

Abstract
Immune surveillance by cytotoxic lymphocytes against cancer has been postulated for decades, but direct evidence for the role of cytotoxic lymphocytes in protecting against spontaneous malignancy has been lacking. As the rejection of many experimental cancers by cytotoxic T lymphocytes and natural killer cells is dependent on the pore-forming protein perforin (pfp), we examined pfp-deficient mice for increased cancer susceptibility. Here we show that pfp-deficient mice have a high incidence of malignancy in distinct lymphoid cell lineages (T, B, NKT), indicating a specific requirement for pfp in protection against lymphomagenesis. The susceptibility to lymphoma was accentuated by simultaneous lack of expression of the p53 gene, mutations in which also commonly predispose to human malignancies, including lymphoma. In contrast, the incidence and age of onset of sarcoma was unaffected in p53-deficient mice. Pfp-deficient mice were at least 1,000-fold more susceptible to these lymphomas when transplanted, compared with immunocompetent mice in which tumor rejection was controlled by CD8+ T lymphocytes. This study is the first that implicates direct cytotoxicity by lymphocytes in regulating lymphomagenesis.
Keywords: tumor immunity, perforin, lymphoma, cytotoxic T lymphocyte, surveillance
Introduction
Burnet first forwarded the concept of tumor immune surveillance 1 and others, such as Thomas 2, have developed this idea. Since then, it has largely been assumed that both innate (including NK cells) and acquired (including CTLs) mechanisms may contribute to host antitumor immunity by destroying transformed cells or creating a local environment that suppresses tumor growth. Although direct cytotoxicity by NK cells and CTLs can be used as one critical indicator of antitumor immunity in experimental cancer models 3, a good correlation is not found in most spontaneously occurring human cancers, or even in human immunotherapy trials designed to stimulate antitumor cytotoxicity 4. Perforin (pfp), exclusively expressed by NK cells and CTLs 5 6, is essential for inducing tumor cell apoptosis in vitro 7 8 and pfp-deficient mice show an increase in the growth 9 10 and metastasis 11 of experimental tumors. Nevertheless, evidence for a role of killer cell cytotoxicity in surveillance against spontaneous cellular transformation is lacking. In addition, none of the tumor models used to study antitumor cytotoxicity have accurately mimicked the clinical setting where tumors arise sporadically after clonal expansion of a single transformed cell.
We chose to address this issue by examining whether pfp-deficient mice have an abnormal incidence or an altered pathological spectrum of cancer over their life-span. This approach presented a potential problem, as it had previously been reported that pfp-deficient mice up to 12 mo of age are not abnormally prone to malignancy 9, indicating that any increased susceptibility to cancer would take considerable time to become apparent. Mutations of p53, a key regulator of apoptosis and cell-cycle arrest in response to DNA damage 12, are found in approximately half of all human cancers 13 14, and inherited mutations exemplified by Li-Fraumeni syndrome are strongly associated with increased susceptibility to epithelial, lymphoid and connective tissue malignancies 15. We thus elected to examine whether tumorigenesis would be accelerated in tumor-prone p53-deficient mice 16 17 that also lacked pfp expression.
Materials and Methods
Mice.
C57BL/6 × 129/Sv pfp-deficient mice (18; courtesy of Dr. William Clark, University of California, Los Angeles, CA) were backcrossed five generations to C57BL/6 mice (purchased from the Walter and Eliza Hall Institute for Medical Research, Parkville, Australia). These mice were then bred to C57BL/6 (B6).p53+/− mice (backcross n = 15) obtained from Dr. Alan Harris (Walter and Eliza Hall Institute for Medical Research) to generate mice that were heterozygous for both the perforin and p53 alleles.
Experimental Carcinogenesis.
A total of 48 pfp+/−p53+/− founder parent pairs were used to generate progeny that expressed all possible combinations of alleles (9 genotypes). Each genotype comprised ∼50% male and 50% female mice. All mice were bred and maintained in a single clean room of the Austin Research Institute Biological Research Laboratories. Soon after birth, the genotype of each mouse was determined from genomic DNA (sampled by tail clipping) using both Southern blot analysis and PCR. The results were recorded and the mice were then raised anonymously in their original litters for the length of the experiment. The PCR method for screening pfp used the following oligomers: 5′-CGT GAG AGG TCA GCA TCC TTC-3′ (sense primer in exon 2); 5′-TGG CCT AGG GTT CAC ATC CAG-3′ (antisense primer in exon 2, downstream of the unique SmaI site into which the neomycin resistance gene cassette was inserted to disrupt the coding sequence); and 5′-ATA TTG GCT GCA GGG TCG CTC-3′ (antisense primer within neomycin resistance gene cassette). The screening of p53 used the following oligomers: 5′-TTA TGA GCC ACC CGA GGT-3′; 5′-TAT ACT CAG AGC CGG CCT-3′; and 5′-TCC TCG TGC TTT ACG GTA TC-3′. All mice were routinely screened for viruses, parasites, and other microbes and tested negative over the entire course of the experiment. Mice within the study were monitored for health and weighed twice weekly. Any mouse with an abnormality (palpable mass, abdominal distension, weight loss >10%, or ruffled fur) was killed, its age was recorded, and a postmortem was performed. Mean age of death ± SEM was calculated and the probability of significance was determined using a nonparametric Mann-Whitney test. The significance of proportions of tumors and in particular disseminated lymphomas was determined by a Fisher's exact test.
Histopathology and Surface Phenotyping of Tumors.
A full autopsy was performed at death, and tumors (macroscopically detected), spleen, liver, thymus, and lymph nodes were routinely examined by histology after fixing these tissues in formalin and on occasion also fresh freezing them. The preparation and staining of sections for histology were carried out by the Department of Anatomical Pathology, Austin and Repatriation Medical Centre. No data from these analyses suggested to us that pfp−/−/p53 mutant mice developed a spectrum of tumors that was distinct from that typically reported and observed in corresponding pfp+/+/p53 mutant mice (i.e., tumors were primarily sarcomas and lymphomas). Lymphomas from B6.pfp−/−p53+/− mice were also assessed for surface phenotype by multiparameter flow cytometric analysis. The following reagents were used: anti–α/β-TCR–FITC (H57-597; PharMingen); anti–NK1.1-PE (PK136; PharMingen); anti–CD4-FITC (CT4; Caltag Laboratories); anti–CD8α-APC (53.6.7; PharMingen); anti–CD8β-biotin (53.5.8; PharMingen); anti–CD11c-FITC (HL3; PharMingen); anti–B220-PE (RA3-6B2; Caltag Laboratories); anti–Thy-1–PE (30-H12; PharMingen); anti–CD45.2-FITC (104; PharMingen); anti–γ/δ-TCR–biotin (clone GL3; PharMingen); anti–Mac-1–biotin (M170; Caltag Laboratories); goat anti–mouse-Ig–FITC (Silenus Laboratories); and anti–Streptavidin-tricolor (Caltag Laboratories). Anti-Fc receptor (2.4G2) was used to prevent nonspecific binding by mAb. Staining was carried out in PBS with 5% FCS and 0.02% sodium azide on ice. Fresh splenocytes from 6-wk-old B6 mice were always used as labeling controls. Analysis was carried out on a FACScalibur™ using CellQuest software (Becton Dickinson).
Tumor Transplantation Experiments.
Nine lymphomas were transplanted directly from B6.pfp−/−p53+/− or pfp−/−p53+/+ mice into B6 and B6.pfp−/− mice and subsequently maintained in B6.pfp−/− mice. Two of these, designated PN53H-1 (B220+ H-2Kb+H-2Db+Ig+) and PN53H-7 (CD8α/β1B220+lin−H-2Kb+ H-2Db+), were randomly chosen to further investigate factors regulating their growth in B6 mice. Groups of five B6 or B6.pfp−/− mice were injected intraperitoneally with increasing numbers of lymphoma cells and observed daily for tumor growth for >100 d. Some groups of B6 mice were depleted of NK1.1+, Thy-1+, or CD8+ cells in vivo, by treatment with 100 μg anti-NK1.1 (PK136), anti–Thy-1 (T24/31.7) mAb, or anti-CD8 (53-6.7) respectively, on days −2 and 0 (day of i.p. tumor inoculation) and weekly thereafter. This protocol has been demonstrated to effectively deplete NK1.1+, Thy-1+, or CD8+ cells 11.
Results and Discussion
Mixed litters comprising the nine resulting genotypes of pfp+/−p53+/− intercrossed mice were monitored for tumor development (Fig. 1). All the offspring were observed for up to 2 yr, under standard (non-pathogen free) boarding conditions.
Figure 1.
Perforin protects mice from spontaneous lymphomas. The appearance of tumors was recorded in mice derived from a pfp+/−p53+/− intercross. Groups of mice (n = 16–34) were evaluated on a weekly basis and when moribund, tumor type (□, thymic lymphoma; ▪, disseminated lymphoma; ▴, sarcoma; ○, other tumors) recorded against the age at time of death/autopsy (in days). The perforin genotype (pfp−/−, pfp+/−, or pfp+/+) is also displayed on the x-axis. (A) p53+/+, (B) p53+/−, or (C) p53−/−. The MA of p53−/− mice developing disseminated lymphoma is depicted by the crossbar in C. All p53−/− mice studied developed tumors and the numbers of p53+/− and p53+/+ mice developing lymphomas are shown in parentheses. In D, the percentage of pfp−/− and pfp+/+ mice free of disseminated lymphoma (from A and B) are plotted against their age at time of death in days, and P values (Fisher's exact test) comparing the proportions of disseminated lymphoma between pfp−/− and pfp+/+ mice are indicated.




Perforin-deficient Mice Develop Spontaneous Late Onset Lymphoma.
As previously reported 9, no pfp−/−p53+/+ mice developed cancer before 12 mo of age. Soon thereafter, however, these mice began to succumb to aggressive disseminated lymphomas affecting the spleen, liver and lymph nodes (between 436 and 635 d, mean age at time of death [MA] = 569 ± 22 d). The difference in lymphoma incidence in pfp−/−p53+/+ (10 out of 20) and pfp+/+p53+/+ (1 out of 16) was highly significant (P = 0.0091 Fisher's exact test; Fig. 1a and Fig. d). As mice heterozygous for pfp expression (pfp+/−p53+/+) did not develop any lymphomas (0 out of 20), <3% (1 out of 36) of the mice expressing at least one perforin allele succumbed to lymphoma. Given the group sizes in our experiments, the collective incidence of nonlymphatic malignancies in these groups of mice (3 out of 56, comprising two sarcomas and one hepatoma) was too low to evaluate whether pfp expression influenced susceptibility to other types of cancer.
p53 Deficiency Accentuates Lymphomagenesis.
The association between pfp deficiency and susceptibility to lymphoma was strongly reiterated in p53+/− mice, and the MA was also hastened by 90 d compared with p53+/+ mice. Pfp−/−p53+/− mice typically developed lymphoma between days 338 and 606 (MA = 569 ± 22 d vs. 479 ± 22 d; P = 0.0264, Mann-Whitney). In agreement with Jacks et al. 16, p53+/− mice expressing wild-type pfp levels rarely developed spontaneous tumors before day 250, and 50% (15 out of 30) had developed tumors by day 650 (Fig. 1b and Fig. d). Strikingly, however, only 2 out of these 15 tumors were disseminated lymphomas. Thus, both the incidence of tumors (23 out of 27; P = 0.0058, Fisher's exact test) and the proportion of lymphomas (16 out of 23; P = 0.0019) were dramatically increased in pfp−/−p53+/− mice. By contrast, the incidence and age of onset of sarcomas or other tumors were not significantly different between the two groups. Overall, we view the effect of p53 deficiency on lymphomagenesis as an amplification of the effects of failed pfp-dependent immune surveillance against transformed lymphocytes. Although additional p53 heterozygosity clearly resulted in an earlier onset of lymphoma in pfp-deficient mice, the incidence of lymphoma was unaffected (16 out of 27 vs. 10 out of 20, respectively; P = 0.2587). Therefore pfp, and not p53, plays an indispensable role against the development of lymphoma in otherwise normal inbred mice.
Also consistent with previous reports 16 17, all pfp+/+ p53−/− mice developed a spectrum of tumors, predominantly thymic lymphomas, disseminated lymphomas, or sarcomas arising in soft tissue, bone, or blood vessel endothelium by day 300 (MA ± SE = 172 ± 8 d, n = 34; Fig. 1 C). The distribution of tumor types was indistinguishable in pfp−/−p53−/− mice, and the MA of this group was not significantly reduced (151 ± 9 d; P = 0.1128, Mann-Whitney). However, when tumor type was taken into account, it was apparent that the MA was once again significantly reduced for disseminated lymphomas (144 ± 15 vs. 197 ± 15; P = 0.0328), but interestingly, not for sarcomas (147 ± 16 vs. 154 ± 12 d; P > 0.05) or thymic lymphomas (150 ± 14 vs. 160 ± 18 days; P > 0.05). Therefore, within the limits of these group sizes, pfp did not appear to control the development of spontaneous thymic lymphomas in p53 mutant mice. This data may be consistent with the fact that mature effector T cells do not typically recirculate through the thymus, and whether immunosurveillance of thymic lymphoid cells fails to occur by other means remains to be demonstrated. To illustrate the significance of the earlier onset of disseminated lymphomas in pfp−/−p53−/− mice, the reduction in MA was comparable to that observed by others when p53−/− mice received added oncogenic stimuli (γ-irradiation as neonates 19 or a coincident deficiency of one Rb 20 or scid 21 allele), or immune compromise due to deficiency of IFN-γR expression 22. In contrast to IFN-γR−/−p53−/− mice in which increased carcinogenesis might be due to a lack of any of the pleiotropic effects of IFN-γ 23, our study with pfp-deficient mice represents the first in which increased cancer susceptibility can be related precisely to a lack of cytolytic lymphocyte effector function.
There was no difference in tumor histology or spread of lymphoma in pfp+/+ and pfp−/− mice (data not shown). Similarly, the proportions of sarcomas that were undifferentiated or arising in blood vessels or bone in pfp−/−p53+/− and pfp+/+p53+/− mice were unaltered. It was noted that pfp+/−p53+/− and pfp+/−p53−/− mice displayed a tumor incidence, spectrum and onset that was not statistically different from pfp+/+ mice sharing the same p53 genotype (data not shown). As pfp+/− mice may have only half the normal level of pfp protein expression, we were also interested to find that pfp+/−p53−/− mice had an intermediate onset of lymphoma (174 ± 15 d) compared with pfp+/+p53−/− (197 ± 15 d) or pfp−/−p53−/− (144 ± 15 d). Although this finding could be consistent with a gene dose effect in lymphoma surveillance, the MA for pfp heterozygotes was not significantly different from either pfp+/+p53−/− or pfp−/− p53−/− mice (P > 0.2, Mann-Whitney). Furthermore, no evidence of a pfp gene dose effect was seen with either the p53+/+ or p53+/− cohorts.
A Variety of Lymphoma Phenotypes.
To further characterize the disseminated lymphomas arising in pfp−/−p53+/− and pfp−/−p53+/+ mice, a total of 11 tumors were passaged intraperitoneally in B6.pfp−/− and/or scid mice. Of these, nine were successfully passaged, and eight were analyzed by flow cytometry. Unlike previously described lymphomas in p53+/− or p53−/− mice (reference 21 and our unpublished data), which were predominantly of T cell origin, the tumors arising in pfp−/−p53+/− and pfp−/−p53+/+ mice included three of B cell origin (B220+sIg+CD4−CD8−TCR−), three lymphomas of uncertain lineage (CD45+lin−CD8α/β1/− B220+/−), one T cell lymphoma (CD8α/β+TCR-α/β+ B220−CD4−), and one NK T cell lymphoma (NK1.1+ TCR-α/β+B220+CD4−CD8−). All lymphomas examined were MHC class I+ (H-2Kb and H-2Db) and CD1d+ and thus potentially capable of presenting tumor antigen to MHC class I–restricted CTLs.
CTL-mediated Rejection of Transplanted Lymphomas.
To establish the malignancy of these tumors and that immune surveillance had failed in pfp-deficient mice, lymphomas were transplanted into wild-type B6 or B6.pfp−/− mice. Two representative experiments of the nine lymphomas that were inoculated are depicted in Fig. 2. Surprisingly, as few as 103 lymphoma cells (PN53H-1, B220+H-2Kb+ H-2Db+Ig+) killed all B6.pfp−/− mice or B6 mice depleted of Thy-1+ or CD8+ cells within 60–80 d of i.p. inoculation (Fig. 2 A). Similarly, only 103 PN53H-7 lymphoma (CD8α/β+B220+lin−H-2Kb+H-2Db+) cells killed all B6.pfp−/− mice or B6 mice depleted of Thy-1+ or CD8+ cells within 25–55 d of i.p. inoculation (Fig. 2 B). When transplanted, these tumors (and the other lymphomas studied) displayed a pattern of organ infiltration similar to that originally observed. In contrast, up to 106 PN53H-1 or PN53H-7 lymphoma cells were effectively cleared in untreated or anti-NK1.1–treated B6 mice (Fig. 2), and the majority of B6 mice receiving even greater doses of inoculum (>107) did not develop tumor (data not shown). Interestingly, one B6 mouse that did succumb to higher doses of PN53H-1 cells developed a lymphoma that appeared to have escaped immune detection by losing expression of MHC class I Kb (Db+Kb−). Upon subsequent transfer, this lymphoma grew in both B6 and B6.pfp−/− mice at doses from 105–107 cells (data not shown). Despite the fact that all lymphomas arising in pfp−/−p53+/− mice were efficiently killed by IL-2–activated NK cells from B6 (data not shown), tumor growth occurred with 104–105 times fewer cells in B6.pfp−/− mice, and T cells, not NK cells, were responsible for lymphoma rejection. To eliminate any possibility that minor 129 alloantigens potentially expressed by the lymphomas may have accounted for their rapid rejection in B6 mice, five of the lymphomas (including PN53H-1) were transplanted (107 cells) into (B6 × 129)F1 mice. The similar rejection of these lymphoma cells in B6 and (B6 × 129)F1 mice confirmed that tumor antigens, not alloantigens, were responsible for these rejection responses.
Figure 2.
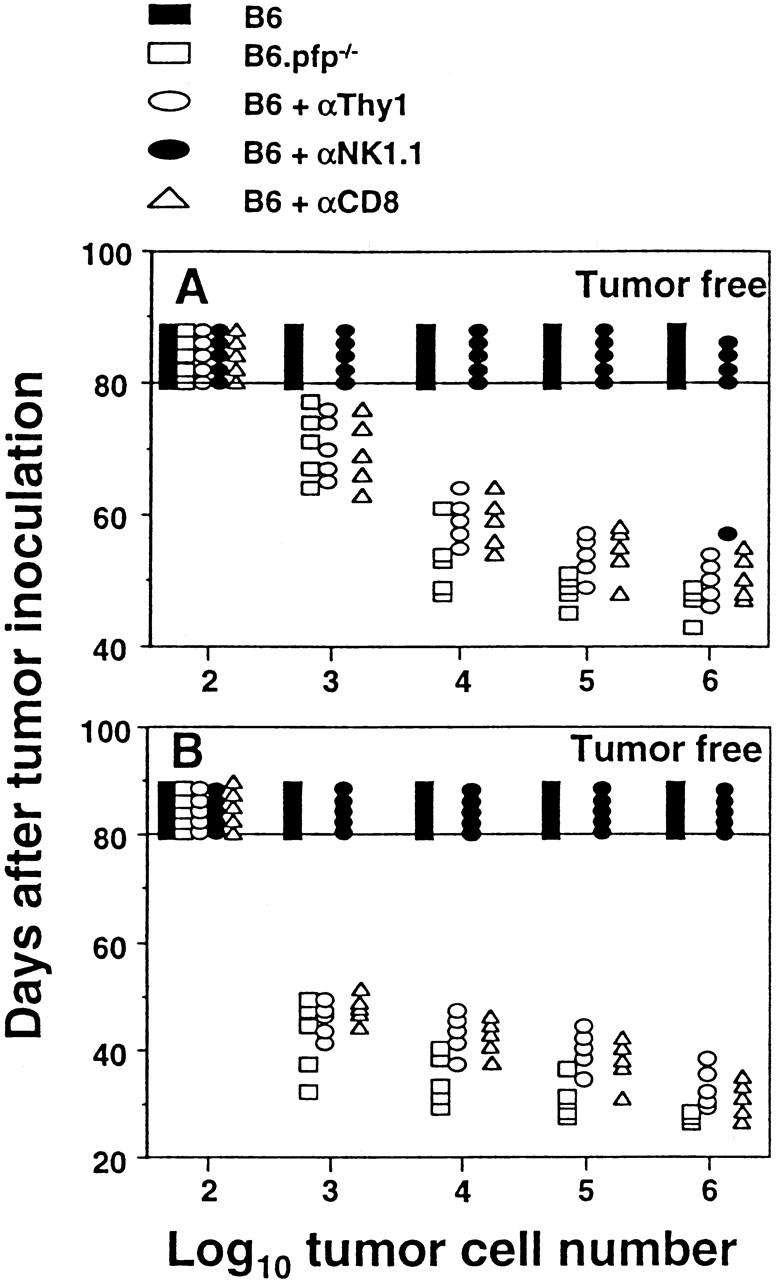
Spontaneous lymphomas arising in pfp−/−p53+/− mice are malignant, immunogenic, and rejected by CD8+ T cells. Elimination of lymphomas transplanted directly intraperitoneally (102–106 cells in 0.2 ml PBS, as indicated) into untreated B6 or B6.pfp−/− mice or B6 mice (5/group) depleted of Thy-1+, NK1.1+, or CD8+ cells using mAb 24. Each symbol depicts an individual mouse. A, PN53H-1; B, PN53H-7.
In conclusion, our collective data strongly indicated that cytotoxic lymphocyte pfp offers protection from disseminated lymphoma. The absence of pfp led to increased lymphomagenesis in several distinct cell lineages and p53 mutation enhanced spontaneous tumor formation. Transplantation of these lymphomas into wild-type and lymphocyte subset–depleted mice revealed that tumor rejection was dependent upon CD8+ T cells. Importantly, this is the first study to demonstrate that lymphocyte-mediated cytotoxicity plays an important role in promoting host resistance to spontaneous tumor formation. As such, it provides encouragement that efforts to promote CTL cytotoxicity directed at tumors have merit.
Notwithstanding the clear differences seen in lymphoma incidence, many additional questions are raised by our studies. The lymphomas arising in pfp-deficient mice appeared to be controlled by CD8+ T cells, yet clearly other innate cell types, such as NK and NKT cells, express pfp and control tumor development and metastasis 11 24. Interestingly, the lymphomas arising in the absence of appreciable NK cytotoxicity apparently experience no selective pressure to lose MHC class I expression when pfp is also absent in host T cells. Given the role of CD8+ T cells demonstrated herein, it is curious that the reported rates of spontaneous tumor development in nu/nu (T cell–deficient) and scid/scid (B and T cell–deficient) mice are very low 25 26. Interestingly, scid/NOD mice (with defective NK cell cytotoxicity) have a high incidence of thymoma 27, and thus innate effector cells using pfp may help maintain immune surveillance despite the lack of functional T cells. Although the bg/bg mutation in NK cell degranulation, predisposes this natural mutant mouse to disseminated malignant lymphoma and increased metastasis 28 29, the relative importance of NK or NKT cells in tumor immune surveillance will be better determined in mice exclusively deficient of each cell type.
The genetic background of mice and their housing conditions may be important factors that determine tumor incidence and type. We are currently assessing pfp deficiency in BALB/c mice and another strain of gene-targeted C57BL/6 mice 10 in the same clean facility. It has been well documented that a large proliferative expansion and persistence of antigen-reactive T and B cells and APCs occurs in pfp-deficient mice challenged with lymphocytic choriomeningitis virus (LCMV). These mice also experience reductions in bone marrow hematopoietic precursor cells, probably due to enhanced hemophagocytosis 10 30 31. This condition bears a striking resemblance to the human autosomal recessive immunodeficiency, familial hemophagocytic lymphohistiocytosis (FHL), which presents in infancy with hepatosplenomegaly due to a pronounced histiocytic infiltrate, hemophagocytosis, and immune suppression characterized by reduced or absent NK cell activity 32. Some of these patients have been shown to lack perforin expression, due to inherited mutations in the perforin gene 32. Although the mice were not exposed to LCMV, nor developed histiocytic infiltrates, in this study, it is possible that other microorganisms might provide antigenic stimulation also leading to lymphoproliferation, extended cell survival, and an increased pool of cells vulnerable to oncogenesis in pfp−/− mice. Thus, the increased lymphoma incidence associated with pfp-deficiency might still have its genesis both in an increased number of premalignant cells and the absence of the cytotoxic mechanism that normally eliminates them. Alternatively, murine lymphomas are often associated with reactivation of endogenous retroviruses during senescence, and thus the antigenic targets of the CTLs controlling these lymphomas might be viral. It is harder to propose this same etiology to the lymphomas arising in much younger p53−/− mice, implying that direct perforin-mediated cytolysis normally eliminates these lymphomas regardless of their genesis.
Given that pfp is also critical in protection from methylcholanthrene-induced fibrosarcoma 9 24, we are left to ponder why the absence of pfp does not additionally sensitize p53+/− mice to spontaneous sarcoma. Clearly, more needs to be understood about the influence different oncogenic hits have on shaping the host immune response to different cell types, and the effector mechanism(s) that are subsequently mobilized. Although the oncogenic events underlying the development of spontaneous lymphomas in pfp−/−p53+/+ and pfp−/−p53+/− mice remain to be determined at the molecular level, it is clear that these tumors present target structure(s) that can be recognized by CTLs, and pfp-dependent granule-mediated cytolysis is protective. Future studies to further evaluate the potential role of pfp in controlling other lymphoid malignancies and spontaneous tumors arising in nonlymphoid tissues will be of great interest.
Acknowledgments
We wish to thank the staff of the Biological Research Facility, Austin Research Institute for skilful animal handling and we also thank Andreas Strasser, Eugene Maraskovsky, and Ian McKenzie for critically reviewing this manuscript.
This work was supported by a project grant from the National Health & Medical Research Council of Australia to M.J. Smyth and J.A. Trapani, and by a Dora Lush Postgraduate Scholarship (NH&MRC) to K.Y.T. Thia.
Footnotes
M.J. Smyth and K.Y.T. Thia contributed equally to this work.
References
- Burnet F.M. The concept of immunological surveillance. Prog. Exp. Tumor Res. 1970;13:1–27. doi: 10.1159/000386035. [DOI] [PubMed] [Google Scholar]
- Thomas L. On immunosurveillance in human cancer. Yale J. Biol. Med. 1982;55:329–333. [PMC free article] [PubMed] [Google Scholar]
- Melief C.J., Kast W.M. Cytotoxic T lymphocyte therapy of cancer and tumor escape mechanisms. Semin. Cancer Biol. 1991;2:347–354. [PubMed] [Google Scholar]
- Whiteside T.L., Vujanovic N.L., Herberman R.B. Natural killer cells and tumor therapy. Curr. Top. Microbiol. Immunol. 1998;230:221–244. doi: 10.1007/978-3-642-46859-9_13. [DOI] [PubMed] [Google Scholar]
- Smyth M.J., Ortaldo J.R., Shinkai Y., Yagita H., Nakata M., Okumura K., Young H.A. Interleukin 2 induction of pore-forming protein gene expression in human peripheral blood CD8+ T cells. J. Exp. Med. 1990;171:1269–1281. doi: 10.1084/jem.171.4.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki A., Shinkai Y., Yagita H., Okumura K. Expression of perforin in murine natural killer cells and cytotoxic T lymphocytes in vivo. Eur. J. Immunol. 1992;22:1215–1219. doi: 10.1002/eji.1830220516. [DOI] [PubMed] [Google Scholar]
- Liu C.C., Walsh C.M., Young J.D. Perforinstructure and function. Immunol. Today. 1995;16:194–201. doi: 10.1016/0167-5699(95)80121-9. [DOI] [PubMed] [Google Scholar]
- Trapani J.A., Sutton V.R., Smyth M.J. CTL granulesevolution of vesicles essential for combating virus infections. Immunol. Today. 1999;20:351–356. doi: 10.1016/s0167-5699(99)01488-7. [DOI] [PubMed] [Google Scholar]
- van den Broek M.E., Kagi D., Ossendorp F., Toes R., Vamvakas S., Lutz W.K., Melief C.J., Zinkernagel R.M., Hengartner H. Decreased tumor surveillance in perforin-deficient mice. J. Exp. Med. 1996;184:1781–1790. doi: 10.1084/jem.184.5.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagi D., Ledermann B., Burki K., Seiler P., Odermatt B., Olsen K.J., Podack E.R., Zinkernagel R.M., Hengartner H. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature. 1994;369:31–37. doi: 10.1038/369031a0. [DOI] [PubMed] [Google Scholar]
- Smyth M.J., Thia K.Y., Cretney E., Kelly J.M., Snook M.B., Forbes C.A., Scalzo A.A. Perforin is a major contributor to NK cell control of tumor metastasis. J. Immunol. 1999;162:6658–6662. [PubMed] [Google Scholar]
- Levine A.J. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- Hollstein M., Rice K., Greenblatt M.S., Soussi T., Fuchs R., Sorlie T., Hovig E., Smith-Sorensen B., Montesano R., Harris C.C. Database of p53 gene somatic mutations in human tumors and cell lines. Nucleic Acids Res. 1994;22:3551–3555. [PMC free article] [PubMed] [Google Scholar]
- Harris C.C., Hollstein M. Clinical implications of the p53 tumor-suppressor gene. N. Engl. J. Med. 1993;329:1318–1327. doi: 10.1056/NEJM199310283291807. [DOI] [PubMed] [Google Scholar]
- Malkin D., Li F.P., Strong L.C., Fraumeni J.F., Jr., Nelson C.E., Kim D.H., Kassel J., Gryka M.A., Bischoff F.Z., Tainsky M.A. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233–1238. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- Jacks T., Remington L., Williams B.O., Schmitt E.M., Halachmi S., Bronson R.T., Weinberg R.A. Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 1994;1:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- Donehower L.A., Harvey M., Slagle B.L., McArthur M.J., Montgomery C.A., Jr., Butel J.S., Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- Walsh C.M., Matloubian M., Liu C.C., Ueda R., Kurahara C.G., Christensen J.L., Huang M.T., Young J.D., Ahmed R., Clark W.R. Immune function in mice lacking the perforin gene. Proc. Natl. Acad. Sci. USA. 1994;91:10854–10858. doi: 10.1073/pnas.91.23.10854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp C.J., Wheldon T., Balmain A. p53-deficient mice are extremely susceptible to radiation-induced tumorigenesis. Nat. Genet. 1994;8:66–69. doi: 10.1038/ng0994-66. [DOI] [PubMed] [Google Scholar]
- Williams B.O., Remington L., Albert D.M., Mukai S., Bronson R.T., Jacks T. Cooperative tumorigenic effects of germline mutations in Rb and p53. Nat. Genet. 1994;7:480–484. doi: 10.1038/ng0894-480. [DOI] [PubMed] [Google Scholar]
- Nacht M., Strasser A., Chan Y.R., Harris A.W., Schlissel M., Bronson R.T., Jacks T. Mutations in the p53 and SCID genes cooperate in tumorigenesis. Genes Dev. 1996;10:2055–2066. doi: 10.1101/gad.10.16.2055. [DOI] [PubMed] [Google Scholar]
- Kaplan D.H., Shankaran V., Dighe A.S., Stockert E., Aguet M., Old L.J., Schreiber R.D. Demonstration of an interferon γ–dependent tumor surveillance system in immunocompetent mice. Proc. Natl. Acad. Sci. USA. 1998;95:7556–7561. doi: 10.1073/pnas.95.13.7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm U., Klamp T., Groot M., Howard J.C. Cellular responses to interferon-γ. Annu. Rev. Immunol. 1997;15:749–795. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- Smyth M.J., Thia K.Y.T., Street S.E., Cretney E., Trapani J.A., Taniguchi M., Kawano T., Pelikan S.B., Crowe N.Y., Godfrey D.I. Differential tumor surveillance by natural killer (NK) and NKT cells. J. Exp. Med. 2000;191:661–668. doi: 10.1084/jem.191.4.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutman O. Spontaneous tumors in nude miceeffect of the viable yellow gene. Exp. Cell Biol. 1979;47:129–135. doi: 10.1159/000162929. [DOI] [PubMed] [Google Scholar]
- Gershwin M.E., Ohsugi Y., Castles J.J., Ikeda R.M., Ruebner B. Anti-mu induces lymphoma in germfree congenitally athymic (nude) but not in heterozygous (nu/+) mice. J. Immunol. 1983;131:2069–2073. [PubMed] [Google Scholar]
- Shultz L.D., Schweitzer P.A., Christianson S.W., Gott B., Schweitzer I.B., Tennent B., McKenna S., Mobraaten L., Rajan T.V., Greiner D.L. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J. Immunol. 1995;154:180–191. [PubMed] [Google Scholar]
- Talmadge J.E., Meyers K.M., Prieur D.J., Starkey J.R. Role of NK cells in tumour growth and metastasis in beige mice. Nature. 1980;284:622–624. doi: 10.1038/284622a0. [DOI] [PubMed] [Google Scholar]
- Haliotis T., Ball J.K., Dexter D., Roder J.C. Spontaneous and induced primary oncogenesis in natural killer (NK)-cell-deficient beige mutant mice. Int. J. Cancer. 1985;35:505–513. doi: 10.1002/ijc.2910350414. [DOI] [PubMed] [Google Scholar]
- Binder D., van den Broek M.F., Kagi D., Bluethmann H., Fehr J., Hengartner H., Zinkernagel R.M. Aplastic anemia rescued by exhaustion of cytokine-secreting CD8+ T cells in persistent infection with lymphocytic choriomeningitis virus. J. Exp. Med. 1998;187:1903–1920. doi: 10.1084/jem.187.11.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matloubian M., Suresh M., Glass A., Galvan M., Chow K., Whitmire J.K., Walsh C.M., Clark W.R., Ahmed R. A role for perforin in downregulating T-cell responses during chronic viral infection. J. Virol. 1999;73:2527–2536. doi: 10.1128/jvi.73.3.2527-2536.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepp S.E., Dufourcq-Lagelouse R., Le Deist F., Bhawan S., Certain S., Mathew P.A., Henter J.I., Bennett M., Fischer A., de Saint Basile G., Kumar V. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;286:1957–1959. doi: 10.1126/science.286.5446.1957. [DOI] [PubMed] [Google Scholar]