

Abstract
Transforming growth factor (TGF)-β1 is a pleiotropic cytokine/growth factor that is thought to play a critical role in the modulation of inflammatory events. We demonstrate that exogenous TGF-β1 can inhibit the expression of the proinflammatory adhesion molecule, E-selectin, in vascular endothelium exposed to inflammatory stimuli both in vitro and in vivo. This inhibitory effect occurs at the level of transcription of the E-selectin gene and is dependent on the action of Smad proteins, a class of intracellular signaling proteins involved in mediating the cellular effects of TGF-β1. Furthermore, we demonstrate that these Smad-mediated effects in endothelial cells result from a novel competitive interaction between Smad proteins activated by TGF-β1 and nuclear factor κB (NFκB) proteins activated by inflammatory stimuli (such as cytokines or bacterial lipopolysaccharide) that is mediated by the transcriptional coactivator cyclic AMP response element–binding protein (CREB)-binding protein (CBP). Augmentation of the limited amount of CBP present in endothelial cells (via overexpression) or selective disruption of Smad–CBP interactions (via a dominant negative strategy) effectively antagonizes the ability of TGF-β1 to block proinflammatory E-selectin expression. These data thus demonstrate a novel mechanism of interaction between TGF-β1–regulated Smad proteins and NFκB proteins regulated by inflammatory stimuli in vascular endothelial cells. This type of signaling mechanism may play an important role in the immunomodulatory actions of this cytokine/growth factor in the cardiovascular system.
Keywords: inflammation, transcription, transforming growth factor, vascular biology, endothelium
Introduction
As the requisite interface between blood and all tissues of the body, vascular endothelium plays an integral role in the process of inflammation. Rather than being simply a passive barrier, it is now clear that this cell type can integrate both blood-borne and tissue-derived signals, and thus function to actively mediate the recruitment of leukocytes to areas of inflammation. A critical event in this process is the regulated endothelial expression of cell surface adhesion molecules such as E-selectin. This protein, which was originally identified as a cytokine-inducible cell surface protein in cultured endothelial cells, has been shown to play an important role in mediating early leukocyte–endothelial interactions such as initial attachment and rolling 1. Thus, the regulated expression of this and other adhesion molecules in vascular endothelium has been an active area of investigation 2. E-selectin expression is regulated primarily at the level of transcriptional activation, a process that is mediated by a series of tandem nuclear factor κB (NFκB) sites in the promoter of this gene. Stimuli such as IL-1β and TNF-α can rapidly induce the expression of the E-selectin gene in endothelial cells by activating NFκB and stimulating the transcription of the E-selectin promoter 2 3 4 5.
The TGF-β superfamily constitutes a large and diverse family of protein cytokines/growth factors that play important roles in processes ranging from differentiation and development to the regulation of cell growth and extracellular matrix biology 6 7. One of the areas in which TGF-β1 (the prototypic member of this family) is thought to play an especially critical role is the modulation of inflammatory responses. TGF-β1 is a powerful and essential immune modulator in the vascular system, where it participates in the regulation of inflammatory events in both leukocytes and vascular endothelial cells 8. This is most dramatically illustrated by the phenotype of mice that lack the gene for the TGF-β1 isoform. These mice die in utero or in the perinatal period because of widespread, uncontrolled inflammation, and this pathological phenotype can be reversed by the systemic administration of active soluble TGF-β1 9. Previous work has demonstrated that TGF-β1 can inhibit cytokine-induced E-selectin expression in cultured endothelial cells (a cardinal sign of endothelial activation in the context of inflammation), suggesting that the endothelium may be an important target for the antiinflammatory actions of TGF-β1 10.
Over the last several years, the general intracellular signaling mechanisms used by the TGF-β superfamily have begun to be elucidated 6 7 11. In the case of TGF-β1, its cellular effects appear to be transduced via at least three types of cell surface receptors (types I, II, III), two of which are serine/threonine kinases. The active form of soluble TGF-β1 binds to the type II receptor at the cell surface, and this complex subsequently interacts with and transphosphorylates the cytoplasmic domain of the type I receptor. This phosphorylation event activates the type I receptor kinase domain, which then propagates downstream signals within the cell. Smad proteins (known previously as MAD-related proteins for mothers against decapentaplegic) are essential components in the signaling pathways of the TGF-β1 type I serine/threonine kinase receptors 12. Smads function as intracellular signaling intermediates by specifically interacting with ligand-activated receptor complexes at the cell surface and subsequently translocating to the nucleus, where they modulate gene expression (a general mechanism of action that is thought to be similar to that of the signal transducer and activator of transcription [STAT] family of signaling molecules). We and others have demonstrated that Smad proteins interact functionally with the transcriptional coactivators CREB-binding protein (CBP)/P300, a process that is essential to their ability to function as transcriptional effectors 13 14 15 16 17.
In this report, we demonstrate that the ability of TGF-β1 to antagonize cytokine-induced E-selectin expression in vascular endothelium is Smad protein dependent and involves a novel “competitive” interaction between Smad proteins and NFκB that is mediated by the transcriptional coactivator CBP. These data thus indicate a new mechanism by which this important class of cytokines/growth factors can interact with and modulate a signal transduction pathway mediating inflammatory events.
Materials and Methods
Reagents, Cell Culture, Transfections, and Reporter Assays.
Human umbilical vein endothelial cells (HUVECs) were isolated from multiple segments of normal term umbilical cords, pooled, and cultured in Medium 199 (GIBCO BRL) supplemented with Endothelial Cell Growth Supplement (50 μg/ml; Collaborative Research, Inc.), heparin (50 μg/ml, porcine intestinal; Sigma-Aldrich), antibiotics (penicillin-G, 100 U/ml and streptomycin, 100 μg/ml; Sigma-Aldrich), and 20% fetal bovine serum. Primary bovine aortic endothelial cells (BAECs) were isolated and cultured as described 18. These were used at passages 3–9. Transient transfections were performed on subconfluent (70%) monolayers of endothelial cells by coincubating the cells with DNA and lipofectamine (GIBCO BRL) for 6 h according to manufacturer's instructions. The DNA/lipofectamine mixture was then removed and replaced with media containing serum. In all cases, the monolayers were allowed to reach confluence for 24–48 h after transfection before subsequent treatment or analysis. A plasmid encoding the cytomegalovirus promoter driving expression of the β-galactosidase gene (CMV-β gal) was routinely cotransfected to control for transfection efficiency and normalize for yield, and individual conditions were performed in duplicate. When multiple plasmids were cotransfected (e.g., reporter and expression plasmids), the total amount of DNA was kept constant by augmenting samples with empty expression vectors. Both luciferase and β-galactosidase activity were measured in the same sample using the Dual-Light system (Tropix). For the transfection experiments, the results shown are reported as ratios of luciferase to β-galactosidase activity and are representative of at least three independent experiments. The error bars displayed represent ± 1 SD of the mean.
The Smad protein expression constructs are all present in pCi-neo (Promega) and encode epitope-tagged versions of the Smad proteins (hemagglutinin or Flag) or are fused in frame to the Gal4 DNA binding domain, and have been described previously 13 19. Smad2*P is a form of Smad2 that has had its COOH-terminal SSXS motif deleted, and Smad4d514 is a truncated form of Smad4 that terminates at amino acid 514. The expression of these proteins in transfection experiments was monitored by Western analyses to maintain equal levels between constructs. The E-selectin promoter was provided by T. Collins (Brigham and Women's Hospital, Boston, MA), and the CBP expression construct was provided by S. Bhattacharya (Dana-Farber Cancer Institute, Boston, MA). The simplified NFκB promoter and the Gal4-Jun fusion constructs were obtained from CLONTECH Laboratories, Inc. The constructs expressing the subdomains of CBP were created by PCR and incorporated a Flag epitope tag at their NH2 terminus, and were cloned into pCi-neo (Promega). All constructs have been sequenced. Active TGF-β1 was obtained from R&D Systems.
Mobility Shift, Western, and Fluorescence Immunoassay Analyses.
Western blots for Smad2, p65, CBP, and the epitope tags (Flag, hemagglutinin, myc) were performed with antisera obtained commercially from Santa Cruz Biotechnology, Inc., Zymed Laboratories, BD PharMingen, and R&D Systems. After separation of proteins on SDS-PAGE gels, they were transferred to nitrocellulose by electroblot and the membranes were incubated with the appropriate antisera. Proteins were detected with the use of horseradish peroxidase–linked secondary antisera and chemiluminescence. Nuclear extracts prepared from HUVEC monolayers were used for the mobility shift analyses and prepared as described 3. The sequences of the double-stranded oligonucleotide probe used was 5′-GCCATTGGGGATTTCCTCTTTACTGGATGT-3′. The specificity of the gel shifts was confirmed by immunodepleting these extracts with the anti-p65 antisera before mobility shift analysis. This was accomplished by incubating the extracts with the appropriate antisera (or nonimmune IgG as the control) overnight at 4°C, then capturing the antibodies with the appropriate antiisoform-coupled beads for 30 min and removing them by centrifugation. Fluorescence immunoassays for cell surface E-selectin were performed by fixing the endothelial monolayers with 2% paraformaldehyde for 2 min, then incubating the monolayers with an antisera that reacts with cell surface E-selectin, as described previously 1. After washing, the monolayers are incubated with a secondary antibody coupled to FITC, then washed and lysed, and the fluorescence was read in a plate reader. This assay is a highly quantitative and reproducible assessment of the level of cell surface E-selectin expression.
In Vivo Experiments.
LPS (Salmonella typhi; Sigma-Aldrich) was administered to experimental rats at 4 mg/kg intraperitoneally, and 6 h later the hearts, lungs, and liver of these animals were harvested, washed in sterile cold PBS, and frozen in liquid nitrogen. For the pretreatment experiments, animals received either vehicle or 20 μg/kg active TGF-β1 30 min before LPS challenge. Total RNA was prepared from tissues by rapid homogenization in lysis solution and extraction (Ambion), and was separated on formaldehyde gels and blotted to nylon for Northern analysis. Extracts of tissues for mobility shift analyses or Western analyses were performed essentially as described 3. In brief, the tissues were ground up under liquid nitrogen, and proteins were extracted in buffer containing 20 mM Hepes, pH 7.4, 10mM KCl, 400 mM NaCl, 0.15 mM EDTA, 1 mM dithiothreitol, and protease and phosphatase inhibitors. ∼2–5 μg of protein extract was used per mobility shift reaction. For the Western analysis, the ground tissues were immediately extracted with buffer containing 1% SDS, 50 mM Tris, pH 7.5, 50mM NaCl, 1 mM EDTA, and protease and phosphatase inhibitors. These extracts were then analyzed as described above.
Results
TGF-β Can Inhibit Inflammatory Cytokine-induced Adhesion Molecule Expression in Endothelial Cells by Transcriptional Mechanism(s) Involving NFκB.
Exogenous TGF-β1 has been reported to inhibit the expression of a variety of inflammatory genes in vascular endothelial cells. One such gene is E-selectin, an inducible cell surface adhesion molecule selectively expressed in vascular endothelium. We chose to examine the cytokine-induced expression of this gene in the presence or absence of pretreatment with soluble active TGF-β1. As demonstrated by a fluorescence immunoassay (Fig. 1 A), E-selectin protein is rapidly induced on the surface of cultured HUVECs by both 5 and 10 U/ml of recombinant human IL-1β. However, when the cells are preincubated in the presence of TGF-β1, this induction can be markedly attenuated. This effect could be seen with a little as 30 min of pretreatment with TGF-β (data not shown). The dose response for the inhibitory effect of exogenous TGF-β1 demonstrated a U-shaped curve whereby intermediate doses of TGF-β1 (0.1–1 ng/ml of active TGF-β1) were maximally effective, whereas higher doses (5–10 ng/ml) were significantly less effective (Fig. 1 A; data not shown). The data displayed in Fig. 1 consist of representative data using low-passage HUVECs at confluence and 6 h of TGF-β1 prestimulation followed by 4 h of rhIL-1β stimulation. These results confirm the findings of several other investigators, demonstrating that TGF-β1 can effectively inhibit endothelial adhesion molecule expression in vitro 10.
Figure 1.
(A) Inducible expression of E-selectin protein on the surface of cultured endothelial cells in response to Il-1β can be inhibited by pretreatment with active TGF-β1. The level of cell surface E-selectin protein was determined by fluorescence immunoassay at baseline and 4 h after treatment with 5 or 10 U/ml of rhIL-1β (left). To assess the effect of TGF-β1 pretreatment, the cells were exposed to the indicated doses of active TGF-β1 for 6 h before cytokine treatment. The right panel demonstrates that pretreatment of HUVECs with intermediate doses of TGF-β1 markedly diminished cytokine-induced E-selectin expression. (B) Pretreatment of TGF-β can inhibit cytokine-induced transcription of the E-selectin promoter and a simplified NFκB-dependent promoter in endothelial cells. Luciferase reporter constructs containing either the E-selectin promoter (left and middle) or a promoter consisting of a series of tandem NFκB sites (right) were transiently transfected into HUVECs. The cells were subsequently stimulated with rhIL-1β for 4 h, and luciferase levels were measured and normalized to β-galactosidase activity (from a cotransfected CMV-β gal reporter vector). As shown in the left panel, both 5 and 10 U/ml of rhIL-1β effectively induce transcription from the E-selectin promoter. In the middle and right panels, the cells were pretreated with the indicated doses of active TGF-β 1 for 6 h before cytokine stimulation (5 U/ml). Intermediate doses of TGF-β1 were effective at inhibiting cytokine-induced, NFκB-mediated transcription.


The inducible expression of E-selectin in endothelial cells is primarily regulated at the level of transcriptional activation. To examine whether the ability of TGF-β1 to inhibit cell surface protein expression resulted from a downregulation of transcriptional activation of the E-selectin gene, we used an E-selectin promoter construct in transient transfection assays. As shown in Fig. 1 B, this construct is significantly induced in the presence of rhIL-1β (as well as TNF-α; data not shown), and preincubation of the transfected cells with TGF-β1 before inflammatory cytokine stimulation resulted in a significant attenuation of the induction of this promoter. The dose response of TGF-β1 for this effect was identical to that observed for E-selectin protein induction, suggesting that the inhibitory mechanism involves a modulation of transcriptional events. The E-selectin promoter is known to be regulated by cis-acting NFκB sites. Thus, we examined whether a simplified promoter construct consisting of tandem NFκB sites would demonstrate a similar response. As shown in Fig. 1 B, the induction of this promoter by rhIL-1β in transfected endothelial cells was also significantly inhibited by pretreatment with TGF-β1. We found essentially identical results if we used TNF-α as the inflammatory stimulus or if we used primary cultures of BAECs that are more readily transfectable than HUVECs, and thus less subject to artifacts resulting from low transfection efficiencies (data not shown). Taken together, these results indicate that TGF-β1 is capable of modulating the transcriptional induction of the E-selectin gene through an NFκB-dependent mechanism.
TGF-β–mediated Inhibition of Inducible E-Selectin Expression Is Smad Protein Dependent.
Smad proteins are a recently identified class of intracellular proteins that have been demonstrated to play essential roles in TGF-β1 superfamily–mediated signal transduction and gene expression 6 7 11. These proteins fall into three general classes. The so-called receptor-activated (or signaling) Smads (Smad2 and Smad3 in the case of TGF-β1) are direct cytoplasmic substrates for the serine/threonine kinase present in activated TGF-β superfamily type 1 receptors. Once phosphorylated by the receptor, these proteins complex with a distinct class of Smad, Smad4, which is not a substrate for the receptor but is required for maximal signaling. This complex then translocates to the nucleus and modulates the expression of a variety of genes. A third class of Smad proteins, termed the inhibitory Smads, Smad6 and Smad7, can inhibit specific steps in this signaling cascade by either interacting with the activated type 1 receptor and blocking its ability to phosphorylate signaling Smads such as Smad2 or Smad3, or by interfering with required Smad–Smad interactions 18 20 21.
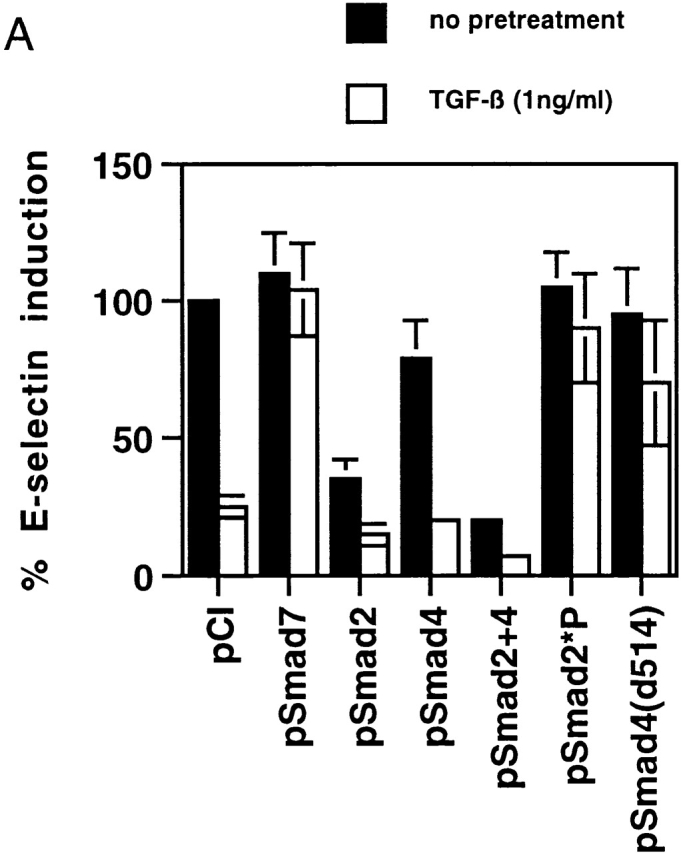
To determine if the inhibitory effects of TGF-β1 on inducible E-selectin gene expression in endothelial cells were Smad dependent, we transfected BAECs with several Smad protein expression constructs and examined the response of a cotransfected E-selectin promoter construct to exogenous TNF-α (both with and without TGF-β pretreatment). As shown in Fig. 2 A, TGF-β1 at 1 ng/ml resulted in a significant inhibition of promoter induction in the presence of a cotransfected empty expression plasmid (pCi), but in the presence of the expression plasmid encoding the inhibitory Smad, Smad7, this TGF-β1–mediated inhibition was completely abolished. Conversely, when Smad2, Smad4, or both Smad2 and Smad4 were overexpressed, there was a reduction in the TNF-α–induced level of E-selectin promoter activity even in the absence of TGF-β1 pretreatment, as well as a significant enhancement of the inhibitory effect of exogenous TGF-β1 (Fig. 2 A). We used two additional constructs: pSmad2*P, which expresses a form of Smad2 that is not a substrate for the activated TGF-β1 type 1 receptor, and a COOH-truncated form of Smad4 (pSmad4d514) that is inactive as a transcriptional activator. When overexpressed, both of these proteins can act as dominant negative regulators of Smad signaling. These constructs significantly ameliorated the ability of TGF-β1 to inhibit inflammatory cytokine–induced E-selectin expression (Fig. 2 A). Taken together, these results indicate that the inhibitory effects of TGF-β1 on inducible E-selectin expression in endothelial cells are Smad protein dependent and can be mimicked by overexpression of Smad2 and -4 and blocked by the inhibitory Smad7.
Figure 2.
(A) TGF-β1–mediated inhibition of IL-1β–induced E-selectin expression in endothelial cells is Smad protein dependent. BAECs were transiently cotransfected with the E-selectin promoter construct and the indicated Smad protein expression constructs or an empty expression vector (pCi). The cells were subsequently stimulated for 4 h with TNF-α, with and without 6 h of pretreatment with TGF-β1 at 1 ng/ml. The level of induction of the E-selectin promoter has been normalized to that seen in the absence of TGF-β1 pretreatment (in the presence of the cotransfected empty expression vector) and expressed as 100%. In the presence of cotransfected empty expression vector (pCI), there is a marked inhibition of E-selectin promoter induction by TGF-β1 pretreatment, as described in the legend to Fig. 1. Coexpression of Smad7, an inhibitory Smad protein, or of Smad2*P or Smad4 (d514), which can act as dominant negative inhibitors, markedly attenuates the TGF-β1–mediated inhibition. In contrast, coexpression of wild-type Smad2, Smad4, or both Smad2 and -4 significantly enhances the level of inhibition seen and is able to induce varying levels of inhibition even in the absence of exogenous TGF-β1 pretreatment. All of the Smad expression constructs encode epitope-tagged species that were expressed at comparable levels as monitored by immunoblot (data not shown). (B) Cytokine-induced activation of NFκB appears preserved in the presence of TGF-β1 pretreatment. HUVEC monolayers were treated with 10 U/ml of rhIL-1β for 30 min and lysed, and a nuclear extract was prepared. Specific NFκB-mediated DNA binding activity was assessed by mobility shift assay and compared with that present in unstimulated HUVEC. As shown in the left panel, there is a specific shifted band present in the rhIL-1β–treated lanes but not in the control (untreated) lane that can be abolished by immunodepletion of the extract with a mixture of anti-p65/antip50 antisera but not nonspecific IgG. As shown in the middle panel, rhIL-1β–induced, NFκB-mediated DNA binding activity (as assessed by this gel shift) appears preserved even in the presence of TGF-β pretreatment. The panels at the right represent immunoblots for IκB and P65 protein levels. The former is markedly depleted by rhIL-1β treatment, even in the presence of TGF-β1 pretreatment. p65 levels appeared stable under these conditions. These findings suggest that IκB degradation and NFκB-mediated DNA binding are not affected by TGF-β1 pretreatment in HUVECs.

The activation of NFκB by inflammatory stimuli in endothelial cells involves a series of events culminating in the phosphorylation and degradation of the inhibitory protein IκB and subsequent nuclear localization and DNA binding of p50/p65 heterodimers to specific DNA sequences 2. To examine the effects of TGF-β1 on these events in endothelial cells, we stimulated confluent HUVECs with rhIL-1β in the presence or absence of TGF-β1 pretreatment, and examined NFκB DNA binding by gel shift analysis and levels of immunoreactive p65 and IKBα by Western blotting. As demonstrated in Fig. 2 B, in extracts of control (unstimulated) HUVECs, there is essentially no detectable gel shift of an NFκB-specific probe. After stimulation with IL-1β, a discrete gel-shifted band is detected. The specificity of this band was confirmed by immunodepleting the extract with an anti-p65 antisera, which eliminated this gel shift. Interestingly, when this analysis was performed on HUVECs that had been preincubated with TGF-β1, there was no detectable difference in NFκB activation as assessed by this gel shift assay (Fig. 2 B). At all doses of TGF-β1 pretreatment, rhIL-1β treatment resulted in the induction of a detectable NFκB-mediated gel shift. When we examined the levels of NFκB proteins by immunoblot, the levels of immunoreactive IκBα were markedly diminished by rhIL-1β treatment, whereas the levels of p65 were not significantly altered whether or not the cells were pretreated with TGF-β1 (Fig. 2 B). These results indicate that under conditions where TGF-β1 can significantly inhibit NFκB-mediated E-selectin expression, activation of NFκB appears to be preserved, and suggest that a step subsequent to NFκB-mediated DNA binding is the site of TGF-β/Smad-mediated inhibition.
TGF-β Can Inhibit Inducible E-Selectin Expression In Vivo.
To determine whether the results in cultured endothelial cells described above could be reproduced in an in vivo model, we examined the ability of systemic TGF-β1 to inhibit LPS-induced E-selectin expression in rats. Previous work has demonstrated the ability of systemically administered TGF-β1 to inhibit the expression of several inflammatory genes, such as inducible nitric oxide synthase II and heme oxygenase, and to ameliorate the course of LPS-induced shock, although the precise mechanism(s) of these effects have not been elucidated 22 23 24. We initially examined the ability of LPS to induce E-selectin mRNA expression in whole organs in vivo. As demonstrated in Fig. 3 A, E-selectin mRNA was not detected in total RNA (15 μg) from the lung, liver, and heart of control rats but is readily detected after 6 h of LPS administration. Given that E-selectin is selectively expressed in vascular endothelium, the varying amounts of E-selectin mRNA detected (in a constant amount of total RNA) probably reflect both the degree to which endothelium contributes the overall RNA content of the individual tissues, and differences in the degree to which E-selectin expression is induced by LPS in these distinct tissue environments. To examine the effects of TGF-β1, a second set of rats received either vehicle or 20 μg/kg of active TGF-β1 intraperitoneally, followed 20 min later by 4 mg/kg of LPS. The lungs, heart, and liver of these animals were harvested 6 h later for RNA and protein analysis. As demonstrated in Fig. 3 A (right), TGF-β1 pretreatment markedly attenuated the level of E-selectin mRNA induced in all three tissues. To examine whether NFκB was being activated to similar extents, we next examined the DNA binding activity of protein lysates from rat tissues. As demonstrated in Fig. 3 B (left), lysates from LPS-treated rat lung tissue demonstrate a gel-shifted band that is not seen in lysates from control lungs and that can be abolished by immunodepletion with an αp65/p50 antisera. When these analyses were performed on lysates from tissues derived from the LPS-treated rats, no detectable difference in NFκB-mediated DNA binding was seen between the sham and TGF-β1–pretreated animals (Fig. 3 B). Fig. 3 C demonstrates that the levels of immunoreactive p65 appear to be equal among the groups and that the levels of IκBα are slightly diminished in the absence of TGF-β1 pretreatment, but not abolished. Taken together, these results indicate that LPS can effectively upregulate E-selectin expression in vivo, and that this effect can be significantly attenuated by systemic pretreatment with TGF-β1. Furthermore, this inhibition can occur in the absence of a detectable decrement in the levels of immunoreactive NFκB or the degree to which NFκB is activated as assessed by specific DNA binding assay.
Figure 3.

(A) Induction of E-selectin mRNA in rat tissues by LPS can be inhibited by systemic pretreatment with TGF-β1. As described in Materials and Methods, rats received 4 mg/kg of LPS intraperitoneally, and 6 h later the indicated tissues were harvested and processed for RNA and protein extracts as described. (A) As shown by Northern analysis (left panel), E-selectin mRNA is not detected in control tissues but is detectable in the lung, liver, and hearts 6 h after LPS administration. In the right panels, tissues from LPS-treated animals that had received either vehicle or TGF-β1 pretreatment were similarly analyzed. Each pair of lanes represents two independently treated animals. Pretreatment with TGF-β1 markedly attenuated E-selectin mRNA induction (upper blots) but did not affect glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA levels (lower blots). (B) NFκB-mediated DNA binding activity appears preserved after TGF-β1 pretreatment in vivo. The lungs and hearts of LPS-treated rats were processed to obtain nuclear extracts for DNA mobility shift analyses with the NFκB-specific oligo probes as described in the legend to Fig. 2. In the left panel, a specific shifted band is detected in lysates from LPS-treated rat lung that is not present in lysates from control lung and that is attenuated by immunodepletion with antip65/p50 antisera but not nonimmune IgG. The middle and right panels are lysates from lung and heart, respectively, from the animals that have received LPS with or without TGF-β1 pretreatment. Each pair of lanes represents two independently treated animals. TGF-β1 pretreatment in vivo does not appear to affect LPS-induced NFκB-mediated DNA binding in these tissues. (C) Western analysis for p65 and IκB levels in tissues from LPS and TGF-β1–pretreated rats. The levels of immunoreactive p65 and IκB in the rat tissues were assessed by immunoblot. Each pair of lanes represents two independently treated animals. Levels of p65 were not significantly altered in response to any of the treatment protocols. The levels of immunoreactive IκB appeared slightly depressed by LPS treatment in the absence of TGF-β1 pretreatment.
TGF-β Inhibition of E-Selectin Expression Is Mediated by Smad–Coactivator (CBP) Interactions.
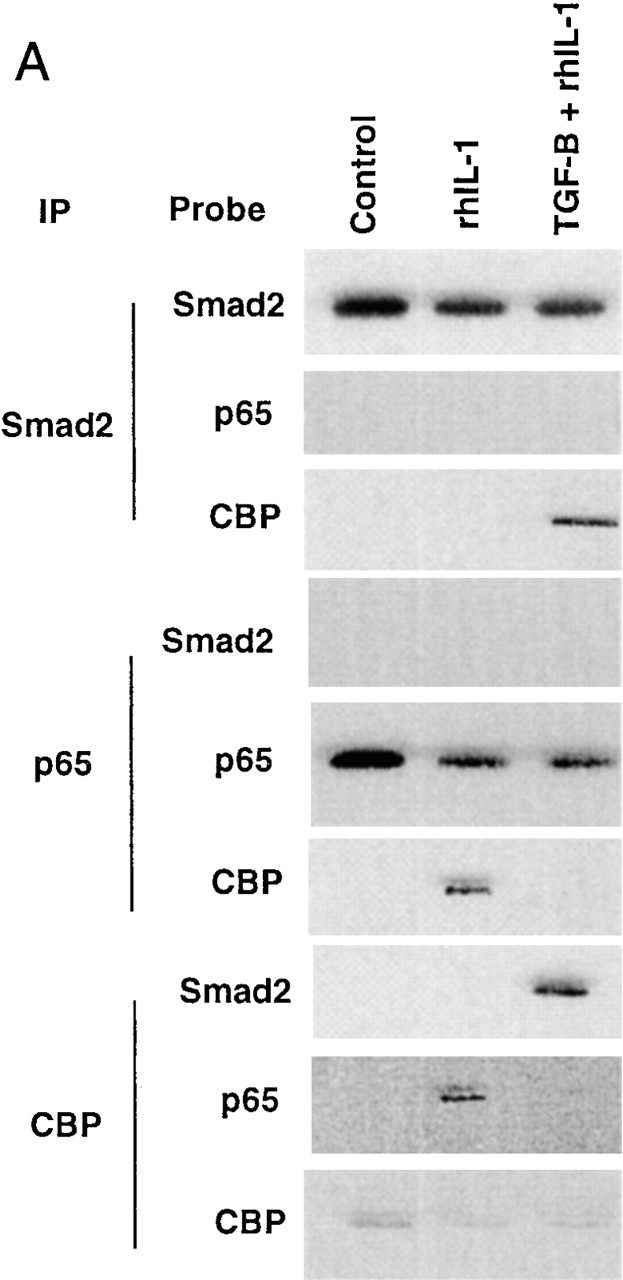
Transcriptional coactivators are a class of proteins that participate in transcriptional regulation by directly interacting with cis-acting transcriptional activators, components of the basal transcriptional apparatus, and chromatin 25 26 27. One member of this class of molecules, CBP, is capable of interacting with many regulated transcriptional effectors, and these interactions have been postulated as important loci of signal integration 28 29 30. Both NFκB and Smad proteins have been demonstrated to interact with CBP, an event that is required for transcriptional activation by these proteins in endothelial cells 13 31. To examine these interactions directly, we subjected lysates from control, rhIL-1β–treated, or TGF-β1–pretreated/rhIL-1β–treated HUVEC monolayers to immunoprecipitation with antisera against Smad2, p65, and CBP and probed the immunoprecipitates for associated proteins by immunoblot. As shown in Fig. 4 A, immunoprecipitations with anti-Smad2 antisera effectively brought down Smad2 protein under all conditions, but only brought down CBP after the cells were treated with TGF-β1, and did not bring down detectable amounts of p65 under any conditions examined. These results are consistent with previous data demonstrating that Smad–CBP interactions are ligand (TGF-β1) dependent 13, and further indicate that p65 and Smad2 do not appear to be present in the same transcriptional complexes in these cells. When these lysates were immunoprecipitated with anti-p65 antisera, CBP only coprecipitated after rhIL-1β treatment. Interestingly, if the cells were pretreated with TGF-β1 before rhIL-1β, then CBP was not found in p65-containing complexes. These results suggest that in the presence of TGF-β1 pretreatment, Smad proteins are able to effectively recruit CBP, whereas p65 is not. These results were confirmed by immunoprecipitating with anti-CBP. This brought down p65 in the rhIL-1β–treated cells; however, in the TGF-β1 pretreated cells, immunoprecipitation with anti-CBP brought down Smad2 but not p65. These results suggest that there is a competition between activated Smads and activated p65 for what may be effectively limiting amounts of CBP in the nuclei of endothelial cells. To examine this hypothesis directly, we overexpressed the full-length CBP protein in BAECs and examined the effects on the ability of TGF-β1 to inhibit NFκB-mediated gene expression. As demonstrated in Fig. 4 B, overexpression of CBP augmented the expression of the NFκB-dependent reporter under all conditions to a modest degree, but completely abolished the ability of TGF-β1 pretreatment to inhibit the TNF-α–induced induction of this promoter. The ability of overexpressed CBP to abolish the inhibitory actions of TGF-β1 appeared dose dependent. As shown in Fig. 4 B (right), increasing ratios of cotransfected CBP expression plasmid resulted in decreasing degrees of inhibition in response to TGF-β1 pretreatment. This effect was not seen with a truncated form of CBP that is not capable of interacting with Smad proteins (data not shown).
Figure 4.
(A) Smad2 but not p65 is recruited into a CBP-containing complex in response to TGF-β1 pretreatment followed by rhIL-1β treatment of HUVECs. HUVEC monolayers were treated with 10 U/ml of rhIL-1β for 30 min with and without pretreatment with TGF-β1 at 1 ng/ml for 6 h. Protein lysates were then prepared and immunoprecipitated with anti-Smad2, anti-p65, or anti-CBP antisera as indicated. These immunoprecipitates were then analyzed by Western blot with the antisera indicated. (B) Overexpression of CBP reverses TGF-β1–mediated inhibition of TNF-α–induced NFκB-mediated transcription. BAEC monolayers were cotransfected with the simplified NFκB-dependent promoter and either empty expression vector or vector encoding wild-type CBP (left). They were then treated with TNF-α (100 U/ml) both with and without TGF-β1 pretreatment (1 ng/ml), as described, and luciferase activity was assayed 12 h later. Overexpression of CBP reversed the ability of TGF-β1 pretreatment to inhibit NFκB-mediated transcription. The right panel shows cells that were transfected with the NFκB-dependent promoter together with increasing ratios of the CBP expression plasmid and subsequently pretreated with TGF-β1 before TNF-α stimulation to demonstrate that this effect is dose dependent. (C) Expression of subdomains of CBP. The region of interaction between CBP and Smad2 was mapped to amino acids 1892–2175, and is shown schematically. Two subdomains of CBP corresponding to amino acids 1459–1891 or 1892–2441, respectively, were subcloned by PCR and expressed as epitope (Flag)-tagged species and detected by anti-Flag immunoblot, as shown. (D) Overexpression of a specific subdomain of CBP can act as selective inhibitor of Smad-dependent transcription when overexpressed. BAECs were transfected with either the TGF-β1 responsive reporter construct p3TP-lux (left), a Gal4-Smad2 fusion protein expression construct together with a Gal4 reporter (middle), or a Gal4-Jun fusion protein expression construct together with the Gal4 reporter (right), and were stimulated by either soluble TGF-β1 (5 U/ml for 18 h) or coexpression of a constitutively active form of mitogen-activated protein kinase kinase 1 (actMEKK1). The effect of overexpression of the subdomains of CBP described in the legend to Fig. 4 C was assessed by cotransfecting these expression constructs as listed. Overexpression of the subdomain corresponding to the Smad interaction domain (1892–2441) was able to inhibit the TGF-β/Smad-dependent promoters but not the Gal4-Jun–mediated promoter. The adjacent subdomain of CBP (1459–1891) did not act as an inhibitor of these promoters. (E) Overexpression of 1892 but not 1459 can reverse TGF-β1–mediated inhibition of NFκB-dependent transcription. BAECs were cotransfected with the simplified NFκB-dependent promoter and the indicated subdomains of CBP (left). The cells were subsequently stimulated with TNF-α with and without TGF-β1 pretreatment, as above. In the presence of the overexpressed 1892 subdomain of CBP, the TGF-β–mediated inhibition of the NFκB-dependent promoter is not observed. The right panel shows cells that were transfected with the NFκB reporter and increasing ratios of the 1892 subdomain expression vector, then pretreated with TGF-β1 before TNF-α stimulation, as above. Increasing doses of the 1892 expression vector result in a progressive decrease in the ability of TGF-β1 pretreatment to inhibit the cytokine induction of this promoter.




To demonstrate that the ability of CBP to reverse TGF-β/Smad-mediated inhibition of NFκB-mediated gene expression was not due to nonspecific induction of general transcription by overexpressed CBP, we employed a dominant negative approach to selectively interfere with Smad–CBP interactions. Using a mammalian two-hybrid strategy, we have mapped the domain of the large CBP protein that interacts with Smad2 in endothelial cells 13 to amino acids 1892–2175 (schematized in Fig. 4 C). We then created vectors to express this domain of CBP (termed 1892) as well as an adjacent domain of CBP corresponding to amino acids 1459–1891 (termed 1459) as epitope-tagged species. The effects of overexpression of these proteins on gene expression in endothelial cells was then examined. As demonstrated in Fig. 4 D, overexpression of 1892 markedly inhibited the TGF-β1–mediated induction of p3Tp-lux, a chimeric TGF-β1 responsive promoter, whereas 1459 had no effect. Similarly, the 1892 protein markedly inhibited the transcriptional activity of a Gal4-Smad2 fusion protein in response to TGF-β1, whereas 1459 had no effect. In contrast, neither protein inhibited the transcriptional activity of a Gal4-Jun fusion protein (stimulated by forced expression of a constitutively active form of the kinase mitogen-activated protein kinase kinase 1 (MEKK-1). These results indicate that overexpression of the 1892–2441 subdomain of CBP can effectively act as a selective inhibitor of TGF-β1–mediated, Smad-2–dependent transcriptional activation in endothelial cells (presumably via a dominant negative mechanism), whereas the 1459–1891 subdomain cannot. We next examined the effects of overexpression of these proteins on the ability of TGF-β1 to inhibit cytokine-induced NFκB-mediated gene expression. As shown in Fig. 4 E, in the presence of overexpressed 1459, pretreatment of the endothelial cells with TGF-β1 was still able to inhibit TNF-α–induced reporter gene expression, whereas overexpression of 1892 completely abolished this inhibitory effect. This effect was also dose dependent in that increasing ratios of the 1892 subdomain expression vector resulted in progressively less inhibition in response to TGF-β1 pretreatment (Fig. 4 E, right). This same pattern of response was seen when the E-selectin promoter was used for these experiments (data not shown). Taken together, these results indicate that Smad–CBP interactions are required for the inhibitory effects of TGF-β1 pretreatment on NFκB-mediated gene expression, and that augmentation of the effectively limited amount of CBP present in endothelial cells is capable of reversing these effects.
Discussion
The ability of TGF-β1 to modulate inflammatory events is thought to be one of the most critical actions of this cytokine/growth factor in postnatal and adult mammals. In this report, we have demonstrated that exogenous active TGF-β1 can effectively inhibit the expression of the proinflammatory adhesion molecule E-selectin in cytokine-stimulated endothelial cells both in vitro and in vivo. This effect occurs at the transcriptional level, as evidenced by reporter gene experiments using the E-selectin promoter, and is mediated by Smad proteins, as overexpression of specific inhibitory or dominant negative Smads can block these actions. Furthermore, we are proposing that this inhibition occurs as a result of a competition between Smad proteins activated by TGF-β1 and NFκB proteins activated by inflammatory cytokines for what appears to be a limited amount of an essential transcriptional coactivator (CBP) present in the nuclei of endothelial cells. This novel mechanism of action for TGF-β1 in endothelial cells is supported by the following findings: (a) under conditions where TGF-β1 pretreatment can effectively inhibit cytokine-induced E-selectin expression in cultured endothelial cells, the levels and state of activation of NFκB appear identical to that seen with cytokine activation alone; (b) although both p65 and Smad2 can be recruited to CBP-containing complexes by either rhIL-1β or TGF-β1, respectively, only Smad2 appears to be effectively recruited to CBP-containing complexes in the presence of both stimuli (i.e., TGF-β1 pretreatment); (c) augmentation (via overexpression) of CBP levels in endothelial cells can effectively reverse TGF-β1–mediated inhibition of cytokine-induced E-selectin expression in endothelial cells; and (d) selective inhibition of Smad–CBP interactions via a dominant negative overexpression strategy effectively blocks TGF-β1–mediated inhibition of E-selectin expression.
Although we have proposed a relatively simple model involving a competition for limiting amounts of CBP, the situation is likely to be more complex. Rather than a simple stoichiometric competition, there may be allosteric changes in the conformation of CBP that are induced by Smad binding and inhibit its ability to interact with NFκB. In addition, there are several additional classes of transcriptional coactivators and corepressors in most cells, and these may be playing a role 32. To investigate this possibility, we have examined the effects of overexpression of p300/CBP-associated factor 1 and steroid receptor coactivator 1, and have not observed any modulation of the effects of TGF-β1 in endothelial cells (data not shown). Thus, in general, it is not yet clear what determines which transcription factors are successful in recruiting CBP, or why other transcriptional complexes are not 30. In our hands, the ability of TGF-β1 pretreatment to effectively inhibit NFκB-mediated E-selectin gene expression could be seen with as little as 30 min of pretreatment with TGF-β1, suggesting that significant alterations in the phenotype of the endothelial cells were not being induced. In addition, because we did not find any evidence that p65 and Smad2 were in the same transcriptional complexes in endothelial cells, it is unlikely that Smad-mediated recruitment of a corepressor to the E-selectin promoter is occurring. Thus, the simplest model that is consistent with the data presented is one where the limited amount of the required coactivator, CBP, present in endothelial cells is selectively recruited to Smad protein–containing complexes, and thus is not available to interact with NFκB even though the latter has been activated. This type of coactivator-mediated signal integration, or “coactivator competition,” has been demonstrated in other systems. For example, the antagonistic interactions between IFN-regulated signal transducer and activator of transcription proteins and the activating protein 1 (AP-1) family of transcriptional activators in macrophages has been postulated to be due to this type of coactivator competition 28. In endothelial cells, the antiinflammatory actions of glucocorticoids may be due to an analogous process involving steroid receptors and NFκB 33. In addition, p53 and NFκB have been reported to mutually repress each other's ability to stimulate transcription by an analogous mechanism involving coactivator competition 34.
Previous work has demonstrated that TGF-β1 can inhibit B cell activation via stabilization and/or induction of IκB, the inhibitory subunit of NFκB 35. Our results in vascular endothelium indicate that the antiinflammatory actions of this growth factor on endothelium are due, at least in part, to a distinct mechanism of action. Under conditions where TGF-β1 pretreatment effectively inhibited cytokine-induced E-selectin expression, we found that IκB levels were markedly diminished, and NFκB activation (as assessed by DNA binding assay) was preserved. Furthermore, our data indicate that TGF-β1–mediated inhibition of inflammatory E-selectin expression can occur in vascular endothelium in vivo. As the data in Fig. 3 indicate, pretreatment of rats with systemic TGF-β1 before LPS challenge can effectively block the induction of E-selectin mRNA in multiple tissues. This effect must be occurring in the vascular endothelium, as this is the only cell type present within these tissues that express this gene 1. In this setting, the activation of NFκB as assessed by gel shift analysis of tissue extracts appears to be preserved.
We assessed the specificity of this effect by specifically interfering with defined Smad–coactivator (CBP) interactions via a dominant negative strategy. By overexpressing a subdomain of CBP that is known to mediate the interactions between this coactivator and Smad2 and -4, we can demonstrate a specific and selective inhibition of Smad-mediated transcriptional events. Under these conditions, TGF-β1 pretreatment of endothelial cells does not inhibit NFκB-mediated gene expression. These results not only demonstrate the requirement for Smad–coactivator interactions in the inhibitory process, but also suggest a novel level at which the actions of these growth factors may be modulated. Reagents designed to specifically interfere with defined transcription factor–coactivator interactions may allow one to selectively modulate the actions of these growth factors at the level of the nucleus.
The recent elucidation of the detailed molecular events by which TGF-β1 and related cytokines/growth factors elicit their cellular effects has resulted in the realization that these pathways can functionally interact with a variety of other intracellular signaling pathways in cells 7. We have demonstrated that TGF-β1 can functionally interact with the NFκB signaling pathway in endothelial cells by a mechanism involving nuclear coactivator interactions. Because the coactivators CBP/P300 interact with a wide variety of transcriptional effectors that are the targets of diverse signaling pathways, these observations may begin to provide a conceptual framework for understanding the complex molecular interactions underlying the pleiotropic, or “context-specific,” actions of this class of cytokines/growth factors.
Acknowledgments
We would like to thank Drs. Charles Homcy and Matt Hart for their critical review of the manuscript.
This work was supported by grants RO1-HL62823-01 (to J.N. Topper) and P01-HL36028 (M.A. Gimbrone, Jr.) from the National Heart, Lung, and Blood Institute, and by a Junior Faculty Award from the Howard Hughes Medical Institute to J.N. Topper.
Footnotes
Abbreviations used in this paper: BAEC, bovine aortic endothelial cell; CBP, cyclic AMP response element–binding protein (CREB)-binding protein; HUVEC, human umbilical vein endothelial cell; NFκB, nuclear factor κB.
References
- Bevilacqua M.P., Stengelin S., Gimbrone M.A.J., Jr., Seed B. Endothelial leukocyte adhesion molecule 1an inducible receptor for neutrophils related to complement regulatory proteins and lectins. Science. 1989;243:1160–1165. doi: 10.1126/science.2466335. [DOI] [PubMed] [Google Scholar]
- Collins T., Read M.A., Neish A.S., Whitley M.Z., Thanos D., Maniatis T. Transcriptional regulation of endothelial cell adhesion moleculesNF-kappa B and cytokine-inducible enhancers. FASEB J. 1995;9:899–909. [PubMed] [Google Scholar]
- Read M.A., Neish A.S., Luscinskas F.W., Palombella V.J., Maniatis T., Collins T. The proteasome pathway is required for cytokine-induced endothelial-leukocyte adhesion molecule expression. Immunity. 1995;2:493–506. doi: 10.1016/1074-7613(95)90030-6. [DOI] [PubMed] [Google Scholar]
- Read M.A., Whitley M.Z., Gupta S., Pierce J.W., Best J., Davis R.J., Collins T. Tumor necrosis factor alpha-induced E-selectin expression is activated by the nuclear factor-kappaB and c-JUN N-terminal kinase/p38 mitogen-activated protein kinase pathways. J. Biol. Chem. 1997;272:2753–2761. doi: 10.1074/jbc.272.5.2753. [DOI] [PubMed] [Google Scholar]
- Whitley M.Z., Thanos D., Read M.A., Maniatis T., Collins T. A striking similarity in the organization of the E-selectin and beta interferon gene promoters. Mol. Cell. Biol. 1994;14:6464–6475. doi: 10.1128/mcb.14.10.6464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J. TGFbeta signalingreceptors, transducers, and Mad proteins. Cell. 1996;85:947–950. doi: 10.1016/s0092-8674(00)81296-9. [DOI] [PubMed] [Google Scholar]
- Piek E., Heldin C.H., Ten Dijke P. Specificity, diversity, and regulation in TGF-beta superfamily signaling. FASEB J. 1999;13:2105–2124. [PubMed] [Google Scholar]
- Metcalfe J.C., Grainger D.J. TGF-betaimplications for human vascular disease. J. Hum. Hypertens. 1995;9:679. [PubMed] [Google Scholar]
- Shull M.M., Ormsby I., Kier A.B., Pawlowski S., Diebold R.J., Yin M., Allen R., Sidman C., Proetzel G., Calvin D. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble J.R., Khew-Goodall Y., Vadas M.A. Transforming growth factor-beta inhibits E-selectin expression on human endothelial cells. J. Immunol. 1993;150:4494–4503. [PubMed] [Google Scholar]
- Wrana J.L. TGF-beta receptors and signalling mechanisms. Miner. Electrolyte Metab. 1998;24:120–130. doi: 10.1159/000057359. [DOI] [PubMed] [Google Scholar]
- Raftery L.A., Twombly V., Wharton K., Gelbart W.M. Genetic screens to identify elements of the decapentaplegic signaling pathway in Drosophila . Genetics. 1995;139:241–254. doi: 10.1093/genetics/139.1.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topper J.N., DiChiara M.R., Brown J.D., Williams A.J., Falb D., Collins T., Gimbrone M.A., Jr. CREB binding protein is a required coactivator for Smad-dependent, transforming growth factor beta transcriptional responses in endothelial cells. Proc. Natl. Acad. Sci. USA. 1998;95:9506–9511. doi: 10.1073/pnas.95.16.9506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janknecht R., Wells N.J., Hunter T. TGF-beta-stimulated cooperation of smad proteins with the coactivators CBP/p300. Genes Dev. 1998;12:2114–2119. doi: 10.1101/gad.12.14.2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X.H., Zhang Y., Wu R.Y., Derynck R. The tumor suppressor Smad4/DPC4 and transcriptional adaptor CBP/p300 are coactivators for smad3 in TGF-beta-induced transcriptional activation. Genes Dev. 1998;12:2153–2163. doi: 10.1101/gad.12.14.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouponnot C., Jayaraman L., Massague J. Physical and functional interaction of SMADs and p300/CBP. J. Biol. Chem. 1998;273:22865–22868. doi: 10.1074/jbc.273.36.22865. [DOI] [PubMed] [Google Scholar]
- Nishihara A., Hanai J.I., Okamoto N., Yanagisawa J., Kato S., Miyazono K., Kawabata M. Role of p300, a transcriptional coactivator, in signalling of TGF-beta. Genes Cells. 1998;3:613–623. doi: 10.1046/j.1365-2443.1998.00217.x. [DOI] [PubMed] [Google Scholar]
- Topper J.N., Cai J., Qiu Y., Anderson K.R., Xu Y.Y., Deeds J.D., Feeley R., Gimeno C.J., Woolf E.A., Tayber O. Vascular MADstwo novel MAD-related genes selectively inducible by flow in human vascular endothelium. Proc. Natl. Acad. Sci. USA. 1997;94:9314–9319. doi: 10.1073/pnas.94.17.9314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J.D., DiChiara M.R., Anderson K.R., Gimbrone M.A., Jr., Topper J.N. MEKK-1, a component of the stress (stress-activated protein kinase/c- Jun N-terminal kinase) pathway, can selectively activate Smad2-mediated transcriptional activation in endothelial cells. J. Biol. Chem. 1999;274:8797–8805. doi: 10.1074/jbc.274.13.8797. [DOI] [PubMed] [Google Scholar]
- Hayashi H., Abdollah S., Qiu Y., Cai J., Xu Y.Y., Grinnell B.W., Richardson M.A., Topper J.N., Gimbrone M.A., Jr., Wrana J.L., Falb D. The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell. 1997;89:1165–1173. doi: 10.1016/s0092-8674(00)80303-7. [DOI] [PubMed] [Google Scholar]
- Hata A., Lagna G., Massague J., Hemmati-Brivanlou A. Smad6 inhibits BMP/Smad1 signaling by specifically competing with the Smad4 tumor suppressor. Genes Dev. 1998;12:186–197. doi: 10.1101/gad.12.2.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellacani A., Wiesel P., Sharma A., Foster L.C., Huggins G.S., Yet S.F., Perrella M.A. Induction of heme oxygenase-1 during endotoxemia is downregulated by transforming growth factor-beta1. Circ. Res. 1998;83:396–403. doi: 10.1161/01.res.83.4.396. [DOI] [PubMed] [Google Scholar]
- Perrella M.A., Yoshizumi M., Fen Z., Tsai J.C., Hsieh C.M., Kourembanas S., Lee M.E. Transforming growth factor-beta 1, but not dexamethasone, down-regulates nitric-oxide synthase mRNA after its induction by interleukin-1 beta in rat smooth muscle cells. J. Biol. Chem. 1994;269:14595–14600. [PubMed] [Google Scholar]
- Perrella M.A., Patterson C., Tan L., Yet S.F., Hsieh C.M., Yoshizumi M., Lee M.E. Suppression of interleukin-1beta-induced nitric-oxide synthase promoter/enhancer activity by transforming growth factor-beta1 in vascular smooth muscle cells. Evidence for mechanisms other than NF- kappaB. J. Biol. Chem. 1996;271:13776–13780. doi: 10.1074/jbc.271.23.13776. [DOI] [PubMed] [Google Scholar]
- Chakravarti D., LaMorte V.J., Nelson M.C., Nakajima T., Schulman I.G., Juguilon H., Montminy M., Evans R.M. Role of CBP/P300 in nuclear receptor signalling. Nature. 1996;383:99–103. doi: 10.1038/383099a0. [DOI] [PubMed] [Google Scholar]
- Kamei Y., Xu L., Heinzel T., Torchia J., Kurokawa R., Gloss B., Lin S.C., Heyman R.A., Rose D.W., Glass C.K., Rosenfeld M.G. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell. 1996;85:403–414. doi: 10.1016/s0092-8674(00)81118-6. [DOI] [PubMed] [Google Scholar]
- Lundblad J.R., Kwok R.P., Laurance M.E., Harter M.L., Goodman R.H. Adenoviral E1A-associated protein p300 as a functional homologue of the transcriptional co-activator CBP. Nature. 1995;374:85–88. doi: 10.1038/374085a0. [DOI] [PubMed] [Google Scholar]
- Horvai A.E., Xu L., Korzus E., Brard G., Kalafus D., Mullen T.M., Rose D.W., Rosenfeld M.G., Glass C.K. Nuclear integration of JAK/STAT and Ras/AP-1 signaling by CBP and p300. Proc. Natl. Acad. Sci. USA. 1997;94:1074–1079. doi: 10.1073/pnas.94.4.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S., Eckner R., Grossman S., Oldread E., Arany Z., D'Andrea A., Livingston D.M. Cooperation of Stat2 and p300/CBP in signalling induced by interferon-alpha. Nature. 1996;383:344–347. doi: 10.1038/383344a0. [DOI] [PubMed] [Google Scholar]
- Kurokawa R., Kalafus D., Ogliastro M.H., Kioussi C., Xu L., Torchia J., Rosenfeld M.G., Glass C.K. Differential use of CREB binding protein-coactivator complexes. Science. 1998;279:700–703. doi: 10.1126/science.279.5351.700. [DOI] [PubMed] [Google Scholar]
- Gerritsen M.E., Williams A.J., Neish A.S., Moore S., Shi Y., Collins T. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc. Natl. Acad. Sci. USA. 1997;94:2927–2932. doi: 10.1073/pnas.94.7.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wotton D., Lo R.S., Lee S., Massague J. A Smad transcriptional corepressor. Cell. 1999;97:29–39. doi: 10.1016/s0092-8674(00)80712-6. [DOI] [PubMed] [Google Scholar]
- Sheppard K.A., Phelps K.M., Williams A.J., Thanos D., Glass C.K., Rosenfeld M.G., Gerritsen M.E., Collins T. Nuclear integration of glucocorticoid receptor and nuclear factor-kappaB signaling by CREB-binding protein and steroid receptor coactivator-1. J. Biol. Chem. 1998;273:29291–29294. doi: 10.1074/jbc.273.45.29291. [DOI] [PubMed] [Google Scholar]
- Wadgaonkar R., Phelps K.M., Haque Z., Williams A.J., Silverman E.S., Collins T. CREB-binding protein is a nuclear integrator of nuclear factor-kappaB and p53 signaling. J. Biol. Chem. 1999;274:1879–1882. doi: 10.1074/jbc.274.4.1879. [DOI] [PubMed] [Google Scholar]
- Arsura M., Wu M., Sonenshein G.E. TGF beta 1 inhibits NF-kappa B/Rel activity inducing apoptosis of B cellstranscriptional activation of I kappa B alpha. Immunity. 1996;5:31–40. doi: 10.1016/s1074-7613(00)80307-6. [DOI] [PubMed] [Google Scholar]