

Abstract
Human macrophages mediate the dissolution of elastic lamina by mobilizing tissue-destructive cysteine proteinases. While macrophage-mediated elastin degradation has been linked to the expression of cathepsins L and S, these cells also express cathepsin K, a new member of the cysteine proteinase family whose elastinolytic potential exceeds that of all known elastases. To determine the relative role of cathepsin K in elastinolysis, monocytes were differentiated under conditions in which they recapitulated a gene expression profile similar to that observed at sites of tissue damage in vivo. After a 12-d culture period, monocyte-derived macrophages (MDMs) expressed cathepsin K in tandem with cathepsins L and S. Though cysteine proteinases are acidophilic and normally confined to the lysosomal network, MDMs secreted cathepsin K extracellularly in concert with cathepsins L and S. Simultaneously, MDMs increased the expression of vacuolar-type H+-ATPase components, acidified the pericellular milieu, and maintained extracellular cathepsin K in an active form. MDMs from a cathepsin K–deficient individual, however, retained the ability to express, process, and secrete cathepsins L and S, and displayed normal elastin-degrading activity. Thus, matrix-destructive MDMs exteriorize a complex mix of proteolytic cysteine proteinases, but maintain full elastinolytic potential in the absence of cathepsin K by mobilizing cathepsins L and S.
Keywords: cysteine proteinases, macrophages, cathepsin K, elastinolysis, pycnodysostosis
Introduction
In inflammatory disease states, the destruction of elastin-rich tissues is associated with the local accumulation of macrophages that express heightened levels of a diverse mix of proteolytic enzymes 1 2 3 4 5 6. Because elastin is resistant to most forms of proteolytic attack and its dissolution can irreversibly modify tissue structure and/or function, increased attention has focused on identifying the subset of macrophage-derived proteinases that participate in the elastinolytic process 1 2 3 4 5 6. Within the papain–cysteine protease family, the only elastinolytic proteinases previously known to be expressed in human macrophages were cathepsins L and S 6 7. However, a new cysteine proteinase, termed cathepsin K, has been identified in human macrophages in vitro as well as in vivo, and the isolated enzyme has been shown to express an elastin-degradative potential that exceeds that of all other mammalian elastases 6 8 9 10 11. Furthermore, as cathepsin K–positive human macrophages have been found in juxtaposition to sheets of fragmented elastin in atherosclerotic lesions in vivo, it has been proposed that inflammatory cells may use this proteinase to degrade the elastic lamina 4 5.
Despite the tissue-destructive potential of cathepsin K, cysteine proteinases are normally routed to acidic endosomal/lysosomal compartments 6. In these intracellular vesicles, cathepsin K, which displays a pH optimum of ∼6.0, expresses maximal enzyme activity and is shielded from denaturation at neutral pH 6 12 13 14. However, given its intracellular trafficking pattern and enzymatic properties, it remains unclear as to whether intact cells can secrete active cathepsin K or use the proteinase to mediate extracellular elastinolysis. To this end, we have characterized cathepsin K expression, processing, and secretion in human monocyte-derived macrophages (MDMs) that have been cultured under conditions in which the fully differentiated cells display a tissue-destructive phenotype similar to that observed in chronic inflammatory sites in vivo. Our findings demonstrate that MDMs can constitutively secrete fully processed cathepsin K into the extracellular space where the proteinase retains enzymic activity. Coincident with the secretion of active cathepsin K, cDNA microarray analysis demonstrated that MDMs upregulated expression of vacuolar-type H+-ATPase components, which allowed the cells to create a pericellular acidic milieu conducive to optimal cysteine proteinase activity. Finally, by taking advantage of the recent observation that patients with an osteochondrodysplastic syndrome, pycnodysostosis, harbor mutations in the cathepsin K gene 15 16, we have generated cathepsin K–deficient MDMs to assess the relative role of this cysteine proteinase in macrophage-mediated elastinolysis.
Materials and Methods
Macrophage Preparation.
Monocytes were adherence purified and cultured in RPMI 1640 (GIBCO BRL) supplemented with either 40% autologous serum or pooled AB serum and 100 U penicillin, 50 U streptomycin, and 2 mM l-glutamine for 12 d as described 7. Where indicated, 12-d-old MDMs were incubated alone or with 0.25 mg/ml zymosan particles (Sigma-Aldrich), 3 μm latex beads (a 1:10 dilution of a 10% suspension; Sigma-Aldrich), 50 ng/ml tetradecanoylphorbol 13-acetate (TPA; Sigma-Aldrich), 5 μM nocodazole, 4 μM taxol, 5 μM cytochalasin D, 5–10 μM bafilomycin A1, or 50 nM folimycin (all from Calbiochem-Novabiochem).
Northern Blot Analysis.
Total RNA was isolated from macrophage cultures using TRIzol reagent (GIBCO BRL) and resolved by 1% agarose gel electrophoresis 7. Cross-linked membranes were then probed for osteopontin, human cartilage glycoprotein (gp)-39, chitotriosidase, cathepsins K, B, S, or L, or glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The following probes were synthesized: osteopontin (bp 371–2709; sequence data available from EMBL/GenBank/DDBJ under accession no. U20758.1), gp-39 (bp 124–1278; accession no. NM-001276.1), chitotriosidase (bp 10–1413; accession no. U29615.1), cathepsin K (bp 139–1134; accession no. S79895.1), cathepsin B (bp 175–1200; accession no. L16510.1), cathepsin S (bp 134–1135; accession no. M90696.1), cathepsin L (bp 131–1138; accession no. M20496.1), and GAPDH (bp 48–1016; accession no. M17851.1).
Western Blot Analysis.
To assess intracellular cysteine protease content, monocyte/MDM extracts were prepared after lysis with 1% Triton X-100 (Sigma-Aldrich) in PBS (GIBCO BRL) supplemented with a protease inhibitor cocktail (Calbiochem-Novabiochem). For secreted proteinases, cultures were switched to serum-free media at indicated times and supernatants were collected after an additional 48-h culture period. During the incubation in serum-free media, MDMs released <0.5% of their total lactate dehydrogenase activity as determined according to the manufacturer's protocol (Sigma-Aldrich). Samples were then treated with the protease inhibitor cocktail and centrifuged (5 min at 10,000 g) to remove cell debris. Equal amounts of protein (10 μg) were loaded under reducing conditions onto a 12.5% SDS-polyacrylamide gel and transferred as described 7. The membranes were then incubated with either cathepsin K mAb 3021 16, mouse anti–human cathepsin B mAb IM-27L (Calbiochem-Novabiochem), rabbit anti–human cathepsin S polyclonal Ab (a gift of H.A. Chapman, University of California at San Francisco, San Francisco, CA), rabbit anti–human cathepsin L (Athens Research & Technology), or rabbit anti–human cathepsin D (Dako). The bound primary Abs were then tagged with horseradish peroxidase–conjugated species-specific secondary Abs (Pierce Chemical Co.) using the SuperSignal West Pico system (Pierce Chemical Co.).
Vacuolar-type H+-ATPase mRNA and Protein Expression.
Labeled cRNA was prepared as described, hybridized on Affymetrix oligonucleotide arrays, and scanned 17. Triton X-100 extracts were immunoblotted with a mouse mAb (3.2 Fl; a gift of M. Forgase, Boston University, Boston, MA) directed against the catalytic A subunit or the vacuolar-type H+-ATPase. Bovine brain extract was used as a positive control.
Detection of Cathepsin K–Cystatin C Complexes and Lysosomal Hydrolase Activity.
To identify cathepsin K–cystatin C complexes, cells were incubated in serum-free media for 24 h in the absence or presence of human cystatin C (100 μg/ml; Calbiochem-Novabiochem) with or without the general cysteine proteinase inhibitor, E-64 (100 μM; Sigma-Aldrich). Supernatants were collected, treated with the protease inhibitor cocktail, and cellular debris was removed by centrifugation (5 min at 10,000 g). The supernatants were precleared (2 h at 4°C) with 20 μl of Protein A/Plus Protein G 50% agarose slurry (Calbiochem-Novabiochem) and then incubated with rabbit anti–cystatin C polyclonal antisera (Calbiochem-Novabiochem) or control antisera for 12 h at 4°C. Cystatin C–anticystatin complexes were immunoprecipitated with 20 μl of Protein A/Plus Protein G 50% agarose slurry (2 h at 4°C) and bound proteins were eluted under nonreducing conditions. The samples were then loaded onto a 12.5% SDS-polyacrylamide gel, resolved under nonreducing conditions, and immunoblotted with anti–cathepsin K mAb.
Electron and Confocal Laser Microscopy.
For scanning electron microscopy, monocyte and/or MDM cultures were fixed in 2% glutaraldehyde and 4% paraformaldehyde in 0.1 M sodium cacodylate buffer (pH 7.4). After postfixation with 1% osmium oxide (Electron Microscopy Sciences), the cells were dehydrated in ethyl alcohol, transferred to hexamethyl-disilizane, and air-dried before gold sputtering. For transmission electron microscopy (TEM), cells were fixed in 2% glutaraldehyde and 1.5% paraformaldehyde in 0.1 M sodium cacodylate buffer (pH 7.4). After postfixation with 1% osmium oxide and dehydration in ethyl alcohol, samples were embedded in epoxy resin and stained with uranyl acetate and lead citrate 18. In selected experiments, the surface plasma membrane of fixed cells was stained with either 0.1% ruthenium red or 0.4 mg/ml cationized ferritin (Sigma-Aldrich), and then processed for TEM 19 20.
To visualize acidified microenvironments, MDMs cultured in the presence or absence of elastin particles were incubated in serum-free media with 10 μg/ml acridine orange (Sigma-Aldrich) for 15 min at 37°C 21 22. In selected experiments, MDM–elastin cocultures were preincubated with 5 μm bafilomycin for 4 h at 37°C to inhibit the vacuolar-type H+-ATPase 22 23 before treatment with acridine orange. Vital confocal fluorescence microscopy was performed on a Bio-Rad MRC 600 using a 488-nm excitation wavelength from an argon ion laser. Extracellular versus intracellular fluorescence was distinguished by using trypan blue as an extracellular quencher 24.
Elastin Degradation.
12-d-old MDMs prepared from normal patients, the haplosufficient parents, or the affected child (for details of the mutations, see reference 16) were incubated with 8 mg 3H-labeled elastin particles (type E60; Elastin Products Co.) in the absence or presence of either 100 μm E-64 7 or 5 μM bafilomycin A1. After a 0–48-h incubation, supernatants were recovered, centrifuged (10 min at 10,000 g), and solubilized elastin was quantified by scintillation counting 7. Results are expressed as micrograms of elastin degraded/106 cells/24 h assuming a specific activity of 1,100 cpm/μg elastin.
Results
Cathepsin K Expression by Human MDMs.
In inflammatory disease states, human macrophages have been shown to differentially express mRNA for several gene products which include not only cathepsin K, but also the matrix protein osteopontin as well as members of the glycosyl hydrolase family (e.g., gp-39 and chitotriosidase [25]). In an attempt to recapitulate a differentiation program similar to that observed at inflammatory sites in vivo, peripheral blood monocytes were cultured for 12 d in 40% autologous serum wherein the cells underwent marked morphologic changes (Fig. 1 A). Coincident with this process, MDMs displayed a profile of increased gene expression that mimicked that observed in vivo with increased expression of osteopontin, gp-39, and chitotriosidase (Fig. 1 B). Likewise, the MDMs markedly increased mRNA expression for cathepsin K (Fig. 2 A). Although MDMs also increased mRNA levels of cathepsins B, S, and L, their patterns of expression were distinct from that of cathepsin K (Fig. 2 B).
Figure 1.
The in vitro differentiation of MDMs. (A) Scanning electron micrographs of differentiating monocytes after 2 h, and 3, 5, 7, and 12 d of culture. Bar, 10 μm. (B) Northern blot analysis of osteopontin, gp-39, and chitotriosidase mRNA expression during MDM differentiation. Results are shown from an experiment performed using total RNA pooled from five donors. Equal loading was determined by GAPDH mRNA hybridization (not shown).


Figure 2.

Northern blot analysis of cysteine proteinase expression during MDM differentiation. (A) Total RNA was extracted at the indicated time points and hybridizations were performed using probes for cathepsins K, B, S, and L. Results are shown from an experiment performed using total RNA pooled from five donors. (B) The levels of mRNA expression of each cathepsin mRNA were compared with those of GAPDH mRNA at the indicated time points. Data are presented as percentage of maximal expression of each cathepsin mRNA: cathepsin K (▪), cathepsin B (▴), cathepsin S (•), and cathepsin L (□).
Cysteine proteinases are synthesized as zymogens and undergo maturation to their enzymatically active forms during transport from the trans-Golgi network to the late endosome/lysosomal compartments 6. In Fig. 3, Western blot analyses of whole cell lysates from monocytes or MDMs identified the cathepsin K proform (∼38 kD) and processed mature form (∼27 kD), as well as an ∼15-kD derivative at day 12 that may represent a degradation product of the mature enzyme. While a similar pattern of expression was noted for cathepsin S, with the pro- and mature forms detected at day 12, the pro- and/or active forms of cathepsins B and L began to accumulate intracellularly within 5 d (Fig. 3).
Figure 3.

Intracellular cysteine proteinase expression during MDM differentiation. Cell lysates (10 μg/lane) were harvested at 2 h, and 3, 5, 7, and 12 d, and immunoblotted with anti–cathepsin K, B, S, and L Abs. The pro- (white arrowheads) and mature (arrows) forms of the enzymes are highlighted. Mature cathepsin B is expressed as a single and two chain active form (M r ∼31 and ∼25 kD, respectively; see reference 7). Results are shown from a single representative experiment of three performed.
MDM Secretion of Pro- and Mature Forms of Cathepsin K.
Cathepsins B, S, and L can be expressed by diverse cell types in vitro or in vivo, but the mature forms of these enzymes are usually stored intracellularly 6 7 26. Nonetheless, analyses of cell-free supernatants from MDMs demonstrate that the cells acquired the ability to secrete both the pro- and mature forms of cathepsin K as well as those of cathepsins B, S, and L, albeit with distinct kinetics (Fig. 4 A). Whereas cathepsins K and L were initially secreted as both pro- and mature forms (at days 5 and 3, respectively), cathepsins B and S were first detected extracellularly as proforms alone (Fig. 4 A). Consistent with reports that lysosomal enzymes may be packaged and mobilized from separate intracellular compartments 27 28 29, the secretion of the lysosomal hydrolase, β-glucosaminidase, was detected in 3-d-old MDMs despite the fact that mature forms of cathepsins K, B, and S were not yet released (Fig. 4B versus A). Likewise, whereas the aspartyl proteinase, procathepsin D, was secreted through day 5, the ∼32-kD mature form of the enzyme 30 was only released from MDMs after 7 d in culture (Fig. 4 C). Thus, while 12-d-old MDMs mobilized mature forms of cysteine proteinases, a lysosomal hydrolase and an aspartyl proteinase, the secretory phenotype for each of these enzymes was regulated differentially.
Figure 4.
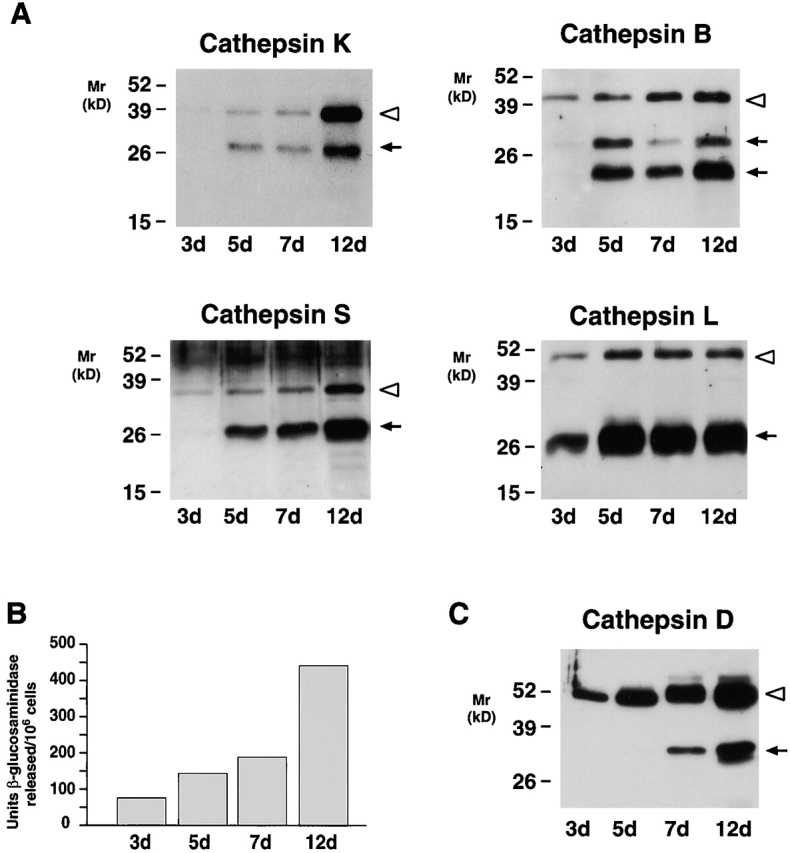
The secretory phenotype of MDMs. (A) Western blot analysis of cathepsins K, B, S, and L secreted by MDMs at the indicated time points. (B) Supernatants collected as above were analyzed for release of β-glucosaminidase or (C) cathepsin D. In A and C, each lane was loaded with 10 μg total protein and the pro- (white arrowheads) and mature (arrows) forms of the enzymes are indicated. Results are shown from a single representative experiment of three performed.
To determine whether cathepsin K secretion could be modulated, MDMs were incubated with either opsonized zymosan particles, latex beads, or TPA 27. However, none of these stimuli affected cathepsin K secretion (Fig. 5A and Fig. C). Similarly, although calcium ionophores have been reported to trigger lysosomal enzyme release in various cell populations 31, no effects were seen on the secretion of either proform or mature cathepsin K (Fig. 5 C). A functional microtubular network has been linked to cathepsin B secretion in tumor cells 32, but neither nocodazole nor taxol affected cathepsin K or B secretion (Fig. 5B and Fig. D). Finally, because the cortical actin network has been proposed to act as a physical barrier to granule docking 33, we finally assessed the role of cytochalasin D on exocytosis. Significantly, MDMs incubated with cytochalasin alone markedly increased cathepsin K as well as B secretion (Fig. 5B and Fig. D; similar results were obtained with cathepsins S and L, data not shown). These findings suggest that MDMs are “primed” for exocytosis, but that the actin network acts as a dominant negative clamp 33.
Figure 5.

Regulation of cathepsin K secretion in MDMs. 12-d-old MDMs were incubated for 48 h in serum-free media alone or in the presence of (A) zymosan (0.25 mg/ml) or latex beads (1:10 dilution of a 10% suspension), (B) nocodazole (5 μM), taxol (4 μM), or cytochalasin D (5 μM), or (C) TPA (50 ng/ml) or ionomycin (1 M). Cathepsin K secretion into the cell-free supernatants was assessed by Western blot analysis in A, B, and C, and cathepsin B in D. The pro- (white arrowheads) and mature (arrows) forms of the enzymes are indicated. Results are shown from a single representative experiment of three performed.
MDMs Generate an Acidic Pericellular Environment during Elastinolysis.
Given the ability of fully differentiated MDMs to constitutively secrete cathepsin K along with a complement of other acidophilic enzymes, we considered the possibility that the macrophages purposely acidify their pericellular space to optimize extracellular proteolytic/hydrolytic activity. As shown in Fig. 6-old MDMs (Fig. 6 A) efficiently blanket large particles of insoluble elastin (Fig. 6 B). TEM analysis demonstrated that the MDM membrane is tightly applied to the surface of the elastin particles (Fig. 6C and Fig. D). To determine whether this interface creates a sequestered environment, MDM–elastin cultures were cooled to 4°C, fixed, and incubated briefly with either a small or large molecular-sized probe (i.e., ruthenium red at ∼ 0.6 kD or cationized ferritin at ∼ 500 kD), and access to the interface was assessed by TEM 19 20. As shown in Fig. 6, the extracellular macrophage–elastin interface was readily stained by ruthenium red (Fig. 6 C). In contrast, cationized ferritin stained the external face of the MDM plasma membrane, but was excluded from the zone of contact (Fig. 6 D).
Figure 6.
Detection of acidic microenvironments in MDM–elastin cocultures. 12-d-old MDMs were either incubated alone (A) or with 2 mg elastin (B) and examined by scanning electron microscopy. (C and D) Cells were incubated with elastin for 24 h and stained with either 0.1% ruthenium red or 0.4 mg/ml cationized ferritin, respectively, and processed for TEM. Micrographs were taken at ×300, ×750, ×10,200, and ×10,100, respectively, with asterisks overlying elastin and arrows pointing to ruthenium red– or ferritin-stained plasma membrane. MDMs incubated alone (E) or with elastin for ∼16 h (F, G, and H) were treated with 10 μg/ml acridine orange and the cells were viewed by confocal laser microscopy. Acidic environments fluoresce orange-red while green fluorescence represents nonspecific association with nucleic acids and unacidified elastin particles (reference 22). The solid white lines outline MDMs; the dotted white lines outline elastin particles in each confocal section. The fluorescence observed surrounding the elastin particles (F) was completely quenched by trypan blue (G). (H) MDMs pretreated with 5 μm bafilomycin A1 (4 h at 37°C) did not acidify cellular compartments as assessed by acridine orange fluorescence. (Inset) Lysates from ∼3 × 104 12-d-old MDMs (lane 1) were immunoblotted with an mAb (3.2 Fl) to the 73-kD catalytic subunit of the vacuolar-type H+-ATPase. Bovine brain extract (lane 2) served as a positive control.


Given the tight cleft formed between the macrophage membrane and the elastin surface, we next determined whether an acidified zone might be visualized with the acidotrophic dye, acridine orange 21 22. As expected, confocal laser micrographs of MDMs incubated with the dye alone displayed the orange-red, intracellular staining pattern characteristic of the acidified endosomal and lysosomal compartments (Fig. 6 E). In the presence of elastin particles, however, additional areas of orange-red fluorescence appeared as lacunar islands surrounding the cell-bound elastin particles (Fig. 6 F). The extracellular localization of these acidified zones was confirmed by the ability of the membrane-impermeable fluorescence quencher, trypan blue, to selectively inhibit lacunar, but not intracellular, fluorescence (Fig. 6 G).
The vacuolar-type H+-ATPase is a multisubunit enzyme system implicated in the acidification of intracellular as well as extracellular compartments in a wide range of cell types 34 35. To determine whether monocyte/macrophage differentiation was associated with the upregulation of vacuolar-type H+-ATPase components, RNA was extracted from monocytes and 1-, 6-, or 12-d-old MDMs, and labeled cRNA was prepared and hybridized to oligonucleotide microarrays 17. As shown in Fig. 7, MDMs increased expression of at least four components of the vacuolar-type H+-ATPase, including the B, F, E, and a subunits. Western blot analysis of fully differentiated MDMs with an mAb directed against the 73-kD catalytic A subunit confirmed the presence of vacuolar-type H+-ATPase components at the protein level (Fig. 6, inset). Consistent with these results, when MDMs were treated with the specific vacuolar-type H+-ATPase inhibitor, bafilomycin A1 22 35, neither the intracellular nor extracellular compartments were acidified as assessed by acridine orange staining (Fig. 6 H). Under these conditions, elastin degradation by 12-d-old MDMs (see below) that had been pretreated with bafilomycin A1 for 4 h was inhibited by 89 ± 2% (mean ± 1 SD; n = 4).
Figure 7.

Vacuolar-type H+-ATPase component expression in MDMs. Total RNA was extracted from monocytes or MDMs at the indicated times and components B (□), a (⋄), E (○), and F (▵) quantified by cDNA microarray analysis. Results are shown as the mean fold increase in mRNA content relative to that expressed by freshly isolated monocytes from a single representative experiment of three performed.
Secretion of Enzymatically Active Cathepsin K by MDMs.
The formation of an acidified pericellular zone could potentially serve to stabilize cathepsin K activity extracellularly. Although we have previously used an active site affinity label to detect active cathepsin B, S, and L secretion from MDMs 7, the diazomethane ketone probe only poorly reacts with active cathepsin K (data not shown). However, MDMs secrete the potent cysteine proteinase inhibitor, cystatin C, which tightly binds active cysteine proteinases 5 6 36. Thus, we reasoned that extracellular cathepsin K–cystatin complexes might be formed if active cathepsin K was excreted. Therefore, MDM supernatants were incubated with anti–cystatin C polyclonal antisera, the immunoprecipitates were resolved by SDS-PAGE, and complexed cathepsin K was probed by Western blot analysis. As shown in Fig. 8 A, mature cathepsin K was readily detected in the cystatin C immunoprecipitates (lane 2, arrowhead). Similarly, cathepsin K complexes were detected when intact MDMs were cultured in the presence of exogenous cystatin C (lane 6). Consistent with the ability of cystatin to form complexes with active cysteine proteinases alone, cathepsin K–cystatin C adducts were not detected when MDMs were cultured in the presence of the cysteine proteinase inhibitor, E-64, either with or without exogenous cystatin C (Fig. 8 A, lanes 8 and 4, respectively). Significantly, E-64 did not inhibit cysteine proteinase secretion per se, as increased quantities of mature cathepsin K (Fig. 8 B, lane 2) or the single chain active form of cathepsin B (Fig. 8 C, lane 2) were detected under these conditions. (The ability of E-64 to inhibit the processing of single chain, active cathepsin B to its two chain form has been described 37.) While the vacuolar-type H+-ATPase inhibitors, bafilomycin A1 or folimycin, would be predicted to affect cathepsin K extracellular activity by disrupting pericellular acidification 22 23 35, intracellular acidification plays a key role in regulating the maturation of cysteine proteinase zymogens, membrane transport processes, as well as their mannose 6-phosphate receptor–mediated trafficking to the lysosomal compartment 6 28 29 35. Consequently, MDMs cultured in the presence of vacuolar-type H+-ATPase inhibitors secreted only proforms of cathepsin K or B extracellularly (Fig. 8B and Fig. C), and effects on the enzymatic activity of mature extracellular cathepsins could not be determined.
Figure 8.

Identification of extracellular cathepsin K–cystatin C complexes in MDM cultures. (A) 12-d-old MDMs were incubated for 48 h in serum-free media (lanes 1–4) or serum-free media supplemented with human cystatin C (100 μg/ml; lanes 5–8). Cystatin C or cathepsin K–cystatin complexes were immunoprecipitated using rabbit preimmune sera (lanes 1, 3, 5, and 7) or a specific anti–cystatin C Ab (lanes 2, 4, 6, and 8). Immunoprecipitates were then resolved by SDS-PAGE, transblotted, and probed with an anti–cathepsin K mAb. In lanes 2 and 6, mature cathepsin K (black arrowhead) was identified as a complex with endogenous or exogenous cystatin C, respectively. When MDMs were incubated with 100 μM E-64 during the 48-h culture period (lanes 3, 4, 7, and 8), secreted cathepsin K was inactivated and did not form complexes with cystatin C. Nonspecific bands are observed as a consequence of a spurious cross-reaction of the secondary Abs with the rabbit polyclonal antisera used to immunoprecipitate cystatin C. Results are shown as a single representative experiment of two performed. (B and C) 12-d-old MDMs were incubated for 48 h in serum-free media alone or in the presence of E-64 (100 μM), bafilomycin A1 (10 μM), or folimycin (50 nM) and the supernatants were examined for cathepsin K (B) or cathepsin B (C) secretion. The pro- (white arrowheads) and mature (arrows) forms of the enzymes are indicated. Results are shown as a single representative experiment of three performed.
Cysteine Proteinase Processing and Elastinolytic Activity in Cathepsin K–deficient Macrophages.
Given the ability of MDMs to secrete active cathepsin K, we next sought to determine whether the proteinase played a necessary role in extracellular elastinolysis by monitoring the functional activity of cathepsin K–deficient macrophages derived from the peripheral blood of a patient with pycnodysostosis 15 16. As the affected individual is a compound heterozygote 16, we first examined cell lysates and supernatants for evidence of residual cathepsin K expression. In 12-d-old MDMs, only trace amounts of procathepsin K can be detected intracellularly or extracellularly relative to controls (Fig. 9 A). Significantly, cathepsin K deficiency did not affect the intracellular content or secretion of cathepsins S or L (Fig. 9 A). We did note, however, that pycnodysostosis-derived MDMs (a) both contained and secreted smaller quantities of cathepsin B and (b) secreted multiple forms of procathepsin L (Fig. 9 A). Because the secretion of the elastinolytic cysteine proteinases, cathepsins S and L, were unaffected by cathepsin K mutations (cathepsin B does not degrade elastin [7]), 12-d-old MDMs derived from a control, a haplosufficient parent with ∼50% normal levels of cathepsin K 16, and the affected kindred were incubated with insoluble elastin in 40% serum and elastinolysis was quantified. Surprisingly, despite the absence of mature cathepsin K in the pyknodysostotic MDMs, elastin degradation was unaffected (Fig. 9 B). Cysteine proteinase–independent pathways had not compensated for cathepsin K deficiency in these cells, as E-64 similarly inhibited elastin degradation by control, carrier, and affected MDMs (Fig. 9 B). Thus, although normal MDMs actively secrete mature cathepsin K, L, and S, the latter two cysteine proteinases alone can allow MDMs to maintain their full elastinolytic potential.
Figure 9.
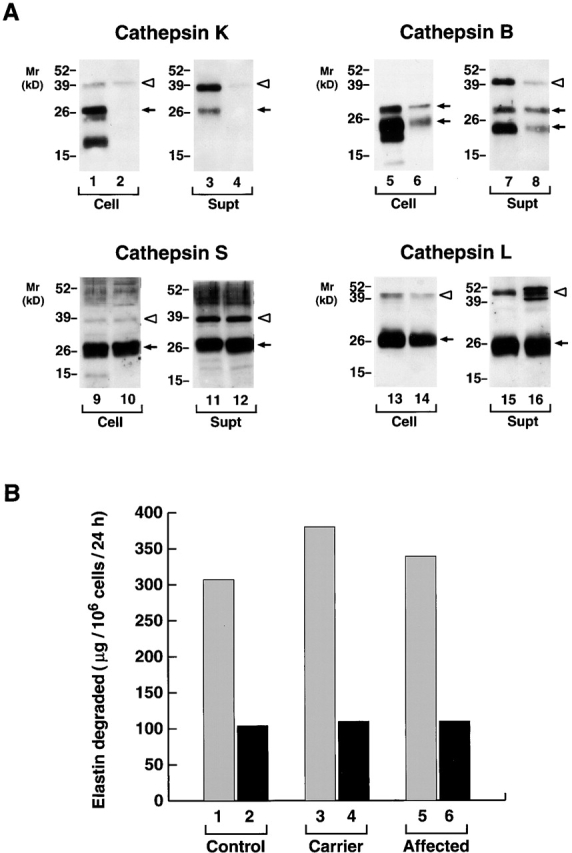
The cysteine proteinase expression and elastinolytic activity of normal versus pyknodysostotic MDMs. (A) Western blot analysis of cathepsin K (lanes 1–4), cathepsin B (lanes 5–8), cathepsin S (lanes 9–12), and cathepsin L (lanes 13–16) expression in 12-d-old MDMs from a normal control (lanes 1, 3, 5, 7, 9, 11, 13, and 15) or a pyknodysostotic donor (lanes 2, 4, 6, 8, 10, 12, 14, and 16). Cell lysates (lanes 1, 2, 5, 6, 9, 10, 13, and 14) or cell-free supernatants (lanes 3, 4, 7, 8, 11, 12, 15, and 16) (all 10 μg/lane) were immunoblotted with the respective Abs. The pro- (white arrowheads) and mature (arrows) forms of cathepsins K, B, S, and L are identified as shown. (B) MDM-mediated 3H-elastin degradation. The elastinolytic activity of cells derived from a healthy donor (lanes 1 and 2), an unaffected heterozygous individual (lanes 3 and 4), or an affected subject (lanes 5 and 6) was evaluated in the absence (lanes 1, 3, and 5) or presence (lanes 2, 4, and 6) of 100 μM E-64. Results are shown from a single experiment.
Discussion
After the initial identification of cathepsin K, it was noted not only that the isolated enzyme could exert a powerful tissue-destructive potential in vitro, but also that its expression in vivo was associated with both physiologic and pathologic matrix remodeling events 4 5 6 9 10 11 12 38 39. Early studies suggested that cathepsin K expression might be restricted to osteoclasts in vivo 40 41, but the cysteine proteinase was subsequently localized to other cell types including macrophages, synovial fibroblasts, smooth muscle cells, pulmonary epithelium, and tumor cells 8 9 10 42 43. Given these findings, it has been largely assumed that normal or neoplastic cells would secrete the cysteine proteinase into the extracellular milieu for the purpose of degrading extracellular matrix components. Interestingly, however, no cell type, including the osteoclast, has been shown to either secrete the fully processed, mature form of cathepsin K or regulate the enzyme's activity extracellularly. Nonetheless, recent reports have documented the expression of cathepsin K in macrophages at sites of elastinolytic damage in atherosclerotic vessels in vivo coupled with an associated perturbation of the cysteine proteinase–cystatin axis 4 5. Hence, we sought to determine (a) the ability of MDMs to express, process, and secrete active cathepsin K and (b) the relative role that cathepsin K plays in MDM-dependent elastinolysis.
To generate MDMs that recapitulate the destructive phenotype associated with inflammatory disease states in vivo, peripheral blood monocytes were cultured in the presence of high concentrations of autologous sera atop a plastic substratum. Under these conditions, macrophages display striking elastinolytic activity while simultaneously displaying a profile of gene products similar to that previously localized to macrophages at sites of tissue damage in vivo 7 25. During the differentiation process, cathepsin K mRNA and protein expression were markedly upregulated with kinetics distinct from that observed with cathepsins B, L, and S. Though the cathepsin K and cathepsin S genes lie within 150 kb of each other on chromosome 1q21, and appear to have arisen from a common ancestral cysteine proteinase, their expression patterns were divergent 44 45. Relevant to macrophage differentiation, the cathepsin K promoter contains AP-1, PU.1, and H-APF-1 regulatory elements 44 45, but which, if any, of the binding sites participate in late onset gene expression remains to be determined.
Coincident with the upregulation of cathepsin K mRNA levels, MDMs began to express the cathepsin K protein. Like the other cysteine proteinases, cathepsin K is synthesized as a proenzyme that undergoes processing to its mature form presumably during transit from the trans-Golgi network to the late endosomal/lysosomal compartments 6 26. At the earliest time points when cathepsin K protein could be detected (i.e., between days 3 and 5), MDMs secreted only small amounts of procathepsin K in concert with the proforms of cathepsins B and S. While cysteine proteinases are primarily routed to the late endosome/lysosomal compartments by both mannose 6-phosphate receptor–dependent and –independent pathways, lysosomal proteinases can escape this delivery process and be secreted as proforms after exit from the trans-Golgi network 6 26 46 47. We cannot, however, rule out the possibility that proforms may be secreted in a regulated fashion from an intracellular compartment (e.g., early endosome or a granule-like vesicle) 28 29 47 48. Interestingly, whereas the cysteine proteinases, cathepsins K, S, B, and the aspartyl proteinase, cathepsin D, initially exited the cell predominately as proforms, cathepsin L was preferentially released as the mature enzyme. These findings support recent reports that the distribution of cysteine proteinases in intracellular compartments varies between early endosome, late endosome, and lysosomal pools 28 29. Furthermore, as the secretion of cysteine proteinases as well as acid hydrolases such as β-glucosaminidase, acid phosphatase, and β-glucuronidase can each be regulated in selective fashion (e.g., 27–29, 49), it appears that functionally distinct pools of lysosomal enzymes may exist in MDMs. We cannot, however, rule out the possibility that cysteine proteinases and lysosomal hydrolases failed to accumulate extracellularly in tandem due to differences in the stability of the released enzymes or their rate of reuptake 6.
Regardless of the intracellular localization of pro- and mature forms of lysosomal hydrolases and proteinases, MDMs ultimately engage a program wherein fully processed acidophilic enzymes are actively exteriorized. In other cell systems, secretion of lysosomal components can be regulated by particulate or soluble stimuli 27 48 49. Intriguingly, none of these agents altered cysteine proteinase secretion despite the fact that these stimuli can affect lysosomal hydrolase and/or matrix metalloproteinase expression (data not shown). Cathepsin K secretion was, however, dramatically upregulated after disassembly of the actin filament network with cytochalasin. As reviewed by Muallem et al., cortical actin networks act as a physical barrier to vesicle docking 33. In fusion-competent cells, the disassembly of the actin network alone is permissive for secretion as we have observed for MDMs. These results contrast with other hematopoietic cell types, including neutrophils, where actin filament depolymerization alone does not trigger exocytosis until the cells are specifically triggered by agonists 33. Apparently, the fully differentiated MDM displays a fusion-competent state wherein the cortical actin network alone may regulate endosome/lysosome–plasma membrane docking events.
Cathepsins K, L, S, B, and D, and lysosomal hydrolase each have acidic pH optima and display limited stability in their mature forms at neutral pH 6 7 11 12. Thus, we sought to determine whether MDMs might acidify their pericellular milieu to optimize the proteolytic activity of lysosomal enzymes. Macrophages can mobilize the vacuolar-type H+-ATPase to their plasma membrane, but primary emphasis has focused on the role of the proton pump in maintaining cytosolic pH in the face of an external acid load (e.g., 35, 50). However, it seems likely that the vacuolar-type H+-ATPase plays an additional role by creating a permissive environment for acidophilic enzymes in a manner similar to that described for osteoclasts 35 51. Indeed, taking advantage of the formation of a sequestered environment at the cell–elastin interface where acridine orange could accumulate, acidified zones were visualized in MDMs surrounding elastin particles. It should be noted that the addition of elastin particles was not required for H+ secretion, as a pericellular pH (monitored by a pH-sensitive microelectrode) of ∼6.0 was detected in either the absence or presence of the particles (our unpublished observation). Likewise, elastin did not alter cysteine proteinase release, thus suggesting that MDMs constitutively cosecrete endosomal/lysosomal contents and pump protons into the extracellular space after the 12-d differentiation process. Consistent with this behavior, active cathepsin K was detected extracellularly as defined by the formation of cystatin C–cathepsin K complexes. Because cysteine proteinases display short, but finite half-lives at neutral pH 6 11 12, we cannot rule out the possibility that a steady state concentration of cathepsin K exists in the pericellular milieu independent of the action of the vacuolar-type H+-ATPase. As noted, bafilomycin A1 and folimycin disrupt lysosomal enzyme trafficking and processing 28 29 35, thus obfuscating our ability to define the role of the extracellular pH alone in regulating cathepsin K activity. Nonetheless, taken in toto, our data indicate that MDMs can mobilize all of the necessary machinery to use cathepsin K as well as cathepsins S and L to express extracellular elastinolytic activity.
Given the ability of MDMs to secrete active cathepsin K as well as the potent elastinolytic potential of the recombinant proteinase 4 5 6 11, it has been assumed that the enzyme would play an important role in the elastinolytic process. However, cathepsin K–deficient MDMs (as well as haplosufficient cells expressing one-half normal levels of cathepsin K) degraded elastin comparably to control cells. Based on the documented ability of cathepsin K to degrade insoluble elastin 6 11, it seems unlikely that the enzyme fails to participate in MDM-mediated elastin degradation. Rather, in the absence of cathepsin K, we conclude that cathepsins L and/or S retain sufficient elastinolytic activity to compensate fully for the loss of one of the three known elastases in this gene family. Alternatively, we cannot rule out the possibility that cathepsin K activity is strictly reserved for the degradation of a target molecule that is uniquely sensitive to its action. In human osteoclasts (which, in contrast to MDMs, express little or no cathepsin B, L, or S, and do not secrete cathepsin D [14, 41]), cathepsin K has been postulated to play a required role in the degradation of type I collagen or other bone matrix–associated proteins 11 12 15 38 39 52. Furthermore, cathepsin K has been shown to degrade type II collagen where it may play a role in the proteolytic degradation of cartilage 9 39. Although our data clearly demonstrate that cathepsin K does not play a required role in MDM-mediated elastinolysis, the secretion of this powerful proteinase could impact on any one of several other extracellular matrix components.
Macrophages have long been known to secrete lysosomal hydrolases both in vitro and in vivo 27 49. However, as the extracellular function of these enzymes has remained largely undefined, the significance of this secretory phenotype was unclear. Furthermore, as demonstrated, lysosomal hydrolase release may not necessarily correlate with cysteine or aspartyl proteinase secretion 28 29. Nonetheless, our data demonstrate that programs for mobilizing the exocytosis of a more complex, and destructive, mix of endosomal/lysosomal contents can converge. Taken together, we posit that the sequestered, acidic microenvironment generated at the MDM–elastin interface optimizes proteolytic activity while limiting access to proteinase inhibitors. Matrix metalloproteinases have also been implicated in elastin turnover 1 3, and we have found that MDMs express several elastinolytic metalloenzymes, including gelatinase B, macrophage metalloelastase, and matrilysin. However, unlike cysteine proteinase–mediated elastinolysis, we have only been able to implicate matrix metalloproteinases in elastin degradation under serum-free conditions (our unpublished observation). In any case, given that cathepsins K, B, L, and D have all been identified in tissues actively undergoing destructive tissue remodeling (e.g., 9–11, 53 54 55 56 57 58), it seems likely that the in vitro MDM phenotype described in our report recapitulates that expressed in vivo. With the recent demonstration that cystatins may be specifically downregulated at sites of tissue damage 5, active cysteine proteinases secreted by macrophages and other cell populations (including endothelial cells, fibroblasts, and tumor cells [55, 56]) may play a more important role in matrix remodeling than previously suspected.
Acknowledgments
We thank Drs. M. Johnson and C. Francomanco (National Institutes of Health) for providing us with access to peripheral blood samples from pycnodysostosis and affected individuals. We also acknowledge with gratitude the assistance of the family members involved.
This work was supported by National Institutes of Health grant AI21301.
Footnotes
Abbreviations used in the paper: GAPDH, glyceraldehyde 3-phosphate dehydrogenase; gp, glycoprotein; MDM, monocyte-derived macrophage; TEM, transmission electron microscopy; TPA, tetradecanoylphorbol 13-acetate.
A. Punturieri's present address is Veteran's Affairs Medical Center, 2215 Fuller Rd., Pulmonary Section (111G), Ann Arbor, MI 48109. V. Reddy's present address is Division of Cardiology, Massachusetts General Hospital, 32 Fruit St., Boston, MA 02114.
References
- Carmeliet P., Moons L., Lijnen R., Baes M., Lemaitre V., Tipping P., Drew A., Eckhout Y., Shapiro S., Lupu F., Collen D. Urokinase-generated plasmin activates matrix metalloproteinases during aneurysm formation. Nat. Genet. 1997;17:439–444. doi: 10.1038/ng1297-439. [DOI] [PubMed] [Google Scholar]
- Weyand C.M., Wagner A.D., Bjornsson J., Goronzy J.J. Correlation of the topographical arrangement and the functional pattern of tissue-infiltrating macrophages in giant cell arteritis. J. Clin. Invest. 1996;98:1642–1649. doi: 10.1172/JCI118959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hautamaki R.D., Kobayashi D.K., Senior R.M., Shapiro S.D. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277:2002–2004. doi: 10.1126/science.277.5334.2002. [DOI] [PubMed] [Google Scholar]
- Sukhova G.K., Shi G.-P., Simon D.I., Chapman H.A., Libby P. Expression of elastolytic cathepsins S and K in human atheroma and regulation of their production in smooth muscle cells. J. Clin. Invest. 1998;102:576–583. doi: 10.1172/JCI181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi G.-P., Sukhova G.K., Grubb A., Ducharme A., Rhode L.H., Lee R.T., Ridker P.M., Libby P., Chapman H.A. Cystatin C deficiency in human atherosclerosis and aortic aneurysms. J. Clin. Invest. 1999;104:1191–1197. doi: 10.1172/JCI7709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman H.A., Riese R.J., Shi G.-P. Emerging roles for cysteine proteases in human biology. Annu. Rev. Physiol. 1997;59:63–88. doi: 10.1146/annurev.physiol.59.1.63. [DOI] [PubMed] [Google Scholar]
- Reddy V.Y., Zhang Q.Y., Weiss S.J. Pericellular mobilization of the tissue-destructive cysteine proteinases, cathepsins B, L, and S, by human monocyte-derived macrophages. Proc. Natl. Acad. Sci. 1995;USA. 92:3849–3853. doi: 10.1073/pnas.92.9.3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi G.-P., Chapman H.A., Bhairi S.M., DeLeeuw C., Reddy V.Y., Weiss S.J. Molecular cloning of human cathepsin O, a novel endoproteinase and homologue of rabbit OC2. FEBS (Fed. Eur. Biochem. Soc.) Lett. 1995;357:129–134. doi: 10.1016/0014-5793(94)01349-6. [DOI] [PubMed] [Google Scholar]
- Hummel K.M., Petrow P.K., Franz J.K., Muller-Ladner U., Aicher W.K., Gay R.E., Bromme D., Gay S. Cysteine proteinase cathepsin K mRNA is expressed in synovium of patients with rheumatoid arthritis and is detected at sites of synovial bone destruction. J. Rheumatol. 1998;25:1887–1894. [PubMed] [Google Scholar]
- Dodds R.A., Connor J.R., Drake F.H., Gowen M. Expression of cathepsin K messenger RNA in giant cells and their precursors in human osteoarthritic synovial tissues. Arthritis Rheum. 1999;42:1588–1593. doi: 10.1002/1529-0131(199908)42:8<1588::AID-ANR4>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Bromme D., Okamoto K., Wang B.B., Biroc S. Human cathepsin O2, a matrix protein-degrading cysteine protease expressed in osteoclasts. Functional expression of human cathepsin O2 in Spodoptera frugiperda and characterization of the enzyme. J. Biol. Chem. 1996;271:2126–2132. doi: 10.1074/jbc.271.4.2126. [DOI] [PubMed] [Google Scholar]
- Bossard M.J., Tomaszek T.A., Thompson S.K., Amegadzie B.Y., Hanning C.R., Jones C., Kurdyla J.T., McNulty D.E., Drake F.H., Gowen M., Levy M.A. Proteolytic activity of human osteoclast cathepsin K. J. Biol. Chem. 1996;271:12517–12524. doi: 10.1074/jbc.271.21.12517. [DOI] [PubMed] [Google Scholar]
- Yamaza T., Goto T., Kamiya T., Kobayashi Y., Sakai H., Tanaka T. Study of immunoelectron microscopic localization of cathepsin K in osteoclasts and other bone cells in the mouse femur. Bone. 1998;23:499–509. doi: 10.1016/s8756-3282(98)00138-0. [DOI] [PubMed] [Google Scholar]
- Xia L., Kilb J., Wex H., Li Z., Lipyansky A., Breuil V., Stein L., Palmer J.T., Dempster D.W., Bromme D. Localization of rat cathepsin K in osteoclasts and resorption pitsinhibition of bone resorption and cathepsin K-activity by peptidyl vinyl sulfones. Biol. Chem. 1999;380:679–687. doi: 10.1515/BC.1999.084. [DOI] [PubMed] [Google Scholar]
- Gelb B.D., Shi G.-P., Chapman H.A., Desnick R.J. Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science. 1996;273:1236–1238. doi: 10.1126/science.273.5279.1236. [DOI] [PubMed] [Google Scholar]
- Ho N., Punturieri A., Wilkin D., Szabo J., Johnson M., Whaley J., Davis J., Clark A., Weiss S., Francomano C. Mutations of CTSK result in pycnodysostosis via a reduction in cathepsin K protein. J. Bone Miner. Res. 1999;14:1649–1653. doi: 10.1359/jbmr.1999.14.10.1649. [DOI] [PubMed] [Google Scholar]
- Lockhart D.J., Dong H., Byrne M.C., Follettie M.T., Gallo M.V., Chee M.S., Mittmann M., Wang C., Kobayashi M., Horton H., Brown E.L. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat. Biotechnol. 1996;14:1675–1680. doi: 10.1038/nbt1296-1675. [DOI] [PubMed] [Google Scholar]
- Hiraoka N., Allen E., Apel I.J., Gyetko M.R., Weiss S.J. Matrix metalloproteinases regulate neovascularization by acting as pericellular fibrinolysins. Cell. 1998;95:365–377. doi: 10.1016/s0092-8674(00)81768-7. [DOI] [PubMed] [Google Scholar]
- Kruth H.S., Skarlatos S.I., Lilly K., Chang J., Ifrim I. Sequestration of acetylated LDL and cholesterol crystals by human monocyte-derived macrophages. J. Cell Biol. 1995;129:133–145. doi: 10.1083/jcb.129.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinstein S., Furuya W. Assessment of Na+-H+ exchange activity in phagosomal membranes of human neutrophils. Am. J. Physiol. 1988;254:C272–C285. doi: 10.1152/ajpcell.1988.254.2.C272. [DOI] [PubMed] [Google Scholar]
- Baron R., Neff L., Louvard D., Courtoy P.J. Cell-mediated extracellular acidification and bone resorptionevidence for a low pH in resorbing lacunae and localization of a 100-kD lysosomal membrane protein at the osteoclast ruffled border. J. Cell Biol. 1985;101:2210–2222. doi: 10.1083/jcb.101.6.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimori T., Yamamoto A., Moriyama Y., Futai M., Tashiro Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H+-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J. Biol. Chem. 1991;266:17707–17712. [PubMed] [Google Scholar]
- Woo J.-T., Shinohara C., Sakai K., Hasumi K., Endo A. Inhibition of the acidification of endosomes and lysosomes by the antibiotic concanamycin B in macrophage J774. Eur. J. Biochem. 1992;207:383–389. doi: 10.1111/j.1432-1033.1992.tb17061.x. [DOI] [PubMed] [Google Scholar]
- Loike J.D., Silverstein S.C. A fluorescence quenching technique using trypan blue to differentiate between attached and ingested glutaraldehyde-fixed red blood cells in phagocytosing murine macrophages. J. Immunol. Methods. 1983;57:373–379. doi: 10.1016/0022-1759(83)90097-2. [DOI] [PubMed] [Google Scholar]
- Boot R.G., van Achterberg T.A.E., van Aken B.E., Renkema G.H., Jacobs M.J.H.M., Aerts J.M.F.G., de Vries C.J.M. Strong induction of members of the chitinase family of proteins in atherosclerosis. Chitotriosidase and human cartilage gp-39 expressed in lesion macrophages. Arterioscler. Thromb. Vasc. Biol. 1999;19:687–694. doi: 10.1161/01.atv.19.3.687. [DOI] [PubMed] [Google Scholar]
- Lingeman R.G., Joy D.S., Sherman M.A., Kane S.E. Effect of carbohydrate position on lysosomal transport of procathepsin L. Mol. Biol. Cell. 1998;9:1135–1147. doi: 10.1091/mbc.9.5.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy K., Musson R.A., Henson P.M. Protein synthesis-dependent and protein synthesis-independent secretion of lysosomal hydrolases from rabbit and human macrophages. J. Reticuloendothel. Soc. 1982;31:131–144. [PubMed] [Google Scholar]
- Claus V., Jahraus A., Tjelle T., Berg T., Kirschke H., Faulstich H., Griffiths G. Lysosomal enzyme trafficking between phagosomes, endosomes, and lysosomes in J774 macrophages. J. Biol. Chem. 1998;273:9842–9851. doi: 10.1074/jbc.273.16.9842. [DOI] [PubMed] [Google Scholar]
- Jahraus A., Tjelle T.E., Berg T., Habermann A., Storrie B., Ullrich O., Griffiths G. In vitro fusion of phagosomes with different endocytic organelles from J774 macrophages. J. Biol. Chem. 1998;273:30379–30390. doi: 10.1074/jbc.273.46.30379. [DOI] [PubMed] [Google Scholar]
- Imort M., Zuhlsdorf M., Feige U., Hasilik A., von Figura K. Biosynthesis and transport of lysosomal enzymes in human monocytes and macrophages. Effects of ammonium chloride, zymosan and tunicamycin. Biochem. J. 1983;214:671–678. doi: 10.1042/bj2140671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A., Webster P., Ortego J., Andrews N.W. Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J. Cell Biol. 1997;137:93–104. doi: 10.1083/jcb.137.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozhin J., Sameni M., Ziegler G., Sloane B.F. Pericellular pH affects distribution and secretion of cathepsin B in malignant cells. Cancer Res. 1994;54:6517–6525. [PubMed] [Google Scholar]
- Muallem S., Kwiatkowska K., Xu X., Yin H.L. Actin filament disassembly is a sufficient final trigger for exocytosis in nonexcitable cells. J. Cell Biol. 1995;128:589–598. doi: 10.1083/jcb.128.4.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B.S., Krits I., Crane-Zelkovic M.K., Gluck S.L. A novel transcription factor regulates expression of the vacuolar H+-ATPase subunit through AP-2 sites during monocytic differentiation. J. Biol. Chem. 1997;272:174–181. [PubMed] [Google Scholar]
- Stevens T.H., Forgac M. Structure, function and regulation of the vacuolar (H+)-ATPase. Annu. Rev. Cell Dev. Biol. 1997;13:779–808. doi: 10.1146/annurev.cellbio.13.1.779. [DOI] [PubMed] [Google Scholar]
- Schick C., Pemberton P.A., Shi G.-P., Kamachi Y., Cataltepe S., Bartuski A.J., Gornstein E.R., Bromme D., Chapman H.A., Silverman G.A. Cross-class inhibition of the cysteine proteinases cathepsins K, L, and S by the serpin squamous cell carcinoma antigen 1a kinetic analysis. Biochemistry. 1998;37:5258–5266. doi: 10.1021/bi972521d. [DOI] [PubMed] [Google Scholar]
- Hara K., Kominami E., Katunuma N. Effect of proteinase inhibitors on intracellular processing of cathepsin B, H and L in rat macrophages. FEBS (Fed. Eur. Biochem. Soc.) Lett. 1988;231:229–231. doi: 10.1016/0014-5793(88)80737-3. [DOI] [PubMed] [Google Scholar]
- Garnero P., Borel O., Byrjalsen I., Ferreras M., Drake F.H., McQueney M.S., Foged N.T., Delmas P.D., Delaisse J.-M. The collagenolytic activity of cathepsin K is unique among mammalian proteinases. J. Biol. Chem. 1998;273:32347–32352. doi: 10.1074/jbc.273.48.32347. [DOI] [PubMed] [Google Scholar]
- Kafienah W., Bromme D., Buttle D.J., Croucher L.J., Hollander A.P. Human cathepsin K cleaves native type I and II collagens at the N-terminal end of the triple helix. Biochem. J. 1998;331:727–732. doi: 10.1042/bj3310727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tezuka K.-I., Tezuka Y., Maejima A., Sato T., Nemoto K., Kamioka H., Hakeda Y., Kumegawa M. Molecular cloning of a possible cysteine proteinase predominantly expressed in osteoclasts. J. Biol. Chem. 1994;269:1106–1109. [PubMed] [Google Scholar]
- Drake F.H., Dodds R.A., James I.E., Connor J.R., Debouck C., Richardson S., Lee-Rykaczewski E., Coleman L., Rieman D., Barthlow R. Cathepsin K, but not cathepsins B, L, or S, is abundantly expressed in human osteoclasts. J. Biol. Chem. 1996;271:12511–12516. doi: 10.1074/jbc.271.21.12511. [DOI] [PubMed] [Google Scholar]
- Buhling F., Gerber A., Hackel C., Kruger S., Kohnlein T., Bromme D., Reinhold D., Ansorge S., Welte T. Expression of cathepsin K in lung epithelial cells. Am. J. Respir. Cell Mol. Biol. 1999;20:612–619. doi: 10.1165/ajrcmb.20.4.3405. [DOI] [PubMed] [Google Scholar]
- Littlewood-Evans A.J., Bilbe G., Bowler W.B., Farley D., Wlodarski B., Kokubo T., Inaoka T., Sloane J., Evans D.B., Gallagher J.A. The osteoclast-associated protease cathepsin K is expressed in human breast carcinoma. Cancer Res. 1997;57:5386–5390. [PubMed] [Google Scholar]
- Gelb B.D., Shi G.-P., Heller M., Weremowicz S., Morton C., Desnick R.J., Chapman H.A. Structure and chromosomal assignment of the human cathepsin K gene. Genomics. 1997;41:258–262. doi: 10.1006/geno.1997.4631. [DOI] [PubMed] [Google Scholar]
- Rood J.A., Van Horn S., Drake F.H., Gowen M., Debouck C. Genomic organization and chromosome localization of the human cathepsin K gene (CTSK) Genomics. 1997;41:169–176. doi: 10.1006/geno.1997.4614. [DOI] [PubMed] [Google Scholar]
- Kasper D., Dittmer F., von Figura K., Pohlmann R. Neither type of mannose 6-phosphate receptor is sufficient for targeting of lysosomal enzymes along intracellular routes. J. Cell Biol. 1996;134:615–623. doi: 10.1083/jcb.134.3.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuliawat R., Klumperman J., Ludwig T., Arvan P. Differential sorting of lysosomal enzymes out of the regulated secretory pathway in pancreatic β-cells. J. Cell Biol. 1997;137:595–608. doi: 10.1083/jcb.137.3.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths G.M. Secretory lysosomes—a special mechanism of regulated secretion in hemopoietic cells. Trends Cell Biol. 1996;6:329–332. doi: 10.1016/0962-8924(96)20031-5. [DOI] [PubMed] [Google Scholar]
- Schnyder J., Baggiolini M. Secretion of lysosomal hydrolases by stimulated and nonstimulated macrophages. J. Exp. Med. 1978;148:435–450. doi: 10.1084/jem.148.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swallow C.J., Grinstein S., Sudsbury R.A., Rotstein O.D. Relative roles of Na+/H+ exchange and vacuolar-type H+-ATPases in regulating cytoplasmic pH and function in murine peritoneal macrophages. J. Cell. Physiol. 1993;157:453–460. doi: 10.1002/jcp.1041570304. [DOI] [PubMed] [Google Scholar]
- Silver I.A., Murrills R.J., Etherington D.J. Microelectrode studies on the acid microenvironment beneath adherent macrophages and osteoclasts. Exp. Cell Res. 1988;175:266–276. doi: 10.1016/0014-4827(88)90191-7. [DOI] [PubMed] [Google Scholar]
- Saftig P., Hunziker E., Wehmeyer O., Jones S., Boyde A., Rommerskirch W., Moritz J.D., Schu P., von Figura K. Impaired osteoclastic bone resorption leads to osteopetrosis in cathepsin-K-deficient mice. Proc. Natl. Acad. Sci. USA. 1998;95:13453–13458. doi: 10.1073/pnas.95.23.13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole A.R., Hembry R.M., Dingle J.T., Pinder I., Ring E.F.J., Cosh J. Secretion and localization of cathepsin D in synovial tissues removed from rheumatoid and traumatized joints. Arthritis Rheum. 1976;19:1295–1307. doi: 10.1002/art.1780190610. [DOI] [PubMed] [Google Scholar]
- Mort J.S., Recklies A.D., Poole A.R. Extracellular presence of the lysosomal proteinase cathepsin B in rheumatoid synovium and its activity at neutral pH. Arthritis Rheum. 1984;27:509–515. doi: 10.1002/art.1780270505. [DOI] [PubMed] [Google Scholar]
- Iwata Y., Mort J.S., Tateishi H., Lee E.R. Macrophage cathepsin L, a factor in the erosion of subchondral bone in rheumatoid arthritis. Arthritis Rheum. 1997;40:499–509. doi: 10.1002/art.1780400316. [DOI] [PubMed] [Google Scholar]
- Creemers L.B., Jansen I.D.C., Hoeben K.A., Beertsen W., Everts V. Involvement of V-ATPase in the digestion of soft connective tissue collagen. Biochem. Biophys. Res. Commun. 1998;251:429–436. doi: 10.1006/bbrc.1998.9357. [DOI] [PubMed] [Google Scholar]
- Demchik L.L., Sameni M., Nelson K., Mikkelsen T., Sloane B.F. Cathepsin B and glioma invasion. Int. J. Dev. Neurosci. 1999;17:483–494. doi: 10.1016/s0736-5748(99)00011-8. [DOI] [PubMed] [Google Scholar]
- Felbor U., Dreier L., Bryant R.A.R., Ploegh H.L., Olsen B.R., Mothes W. Secreted cathepsin L generates endostatin from collagen XVIII. EMBO (Eur. Mol. Biol. Organ.) J. 2000;19:1187–1194. doi: 10.1093/emboj/19.6.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]