

Abstract
Experimental autoimmune encephalomyelitis (EAE) is a CD4+ T lymphocyte–mediated disease of the central nervous system (CNS) characterized by mononuclear cell infiltration, demyelination, and paralysis. We previously demonstrated a role for chemokines in acute and relapsing EAE pathogenesis. Presently, we investigated the role of CC chemokine receptor 2 (CCR2) in acute EAE. CCR2−/− mice did not develop clinical EAE or CNS histopathology, and showed a significant reduction in T cell– and CNS-infiltrating CD45highF4/80+ monocyte subpopulations. Peripheral lymphocytes from CCR2−/− mice produced comparable levels of interferon-gamma (IFN-γ) and interleukin (IL)-2 in response to antigen-specific restimulation when compared with control mice. Adoptively transferred myelin oligodendrocyte glycoprotein 35-55–specific T cells lacking expression of CCR2 were able to induce EAE, whereas CCR2−/− recipients of wild-type T cells failed to develop disease. These results suggest that CCR2 expression on host-derived mononuclear cells is critical for disease induction.
Keywords: multiple sclerosis, experimental autoimmune encephalomyelitis, CC chemokine receptor 2, knockout, CCL2
Introduction
Experimental autoimmune encephalomyelitis (EAE) is a CD4+ T cell–mediated demyelinating disease of the central nervous system (CNS) that serves as a model for multiple sclerosis (MS) 1. The pathogenic mechanisms of disease development include antigen-specific T cell activation and Th1 differentiation 2 followed by T cell 3 and macrophage migration into CNS with little or no polymorphonuclear cell infiltration 4. The mechanism by which cells traffic to the CNS and accumulate before and during clinical disease is not well understood. However, entry of activated T cells into tissue compartments is a process governed by integrins 5 6 7.
An additional essential step during pathogenesis of tissue specific inflammatory diseases is chemokine-induced recruitment and/or accumulation of leukocytes within tissue sites. Chemokines are potent leukocyte chemoattractants that can be divided into four highly conserved families: the C-x-C, C-C, C, and C-x3-C families, based on the position of the first two cysteines in the NH2 terminus 8. CC chemokine family members have been implicated as functional mediators of immunopathology in EAE (reviewed in reference 9) and are ligands for seven transmembrane-spanning receptors that induce signaling through a G-protein–linked pathway resulting in cytoskeletal rearrangement, intracellular calcium increases, adhesion to endothelium or extracellular matrix proteins, Th1/Th2 differentiation, costimulation, IL-2 production, proliferation, and chemotaxis 10.
Our previous observations showing differential expression and function of CC chemokines in development of EAE 11 led us to hypothesize that infiltrating mononuclear cells require CC chemokine receptor (CCR) expression corresponding to the chemokine ligands produced during disease pathogenesis in order for mononuclear cells to accumulate in the CNS and induce subsequent disease. In this study, we investigated the role of CCR2 during induction of acute EAE in an effort to understand mononuclear cell migration and/or accumulation during the development of EAE.
Materials and Methods
Animals.
Control female B6129PF2/J and C57BL/6 mice were purchased from The Jackson Laboratory. CCR2−/− mice were on an outbred C57BL/6J × 129/Ola genetic background 12. Mice were born and bred under specific pathogen-free conditions and subsequently maintained at Northwestern University. Heterozygous CCR2+/−5+/− littermate controls were generated by crossing the CCR2−/− and CCR5−/− 12 homozygote mice. C57BL/6J mice were subsequently used as wild-type controls for the CCR2−/− mice as there were no differences between the CCR2+/−5+/− heterozygous control mice, B6129PF2/J, or C57BL/6J mice. Animal care and use was performed in accordance with Northwestern University and National Institutes of Health guidelines.
Antigens, Abs, and Flow Cytometry.
Myelin oligodendrocyte glycoprotein (MOG)35–55 (MEVGWYRSPFSRVVHLYRNGK) was purchased from Peptides International, amino acid composition was verified by mass spectrometry, and purity was >98%. mAbs to CD4 (RM4-5), CD8a (Ly-2), CD19 (1D3), CD45 (Ly-5), purified Fc Block (2.4G2), F4/80, and isotype control Abs were obtained commercially (BD PharMingen and Caltag Laboratories). CNS mononuclear cells were isolated by centrifugation (500 g) at 24°C from the interface of a 30–70% discontinuous Percoll (Amersham Pharmacia Biotech) gradient and 106 cells were preincubated with Fc Block for 15 min at 4°C in PBS containing 0.1% NaN3 and 2% BSA (Sigma-Aldrich), followed by specific and isotype control Ab staining. Data collection and analysis were performed on a FACSCalibur™ flow cytometer using CELLQuest™ software (Becton Dickinson).
Cell Culture.
Lymphocytes were cultured in DMEM (GIBCO BRL) as described previously 13. Adoptive transfer cells were cultured at 106 cells/ml in DMEM for 72 h with 30 μg/ml MOG35–55, 20 ng/ml rmIL-12 (R&D Systems), and γ-irradiated (3000 rad) splenocytes (5 × 106 cells/ml).
Disease Induction and Clinical Evaluation.
For actively induced EAE, mice were immunized with 200 μg of MOG35–55 emulsified in CFA containing 4 mg/ml Mycobacterium tuberculosis (Difco) subcutaneously and given 200 ng of pertussis toxin (List Biological Laboratories) intravenously on the day of and 2 d after immunization. For adoptively induced EAE, donor wild-type and CCR2−/− mice were immunized with MOG35–55 in CFA. 10 d after immunization, draining LNs were harvested and cultured as described above. 5 × 106 CD4+ T cells transferred intravenously to recipients who received 100 ng of pertussis toxin intravenously on the day of and 2 d after cell transfer. Individual animals were graded according to clinical severity as follows: grade 0, no abnormality; grade 1, limp tail; grade 2, limp tail and hind limb weakness; grade 3, partial hind limb paralysis; grade 4, complete hind limb paralysis.
CNS Chemokine and CCR Reverse Transcriptase–PCR.
Total RNA was isolated from spinal cords using TRIzol reagent (GIBCO BRL), DNase treated (Promega), and converted to cDNA (CLONTECH Laboratories, Inc.). Chemokine reverse transcriptase (RT)–PCR was performed as described previously 14. PCR conditions were: 94°C for 3 min, followed by 40 cycles of 30 sec at 94°C, 30 sec at 60°C, and 1 min 30 sec at 72°C, with a final extension at 72°C for 3 min. CCR primer sequences used include: CCR1, 15; CCR2, sense 5′-GGTCATGATCCCTATGTGG-3′, antisense 5′-CTGGGCACCTGATTTAAAGG-3′; CCR5 sense 5′-GCTGAAGAGCGTGACTGATA-3′, antisense 5′-GAGGACTGCATGTATAATGA-3′; CCR7 sense 5′-ACAGCGGCCTCCAGAAGAACAGCGG-3′, antisense 5′-TGACGTCATAGGCAATGTTGAGCTG-3′. PCR products were visualized by ethidium bromide 2% agarose gel electrophoresis.
Cytokine and Chemokine ELISA.
Assessment of cytokine production was determined from LN culture supernatants harvested after a 48-h stimulation with MOG35–55 peptide as described previously 13. Assessment of chemokine expression was performed from tissue samples using described previously noncommercial ELISAs 11.
Statistical Analysis.
Comparisons of disease incidence were analyzed by the X2 analysis, using Fisher's exact probability test. Statistical significance of cytokine levels, disease onset, and disease severity was analyzed using the Student's t test for comparisons of two means. Values of P ≤ 0.05 were considered significant.
Results and Discussion
Chemokine Expression in the Spinal Cord during Acute EAE.
To understand the role for chemokine receptors in EAE pathogenesis, we first examined the CNS for chemokine receptor and ligand mRNA expression by RT–PCR including monocyte chemotactic protein-1 (CCL2), macrophage inflammatory protein-1α (CCL3), macrophage inflammatory protein-1β (CCL4), RANTES (CCL5 16), CCR1, CCR2, CCR5, and CCR7. Spinal cords from three different time points corresponding to preclinical when no animals were exhibiting disease symptoms, disease onset when animals showed first symptoms of EAE, and peak EAE were evaluated. The results shown in Fig. 1 A demonstrate CCL2, CCL4, and CCL5 CNS chemokine mRNA expression in wild-type CNS at all three time points examined. CCL3 expression was not detected until clinical onset of disease and remained expressed through acute clinical disease. The results shown in Fig. 1 A also demonstrate CCR2 and 5 mRNA expression at all three time points measured. Unlike CCR2 and 5 expression, CCR1 and 7 expression was not detected until peak acute EAE.
Figure 1.
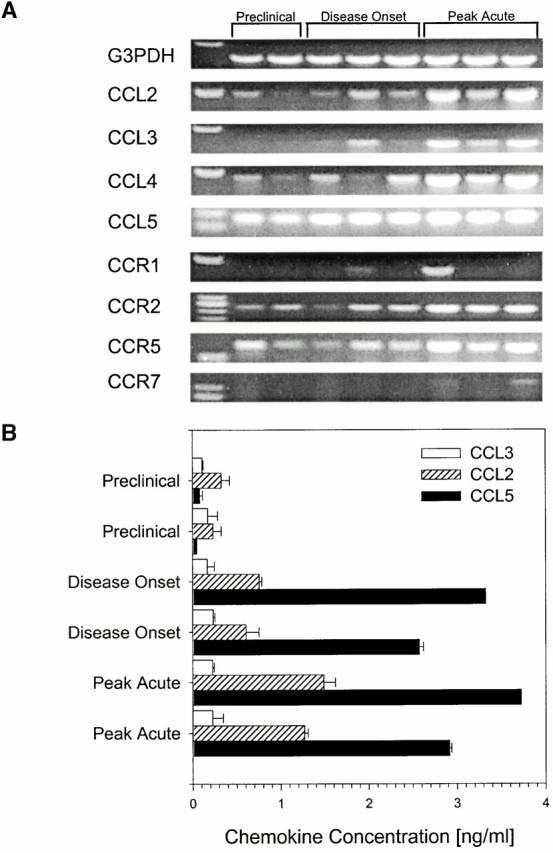
CNS chemokine and chemokine receptor expression. (A) CCL2, CCL4, CCL5, and chemokine receptor mRNA was detected in CNS tissue samples using RT–PCR at all three time points examined after EAE induction. (B) CNS chemokine protein expression for CCL2, CCL3, CCL4, and CCL5 was quantitated from tissue samples using specific ELISA. The results from two representative mice from each time point are shown. In all cases the data shown are representative of two independent experiments.
CNS CCL2 protein production during acute EAE was also examined based on its known role in other EAE susceptible mouse strains 11 and its role as a ligand for CCR2 17. The results shown in Fig. 1 B demonstrate low levels of CNS CCL2 and CCL5 in preclinical mice. As disease progresses though initial clinical symptoms to peak acute disease, levels of CCL2 and CCL5 increase dramatically. CCL3 protein expression was lower compared with CCL2 and CCL5 levels and did not correlate with increasing disease severity (Fig. 1 B). There was no CCL4 protein detected in any spinal cords examined (data not shown). Taken together, these data demonstrate the CNS presence of both CCR2 and its ligand, CCL2, during EAE development.
CCR2−/− Mice Are Protected from Developing Clinical EAE.
To determine if CCR2 was required for acute EAE, we used CCR2−/− mice for active disease induction. B6129PF2, C57BL/6, CCR2+/−5+/− (control groups), and CCR2−/− mice were induced to develop active EAE and monitored for the development of clinical disease. The results shown in Fig. 2 demonstrate that control mice developed typical disease whereas CCR2−/− mice showed a significant reduction (P < 0.001) in acute EAE, with only one mouse showing very mild disease. Spinal cord sections from controls showed extensive meningial, perivascular, and parenchymal infiltrates, whereas sections from CCR2−/− mice showed no mononuclear cell infiltration (data not shown).
Figure 2.

CCR2-deficient mice do not develop clinical EAE. Mice were monitored for development of clinical disease. The data are expressed as the mean clinical disease score for all mice in each group as a function of days after immunization with disease incidence indicated in parentheses. Both mean clinical disease severity and disease incidence were significantly decreased in CCR2−/− animals relative to C57BL/6, B6129PF2, and CCR2+/−5+/− controls (P < 0.001) over the entire course of the experiment. The data shown are representative of four independent experiments.
Flow Cytometric Analysis of CNS-infiltrating Cells.
To discern any differences in infiltrating cell composition we performed flow cytometric analysis on CNS mononuclear cells. Spinal cord mononuclear cells were isolated from representative mice when control groups reached peak acute clinical disease. The immunophenotyping results are shown in Table and reveal that control mouse spinal cord infiltrate was composed of T cells (CD4+ and CD8+) and F4/80+ cells. Not only was there a decrease in the total number of CNS mononuclear cells in the CCR2−/− spinal cords, but there was also a decrease in the percentage of infiltrating T cells (<0.1%). Differential CD45 staining intensity coupled with F4/80 staining has been used to distinguish between infiltrating macrophages and resident microglia. Infiltrating macrophages are CD45highF4/80+, while resident microglial cells are CD45lowF4/80+ 18. Our results indicated that most of the total CNS mononuclear cells from the spinal cords of CCR2−/− mice were resident microglial cells, not infiltrating macrophages. In contrast, ∼50% of the total CNS mononuclear cells from the control mice were CD45 bright, corresponding to infiltrating macrophages.
Table 1.
Decreased Recruitment/Accumulation of T Cells and Infiltrating Monocytes in CCR2−/− Mice
| F4/80 | |||||||
|---|---|---|---|---|---|---|---|
| Disease | Cell recovery | CD4 | CD8 | CD19 | CD45high | CD45low | |
| Experiment 1 | |||||||
| C57BL/6 | Yes | 1.6 × 105 | 20 | 8 | <1 | 54 | 20 |
| B6129PF2 | Yes | 1.7 × 105 | 26 | 9 | <1 | 48 | 19 |
| CCR2−/− | No | 1.1 × 104 | <1 | <1 | <1 | 1 | 85 |
| Experiment 2 | |||||||
| Donor → recipient mice | |||||||
| C57BL/6 CD4+ T cells → C57BL/6 | Yes | 9.5 × 104 | 15 | 11 | 1 | 20 | 55 |
| C57BL/6 CD4+ T cells → CCR2−/− | No | 2.8 × 104 | 4 | 1 | 22 | 4 | 59 |
| CCR2−/− CD4+ T cells → C57BL/6 | Yes | 9.5 × 104 | 11 | 13 | 3 | 11 | 59 |
| CCR2−/− CD4+ T cells → CCR2−/− | No | 1.8 × 104 | 4 | 2 | 20 | 5 | 62 |
Peripheral Antigen Recall Immune Responses.
Disease protection in CCR2−/− mice could result from a defect in IFN-γ responses as described previously 19. When controls were showing peak clinical EAE, representative animals were killed and draining LN cells and splenocytes were stimulated in vitro with MOG35–55 to determine their ability to produce cytokines. The results in Fig. 3 indicate that there was no defect in CCR2−/− cells from draining LNs (A and B) or spleen (C and D) to produce IFN-γ or IL-2 compared with control groups. In addition to producing proinflammatory cytokines, CCR2−/− T cells showed MOG35–55 dose-dependent proliferation responses and no IL-4 expression when compared with all controls (data not shown).
Figure 3.

Lymphocytes from CCR2−/− mice produce proinflammatory cytokines upon antigen restimulation. B6129PF2, C57BL/6, and CCR2−/− mice were monitored for the development of clinical EAE. At the time B6129PF2 mice showed peak acute clinical disease, draining LN (A and B) and splenic cells (C and D) pooled from six representative mice for each group were restimulated with the immunizing peptide in vitro. Cultured supernatant was harvested at 48 h and measured for the production of IL-2 and IFN-γ by specific ELISA. CCR2−/− lymphocytes produced comparable levels of IFN-γ and IL-2 to control C57BL/6 lymphocytes in response to specific peptide challenge. The data shown are representative of three independent experiments.
CCR2−/− Adoptive Transfer Recipients Do Not Develop Clinical EAE.
To help understand disease protection in CCR2−/− mice we performed adoptive transfer EAE studies. The rationale behind this approach was to determine if CCR2 expression was necessary on disease-inducing CD4+ T cells or whether host-derived infiltrating T cells and/or monocytes required CCR2 expression for disease development. Sorted, antigen-restimulated CD4+ T cells from wild-type donors were transferred intravenously to either wild-type or CCR2−/− recipients, and sorted, antigen-restimulated CD4+ T cells from CCR2−/− donors were transferred intravenously to either wild-type or CCR2−/− recipients. The results of this experiment shown in Fig. 4 demonstrate that wild-type recipients of wild-type MOG-specific CD4+ T cells developed acute EAE whereas wild-type recipients of MOG-specific CD4+ T cells from CCR2−/− donors developed a significantly less severe acute EAE (P < 0.05). In contrast, CCR2−/− recipient mice, regardless of whether they received MOG-specific CD4+ T cells from wild-type or CCR2−/− donors, failed to develop EAE. These results suggest that encephalitogenic T cell expression of CCR2 is not a critical factor in development of EAE; rather, expression on host-derived cells is critical for disease induction.
Figure 4.

CCR2−/− recipients do not develop clinical EAE. Mice were monitored for the development of clinical disease after the adoptive transfer of MOG35–55-reactivated CD4+ T cells. The data are expressed as the mean clinical disease score for all mice in each group as a function of days after adoptive transfer with disease incidence in parentheses. Both mean clinical disease severity and disease incidence were significantly decreased in both CCR2−/− recipient groups compared with the C57BL/6 wild-type recipient group receiving CD4+ T cells from wild-type C57BL/6 donors by the Student's t test and X2 analysis over the entire course of the experiment. The C57BL/6 recipient mice receiving CCR2−/− CD4+ T cells was significantly reduced when compared with the wild-type C57BL/6 to C57BL/6 recipient group at the days marked (*; P < 0.05). The data shown are representative of two independent experiments.
CNS Mononuclear Cell Infiltration in Adoptive Transfer Recipients.
To determine the mechanism of disease inhibition seen in CCR2−/− recipients of either wild-type or CCR2−/− CD4+ T cells (Fig. 4), we examined CNS infiltrating mononuclear cells by flow cytometry. When wild-type recipients of wild-type CD4+ T cells showed peak clinical disease, the immunophenotype of the CNS infiltrating cells for all groups was determined. The results shown in Table demonstrate that spinal cord infiltrates from wild-type recipients of wild-type MOG-specific CD4+ T cells were composed of T cells, few B cells, CD45highF4/80+ monocytes, and CD45lowF4/80+ microglial cells. Spinal cord infiltrates from wild-type recipients of CCR2−/− MOG-specific CD4+ T cells exhibited a similar cellular composition. Both of these groups developed EAE (Fig. 4). There was a dramatic reduction in the presence of both CD4+ and CD8+ T cells and CD45highF4/80+ monocytes in both CCR2−/− recipient groups regardless of whether they received MOG-specific CD4+ T cells from CCR2−/− or wild-type donors (Table ). Both of these CCR2−/− recipient groups also failed to develop EAE. Interestingly, in the absence of a large T cell and monocyte infiltrate in the CCR2−/− recipients, the percentage of CD19+ B cells was dramatically increased. This is not an absolute increase in B cells as can be seen by the dramatic decrease in total cell recovery from the spinal cords of both CCR2−/− recipient groups; rather, it is most likely a relative increase due to the decrease in the presence of both T cells and monocytes. These results from this experiment demonstrate that CCR2−/− recipient mice contain very few CNS infiltrating mononuclear cells and that they are resident microglial cells (CD45lowF4/80+; Table ). For a comparison, when the CNS of normal wild-type mice that have never been immunized with MOG35–55 or never received MOG35–55-specific T cells was analyzed for the presence of mononuclear cells by flow cytometry, only CD45lowF4/80+ microglia were found (data not shown). Collectively, these data demonstrate that CCR2 expression is a critical factor for EAE induction by regulating the accumulation of host-derived CNS mononuclear cells.
At least five distinct possibilities exist to potentially explain the current findings. (a) T cells from CCR2−/− mice lack appropriate adhesion molecules known to be important in initial migration from blood to CNS. We found that VLA-4 5 and CD44 20 were expressed on activated T cells in the CCR2−/− mice (data not shown) and therefore considered this possibility unlikely. (b) There was an altered Th1 phenotype in the CCR2−/− mice 19 which contributed to the lack of disease induction. Our findings indicated that CCR2−/− mice mounted a similar MOG35–55-specific Th1 response as measured by IFN-γ expression compared with the control groups. (c) CCR2 expression on T cells was required for disease development, presumably by regulating migration to the CNS. To address this possibility we performed adoptive transfer EAE induction and demonstrated that wild-type recipients of MOG35–55-specific CCR2−/− T cells developed EAE (Fig. 4) and had significant numbers of T cells in the CNS when compared with controls (Table ). Therefore, it does not appear likely that CCR2 expression on MOG35–55-specific, disease-inducing T cells is necessary for EAE development. (d) CCR2 expression is required on monocytes/macrophages for CNS migration and/or accumulation. We favor this hypothesis based on our results demonstrating that CCR2−/− recipients of MOG35–55-specific wild-type CD4+ T cells fail to develop EAE (Fig. 4) as well as fail to show appreciable monocyte (CD45highF4/80+) accumulation in the CNS compared with the positive control (Table ). From these results, however, we are unable to rule out the possibility that CCR2 expression is required on host-derived T cells for EAE development. We do not think that host-derived T cells are required for EAE since disease can be adoptively induced in SCID mice lacking bystander T cells 21. (e) There was a deficiency in CNS microglia and/or an inability of these cells to migrate within the CNS during disease development. Similar numbers of microglia (CD45lowF4/80+) were found in wild-type and CCR2−/− mice, indicating that there is no deficiency in the numbers of microglia (Table ). However, our results cannot rule out the possibility that microglia in CCR2−/− mice have a defect in their ability to migrate within the CNS and contribute to EAE pathogenesis.
Several other inflammatory models have recently identified critical roles for CCR2 expression on a variety of cell types, including monocytes. CCR2−/− mice have impaired monocytic responses, including adhesion, trafficking, and granuloma formation 22 23. CCR2−/− mice were unable to clear infection by Listeria monocytogenes, suggesting a defect in the macrophage-mediated clearance mechanism 24. CCR2 was also shown to be important in a mouse model of hyperreactive airway disease mediated through MCP-1 25. Furthermore, CCR2 mice have reduced atherosclerosis due to a defect in monocyte migration 26.
Our present results lead us to postulate a model for chemokines, especially CCL2, and its receptor, CCR2, in pathogenesis of CNS autoimmune demyelinating disease. Initial activated CD4+ T cell CNS entry is mediated by interactions of VLA-4 and VCAM-1, and perhaps constitutively expressed chemokines such as fractalkine (CX3CL1 27). Once lymphocytes are in the perivascular space, they presumably recognize antigen presented by local APCs, leading to increased chemokine and inflammatory cytokine expression by T cells, and resulting in further induction of chemokine expression by glial cells. Indeed, astrocytes have been shown to express CCL2 and may participate in the recruitment of CCR2+ monocytes during disease development 28. However, in the absence of CCR2 expression, monocytes would fail to accumulate in the CNS in response to CCL2 expression despite the presence of T cells and sufficient numbers would not be present to serve as additional sources of chemokines and/or end stage effector cells involved in tissue damage, demyelination, and subsequent clinical disease. Our data demonstrate an important role for specific chemokine receptor expression and in vivo cellular recruitment during the EAE pathogenesis and open the possibility of using CCR2 receptor antagonists for tissue specific autoimmune disease therapy.
Acknowledgments
This work was supported by National Institutes of Health Training Grant AI07476 (B.T. Fife) and National Institutes of Health Grant NS34510 (W.J. Karpus).
References
- Hohlfeld R. Biotechnological agents for the immunotherapy of multiple sclerosis. Principles, problems, and perspectives. Brain. 1997;120:865–916. doi: 10.1093/brain/120.5.865. [DOI] [PubMed] [Google Scholar]
- Krakowski M.L., Owens T. The central nervous system environment controls effector CD4+ T cell cytokine profile in experimental allergic encephalomyelitis. Eur. J. Immunol. 1997;27:2840–2847. doi: 10.1002/eji.1830271115. [DOI] [PubMed] [Google Scholar]
- Hickey W.F., Hsu B.L., Kimura H. T-lymphocyte entry into the central nervous system. J. Neurosci. Res. 1991;28:254–260. doi: 10.1002/jnr.490280213. [DOI] [PubMed] [Google Scholar]
- Cross A.H., Cannella B., Brosnan C.F., Raine C.S. Homing to central nervous system vasculature by antigen-specific lymphocytes. I. Localization of 14C-labeled cells during acute, chronic, and relapsing experimental allergic encephalomyelitis. Lab. Invest. 1990;63:162–170. [PubMed] [Google Scholar]
- Yednock T.A., Cannon C., Fritz L.C., Sanchez-Madrid F., Steinman L., Karin N. Prevention of experimental autoimmune encephalomyelitis by antibodies against α4β1 integrin. Nature. 1992;356:63–66. doi: 10.1038/356063a0. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y., Kawai K., Honda H., Tomida S., Niimi N., Tamatani T., Miyasaka M., Yoshikai Y. Antibodies against leukocyte function-associated antigen- 1 and against intercellular adhesion molecule-1 together suppress the progression of experimental allergic encephalomyelitis. Cell. Immunol. 1995;164:295–305. doi: 10.1006/cimm.1995.1173. [DOI] [PubMed] [Google Scholar]
- Morrissey S.P., Deichmann R., Syha J., Simonis C., Zettl U., Archelos J.J., Jung S., Stodal H., Lassmann H., Toyka K.V. Partial inhibition of AT-EAE by an antibody to ICAM-1clinico-histological and MRI studies. J. Neuroimmunol. 1996;69:85–93. doi: 10.1016/0165-5728(96)00064-1. [DOI] [PubMed] [Google Scholar]
- Murphy P.M., Baggiolini M., Charo I.F., Hebert C.A., Horuk R., Matsushima K., Miller L.H., Oppenheim J.J., Power C.A. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol. Rev. 2000;52:145–176. [PubMed] [Google Scholar]
- Karpus W.J., Ransohoff R.M. Chemokine regulation of experimental autoimmune encephalomyelitistemporal and spatial expression patterns govern disease pathogenesis. J. Immunol. 1998;161:2667–2671. [PubMed] [Google Scholar]
- Ward S.G., Bacon K., Westwick J. Chemokines and T lymphocytesmore than an attraction. Immunity. 1998;9:1–11. doi: 10.1016/s1074-7613(00)80583-x. [DOI] [PubMed] [Google Scholar]
- Kennedy K.J., Strieter R.M., Kunkel S.L., Lukacs N.W., Karpus W.J. Acute and relapsing experimental autoimmune encephalomyelitis are regulated by differential expression of the CC chemokines macrophage inflammatory protein-1α and monocyte chemotactic protein-1. J. Neuroimmunol. 1998;92:98–108. doi: 10.1016/s0165-5728(98)00187-8. [DOI] [PubMed] [Google Scholar]
- Sato N., Kuziel W.A., Melby P.C., Reddick R.L., Kostecki V., Zhao W., Maeda N., Ahuja S.K., Ahuja S.S. Defects in the generation of IFN-gamma are overcome to control infection with Leishmania donovani in CC chemokine receptor (CCR) 5-, macrophage inflammatory protein-1 alpha-, or CCR2-deficient mice. J. Immunol. 1999;163:5519–5525. [PubMed] [Google Scholar]
- Karpus W.J., Peterson J.D., Miller S.D. Anergy in vivodown-regulation of antigen-specific CD4+ Th1 but not Th2 cytokine responses. Int. Immunol. 1994;6:721–730. doi: 10.1093/intimm/6.5.721. [DOI] [PubMed] [Google Scholar]
- Hoffman L.M., Fife B.T., Begolka W.S., Miller S.D., Karpus W.J. Central nervous system chemokine expression during Theiler's virus-induced demyelinating disease. J. Neurovirol. 1999;5:635–642. doi: 10.3109/13550289909021292. [DOI] [PubMed] [Google Scholar]
- Tanabe S., Heesen M., Berman M.A., Fischer M.B., Yoshizawa I., Luo Y., Dorf M.E. Murine astrocytes express a functional chemokine receptor. J. Neurosci. 1997;17:6522–6528. doi: 10.1523/JNEUROSCI.17-17-06522.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlotnik A., Yoshie O. Chemokinesa new classification system and their role in immunity. Immunity. 2000;12:121–127. doi: 10.1016/s1074-7613(00)80165-x. [DOI] [PubMed] [Google Scholar]
- Kurihara T., Bravo R. Cloning and functional expression of mCCR2, a murine receptor for the C-C chemokines JE and FIC. J. Biol. Chem. 1996;271:11603–11607. doi: 10.1074/jbc.271.20.11603. [DOI] [PubMed] [Google Scholar]
- Ford A.L., Goodsall A.L., Hickey W.F., Sedgwick J.D. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J. Immunol. 1995;154:4309–4321. [PubMed] [Google Scholar]
- Boring L., Gosling J., Chensue S.W., Kunkel S.L., Farese R.V.J., Broxmeyer H.E., Charo I.F. Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J. Clin. Invest. 1997;100:2552–2561. doi: 10.1172/JCI119798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocke S., Piercy C., Steinman L., Weissman I.L., Veromaa T. Antibodies to CD44 and integrin alpha4, but not L-selectin, prevent central nervous system inflammation and experimental encephalomyelitis by blocking secondary leukocyte recruitment. Proc. Natl. Acad. Sci. USA. 1999;96:6896–6901. doi: 10.1073/pnas.96.12.6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones R.E., Bourdette D.N., Whitham R.H., Offner H., Vandenbark A.A. Induction of experimental autoimmune encephalomyelitis in severe combined immunodeficient mice reconstituted with allogeneic or xenogeneic hematopoietic cells. J. Immunol. 1993;150:4620–4629. [PubMed] [Google Scholar]
- Kuziel W.A., Morgan S.J., Dawson T.C., Griffin S., Smithies O., Ley K., Maeda N. Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc. Natl. Acad. Sci. USA. 1997;94:12053–12058. doi: 10.1073/pnas.94.22.12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warmington K.S., Boring L., Ruth J.H., Sonstein J., Hogaboam C.M., Curtis J.L., Kunkel S.L., Charo I.R., Chensue S.W. Effect of C-C chemokine receptor 2 (CCR2) knockout on type-2 (Schistosomal antigen-elicited) pulmonary granuloma formationanalysis of cellular recruitment and cytokine responses. Am. J. Pathol. 1999;154:1407–1416. doi: 10.1016/S0002-9440(10)65394-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara T., Warr G., Loy J., Bravo R. Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J. Exp. Med. 1997;186:1757–1762. doi: 10.1084/jem.186.10.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell E.M., Charo I.F., Kunkel S.L., Strieter R.M., Boring L., Gosling J., Lukacs N.W. Monocyte chemoattractant protein-1 mediates cockroach allergen-induced bronchial hyperreactivity in normal but not CCR2−/− micethe role of mast cells. J. Immunol. 1999;163:2160–2167. [PubMed] [Google Scholar]
- Dawson T.C., Kuziel W.A., Osahar T.A., Maeda N. Absence of CC chemokine receptor-2 reduces atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis. 1999;143:205–211. doi: 10.1016/s0021-9150(98)00318-9. [DOI] [PubMed] [Google Scholar]
- Bazan J.F., Bacon K.B., Hardiman G., Wang W., Soo K., Rossi D., Greaves D.R., Zlotnik A., Schall T.J. A new class of membrane-bound chemokine with a CX3C motif. Nature. 1997;385:640–644. doi: 10.1038/385640a0. [DOI] [PubMed] [Google Scholar]
- Ransohoff R.M., Hamilton T.A., Tani M., Stoler M.H., Shick H.E., Major J.A., Estes M.L., Thomas D.M., Tuohy V.K. Astrocyte expression of mRNA encoding cytokines IP-10 and JE/MCP-1 in experimental autoimmune encephalomyelitis. FASEB (Fed. Am. Soc. Exp. Biol.) J. 1993;7:592–600. doi: 10.1096/fasebj.7.6.8472896. [DOI] [PubMed] [Google Scholar]