

Abstract
Hyaluronan (HA), a high molecular weight glycosaminoglycan, is expressed abundantly in the extracellular matrix and on cell surfaces. Although HA is known to bind many adhesion molecules, little information has been available with respect to its direct physiological role. In this study, we developed a novel 12-mer (GAHWQFNALTVR) peptide inhibitor of HA, termed “Pep-1,” by using phage display technology. Pep-1 showed specific binding to soluble, immobilized, and cell-associated forms of HA, and it inhibited leukocyte adhesion to HA substrates almost completely. Systemic, local, or topical administration of Pep-1 inhibited the expression of contact hypersensitivity responses in mice by blocking skin-directed homing of inflammatory leukocytes. Pep-1 also inhibited the sensitization phase by blocking hapten-triggered migration of Langerhans cells from the epidermis. These observations document that HA plays an essential role in “two-way” trafficking of leukocytes to and from an inflamed tissue, and thus provide technical and conceptual bases for testing the potential efficacy of HA inhibitors (e.g., Pep-1) for inflammatory disorders.
Keywords: glycosaminoglycan, leukocyte homing, contact hypersensitivity, Langerhans cell, phage display
Introduction
Extracellular matrix molecules (e.g., fibronectin, laminin, collagens, proteoglycans, and glycosaminoglycans) bind specific cell surface receptors via protein–protein and protein–carbohydrate interactions. Hyaluronan (HA), a large glycosaminoglycan consisting of repeating disaccharide units of N-acetyl glucosamine and glucuronic acid, is expressed abundantly in the extracellular matrix as well as on cell surfaces. HA has been shown to bind several different molecules, including CD44 1 2, the receptor for HA-mediated motility (RHAMM 3), link protein 4, aggrecan 5, versican 6, hyaluronectin 7, neurocan 8, liver sinusoidal endothelial HA receptor 9, inter-α-trypsin inhibitor–related proteins 10, BEHAB (brain-enriched HA binding; reference 11), CD38 12, lymphatic vessel endothelial HA receptor 1 13, and white fat/bone marrow/osteoblast HA binding proteins 14. Conversely, CD44 binds not only HA, but also collagens 15 16, fibronectin 17, chondroitin sulfates 18, heparin 18, heparin sulfate 18, and serglycins 19. Thus, although CD44 (or HA) is generally considered to be a primary HA receptor (or a principal CD44 ligand), HA–CD44 interaction represents one of the multiple mechanisms by which HA and CD44 may regulate cellular activities.
Previous studies using CD44 inhibitors (e.g., anti-CD44 antibodies and fusion proteins containing extracellular domains of CD44) have established the concept that CD44 is involved in diverse cellular functions such as cell adhesion and migration 2 20 21, expression of other adhesion molecules 22 23, leukocyte activation 24 25, production of cytokines and chemokines 26 27 28, apoptosis 29 30, and tumor metastasis 31 32 33. The same inhibitors have also exhibited potent pharmacological activities in animal models of T cell–mediated inflammatory diseases 21 34. These observations may initially appear to support the notion that HA–CD44 interaction participates in the rolling of leukocytes over endothelial cells, thus promoting leukocyte homing 35 36. However, it should be pointed out that those CD44 inhibitors (which potentially interfere with the binding of CD44 to its multiple ligands) revealed the function of CD44, but not the role for HA. We reasoned that reagents designed to block the function of HA would provide a unique tool for studying physiological roles of HA. Here, we report the biochemical and biological properties of a newly developed HA inhibitor.
Materials and Methods
Animals and Cell Lines.
Female BALB/c mice (6–8 wk) were purchased from The Jackson Laboratory. All animal experiments were approved by the Institutional Review Board. The SVEC endothelial cell, Pam 212 keratinocyte, and XS106 Langerhans cell lines were maintained as described previously 37 38 39 40. The BW5147 thymoma line was purchased from American Type Culture Collection, and T cells were isolated from BALB/c mice and from human peripheral blood by using T cell enrichment columns (R&D Systems).
Isolation of HA-binding Peptides by Phage Display.
The M13 phage library (Ph.D.-12™ kit) expressing random 12-mer peptides fused to pIII proteins with the complexity of ∼109 was purchased from New England Biolabs, Inc. Falcon 35-mm tissue culture dishes (Becton Dickinson) were incubated overnight at 4°C with 2.5 mg/ml of sodium HA (>95% pure preparation derived from bacterial fermentation; Acros Organics) in distilled water. In the fourth cycle of panning, the HA concentration was reduced to 250 μg/ml. After extensive washing with PBS, the HA-coated plates were countercoated with 1% BSA in PBS containing 0.1% Tween 20 for 2 h at 37°C. Phage with titers of 1010 PFU (first cycle) or 1011 PFU (subsequent cycles) suspended in 0.01% BSA in PBS were incubated on the HA-coated plate for 30 min at room temperature with gentle rocking. Subsequently, the plate was washed 10 times with 0.1% Tween 20 in PBS to remove unbound phage, then incubated for 2 h at 37°C with 330 U/ml hyaluronidase (HAase; Sigma-Aldrich) in enzyme buffer (20 mM NaH2PO4, 77 mM NaCl, 0.01% BSA, pH 6.0). The phages that were eluted by the HAase treatment were amplified in Escherichia coli, titrated, and then subjected to the next cycle of panning. After the second panning against HA-coated plates, the phages were incubated on the plate coated with 2.5 mg/ml chondroitin sulfate A (CSA; Sigma-Aldrich) to remove the clones that bound nonspecifically to glycosaminoglycans. After four cycles of positive selection over HA and an additional cycle of negative selection over CSA, 19 independent phage plaques were picked randomly, and single-stranded DNAs isolated from these clones were sequenced using the primer 5′-CCCTCATAGTTAGCGTAACG-3′. Deduced amino acid sequences were then searched for their molecular identities and homologies using advanced BLAST search (EMBL/GenBank/DDBJ) and AA CompIdent tool (SWISS-PROT and TrEMBL).
Synthesis and Labeling of Pep-1 and Other Peptides.
Pep-1 and other control peptides were synthesized in house or by Research Genetics using standard fMOC chemistry. The amidated G-G-G-S linker sequence was included at the COOH terminus of each peptide. Each synthetic peptide was analyzed by reverse-phase HPLC and mass spectrometry to assure the purity and identity. Biotinylated peptides were synthesized by the addition of an amidated Lys residue at the COOH terminus with biotin covalently linked to the ε-amine. Peptide stock solutions were prepared immediately before use by dissolution in DMSO followed by dilution in PBS to achieve 1 or 2 mg/ml concentration in 2% DMSO. Peptide solutions (1 mg/ml) were iodinated by a 3-h incubation on ice with 1 mM sulfosuccimidyl-3-(4-hydroxyphenyl) proprinate (Pierce Chemical Co.), followed by labeling with 10 μCi/ml of 125I (ICN Biomedicals) using IODO-beads (Pierce Chemical Co.).
Pep-1 Binding to HA-coated Beads.
Uncoated paramagnetic beads (Dynal) were labeled with 2 mg/ml HA according to the protocol from the manufacturer, and then were countercoated with 3% BSA in PBS overnight at 4°C. To test binding, 125I-labeled peptides in 3% BSA in PBS were incubated with the HA-coated beads for 1 h at 4°C, washed three times, and then counted for radioactivities. HA-specific binding was determined by subtracting the baseline cpm that bound to noncoated beads from the cpm associated with the HA-coated beads. Specific binding was then calculated from the specific activity (∼3.45 × 107 cpm/μg) and the number of beads added to the assay.
Cellular Binding of Biotinylated Pep-1 and FITC-conjugated HA.
The SVEC endothelial cells were incubated for 30 min on ice with biotinylated peptides (100 μg/ml), washed extensively, labeled with FITC-conjugated streptavidin (Jackson ImmunoResearch Laboratories), and then analyzed by FACSCalibur™ (Becton Dickinson). Some samples were pretreated with 330 U/ml HAase before incubation with peptides to assess HA specificity. The BW5147 thymoma cells were incubated for 30 min on ice with FITC-conjugated HA (0.4 μg/ml; reference 41), washed extensively, and then analyzed by FACS®. In some experiments, FITC-conjugated HA was first incubated with Pep-1 or control peptides for 2 h at 4°C before the binding assay.
Cell Adhesion Assays.
HA solutions (100 μl/well) were added to Amine CovaLink 96-well plates (Nunc), followed by addition of 50 μl of 0.1 N HCl and 50 μl of 200 μM 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (Pierce Chemical Co.). After overnight incubation at room temperature, the wells were washed three times with PBS containing 2 M NaCl and 40 μM MgSO4, followed by two additional washes with PBS alone. The HA-coated plates were then countercoated with 5% FCS in PBS for 2 h at 37°C. Cells were radiolabeled with 30 μCi/ml of [35S] methionine/cysteine (ICN Biomedicals) for 2 h (BW5147 cells) or overnight (other cell types), washed extensively, and incubated in PBS containing 5% FCS and 2 mM CaCl2 on the HA-coated plates for 30 min at room temperature. After removal of nonadherent cells, the adherent cells were solubilized in 1% SDS and counted for radioactivities. The percentages of adherent cells were calculated by dividing the recovered cpm by the total cpm added to each well. In some experiments, HA-coated wells were pretreated with Pep-1 or control peptides in PBS containing 5% FCS and 2 mM CaCl2 for 2 h at room temperature. Alternatively, the cells were preincubated with 70 μg/ml of anti-CD44 mAb KM81 (American Type Culture Collection) or isotype-matched control IgG.
Langerhans Cell Migration Assay.
BALB/c mice received topical application of 0.5% dinitrofluorobenzene (DNFB) on the right ears and vehicle alone on the left ears. Ear skin samples were harvested 24 h later and processed for whole mount staining for Langerhans cells as before 42. In brief, the epidermis was separated as an intact sheet from the remaining dermis after a 20-min incubation in 0.5 M ammonium thiocyanate at 37°C, fixed in acetone, and then stained with FITC-conjugated anti-IA mAb 2G9 (BD PharMingen). Numbers of IA+ epidermal cells (i.e., Langerhans cells) were counted under an Olympus BX60 fluorescence microscope (Olympus) equipped with a Sensys digital camera system (Photometrics) and Metaview software (Universal Imaging Corp.) at ×400 magnification in 10 different fields per sample.
Contact Hypersensitivity Assays.
Contact hypersensitivity reactions were examined as before 40. In brief, mice were sensitized on shaved abdominal skin with 0.5% DNFB and challenged 7 d later with 0.2% DNFB on the right ears. The left ears were challenged with vehicle alone to serve as controls. In the “double-sensitization” experiments, mice were sensitized by topical application of 0.5% DNFB on the left ears (or the abdomen) and 1.25% oxazolone (OX) on the abdomen (or the left ears); these mice were challenged with 0.2% DNFB or 0.5% OX on the right ears. To induce chronic inflammation, mice were sensitized with 0.5% DNFB on the ear skin and challenged repeatedly by the applications of 0.2% DNFB on the same site every other day 43. The ear thickness was measured at 24 h (for OX), 48 h (for DNFB), or every 24 h (in the chronic model) after challenge by a third experimenter “blinded” to sample identity, using an engineer's micrometer. The ear swelling response was calculated as the thickness of the right ear (painted with a hapten) minus the baseline thickness of the left ear (painted with vehicle alone), except for the double-sensitization and chronic inflammation experiments, in which the thickness measured immediately before challenge served as the baseline.
Pep-1 Treatment Protocols.
Pep-1 or control peptides were administered into animals with three different protocols: (a) subcutaneous injection into ears (40 μg/ear/injection), (b) intravenous injection (1 mg/animal), and (c) topical application on ears (40 μg/ear) after removal of barrier function of stratum corneum by soaking ear skin twice with acetone 42. Mice were treated with one of these protocols before or after sensitization as described in the figure legends.
Statistical Analyses.
Animal experiments were conducted with 5–15 mice per group, and in vitro experiments were performed with triplicate samples. Comparisons between two groups were performed with a two-tailed Student's t-test, and more than two groups were compared by analysis of variance (ANOVA). Each experiment was repeated two to five times to assess reproducibility.
Results
Isolation of HA-binding Peptides.
Although the phage display technology has been used almost exclusively to isolate peptide fragments that bind to polypeptide targets, we reasoned that the same technology might be applicable to isolate HA-binding peptides. The M13 phage library expressing random 12-mer peptides fused to the pIII minor coat protein (with the complexity of ∼109) was incubated on polystyrene plates that had been coated with HA and countercoated with BSA. After removal of unbound phage, HAase was added to the panning plates to elute only those phage clones that had bound to HA, but not to polystyrene surfaces or BSA. After four cycles of panning over HA-coated plates, we isolated and sequenced 19 independent phage clones. Strikingly, despite a theoretical complexity of 109 in the starting population, an overwhelming majority (13/19) of the isolated clones expressed an identical peptide motif of G-A-H-W-Q-F-N-A-L-T-V-R, which was designated as “Pep-1.” We also identified two motifs that were each displayed by two independent phage clones (T-S-Y-G-R-P-A-L-L-P-A-A; Pep-2, and M-D-H-L-A-P-T-F-R-P-A-I; Pep-3) and two motifs that were expressed by single clones (T-L-R-A-I-W-P-M-W-M-S-S; Pep-4, and I-P-L-T-A-N-Y-Q-G-D-F-T; Pep-5). None of these peptide sequences showed significant (>25%) identities to the known HA-binding domain sequences of CD44, RHAMM, or link protein 44, or to any of the polypeptides currently registered in the EMBL/GenBank/DDBJ, SWISS-PRO, and TrEMBL databases. Nor did they contain a consensus motif, B(X)7B, known to be conserved among the above three HA receptors, where B represents a basic amino acid residue and X represents any nonacidic residue 44. Moreover, none of the isolated peptides contained “diagnostic” sequences that are commonly found in peptides that bind nonspecifically to the polystyrene surface (45; information from New England Biolabs, Inc.).
HA-binding Capacity of Pep-1.
We synthesized Pep-1 through Pep-4 to test their HA-binding capacities. As a control, we synthesized a 12-mer peptide (S-A-T-P-A-S-A-P-Y-P-L-A), termed the “random peptide” (RP), based on the sequence expressed by a phage clone randomly selected from the original library. We designed all the synthetic peptides to include the spacer sequence G-G-G-S at the COOH terminus to mimic the original configuration of the peptide moieties fused to the pIII proteins and displayed on phage surfaces. These peptides were 125I labeled and examined for binding to HA-coated beads. As shown in Fig. 1 A, Pep-1, but not Pep-2 through Pep-4, showed significantly higher binding to HA than did the RP control. Specificity of this binding was supported by the observations that: (a) Pep-1 bound to the HA-coated beads in a dose-dependent manner with an apparent affinity of Kd = 1.65 μM (Fig. 1B and Fig. C); (b) Pep-1 showed significantly higher binding to the HA-coated beads than to the beads coated with a control glycosaminoglycan CSA (Fig. 1 D); (c) HAase treatment of the HA-coated beads abrogated their Pep-1 binding capacity (Fig. 1 D); and (d) binding of 125I-labeled Pep-1 to HA was inhibited in a dose-dependent fashion by addition of “cold” Pep- 1 (Fig. 1 E).
Figure 1.

Binding of Pep-1 to HA-coated beads. (A) The indicated peptide was labeled with 125I and tested at 50 μg/ml for the binding to HA-coated beads. (B) 125I-labeled Pep-1 was tested at the indicated concentrations for the binding to HA-coated beads. (C) Binding data from an independent 125I–Pep-1 binding experiment were subjected to Scatchard analysis to estimate relative affinity of Pep-1 binding to HA. (D) 125I-labeled Pep-1 (50 μg/ml) was tested for the binding to HA- or CSA-coated beads in the presence or absence of HAase pretreatment. (E) HA-coated beads were first incubated with the indicated concentrations of nonlabeled Pep-1 for 60 min, followed by an additional 60-min incubation with 125I-labeled Pep-1 (50 μg/ml). All the data in this figure are representative of at least three independent experiments, showing the means ± SD from triplicate samples. Asterisks indicate statistically significant differences (**P < 0.01) assessed by ANOVA (A) or Student's t test (D).
On the other hand, we observed no saturation in terms of the binding of 125I-labeled Pep-1 to HA-coated beads in the direct binding assay (Fig. 1 B), and only partial (up to 45%) inhibition by addition of excess amounts of cold Pep-1 in the competition assay (Fig. 1 E). The inability to saturate and achieve complete inhibition may first appear to suggest nonspecificity of Pep-1 binding to HA. However, saturation and complete inhibition are the features expected only for the binding of a ligand to a receptor that has discrete and isolated binding sites. HA is a long, homogeneous polymer consisting of repeating disaccharide units of N-acetyl glucosamine and glucuronic acid, and thus should be considered to be a lattice with nondiscrete and overlapping binding sites. Under this circumstance, binding of a ligand becomes entropically unfavorable for saturation of the receptor. In other words, the lattice thermodynamically resists being saturated, and thus, full saturation (and complete inhibition) is only achievable by increasing the free ligand concentration to a practically unattainable level 46 47 48. Therefore, our results are in complete agreement with the model in which multiple Pep-1 molecules bind to HA in a specific and nondiscrete fashion. From the same point of view, it should be emphasized that the above apparent Kd value is a crude estimate based on the simplest model of discrete binding and may not represent the absolute affinity.
We next examined whether Pep-1 would also bind to HA molecules expressed on cell surfaces. As shown in Fig. 2 A, biotinylated Pep-1 showed significantly higher binding than did the biotinylated RP control to the SVEC endothelial cell line, which is known to express HA abundantly on the surface 49. Importantly, this binding was diminished significantly by HAase pretreatment of the SVEC cells, indicating that Pep-1 bound primarily to HA among many surface molecules (Fig. 2 B). Consistent with the observations in the bead binding assays, Pep-2 through Pep-4 showed no specific binding to the SVEC cells (Fig. 2 A). The Pam 212 keratinocyte line also showed significant binding of biotinylated Pep-1, but not Pep-2 through Pep-4, and this binding was abrogated by HAase treatment (data not shown). These observations demonstrated the ability of Pep-1 to bind not only to the HA-coated substrate, but also to the HA molecules expressed on the surfaces of endothelial cells and keratinocytes.
Figure 2.

Binding of Pep-1 to surface HA expressed on endothelial cells. (A) SVEC endothelial cells were harvested from the culture flask by a 2-min exposure to 0.25% (wt/vol) trypsin/1 mM EDTA at room temperature. The cells were then incubated for 30 min with biotinylated Pep-1 through Pep-4 (100 μg/ml; closed histograms) or biotinylated RP control (open histograms). After extensive washing, the cells were incubated with FITC-conjugated streptavidin and subjected to FACS® analyses. Data shown are representative of three independent experiments. (B) SVEC cells were treated for 1 h at 37°C with HAase (330 U/ml; closed histogram) or mock treated (open histogram with broken lines) before incubation with biotinylated Pep-1 (100 μg/ml) or with biotinylated RP control (open histogram with a solid line). Data shown are representative of three independent experiments.
In Vitro Capacity of Pep-1 to Inhibit HA Function.
To test the potential of Pep-1 to block the function of HA, we employed the BW5147 thymoma cell line, which is known to express CD44 abundantly and bind to HA 25. FITC-conjugated HA bound to the BW5147 cell surfaces (Fig. 3 A), and this binding was inhibited by Pep-1 in a dose-dependent fashion, with ∼75% inhibition achieved at 500 μg/ml (Fig. 3B and Fig. C). By contrast, the RP control showed no inhibitory effects at any tested concentrations. The same cell line showed significant adhesion onto HA-coated plates (Fig. 4 A), and the adhesion was blocked almost completely by anti-CD44 mAb (Fig. 4 B), indicating that CD44 served as a primary adhesion receptor of HA for this cell type. Importantly, Pep-1, but not the RP control, inhibited the CD44-dependent adhesion to HA substrates in a dose-dependent manner, with almost complete inhibition observed at 500 μg/ml (Fig. 4B and Fig. C). These results indicated the ability of Pep-1 to inhibit the function of both soluble and immobilized forms of HA.
Figure 3.

Pep-1 inhibits the binding of soluble HA to leukocyte surfaces. (A) BW5147 thymoma cells were incubated with FITC-conjugated HA at the indicated concentrations, washed extensively, and analyzed by FACS® for the mean fluorescence intensities (MFI). Data were plotted linearly and evaluated with a hyperbolic fit (half saturation = 1.4 μg/ml; saturation = 279.4 μg/ml). (B) FITC-conjugated HA (0.4 μg/ml) was tested for the binding to BW5147 cells before (open histograms) or after preincubation of FITC-HA with either Pep-1 or RP (500 μg/ml; closed histograms). Broken lines indicate control samples incubated with PBS alone. (C) FITC-conjugated HA (0.4 μg/ml) was incubated with the indicated concentrations of Pep-1 (•) or RP (○), and then tested for the binding to BW5147 cells. All data are representative of at least three independent experiments.
Figure 4.

Pep-1 inhibits CD44-dependent leukocyte adhesion to HA substrates. (A) 35S-labeled BW5147 cells were incubated for 30 min at room temperature in the plates coated with the indicated concentrations of HA. The data shown are the means ± SD of percentage binding from triplicate samples. (B) Binding of 35S-labeled BW5147 cells to the HA-coated plates (0.1 μg/ml) was tested after pretreatment of the cells with 70 μg/ml of anti-CD44 mAb (KM81) or control IgG, or after pretreatment of the substrate with Pep-1 or RP (500 μg/ml). Statistically significant differences (Student's t test) are indicated with asterisks (**P < 0.01). (C) HA-coated plates (0.1 μg/ml) were pretreated with the indicated concentrations of Pep-1 (•) or RP (○). 35S-labeled BW5147 cells were then tested for the binding to these plates. (D) Wild-type Pep-1, RP, and Ala-substituted mutants of Pep-1 were tested at 500 μg/ml for their ability to inhibit the adhesion of 35S-labeled BW5147 cells to HA-coated plates. Asterisks indicate statistically significant differences (**P < 0.01) compared with the RP peptide control. (E) Composition-matched, sequence-disparate peptides were synthesized by scrambling the Pep-1 sequence randomly. The original Pep-1, RP, and scrambled peptides with the indicated sequences were tested at 500 μg/ml for their ability to inhibit the adhesion of 35S-labeled BW5147 cells to HA-coated plates. Asterisks indicate statistically significant differences (**P < 0.01) compared with the RP peptide control. (F) Murine splenic T cells, human peripheral blood T cells, or murine Langerhans cell line XS106 were 35S-labeled and tested for their adhesion to HA-coated plates in the presence or absence of the indicated pretreatment. Asterisks indicate statistically significant differences (**P < 0.01). Data in this figure are representative of two or three independent experiments.
To determine which amino acid residue(s) in Pep-1 were critical, we tested biological activities of Ala-substituted mutants of Pep-1. Once again, the wild-type Pep-1 inhibited BW5147 cell adhesion to HA-coated plates (Fig. 4 D). This activity was maintained after Ala substitution of the His residue at position 3, Asn residue at position 7, or Arg residue at position 12. By contrast, Ala substitution at position 4, 5, 6, 9, 10, or 11 abrogated the biological activity almost completely. Interestingly, the replaceable residues were all charged amino acids, whereas the irreplaceable ones were either aliphatic or polar aliphatic amino acids, implying that Pep-1 might bind to HA by a hydrophobic–hydrophobic interaction. A next question concerned whether the amino acid composition would be the sole determinant for the biological activity observed with Pep-1. To test this question, we scrambled the Pep-1 sequence randomly and synthesized four composition-matched, sequence-disparate peptides. None of these scrambled peptides blocked the adhesion of BW5147 cells to the HA-coated plates, whereas almost complete inhibition was achieved with the original Pep-1 (Fig. 4 E). These observations illustrate the uniqueness of the Pep-1 in terms of having proper amino acid residues in the appropriate positions for exhibiting its biological activity.
Consistent with the concept that HA–CD44 interaction mediates the migration and homing of inflammatory leukocytes 35 36, mitogen-activated T cells from mouse spleens and from human peripheral blood both showed significant adhesion to the HA-coated plates, and anti–mouse CD44 mAb KM81 markedly (75%) blocked mouse T cell adhesion to HA (Fig. 4 F). The murine epidermal-derived dendritic cell line (XS106), which exhibits many features of mature Langerhans cells 39 40, also bound significantly to the HA-coated plates, whereas this binding was blocked only partially by anti-CD44 mAb. The extent of inhibition remained <60% even at higher concentrations of mAb (data not shown), suggesting that CD44 (or the epitope recognized by our anti-CD44 mAb KM81) was one, but not the only receptor mediating the adhesion of this cell type to HA. Importantly, Pep-1 inhibited the adhesion of all the tested leukocyte preparations to HA-coated plates almost completely (90–100%), whereas the RP control showed no significant effect. These observations validated our subsequent use of Pep-1 to study physiological functions of HA in animals.
In Vivo Impact of Pep-1 on Langerhans Cell Migration.
To test pharmacological activities of Pep-1 in vivo, we have chosen the skin as a target organ, because especially large amounts of HA are present in the skin 50. Moreover, CD44 has been shown to mediate Langerhans cell migration from the epidermis after inflammatory stimuli 51 and skin-directed homing of pro-inflammatory leukocytes under pathogenic conditions 21 51 52. Thus, we thought that skin inflammation would provide a unique opportunity to study the in vivo impact of Pep-1 on the “two-way trafficking” of leukocytes from and to the inflamed tissue. Consistent with a previous report 53, epidermal densities of Langerhans cells were reduced significantly (∼35%) 24 h after topical application of a reactive hapten DNFB, reflecting their migration into afferent lymphatics (Fig. 5 A). Two local injections of Pep-1 before DNFB application prevented Langerhans cell migration almost completely (>80% inhibition in seven independent experiments). By contrast, neither Pep-2 nor RP showed any significant effects. In the absence of DNFB painting, Langerhans cell densities were not affected by Pep-1 or other control peptides. These results documented the in vivo ability of Pep-1 to inhibit hapten-triggered Langerhans cell emigration from the epidermis.
Figure 5.

Impact of Pep-1 on hapten-triggered Langerhans cell migration. (A) BALB/c mice received two subcutaneous injections (40 μg/ear/injection) of the indicated peptides in both ears 24 and 1 h before topical application of 0.5% DNFB (right ears, black bars) or vehicle alone (left ears, white bars). Ear skin samples were harvested 24 h after DNFB painting, and were examined for the surface densities of IA+ epidermal Langerhans cells. Data shown are the means ± SEM (n = 5), representative of three independent experiments. Statistically significant differences are indicated with asterisks (**P < 0.01; Student's t test). (B) BALB/c mice received a single subcutaneous injection (40 μg/injection) of Pep-1 at the indicated concentrations 24 h before DNFB application. Data shown are the means ± SEM (n = 10) of percentage of Langerhans cell emigration at 24 h after DNFB application, i.e., the extent of DNFB-induced reduction in Langerhans cell density as calculated by the following formula: [(Langerhans cell number in vehicle painted ear − Langerhans cell number in DNFB painted ear)/Langerhans cell number in vehicle painted ear] × 100. Statistically significant differences compared with the PBS alone group (Pep-1 dose = 0 μg/ear) are indicated with asterisks (*P < 0.05; **P < 0.01). (C) Pep-1 (40 μg/ear) or PBS alone was injected subcutaneously into both ears at 3, 2, or 1 d before topical application of 0.5% DNFB (right ears, black bars) or vehicle alone (left ears, white bars). Data shown are the means ± SEM (n = 5) of Langerhans cell densities at 24 h after DNFB application. Statistically significant differences are indicated with asterisks (**P < 0.01).
The Ala-substituted Pep-1 mutant (His3→Ala), which inhibited almost completely the adhesion of BW5147 cells to the HA substrate, was found as efficient as the wild-type Pep-1 in its capacity to prevent Langerhans cell migration. By contrast, Ala substitution of Trp residue at position 4, one of the irreplaceable residues of Pep-1, abrogated not only the in vitro ability to inhibit cell adhesion, but also the in vivo ability to prevent Langerhans cell migration (Fig. 5 A). In dose response experiments, Langerhans cell migration was inhibited partially by a single injection of 13.3 μg/ear Pep-1 before DNFB application, with more prominent inhibition achieved at 40 μg/ear (Fig. 5 B). By altering the timing of Pep-1 administration, we found that DNFB-triggered Langerhans cell migration was blocked completely by a single injection of Pep-1 at 1 or 2 d before DNFB application, whereas Langerhans cells began to migrate from the epidermis when Pep-1 was administered 3 d before DNFB painting (Fig. 5 C).
In Vivo Efficacy of Pep-1 to Inhibit Contact Hypersensitivity Responses.
Migration of hapten-pulsed Langerhans cells to the draining lymph node (where antigen presentation takes place) is the first event for sensitization, i.e., clonal expansion of hapten-reactive T cells. Thus, it was of particular interest to determine the impact of Pep-1 on the sensitization phase of contact hypersensitivity responses. BALB/c mice received two local injections of Pep-1 or RP control into the left ears before sensitization; these mice were then “double sensitized” by topical application of DNFB onto the peptide-injected sites (left ear) and a second hapten OX onto irrelevant sites (abdomen). When challenged 1 wk later with DNFB on the nontreated right ears, the Pep-1 group showed significantly reduced swelling responses relative to the RP control group, indicating that Pep-1 suppressed contact hypersensitivity responses at the sensitization phase (Fig. 6 A, upper panels). On the other hand, swelling responses to the second hapten OX were comparable between the Pep-1 group and the RP group, with the implication that locally administered Pep-1 did not affect the sensitization process that took place in a distant site in the same animals. Conversely, the animals receiving Pep-1 injections and OX sensitization on the left ears and DNFB sensitization on the abdomen showed significantly reduced swelling responses only to OX (Fig. 6 A, lower panels). The Pep-1 group and the RP group did not differ significantly when they were resensitized to and rechallenged with the same hapten, indicating that Pep-1 did not cause long-term unresponsiveness (Fig. 6 B). In summary, contact hypersensitivity responses were suppressed locally and temporally by local administration of Pep-1 before sensitization.
Figure 6.

Impact of Pep-1 on the sensitization phase of contact hypersensitivity responses. (A) BALB/c mice received subcutaneous injections of Pep-1 or RP (40 μg/ear/injection) 24 and 1 h before sensitization on the left ears. The same mice were double sensitized by topical application of 0.5% DNFB and 1.25% OX on the indicated skin sites (either left ears or abdominal skin) and challenged 7 d later on the right ears with 0.2% DNFB or 0.5% OX as indicated. The data are representative of two independent experiments, showing the means ± SEM (n = 10) of ear swelling responses at 24 h for OX and 48 h for DNFB, when maximal responses were observed for each hapten. Asterisks indicate statistically significant differences (*P < 0.05; **P < 0.01) assessed by Student's t test. (B) The mice that had received Pep-1 or RP injections and DNFB sensitization on the left ears (shown in A; day 0) were resensitized by topical application of 0.5% DNFB on day 7 and rechallenged with 0.2% DNFB on day 14. The results shown are the means ± SEM (n = 10) of the ear swelling responses. Statistically significant differences are indicated with asterisks (**P < 0.01).
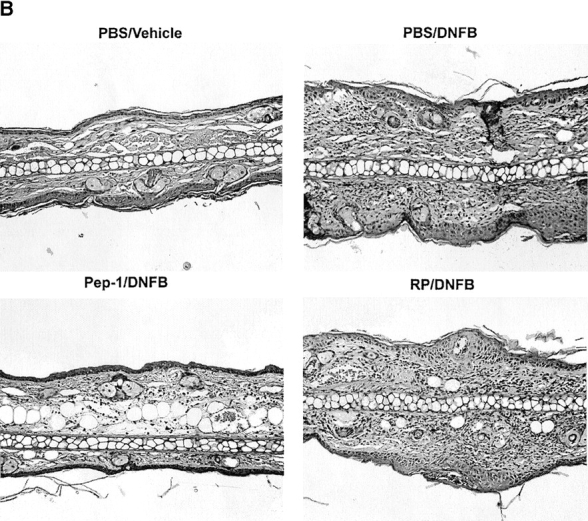
To test the impact on the elicitation phase, DNFB-sensitized animals received two local injections of Pep-1 into the right ears before DNFB challenge. As shown in Fig. 7 A (left), the Pep-1 group showed significantly reduced swelling responses relative to the control groups receiving RP, Pep-2, or PBS alone. Elicitation responses were also inhibited significantly by a single intravenous injection of Pep-1 or even by topical application of Pep-1 on the ear skin before DNFB challenge (Fig. 7 A, middle and right). Histological examination revealed that locally administered Pep-1 reduced the extent of edema and the degree of leukocyte infiltration compared with RP, Pep-2, or PBS controls (Fig. 7 B). These observations were validated statistically by measuring the skin thickness and counting the number of inflammatory leukocytes in the histology sections (Fig. 7 C). Thus, the expression of contact hypersensitivity responses was inhibited at the elicitation phase by systemic, local, or even topical application of Pep-1 in already sensitized animals.
Figure 7.
Impact of Pep-1 on the expression of contact hypersensitivity responses. (A) BALB/c mice were sensitized with 0.5% DNFB on abdominal skin and challenged 7 d later with 0.2% DNFB on the right ears and vehicle alone on the left ears. These mice received the following: (a) two subcutaneous injections (40 μg/ear/injection) of the indicated peptides into both ears 24 and 1 h before challenge (n = 5); (b) a single intravenous injection (1 mg/animal) at 1 h before challenge (n = 10); or (c) a single topical application (40 μg/ear) at 1 h before challenge (n = 10). The data are representative of two independent experiments, showing the means ± SEM of the ear swelling responses at 48 h. Statistically significant differences (ANOVA for the subcutaneous injection protocol and Student's t test for other protocols) are indicated with asterisks (*P < 0.05; **P < 0.01). (B) Ear skin samples harvested from the animals receiving the subcutaneous injections of Pep-1 or RP (left columns in A) were processed for hematoxylin and eosin staining. Data shown are the representative fields (original magnification: ×100). (C) Histological samples (n = 10) were examined by a third individual for the ear thickness and the number of skin-infiltrating leukocytes under a microscope. Asterisks indicate statistically significant differences (**P < 0.01) assessed by ANOVA. (D) BALB/c mice were painted on both ears with 0.5% DNFB on day 0 and with 0.2% DNFB on days 2 and 4 (as indicated by closed triangles). Pep-1 (•) or RP (○) (40 μg/ear/injection) was injected locally in the left or right ears of the same animals, respectively, on days 0, 1, 3, and 5 (as indicated by ▿). The data shown are representative of two independent experiments, showing the means ± SEM (n = 15) of ear swelling responses. Statistically significant differences between the Pep-1 group and the RP group are indicated with asterisks (**P < 0.01; Student's t test).


To study the efficacy of Pep-1 in chronic skin inflammation, we employed a Th2-mediated chronic dermatitis model developed by Kitagaki et al. 43. As shown in Fig. 7 D, repeated applications of DNFB on the same ears caused progressive and remarkable swelling responses; for example, the extent of ear swelling on day 6 was more than threefold greater than the maximal responses observed in the acute contact hypersensitivity model. Local injections of Pep-1 immediately before and during DNFB applications diminished the swelling responses significantly at each time point tested compared with the RP control. These observations implied that Pep-1 might be applicable for the treatment of acute as well as chronic inflammatory skin disorders. With respect to the relative safety, none of the mice (> 500 in total) that had received Pep-1 treatments in various protocols died during the experimental periods of 1–6 wk. Moreover, none of the three BALB/c mice that had received three weekly intravenous injections of Pep-1 (1 mg/injection/animal) developed any apparent adverse effects during the 3-mo follow-up period in terms of changes in body weight and serum levels of aspartate aminotransferase, alanine aminotransferase, γ-glutamyl transpeptidase, and total bilirubin (data not shown). Nor did they develop detectable humoral immune responses to Pep-1 (tested by ELISA) or cellular responses (tested by proliferative responses of splenic T cells to Pep-1; data not shown). These results suggested that pharmacological doses of Pep-1 could be administered repeatedly into living animals without causing apparent acute side effects.
Discussion
By using the phage display technology, we have isolated a 12-mer peptide that binds to soluble, immobilized, and cell-associated forms of HA and inhibits the function of HA. To recapitulate the essence of our findings, Pep-1 blocked (a) in vitro adhesion of the BW5147 thymoma line, XS106 Langerhans cell line, and murine and human T cells to HA-coated plates; (b) hapten-triggered migration of Langerhans cells from the epidermis; (c) contact hypersensitivity responses at sensitization and elicitation phases; and (d) chronic skin inflammation induced by repeated hapten applications.
Langerhans cells reside normally in the epidermis, being surrounded by keratinocytes, and they migrate to draining lymph nodes in response to inflammatory stimuli. Homophilic interaction of E-cadherin, which is expressed by both populations, is thought to mediate the retention of Langerhans cells within the epidermis 54, and α6 integrins appear to mediate Langerhans cell migration through the dermoepidermal junction 53. Our results now document a critical role played by HA in their migration out of the epidermis. In this regard, other investigators have reported that (a) HA is expressed abundantly on keratinocyte surfaces 55; (b) CD44 expression by Langerhans cells is upregulated by inflammatory stimuli in vitro 56; (c) Langerhans cells begin to express CD44v4, v5, v6, and v9 isoforms in vivo after hapten painting 51; and (d) anti-CD44v6 mAb blocks Langerhans cell emigration in the skin organ culture system 51. Thus, it is conceivable that Pep-1 prevented Langerhans cell migration by interfering with molecular interaction between HA (expressed on keratinocytes and in the dermal matrix) and specific CD44 isoforms (expressed on activated Langerhans cells). We have also observed that Pep-1 inhibits contact hypersensitivity responses at both sensitization and elicitation phases. In this regard, other investigators have reported that (a) HA expression on endothelial cells is upregulated by inflammatory stimuli 57; (b) skin-infiltrating T cells in inflammatory diseases selectively express CD44v10 and CD44v3 isoforms 52; and (c) anti-CD44 mAb reduces the extent of edema and leukocyte infiltration of skin, thereby inhibiting the expression of contact hypersensitivity responses 21 51. Thus, it is likely that Pep-1 prevents skin inflammation by blocking the interaction between HA (expressed on endothelial cells and in the extracellular matrix) and specific CD44 isoforms (expressed on skin-homing leukocytes). In summary, by using a specific inhibitor of HA, we have identified HA to function as a primary adhesive substrate for CD44-mediated migration of Langerhans cells and inflammatory leukocytes from and to the inflamed skin, respectively.
Pep-1 differs from conventional CD44 inhibitors (e.g., anti-CD44 mAb and CD44-Ig fusion proteins) in many respects. First, they differ in target specificity. Both may be equally effective in blocking chemical HA–CD44 interaction by itself, as has been observed for CD44-dependent adhesion of BW5147 cells and T cells to HA-coated plates. On the other hand, Pep-1 was more efficient than anti-CD44 mAb in blocking the adhesion of XS106 cells to HA-coated plates, perhaps reflecting the fact that CD44 is one of the multiple HA receptors. Similar observations have been reported by other investigators. For example, although HA stimulated macrophages to elaborate IL-1β and IL-6, anti-CD44 mAb blocked only IL-1β secretion 26. Likewise, antibodies against RHAMM (a second cell surface receptor of HA) but not against CD44 inhibited HA-mediated locomotion of ras-transformed fibroblasts, although they expressed both RHAMM and CD44 58. Thus, Pep-1 represents an entirely new class of inhibitors designed to block the function of HA (instead of its receptor). Secondly, CD44 represents a family of many different isoforms resulting from alternative splicing and posttranslational modifications (including glycosylation, phosphorylation, sulfation, and receptor clustering; references 49, 59 60 61 62), whereas HA is a simple glycosaminoglycan consisting of repeating disaccharide units, thereby providing a more universally accessible target for inhibitors. In fact, Pep-1 bound to and inhibited the function of all tested forms of HA molecules. Finally, the molecular size (a 12-mer peptide) of Pep-1 may be an advantage over conventional CD44 inhibitors (antibodies and fusion proteins); Pep-1, indeed, exhibited significant pharmacological activities even after topical application.
Preclinical efficacies have been documented for CD44 inhibitors in many inflammatory disease models, such as allergic contact dermatitis 21 51, collagen-induced arthritis 34, autoimmune type I diabetes 63, experimental autoimmune encephalomyelitis 64, and allogeneic skin graft rejection 65. Moreover, these inhibitors prevented tumor metastasis efficiently 31 32 33, reflecting the fact that CD44 not only mediates the adhesion and migration of tumor cells, but also regulates their growth 32, differentiation 66, apoptosis 67, expression of other adhesion molecules 68, and metalloproteinase activity 69. We believe that Pep-1 will provide a unique opportunity to study potential roles of HA in the above inflammatory diseases and tumor metastasis. Studies are in progress in our laboratory to determine the impact of Pep-1 on skin graft rejection and experimental lung metastasis of B16-F10 melanoma cells.
A striking observation from a chemical standpoint was that an overwhelming majority of the peptides isolated by phage display encoded an identical amino acid sequence (i.e., Pep-1). This documents an exceptionally high efficiency of our panning protocol, most likely owing to the elution step with HAase treatment. We believe that this is the first demonstration that phage display technology can be used to isolate peptides that bind to a given carbohydrate molecule. Even more strikingly, Pep-1 lost its biological activity by Ala substitution of any single residue, except for His3, Asn7, or Arg12 residue. Although the structural basis for Pep-1 binding to HA is presently unknown, we speculate, based on the binding profile of Pep-1 mutants and the known secondary and tertiary structures of HA 70 71, that hydrophobic and/or polar aliphatic residues of Pep-1 may function as primary binding sites for polar, hydrophobic, or formally charged groups on HA. We anticipate that Pep-1 and its mutants will provide unique tools for studying the physical and chemical interaction of HA to its ligands. Such analyses may, in turn, lead to the development of new Pep-1 derivatives with improved affinities to HA.
Acknowledgments
We thank Lesa Ellinger and Dale Edelbaum for their technical assistance, and Pat Adcock for her secretarial assistance.
This study was supported by National Institutes of Health grants RO3-AR47402, RO1-AR35068, RO1-AR43777, and RO1-AI43262, and by a Centre de Recherches et Investigations Epidermiques et Sensorielles (CE.R.I.E.S.) Award (to A. Takashima).
Footnotes
M. Mohamadzadeh's present address is Baylor Institute for Immunology Research, Sammons Cancer Center, Dallas, TX 75204.
Abbreviations used in this paper: ANOVA, analysis of variance; CSA, chondroitin sulfate A; HA, hyaluronan; HAase, hyaluronidase; OX, oxazolone; RHAMM, receptor for hyaluronan-mediated motility; RP, random peptide.
References
- Aruffo A., Stamenkovic I., Melnick M., Underhill C.B., Seed B. CD44 is the principal cell surface receptor for hyaluronate. Cell. 1990;61:1303–1313. doi: 10.1016/0092-8674(90)90694-a. [DOI] [PubMed] [Google Scholar]
- Miyake K., Underhill C.B., Lesley J., Kincade P.W. Hyaluronate can function as a cell adhesion molecule and CD44 participates in hyaluronate recognition. J. Exp. Med. 1990;172:69–75. doi: 10.1084/jem.172.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick C., Hoare K., Owens R., Hohn H.P., Hook M., Moore D., Cripps V., Austen L., Nance D.M., Turley E.A. Molecular cloning of a novel hyaluronan receptor that mediates tumor cell motility. J. Cell. Biol. 1992;117:1343–1350. doi: 10.1083/jcb.117.6.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham T.E. The role of link-protein in the structure of cartilage proteoglycan aggregates. Biochem. J. 1979;177:237–247. doi: 10.1042/bj1770237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H., Cheung S.C., Itano N., Kimata K., Yamada Y. Identification of hyaluronan-binding domains of aggrecan. J. Biol. Chem. 1997;272:28057–28065. doi: 10.1074/jbc.272.44.28057. [DOI] [PubMed] [Google Scholar]
- LeBaron R.G., Zimmermann D.R., Ruoslahti E. Hyaluronate binding properties of versican. J. Biol. Chem. 1992;267:10003–10010. [PubMed] [Google Scholar]
- Delpech B., Halavent C. Characterization and purification from human brain of a hyaluronic acid-binding glycoprotein, hyaluronectin. J. Neurochem. 1981;36:855–859. doi: 10.1111/j.1471-4159.1981.tb01672.x. [DOI] [PubMed] [Google Scholar]
- Rauch U., Karthikeyan L., Maurel P., Margolis R.U., Margolis R.K. Cloning and primary structure of neurocan, a developmentally regulated, aggregating chondroitin sulfate proteoglycan of brain. J. Biol. Chem. 1992;267:19536–19547. [PubMed] [Google Scholar]
- Yannariello-Brown J., Frost S.J., Weigel P.H. Identification of the Ca2+-independent endocytic hyaluronan receptor in rat liver sinusoidal endothelial cells using a photoaffinity cross-linking reagent. J. Biol. Chem. 1992;267:20451–20456. [PubMed] [Google Scholar]
- Zhao M., Yoneda M., Ohashi Y., Kurono S., Iwata H., Ohnuki Y., Kimata K. Evidence for the covalent binding of SHAP, heavy chains of inter-alpha-trypsin inhibitor, to hyaluronan. J. Biol. Chem. 1995;270:26657–26663. doi: 10.1074/jbc.270.44.26657. [DOI] [PubMed] [Google Scholar]
- Jaworski D.M., Kelly G.M., Hockfield S. BEHAB, a new member of the proteoglycan tandem repeat family of hyaluronan-binding proteins that is restricted to the brain. J. Cell. Biol. 1994;125:495–509. doi: 10.1083/jcb.125.2.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishina H., Inageda K., Takahashi K., Hoshino S., Ikeda K., Katada T. Cell surface antigen CD38 identified as ecto-enzyme of NAD glycohydrolase has hyaluronate-binding activity. Biochem. Biophys. Res. Commun. 1994;203:1318–1323. doi: 10.1006/bbrc.1994.2326. [DOI] [PubMed] [Google Scholar]
- Banerji S., Ni J., Wang S.X., Clasper S., Su J., Tammi R., Jones M., Jackson D.G. LYVE-1, a new homologue of the CD44 glycoprotein, is a lymph-specific receptor for hyaluronan. J. Cell. Biol. 1999;144:789–801. doi: 10.1083/jcb.144.4.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsifrina E., Ananyeva N.M., Hastings G., Liau G. Identification and characterization of three cDNAs that encode putative novel hyaluronan-binding proteins, including an endothelial cell-specific hyaluronan receptor. Am. J. Pathol. 1999;155:1625–1633. doi: 10.1016/S0002-9440(10)65478-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehnis T., Dieterich W., Bauer M., Lampe B., Schuppan D. A chondroitin/dermatan sulfate form of CD44 is a receptor for collagen XIV (undulin) Exp. Cell Res. 1996;229:388–397. doi: 10.1006/excr.1996.0384. [DOI] [PubMed] [Google Scholar]
- Knutson J.R., Iida J., Fields G.B., McCarthy J.B. CD44/chondroitin sulfate proteoglycan and α2β1 integrin mediate human melanoma cell migration on type IV collagen and invasion of basement membranes. Mol. Biol. Cell. 1996;7:383–396. doi: 10.1091/mbc.7.3.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalkanen S., Jalkanen M. Lymphocyte CD44 binds the COOH-terminal heparin-binding domain of fibronectin. J. Cell Biol. 1992;116:817–825. doi: 10.1083/jcb.116.3.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleeman J.P., Kondo K., Moll J., Ponta H., Herrlich P. Variant exons v6 and v7 together expand the repertoire of glycosaminoglycans bound by CD44. J. Biol. Chem. 1997;272:31837–31844. doi: 10.1074/jbc.272.50.31837. [DOI] [PubMed] [Google Scholar]
- Toyama-Sorimachi N., Kitamura F., Habuchi H., Tobita Y., Kimata K., Miyasaka M. Widespread expression of chondroitin sulfate-type serglycins with CD44 binding ability in hematopoietic cells. J. Biol. Chem. 1997;272:26714–26719. doi: 10.1074/jbc.272.42.26714. [DOI] [PubMed] [Google Scholar]
- Koshiishi I., Shizari M., Underhill C.B. CD44 can mediate the adhesion of platelets to hyaluronan. Blood. 1994;84:390–396. [PubMed] [Google Scholar]
- Camp R.L., Scheynius A., Johansson C., Puré E. CD44 is necessary for optimal contact allergic responses but is not required for normal leukocyte extravasation. J. Exp. Med. 1993;178:497–507. doi: 10.1084/jem.178.2.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama-Sorimachi N., Miyake K., Miyasaka M. Activation of CD44 induces ICAM-1/LFA-1-independent, Ca2+ Mg2+-independent adhesion pathway in lymphocyte-endothelial cell interaction. Eur. J. Immunol. 1993;23:439–446. doi: 10.1002/eji.1830230221. [DOI] [PubMed] [Google Scholar]
- Fujii K., Tanaka Y., Hubscher S., Saito K., Ota T., Eto S. Cross-linking of CD44 on rheumatoid synovial cells up-regulates VCAM-1. J. Immunol. 1999;162:2391–2398. [PubMed] [Google Scholar]
- Guo Y.J., Ma J., Wong J.H., Lin S.C., Chang H.C., Bigby M., Sy M.S. Monoclonal anti-CD44 antibody acts in synergy with anti-CD2 but inhibits anti-CD3 or T cell receptor-mediated signaling in murine T cell hybridomas. Cell. Immunol. 1993;152:186–199. doi: 10.1006/cimm.1993.1278. [DOI] [PubMed] [Google Scholar]
- Bourguignon L.Y., Lokeshwar V.B., Chen X., Kerrick W.G. Hyaluronic acid-induced lymphocyte signal transduction and HA receptor (GP85/CD44)-cytoskeleton interaction. J. Immunol. 1993;151:6634–6644. [PubMed] [Google Scholar]
- Khaldoyanidi S., Moll J., Karakhanova S., Herrlich P., Ponta H. Hyaluronate-enhanced hematopoiesistwo different receptors trigger the release of interleukin-1β and interleukin-6 from bone marrow macrophages. Blood. 1999;94:940–949. [PubMed] [Google Scholar]
- Horton M.R., Burdick M.D., Strieter R.M., Bao C., Noble P.W. Regulation of hyaluronan-induced chemokine gene expression by IL-10 and IFN-γ in mouse macrophages. J. Immunol. 1998;160:3023–3030. [PubMed] [Google Scholar]
- Fitzgerald K.A., O'Neill L.A. Characterization of CD44 induction by IL-1a critical role for Egr-1. J. Immunol. 1999;162:4920–4927. [PubMed] [Google Scholar]
- Henke C., Bitterman P., Roongta U., Ingbar D., Polunovsky V. Induction of fibroblast apoptosis by anti-CD44 antibodyimplications for the treatment of fibroproliferative lung disease. Am. J. Pathol. 1996;149:1639–1650. [PMC free article] [PubMed] [Google Scholar]
- Ayroldi E., Cannarile L., Migliorati G., Bartoli A., Nicoletti I., Riccardi C. CD44 (Pgp-1) inhibits CD3 and dexamethasone-induced apoptosis. Blood. 1995;86:2672–2678. [PubMed] [Google Scholar]
- Cannistra S.A., Kansas G.S., Niloff J., DeFranzo B., Kim Y., Ottensmeier C. Binding of ovarian cancer cells to peritoneal mesothelium in vitro is partly mediated by CD44H. Cancer Res. 1993;53:3830–3838. [PubMed] [Google Scholar]
- Bartolazzi A., Peach R., Aruffo A., Stamenkovic I. Interaction between CD44 and hyaluronate is directly implicated in the regulation of tumor development. J. Exp. Med. 1994;180:53–66. doi: 10.1084/jem.180.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahalka M.A., Okon E., Gosslar U., Holzmann B., Naor D. Lymph node (but not spleen) invasion by murine lymphoma is both CD44- and hyaluronate-dependent. J. Immunol. 1995;154:5345–5355. [PubMed] [Google Scholar]
- Mikecz K., Brennan F.R., Kim J.H., Glant T.T. Anti-CD44 treatment abrogates tissue oedema and leukocyte infiltration in murine arthritis. Nat. Med. 1995;1:558–563. doi: 10.1038/nm0695-558. [DOI] [PubMed] [Google Scholar]
- DeGrendele H., Estess P., Picker L.J., Siegelman M.H. CD44 and its ligand hayluronate mediate rolling under physiologic flowa novel lymphocyte-endothelial cell primary adhesion pathway. J. Exp. Med. 1996;183:1119–1130. doi: 10.1084/jem.183.3.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark R.A., Alon R., Springer T.A. CD44 and hyaluronan-dependent rolling interactions of lymphocytes on tonsillar stroma. J. Cell. Biol. 1996;134:1075–1087. doi: 10.1083/jcb.134.4.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminski M.J., Cruz P.D., Jr., Bergstresser P.R., Takashima A. Killing of skin-derived tumor cells by mouse dendritic epidermal T cells. Cancer Res. 1993;53:4014–4019. [PubMed] [Google Scholar]
- Matsue H., Bergstresser P.R., Takashima A. Keratinocyte-derived IL-7 serves as a growth factor for dendritic epidermal T cells in mice. J. Immunol. 1993;151:6012–6019. [PubMed] [Google Scholar]
- Timares L., Takashima A., Johnston S.A. Quantitative analysis of the immunopotency of genetically transfected dendritic cells. Proc. Natl. Acad. Sci. USA. 1998;95:13147–13152. doi: 10.1073/pnas.95.22.13147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsue H., Matsue K., Walters M., Okumura K., Yagita H., Takashima A. Induction of antigen-specific immunosuppression by CD95L cDNA-transfected “killer” dendritic cells. Nat. Med. 1999;5:930–937. doi: 10.1038/11375. [DOI] [PubMed] [Google Scholar]
- de Belder A.N., Wik O. Preparation and properties of fluorescein-labelled hyaluronate. Carbohydr. Res. 1975;44:251–257. doi: 10.1016/s0008-6215(00)84168-3. [DOI] [PubMed] [Google Scholar]
- Abeyama K., Eng W., Jester J.V., Vink A.A., Edelbaum D., Cockerell C.J., Bergstresser P.R., Takashima A. A role for NFκB-dependent gene transactivation in sunburn. J. Clin. Invest. 2000;105:1751–1759. doi: 10.1172/JCI9745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagaki H., Fujisawa S., Watanabe K., Kayakawa K., Shiohara T. Immediate-type hypersensitivity response followed by late reaction is induced by repeated epicutaneous application of contact sensitizing agents in mice. J. Invest. Dermatol. 1995;105:749–755. doi: 10.1111/1523-1747.ep12325538. [DOI] [PubMed] [Google Scholar]
- Yang B., Yang B.L., Savani R.C., Turley E.A. Identification of a common hyaluronan binding motif in the hyaluroana binding proteins RHAMM, CD44 and link protein. EMBO (Eur. Mol. Biol. Organ.) J. 1994;13:286–296. doi: 10.1002/j.1460-2075.1994.tb06261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adey N.B., Mataragnon A.H., Rider J.E., Carter W.G., Kay B.K. Characterization of phage that bind plastic from phage-displayed random peptide libraries. Gene. 1995;156:27–31. doi: 10.1016/0378-1119(95)00058-e. [DOI] [PubMed] [Google Scholar]
- Silberberg A. The adsorption of flexible macromolecules. Part II. The shape of the adsorbed molecule; the adsorption isotherm surface tension, and pressure. J. Phys. Chem. 1962;66:1884–1907. [Google Scholar]
- Latt S.A., Sober H.A. Protein-nucleic acid interactions. II. Oligopeptide-polyribonucleotide binding studies. Biochemistry. 1967;6:3293–3306. doi: 10.1021/bi00862a040. [DOI] [PubMed] [Google Scholar]
- McGhee J.D., von Hippel P.H. Theoretical aspects of DNA-protein interactionsco-operative and non-co-operative binding of large ligands to a one-dimensional homogeneous lattice. J. Mol. Biol. 1974;86:469–489. doi: 10.1016/0022-2836(74)90031-x. [DOI] [PubMed] [Google Scholar]
- Maiti A., Maki G., Johnson P. TNA-α induction of CD44-mediated leukocyte adhesion by sulfation. Science. 1998;282:941–943. doi: 10.1126/science.282.5390.941. [DOI] [PubMed] [Google Scholar]
- Knudson C.B., Knudson W. Hyaluronan-binding proteins in development, tissue, homeostasis, and disease. FASEB J. 1993;7:1233–1241. [PubMed] [Google Scholar]
- Weiss J.M., Sleeman J., Renkl A.C., Dittmar H., Termeer C.C., Taxis S., Howells N., Schöpf E., Ponta H., Herrlich P., Simon J.C. An essential role for CD44 variant isoforms in epidermal Langerhans cell and blood dendritic cell function. J. Cell. Biol. 1997;137:1137–1147. doi: 10.1083/jcb.137.5.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner S.N., Wagner C., Reinhold U., Funk R., Zöller M., Goos M. Predominant expression of CD44 splice variant v10 in malignant and reactive human skin lymphocytes. J. Invest. Dermatol. 1998;111:464–471. doi: 10.1046/j.1523-1747.1998.00302.x. [DOI] [PubMed] [Google Scholar]
- Price A.A., Cumberbatch M., Kimber I., Ager A. α6 integrins are required for Langerhans cell migration from the epidermis. J. Exp. Med. 1997;186:1725–1735. doi: 10.1084/jem.186.10.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang A., Amagai M., Granger L.G., Stanley J.R., Udey M.C. Adhesion of epidermal Langerhans cells to keratinocytes mediated by E-cadherin. Nature. 1993;361:82–85. doi: 10.1038/361082a0. [DOI] [PubMed] [Google Scholar]
- Tammi R., MacCallum D., Hascall V.C., Pienimaki J.P., Hyttinen M., Tammi M. Hyaluronan bound to CD44 on keratinocytes is displaced by hyaluronan decasaccharides and not hexasaccharides. J. Biol. Chem. 1998;273:28878–28888. doi: 10.1074/jbc.273.44.28878. [DOI] [PubMed] [Google Scholar]
- Osada A., Nakashima H., Furue M., Tamaki K. Up regulations of CD44 expression by tumor necrosis factor-α is neutralized by interleukin 10 in Langerhans cells. J. Invest. Dermatol. 1995;105:124–127. doi: 10.1111/1523-1747.ep12313437. [DOI] [PubMed] [Google Scholar]
- Mohamadzadeh M., DeGrendele H., Arizpe H., Estess P., Siegelman M. Proinflammatory stimuli regulate endothelial hyaluronan expression and CD44-HA-dependent primary adhesion. J. Clin. Invest. 1998;101:97–108. doi: 10.1172/JCI1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turley E.A., Austen L., Moore D., Hoare K. Ras-transformed cells express both CD44 and RHAMM hyaluronan receptorsonly RHAMM is essential for hyaluronan-promoted locomotion. Exp. Cell Res. 1993;207:277–282. doi: 10.1006/excr.1993.1194. [DOI] [PubMed] [Google Scholar]
- Katoh S., Miyagi T., Taniguchi H., Matsubara Y., Kadota J., Tominaga A., Kincade P.W., Matsukura S., Kohno S. Cutting edgean inducible sialidase regulates the hyaluronic acid binding ability of CD44-bearing human monocytes. J. Immunol. 1999;162:5058–5061. [PubMed] [Google Scholar]
- Lesley J., Kincade P.W., Hyman R. Antibody-induced activation of the hyaluronan receptor function of CD44 requires multivalent binding by antibody. Eur. J. Immunol. 1993;23:1902–1909. doi: 10.1002/eji.1830230826. [DOI] [PubMed] [Google Scholar]
- Sleeman J., Rudy W., Hofmann M., Moll J., Herrlich P., Ponta H. Regulated clustering of variant CD44 proteins increases their hyaluronate binding capacity. J. Cell. Biol. 1996;135:1139–1150. doi: 10.1083/jcb.135.4.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neame S.J., Isacke C.M. Phosphorylation of CD44 in vivo requires both Ser323 and Ser325, but does not regulate membrane localization or cytoskeletal interaction in epithelial cells. EMBO (Eur. Mol. Biol. Organ.) J. 1992;11:4733–4738. doi: 10.1002/j.1460-2075.1992.tb05578.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss L., Slavin S., Reich S., Cohen P., Shuster S., Stern R., Kaganovsky E., Okon E., Rubinstein A.M., Naor D. Induction of resistance to diabetes in non-obese diabetic mice by targeting CD44 with specific monoclonal antibody. Proc. Natl. Acad. Sci. USA. 2000;97:285–290. doi: 10.1073/pnas.97.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocke S., Piercy C., Steinman L., Weissman I.L., Veromaa T. Antibodies to CD44 and integrin α4, but not L-selectin, prevent central nervous system inflammation and experimental encephalomyelitis by blocking secondary leukocyte recruitment. Proc. Natl. Acad. Sci. USA. 1999;96:6896–6901. doi: 10.1073/pnas.96.12.6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiter S., Weber B., Tilgen W., Zöller M. Down-modulation of host reactivity by anti-CD44 in skin transplantation. Transplantation. 1998;66:778–791. doi: 10.1097/00007890-199809270-00013. [DOI] [PubMed] [Google Scholar]
- Charrad R.-S., Li Y., Delpech B., Balitrand N., Clay D., Jasmin C., Chomienne C., Smadja-Joffe F. Ligation of the CD44 adhesion molecule reverses blockage of differentiation in human acute myeloid leukemia. Nat. Med. 1999;5:669–676. doi: 10.1038/9518. [DOI] [PubMed] [Google Scholar]
- Yu Q., Toole B.P., Stamenkovic I. Induction of apoptosis of metastatic mammary carcinoma cells in vivo by disruption of tumor cell surface CD44 function. J. Exp. Med. 1997;186:1985–1996. doi: 10.1084/jem.186.12.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujisaki T., Tanaka Y., Fujii K., Mine S., Saito K., Yamada S., Yamashita U., Irimura T., Eto S. CD44 stimulation induces integrin-mediated adhesion of colon cancer cell lines to endothelial cells by up-regulation of integrins and c-Met and activation of integrins. Cancer Res. 1999;59:4427–4434. [PubMed] [Google Scholar]
- Yu Q., Stamenkovic I. Localization of matrix metalloproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes Dev. 1998;13:35–48. doi: 10.1101/gad.13.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heatley F., Scott J.E. A water molecule participates in the secondary structure of hyaluronan. Biochem. J. 1988;254:489–493. doi: 10.1042/bj2540489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott J.E., Heatley F. Hyaluronan forms specific stable tertiary structures in aqueous solutiona 13C NMR study. Proc. Natl. Acad. Sci. USA. 1999;96:4850–4855. doi: 10.1073/pnas.96.9.4850. [DOI] [PMC free article] [PubMed] [Google Scholar]