after 2 wk of enzyme therapy (n = 4, stippled bars). Mean values are given as total cells ± SE. Statistical significance was determined using Student's t test analysis; *P ≤ 0.02.
ADA-deficient mice are lymphopenic, showing a two- to threefold reduction in the number of circulating lymphocytes 15. Complete blood cell counts were assessed in ADA-deficient mice and ADA-deficient mice treated with PEG-ADA to determine if the improvement in lung inflammation was related to an improvement in lymphocyte counts. There was a twofold decrease in the number of lymphocytes seen in the periphery of ADA-deficient mice (Table ); however, there was not a significant improvement in the number of lymphocytes found in the circulation after PEG-ADA treatment. There was a twofold elevation in the number of peripheral eosinophils in ADA-deficient mice, and peripheral eosinophil numbers were normalized after PEG-ADA treatment. These data suggested that the PEG-ADA treatments used in this study did not have a significant impact on the lymphopenia seen in ADA-deficient mice, but were capable of reducing elevations in lung and circulating eosinophils.
The amount of alveolar and bronchial tissue damage was quantitated before and after PEG-ADA treatments to determine what effect PEG-ADA treatments had on this damage. The severe alveolar defect seen in ADA-deficient mice was not significantly improved 72 h after treatment with PEG-ADA (Fig. 6 a). In contrast, the mucus production seen in the bronchial airways was substantially lower 72 h after PEG-ADA treatments (Fig. 6 b). Collectively, these results suggested that restoring ADA enzymatic activity to ADA-deficient mice decreased the number of eosinophils found in BALF and attenuated mucus production in bronchial airways, but did not reverse the abnormal lung structure resulting from abnormal alveogenesis and lung inflammation.
Figure 6.

Quantification of lung histopathology in ADA-deficient mice and ADA-deficient mice treated with PEG-ADA. (a) Mean cord lengths of alveolar airways were determined in lungs of 18-d-old control and ADA-deficient mice, and lungs of 21-d-old mice 72 h after treatment with PEG-ADA. Values are given as mean cord lengths in μm ± SE, n = 5 for each condition. *P ≤ 0.02 for increases in mean cord lengths compared with controls. (b) The degree of mucus production was determined in the bronchial airways of 18-d-old control and ADA-deficient mice, and 21-d-old ADA-deficient mice 3 d after treatment with PEG-ADA. Values are given as mean airway mucus indices in arbitrary pixel units ± SE, n = 5 for each condition. Statistical significance was determined using Student's t test analysis; *P ≤ 0.01 for an increase in mucus index between control and ADA-deficient samples, **P ≤ 0.02 for a decrease in mucus index between ADA-deficient samples and PEG-ADA–treated ADA-deficient samples.
Treatment of ADA-deficient Mice with PEG-ADA Results in Normalization of Lung Adenine Nucleoside Levels.
The elevated levels of adenosine, and to a lesser extent 2′-deoxyadenosine, in the lungs of ADA-deficient mice 15 suggested that pulmonary inflammation observed in these mice was due to disturbances in adenine metabolism. The rapid reversal of respiratory distress and lung eosinophilia after PEG-ADA enzyme therapy further suggested that the efficacy of this treatment was a consequence of lowering adenosine and 2′-deoxyadenosine levels. This was confirmed by examining the levels of ADA substrates in ADA-deficient lungs after PEG-ADA treatments (Fig. 7). The levels of adenosine in control lungs at day 18 were <0.2 nmol/mg protein, whereas adenosine levels in the lungs of ADA-deficient mice were elevated >20-fold (6.3 nmol/mg protein). 2′-Deoxyadenosine was not detected in control lungs and was elevated to 0.05 nmol/mg protein in ADA-deficient lungs. Strikingly, 72 h after treatment with PEG-ADA, adenosine and 2′-deoxyadenosine levels were lowered to near control levels in ADA-deficient lungs. These studies demonstrated that PEG-ADA enzyme therapy could efficiently remove adenosine and 2′-deoxyadenosine from the lungs of ADA-deficient mice, and suggested that accumulations of these nucleosides may play a role in regulating the lung eosinophilia and mucus production seen.
Figure 7.

PEG-ADA treatments reverse accumulations of adenosine and 2′-deoxyadenosine in the lungs of ADA-deficient mice. Adenosine and 2′-deoxyadenosine levels were quantitated in the lungs of 18-d-old control mice (n = 5) and ADA-deficient mice (n = 4), and 21-d-old ADA-deficient mice 3 d after treatment with PEG-ADA (n = 4). Mean values are given as nmol/mg protein ± SE. Statistical significance was determined using Student's t test analysis; *P ≤ 0.002. nd, not detectable at a minimal detection limit of 0.001 nmol/mg protein.
Genetic Replacement of ADA Prevents Lung Metabolic Disturbances and the Development of Histopathologies.
The results obtained using PEG-ADA provided compelling evidence that adenine metabolic disturbances were associated with the onset and progression of the pulmonary changes occurring in ADA-deficient mice. Therefore, we hypothesized that genetically providing an enriched source of ADA to these mice would prevent metabolic disturbances and pulmonary changes. To this end, we examined the status of lung structure, inflammation, and metabolic disturbances in ADA-deficient mice in which expression of ADA was specifically targeted to the forestomach 19. The forestomach was chosen because it is an enriched site of ADA production, and gene regulatory elements necessary for forestomach-specific expression were available 41. Transgenic expression of ADA in the forestomach of otherwise ADA-deficient mice (Fig. 8 e) prevented respiratory distress, allowing the animals to live a normal life span. Examination of lung histology revealed that these mice were comparable to control animals, and did not exhibit signs of abnormal alveogenesis, or increases in alveolar macrophages or eosinophils (Fig. 8, a–c; BALF data not shown). Correspondingly, adenosine and 2′-deoxyadenosine levels in the lungs of these animals were not significantly elevated (Fig. 8 d). Collectively, these data suggested that enriched expression of ADA in the forestomach of ADA-deficient mice could prevent the accumulation of adenosine and 2′-deoxyadenosine in the lung as well as pulmonary histopathologies and inflammation associated with these accumulations.
Figure 8.

Transgenic expression of ADA in the forestomach of ADA-deficient mice prevents adenosine and 2′-deoxyadenosine accumulation, lung inflammation, and lung histopathologies. (a) H&E-stained section of an 18-d-old control lung. (b) H&E-stained section of a 21-d-old ADA-deficient lung. (c) H&E-stained section of a 21-d-old ADA-deficient lung of a mouse expressing an ADA minigene in its forestomach. Bars, (a–c) 250 μm. (d) Adenosine and 2′-deoxyadenosine levels were quantitated in the lungs of 21-d-old control, ADA-deficient, or ADA-deficient mice expressing ADA in their forestomach (forestomach rescue). Mean values are given as nmol/mg protein ± SE; n = 3 for each. Statistical significance was determined using Student's t test analysis; *P ≤ 0.002. nd, not detected at a minimal detection limit of 0.001 nmol/mg protein. (e) Zymogram analysis showing the level of ADA enzymatic activity in the lung (L), blood (Bl), and forestomach (FS) of 21-d-old control, ADA-deficient, and ADA-deficient mice expressing ADA in their forestomach. Purine nucleoside phosphorylase (PNP) was used as a positive control.
Discussion
Results presented in this study demonstrate that the metabolic disturbances associated with ADA deficiency in mice result in abnormal lung development and the promotion of lung inflammation and damage. ADA-deficient mice exhibited alveolar defects that were overcome by genetically restoring ADA enzymatic activity to these animals. In addition, lowering ADA substrates in the lung using enzyme therapy reversed lung eosinophilia and mucus production. The ADA substrates adenosine and 2′-deoxyadenosine both have potent cellular signaling properties, some of which have been implicated to play a role in lung inflammation. These mice will therefore serve as a useful in vivo model system in which to study the role of purinergic signaling in aspects of lung development and disease.
Defects in alveogenesis have been noted in mice deficient in various growth factor signaling pathways, including fibroblast growth factor 42, platelet-derived growth factor 43, and transforming growth factor β 44 signaling pathways. In addition, overexpression of cytokines such as IL-11 45 and IL-13 46 in the lungs of mice results in defects in alveogenesis. These findings suggest that this stage of lung development is influenced by complex signaling pathways. Developmental analysis of lung structure in ADA-deficient mice revealed a defect in alveolarization. This defect preceded the onset of lung inflammation. The pronounced elevations of adenosine in ADA-deficient lungs suggested that perturbations in adenosine signaling may play a role in the alveolar defect seen. Nothing is known with regard to the expression of adenosine receptors during lung development, and examining the expression of adenosine receptors in normal and ADA-deficient lungs will help clarify the role of adenosine signaling during normal and abnormal alveogenesis.
ADA-deficient mice develop and succumb to severe pulmonary inflammation and lung damage by 3 wk of age 15. Characterization of the inflammation revealed a large accumulation of activated macrophages throughout the lungs and an infiltration of eosinophils around pulmonary blood vessels and bronchial airways. The inflammation was progressive, with no inflammation evident until postpartum day 10, after which lung inflammation increased in severity. This inflammation was associated with a pronounced increase in mucus production and a marked increase in lung adenosine levels. The observation that lowering adenosine levels improved pulmonary inflammation and mucus production suggested that adenosine may mediate these processes. Consistent with this suggestion is the extensive literature base showing that adenosine plays a role in inflammatory lung diseases such as asthma and chronic obstructive pulmonary disease (for a review, see reference 20). The exact functions that this signaling nucleoside plays in lung disease are not known, but they likely depend on the type of inflammatory cells present and the distribution of adenosine receptors on these cells. The ability to control adenosine levels in ADA-deficient mice using enzyme replacement therapy will provide a useful model for examining the influence of adenosine on different inflammatory cells in vivo.
Eosinophils have emerged as a major inflammatory cell type in asthma, and an increase in eosinophils is often observed in the lungs of asthmatics 47. These cells can release mediators that contribute to the airway damage often associated with asthma such as bronchial epithelial cell damage and the stimulation of mucus production 48 49. The accumulation of eosinophils in the lungs of ADA-deficient mice may be responsible for the increased mucus production seen. This is supported by the observation that decreasing the number of eosinophils in the lungs of ADA-deficient mice using ADA enzyme therapy also resulted in decreased mucus production. Alternatively, the decreased mucus production may be a direct effect of lowering lung adenosine levels since adenosine signaling has been demonstrated to increase mucus secretion in a canine mucus model 50. Increased mucus production in ADA-deficient mice was not associated with an increase in IL-4 in the BALF, suggesting the production of mucus in this model was not IL-4 dependent. Whether the mucus production was mediated by other Th2 cytokines such as IL-13 or IL-9, or by eosinophil-derived mediators or adenosine itself, remains to be determined.
The involvement of adenosine signaling in eosinophil biology has been demonstrated. The A3 adenosine receptor is expressed on human eosinophils that accumulate in the lung 23, and engagement of this receptor on eosinophils is thought to mediate the release of Ca2+ from intracellular stores 29, inhibit superoxide release 28, and inhibit eosinophil chemotaxis, which may serve a pro- or antiinflammatory role 23 27. Whether or not the A3 receptor is expressed in murine eosinophils and in the lungs of ADA-deficient mice is currently under investigation. However, the large increase in lung eosinophils in ADA-deficient mice and the ability to rapidly reverse this eosinophilia by lowering adenosine concentrations suggest that adenosine signaling may be mediating the lung eosinophilia occurring in these mice.
In addition to an increase in eosinophils, the number and activation of alveolar macrophages were greatly increased in the lungs of ADA-deficient mice. Engagement of adenosine receptors on macrophages elicits both pro- and antiinflammatory events, including the inhibition of TNF-α expression 51 52 and nitric oxide production 51, increased production of IL-10 51, increased differentiation of monocytes into macrophages 31 32 53, increased rates of phagocytosis 32, and stimulation of giant cell formation 30. Therefore, the increased number and activity of alveolar macrophages and giant cells in ADA-deficient mice may result from aberrant adenosine signaling brought about by persistent elevations in lung adenosine levels. Activated macrophages can contribute to alveolar airway damage 54. The enlargement of the alveolar airways in ADA-deficient mice is associated with a defect in alveogenesis. However, the enlargement of these airways is progressive from postpartum day 15 to 18, suggesting that damage to these airways is also occurring. The large number of activated macrophages found in the alveolar airways of these mice may contribute to the increased damage seen. The determination of proteolytic enzyme production by these macrophages and the influence of adenosine signaling on this process will help clarify the role of activated macrophages in this model. The number of macrophages found in the lungs of ADA-deficient mice was not altered 72 h after PEG-ADA treatments, nor was there any improvement in the alveolar damage seen. The persistence of macrophages may indicate that these cells are actively involved in the clearance of cellular debris resultant of the severe eosinophilia and tissue damage seen. The ability to control adenosine levels using varying doses of PEG-ADA will provide a useful tool to explore the involvement of adenosine signaling in both eosinophil and macrophage function.
ADA deficiency in humans is most commonly associated with a combined immunodeficiency 7. However, additional phenotypes have been described, including bone and renal abnormalities 12, hepatocellular damage 13, neurological disorders, and pulmonary insufficiencies 7. Although the treatment of ADA-deficient patients with PEG-ADA has rapid beneficial effects on some of these phenotypes 14 55, it is still not clear whether they are a primary consequence of the ADA deficiency. Here, we demonstrate that ADA enzyme therapy can rapidly reverse respiratory distress in ADA-deficient mice in conjunction with lowering lung adenosine and 2′-deoxyadenosine levels, suggesting that the respiratory distress seen in this model is a direct consequence of ADA deficiency. This suggestion is supported by observations that the ADA enzyme therapy protocol used did not improve the immune status in these animals. Pulmonary insufficiency is common in ADA-deficient patients, and these insufficiencies are most often attributed to bacterial or viral pneumonia that arises from a compromised immune system. However, in many cases of interstitial pneumonia an organism cannot be isolated 7. Our observations in ADA-deficient mice suggest that it is possible that the adenine metabolic disturbances in ADA-deficient patients may directly contribute to the pulmonary insufficiency occurring in this population.
Some ADA-deficient patients have been shown to have elevated levels of IgE, eosinophilia, and an increased incidence of asthma 8 38 39. These individuals are typically patients with delayed or late onset ADA deficiency and thus have milder forms of immunodeficiency 7 8. ADA-deficient mice exhibited an increase in serum IgE, eosinophilia, and developed lung inflammatory changes, suggesting that they resemble patients with a less severe form of ADA deficiency. Consistent with this is the observation that the immunodeficiency seen in ADA-deficient mice is not as severe as that seen in completely ADA-deficient humans 15. However, the immunodeficiency seen in these animals must be considered when trying to understand the nature of the lung inflammation seen. Lung eosinophilia is often associated with a Th2 cytokine profile 56. However, there was not a robust Th2 cytokine profile in BALF collected from ADA-deficient mice. Since Th2 cytokines such as IL-4 are produced largely by CD4 T cells, the immunodeficiency seen in ADA-deficient mice may impact the relative capability to generate Th2 cytokines. Alternatively, the absence of a robust Th2 response suggests that other signaling pathways are involved in mediating the lung eosinophilia seen.
In conclusion, by deleting the enzyme responsible for controlling the levels of adenosine and 2′-deoxyadenosine, we have generated animals that exhibit adenine metabolic disturbances in association with alveolar defects and the development of severe pulmonary inflammation. Lung eosinophilia was reduced and the animals were rescued from respiratory distress by lowering adenosine and 2′-deoxyadenosine levels using ADA enzyme therapy. Although 2′-deoxyadenosine–mediated effects on this phenotype cannot be ruled out, there is substantial evidence to suggest adenosine signaling may be playing an important role in the type of inflammation and tissue damage seen 20. Defining the adenosine receptors expressed on eosinophils, macrophages, and in the lungs of ADA-deficient mice, and using pharmacological and genetic technologies to assess their function, will help us to understand how adenosine influences lung inflammation in this model. This may in turn help guide new therapies for the treatment of lung conditions in which eosinophils and macrophages are thought to mediate damage, including asthma, idiopathic eosinophilic lung inflammation, chronic obstructive pulmonary disease, and emphysema. The correlation of increased lung adenosine and asthma 21 and the ability to relieve lung eosinophilia in mice by lowering adenosine levels raise the possibility that ADA enzyme therapy may be beneficial in the treatment of eosinophilic lung inflammation. Using ADA-deficient mice as a testing ground to understand the basis for adenosine-dependent lung eosinophilia will aid in evaluating the efficacy of such therapies.
Acknowledgments
We thank Drs. Michael S. Hershfield and Giuseppe N. Colasurdo for their helpful discussions and review of the manuscript.
This work was supported by National Institutes of Health grants AI43572 and HL61888 (both to M.R. Blackburn), DK46207, and DK54443 (R.E. Kellems), and Texas Higher Education Coordinating Board Applied Technology Grant 011618-060 (M.R. Blackburn). Funds for this work were also provided by the Mayo Foundation (J.J. Lee).
Footnotes
Abbreviations used in this paper: ADA, adenosine deaminase; BALF, bronchial alveolar lavage fluid; H&E, hematoxylin and eosin; mMBP-1, murine eosinophil granule major basic protein 1; PAS, periodic acid-Schiff; PEG, polyethylene glycol.
References
- Frederiksen S. Specificity of adenosine deaminase toward adenosine and 2′-deoxyadenosine analogues. Arch. Biochem. Biophys. 1966;113:383–388. doi: 10.1016/0003-9861(66)90202-5. [DOI] [PubMed] [Google Scholar]
- Olah M.E., Stiles G.L. Adenosine receptor subtypescharacterization and therapeutic regulation. Annu. Rev. Pharmacol. Toxicol. 1995;35:581–606. doi: 10.1146/annurev.pa.35.040195.003053. [DOI] [PubMed] [Google Scholar]
- Hershfield M.S., Kredich N.M., Ownby D.R., Ownby H., Buckley R. In vivo inactivation of erythrocyte S-adenosylhomocysteine hydrolase by 2′-deoxyadenosine in adenosine deaminase–deficient patients. J. Clin. Invest. 1979;63:807–811. doi: 10.1172/JCI109367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benveniste P., Zhu W., Cohen A. Interference with thymocyte differentiation by an inhibitor of S-adenosylhomocysteine hydrolase. J. Immunol. 1995;155:536–544. [PubMed] [Google Scholar]
- Benveniste P., Cohen A. p53 expression is required for thymocyte apoptosis induced by adenosine deaminase deficiency. Proc. Natl. Acad. Sci. USA. 1995;92:8373–8377. doi: 10.1073/pnas.92.18.8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Kim C.N., Yang J., Jemmerson R., Wang X. Induction of apoptotic program in cell-free extractsrequirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Hershfield M.S., Mitchell B.S. Immunodeficiency diseases caused by adenosine deaminase deficiency and purine nucleoside phosphorylase deficiency. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. The Metabolic and Molecular Basis of Inherited Disease., Vol. 1. 7th ed. McGraw-Hill, Inc.; New York: 1995. pp. 1725–1768. [Google Scholar]
- Hirschhorn R. Immunodeficiency disease due to deficiency of adenosine deaminase. In: Ochs H.D., Smith C.I.E., Puck J.M., editors. Primary Immunodeficiency DiseaseA Molecular and Genetic Approach. Oxford University Press; New York: 1999. pp. 121–138. [Google Scholar]
- Donofrio J., Coleman M.S., Hutton J.J., Daoud A., Lampkin B., Dyminski J. Overproduction of adenine deoxynucleosides and deoxynucleotides in adenosine deaminase deficiency with severe combined immunodeficiency disease. J. Clin. Invest. 1978;62:884–887. doi: 10.1172/JCI109201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizaki H., Suzuki K., Tadakuma T., Ishimura Y. Adenosine receptor-mediated accumulation of cyclic AMP-induced T-lymphocyte death through internucleosomal DNA cleavage. J. Biol. Chem. 1990;265:5280–5284. [PubMed] [Google Scholar]
- Huang S., Apasov S., Koshiba M., Sitkovsky M. Role of A2a extracellular adenosine receptor-mediated signaling in adenosine-mediated inhibition of T-cell activation and expansion. Blood. 1997;90:1600–1610. [PubMed] [Google Scholar]
- Ratech H., Greco M.A., Gallo G., Rimoin D.L., Kamino H., Hirschhorn R. Pathologic findings in adenosine deaminase-deficient severe combined immunodeficiency. I. Kidney, adrenal, and chondro-osseous tissue alterations. Am. J. Pathol. 1985;120:157–169. [PMC free article] [PubMed] [Google Scholar]
- Bollinger M.E., Arredondo-Vega F.X., Santisteban I., Schwarz K., Hershfield M.S., Lederman H.M. Brief reporthepatic dysfunction as a complication of adenosine deaminase deficiency. N. Engl. J. Med. 1996;334:1367–1371. doi: 10.1056/NEJM199605233342104. [DOI] [PubMed] [Google Scholar]
- Hirschhorn R., Paageorgiou P.S., Kesarwala H.H., Taft L.T. Amelioration of neurologic abnormalities after “enzyme replacement” in adenosine deaminase deficiency. N. Engl. J. Med. 1980;303:377–380. doi: 10.1056/NEJM198008143030706. [DOI] [PubMed] [Google Scholar]
- Blackburn M.R., Datta S.K., Kellems R.E. Adenosine deaminase-deficient mice generated using a two-stage genetic engineering strategy exhibit a combined immunodeficiency. J. Biol. Chem. 1998;273:5093–5100. doi: 10.1074/jbc.273.9.5093. [DOI] [PubMed] [Google Scholar]
- Wakamiya M., Blackburn M.R., Jurecic R., McArthur M.J., Geske R.S., Cartwright J., Jr., Mitani K., Vaishnav S., Belmont J.W., Kellems R.E. Disruption of the adenosine deaminase gene causes hepatocellular impairment and perinatal lethality in mice. Proc. Natl. Acad. Sci. USA. 1995;92:3673–3677. doi: 10.1073/pnas.92.9.3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migchielsen A.A., Breuer M.L., van Roon M.A., te Riele H., Zurcher C., Ossendorp F., Toutain S., Hershfield M.S., Berns A., Valerio D. Adenosine-deaminase-deficient mice die perinatally and exhibit liver-cell degeneration, atelectasis and small intestinal cell death. Nat. Genet. 1995;10:279–287. doi: 10.1038/ng0795-279. [DOI] [PubMed] [Google Scholar]
- Blackburn M.R., Wakamiya M., Caskey C.T., Kellems R.E. Tissue-specific rescue suggests that placental adenosine deaminase is important for fetal development in mice. J. Biol. Chem. 1995;270:23891–23894. doi: 10.1074/jbc.270.41.23891. [DOI] [PubMed] [Google Scholar]
- Blackburn M.R., Datta S.K., Wakamiya M., Vartabedian B.S., Kellems R.E. Metabolic and immunologic consequences of limited adenosine deaminase expression in mice. J. Biol. Chem. 1996;271:15203–15210. doi: 10.1074/jbc.271.25.15203. [DOI] [PubMed] [Google Scholar]
- Jacobson M.A., Bai T.R. The role of adenosine in asthma. In: Jacobson K.A., Jarvis M.F., editors. Purinergic Approaches in Experimental Therapeutics. Wiley-Liss, Inc.; Danvers, MA: 1997. pp. 315–331. [Google Scholar]
- Driver A.G., Kukoly C.A., Ali S., Mustafa S.J. Adenosine in bronchoalveolar lavage fluid in asthma. Am. Rev. Respir. Dis. 1993;148:91–97. doi: 10.1164/ajrccm/148.1.91. [DOI] [PubMed] [Google Scholar]
- Cushley M.J., Tattersfield A.E., Holgate S.T. Inhaled adenosine and guanosine on airway resistance in normal and asthmatic subjects. Br. J. Clin. Pharmacol. 1983;15:161–165. doi: 10.1111/j.1365-2125.1983.tb01481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker B.A., Jacobson M.A., Knight D.A., Salvatore C.A., Weir T., Zhou D., Bai T.R. Adenosine A3 receptor expression and function in eosinophils. Am. J. Respir. Cell Mol. Biol. 1997;16:531–537. doi: 10.1165/ajrcmb.16.5.9160835. [DOI] [PubMed] [Google Scholar]
- Barnes P.J. Current therapies for asthma. Promise and limitations. Chest. 1997;111:17S–26S. doi: 10.1378/chest.111.2_supplement.17s. [DOI] [PubMed] [Google Scholar]
- Marquardt D.L., Parker C.W., Sullivan T.J. Potentiation of mast cell mediator release by adenosine. J. Immunol. 1978;120:871–878. [PubMed] [Google Scholar]
- Fozard J.R., Pfannkuche H.J., Schuurman H.J. Mast cell degranulation following adenosine A3 receptor activation in rats. Eur. J. Pharmacol. 1996;298:293–297. doi: 10.1016/0014-2999(95)00822-5. [DOI] [PubMed] [Google Scholar]
- Knight D., Zheng X., Rocchini C., Jacobson M., Bai T., Walker B. Adenosine A3 receptor stimulation inhibits migration of human eosinophils. J. Leukoc. Biol. 1997;62:465–468. doi: 10.1002/jlb.62.4.465. [DOI] [PubMed] [Google Scholar]
- Ezeamuzie C.I., Philips E. Adenosine A3 receptors on human eosinophils mediate inhibition of degranulation and superoxide anion release. Br. J. Pharmacol. 1999;127:188–194. doi: 10.1038/sj.bjp.0702476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno Y., Ji X., Mawhorter S.D., Koshiba M., Jacobson K.A. Activation of A3 adenosine receptors on human eosinophils elevates intracellular calcium. Blood. 1996;88:3569–3574. [PMC free article] [PubMed] [Google Scholar]
- Merrill J.T., Shen C., Schreibman D., Coffey D., Zakharenko O., Fisher R., Lahita R.G., Salmon J., Cronstein B.N. Adenosine A1 receptor promotion of multinucleated giant cell formation by human monocytesa mechanism for methotrexate-induced nodulosis in rheumatoid arthritis. Arthritis Rheum. 1997;40:1308–1315. doi: 10.1002/1529-0131(199707)40:7<1308::AID-ART16>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Najar H.M., Ruhl S., Bru-Capdeville A.C., Peters J.H. Adenosine and its derivatives control human monocyte differentiation into highly accessory cells versus macrophages. J. Leukoc. Biol. 1990;47:429–439. doi: 10.1002/jlb.47.5.429. [DOI] [PubMed] [Google Scholar]
- Eppell B.A., Newell A.M., Brown E.J. Adenosine receptors are expressed during differentiation of monocytes to macrophages in vitro. Implications for regulation of phagocytosis. J. Immunol. 1989;143:4141–4145. [PubMed] [Google Scholar]
- Revan S., Montesinos M.C., Naime D., Landau S., Cronstein B.N. Adenosine A2 receptor occupancy regulates stimulated neutrophil function via activation of a serine/threonine protein phosphatase. J. Biol. Chem. 1996;271:17114–17118. doi: 10.1074/jbc.271.29.17114. [DOI] [PubMed] [Google Scholar]
- Walker B.A., Rocchini C., Boone R.H., Ip S., Jacobson M.A. Adenosine A2a receptor activation delays apoptosis in human neutrophils. J. Immunol. 1997;158:2926–2931. [PubMed] [Google Scholar]
- Lee J.J., McGarry M.P., Farmer S.C., Denzler K.L., Larson K.A., Carrigan P.E., Brenneise I.E., Horton M.A., Haczku A., Gelfand E.W. Interleukin-5 expression in the lung epithelium of transgenic mice leads to pulmonary changes pathognomonic of asthma. J. Exp. Med. 1997;185:2143–2156. doi: 10.1084/jem.185.12.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escolar J.D., Gallego B., Tejero C., Escolar M.A. Changes occurring with increasing age in the rat lungmorphometrical study. Anat. Rec. 1994;239:287–296. doi: 10.1002/ar.1092390307. [DOI] [PubMed] [Google Scholar]
- Knudsen T.B., Winters R.S., Otey S.K., Blackburn M.R., Airhart M.J., Church J.K., Skalko R.G. Effects of (R)-deoxycoformycin (pentostatin) on intrauterine nucleoside catabolism and embryo viability in the pregnant mouse. Teratology. 1992;45:91–103. doi: 10.1002/tera.1420450109. [DOI] [PubMed] [Google Scholar]
- Levy Y., Hershfield M.S., Fernandez-Mejia C., Polmar S.H., Scudiery D., Berger M., Sorensen R.U. Adenosine deaminase deficiency with late onset of recurrent infectionsresponse to treatment with polyethylene glycol-modified adenosine deaminase. J. Pediatr. 1988;113:312–317. doi: 10.1016/s0022-3476(88)80271-3. [DOI] [PubMed] [Google Scholar]
- Shovlin C.L., Hughes J.M., Simmonds H.A., Fairbanks L., Deacock S., Lechler R., Roberts I., Webster A.D. Adult presentation of adenosine deaminase deficiency. Lancet. 1993;341:1471. doi: 10.1016/0140-6736(93)90910-9. [DOI] [PubMed] [Google Scholar]
- Hershfield M.S., Chaffee S., Sorensen R.U. Enzyme replacement therapy with polyethylene glycol-adenosine deaminase in adenosine deaminase deficiencyoverview and case reports of three patients, including two now receiving gene therapy. Pediatr. Res. 1993;33:S42–S48. doi: 10.1203/00006450-199305001-00236. [DOI] [PubMed] [Google Scholar]
- Xu P.A., Winston J.H., Datta S.K., Kellems R.E. Regulation of forestomach-specific expression of the murine adenosine deaminase gene. J. Biol. Chem. 1999;274:10316–10323. doi: 10.1074/jbc.274.15.10316. [DOI] [PubMed] [Google Scholar]
- Weinstein M., Xu X., Ohyama K., Deng C.X. FGFR-3 and FGFR-4 function cooperatively to direct alveogenesis in the murine lung. Development. 1998;125:3615–3623. doi: 10.1242/dev.125.18.3615. [DOI] [PubMed] [Google Scholar]
- Bostrom H., Willetts K., Pekny M., Leveen P., Lindahl P., Hedstrand H., Pekna M., Hellstrom M., Gebre-Medhin S., Schalling M. PDGF-A signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell. 1996;85:863–873. doi: 10.1016/s0092-8674(00)81270-2. [DOI] [PubMed] [Google Scholar]
- Kaartinen V., Voncken J.W., Shuler C., Warburton D., Bu D., Heisterkamp N., Groffen J. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat. Genet. 1995;11:415–421. doi: 10.1038/ng1295-415. [DOI] [PubMed] [Google Scholar]
- Ray P., Tang W., Wang P., Homer R., Kuhn C., III, Flavell R.A., Elias J.A. Regulated overexpression of interleukin 11 in the lung. Use to dissociate development-dependent and -independent phenotypes. J. Clin. Invest. 1997;100:2501–2511. doi: 10.1172/JCI119792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z., Homer R.J., Wang Z., Chen Q., Geba G.P., Wang J., Zhang Y., Elias J.A. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J. Clin. Invest. 1999;103:779–788. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strek M.K., Leff A.R. Eosinophils. In: Barnes P.J., Leff A.R., Grunstein M.M., Woolcock A.J., editors. Asthma. . Vol. 1. Lippincott-Raven; Philadelphia: 1997. pp. 353–365. [Google Scholar]
- Leff A.R. Inflammatory mediation of airway hyperresponsiveness by peripheral blood granulocytes. The case for the eosinophil. Chest. 1994;106:1202–1208. doi: 10.1378/chest.106.4.1202. [DOI] [PubMed] [Google Scholar]
- Gleich G.J. The eosinophil and bronchial asthmacurrent understanding. J. Allergy Clin. Immunol. 1990;85:422–436. doi: 10.1016/0091-6749(90)90151-s. [DOI] [PubMed] [Google Scholar]
- Johnson H.G., McNee M.L. Adenosine-induced secretion in the canine tracheamodification by methylxanthines and adenosine derivatives. Br. J. Pharmacol. 1985;86:63–67. doi: 10.1111/j.1476-5381.1985.tb09435.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasko G., Szabo C., Nemeth Z.H., Kvetan V., Pastores S.M., Vizi E.S. Adenosine receptor agonists differentially regulate IL-10, TNF-alpha, and nitric oxide production in RAW 264.7 macrophages and in endotoxemic mice. J. Immunol. 1996;157:4634–4640. [PubMed] [Google Scholar]
- Sajjadi F.G., Takabayashi K., Foster A.C., Domingo R.C., Firestein G.S. Inhibition of TNF-alpha expression by adenosinerole of A3 adenosine receptors. J. Immunol. 1996;156:3435–3442. [PubMed] [Google Scholar]
- Salmon J.E., Brogle N., Brownlie C., Edberg J.C., Kimberly R.P., Chen B.X., Erlanger B.F. Human mononuclear phagocytes express adenosine A1 receptors. A novel mechanism for differential regulation of Fc gamma receptor function. J. Immunol. 1993;151:2775–2785. [PubMed] [Google Scholar]
- Abboud R.T., Ofulue A.F., Sansores R.H., Muller N.L. Relationship of alveolar macrophage plasminogen activator and elastase activities to lung function and CT evidence of emphysema. Chest. 1998;113:1257–1263. doi: 10.1378/chest.113.5.1257. [DOI] [PubMed] [Google Scholar]
- Bollinger M.E., Arredondo-Vega F.X., Santisteban I., Schwarz K., Hershfield M.S., Lederman H.M. Hepatic dysfunction as a complication of adenosine deaminase deficiency. N. Engl. J. Med. 1996;334:1367–1371. doi: 10.1056/NEJM199605233342104. [DOI] [PubMed] [Google Scholar]
- Barnes P.J., Holgate S.T., Laitinen L.A., Pauwels R. Asthma mechanisms, determinants of severity and treatmentthe role of nedocromil sodium. Report of a workshop held in Whistler, British Columbia, Canada, 18–19 May 1995. Clin. Exp. Allergy. 1995;25:771–787. doi: 10.1111/j.1365-2222.1995.tb00016.x. [DOI] [PubMed] [Google Scholar]