

Abstract
Vitamin A and its biologically active derivatives, the retinoids, are recognized as key regulators of vertebrate development, cell growth, and differentiation. Although nuclear receptors have held the attention since their discovery a decade ago, we report here on serine/threonine kinases as a new class of retinoid receptors. The conserved cysteine-rich domain of the NH2-terminal regulatory domains of cRaf-1, as well as several select domains of the mammalian protein kinase C (PKC) isoforms α, δ, ζ, and μ, the Drosophila and yeast PKCs, were found to bind retinol with nanomolar affinity. The biological significance was revealed in the alternate redox activation pathway of these kinases. Retinol served as a cofactor to augment the activation of both cRaf and PKCα by reactive oxygen, whereas the classical receptor-mediated pathway was unaffected by the presence or absence of retinol. We propose that bound retinol, owing to its electron transfer capacity, functions as a tag to enable the efficient and directed redox activation of the cRaf and PKC families of kinases.
Keywords: vitamin A, redox regulation, protein kinase C, Raf kinase, retinoid receptors
Introduction
In the World Health Organization's estimate, vitamin A deficiency causes blindness in 1.5 million children and clinical xerophthalmia in a 10-fold larger cohort. Both groups suffer from immune dysfunction, commonly associated with vitamin A deficiency, which puts these children at high risk for their health and normal development 1. The adverse consequences of vitamin A deficiency on immune system development were first described over 70 years ago 2. The underlying mechanism was studied extensively in the following decades 3 but has remained hidden. As retinoic acid was not capable of completely reversing the immune deficiency, the involvement of other vitamin A metabolites has been postulated. In the 1990s, while studying an intro model of vitamin A deficiency 4, we discovered 14-hydroxy-retro-retinol (14HRR) and 13,14-dihydroxy-retinol (DHR) as the likely mediators of normal immune function, since both compounds, but not retinoic acid, substituted for vitamin A as regulators of growth and survival of lymphocytes 5 6. A strong lead on the mechanism of action emerged with the realization that lack of vitamin A led to programmed cell death in numerous tissues. Likewise, the action of anhydroretinol proved antagonistic to the survival-promoting potential of 14HRR and retinol, by causing apoptosis 7. Because apoptosis occurred unusually promptly and without apparent nuclear involvement, and because known retinoic acid receptors 8 9 neither bound nor responded to 14HHR, a new class of receptors of cytoplasmic vitamin A and 14HRR was postulated 10. Results from experiments with kinase blocking drugs further narrowed the choices and pointed to signaling molecules as direct targets of retinoid action 10. When finally identified, the surprise was that the receptors comprised numerous members of well-known signaling molecules, notably the serine/threonine kinases, protein kinase C (PKC) and cRaf, as detailed in this report.
The family of serine/threonine kinases functions as a second tier of signal integration and amplification, connecting receptor kinases to various effector pathways, including transcription, cell cycle control, cell survival, cell movement, and structural remodeling. The serine/threonine kinases exist in inactive form as globular proteins in the cytoplasm in complex with several regulatory proteins. The cRaf signaling module comprises heat shock proteins, immunophilins 11, and 14-3-3 12. The activation mechanism of cRaf is not fully understood, but includes conventional phosphorylation by upstream kinases, translocation from cytosol to membranes, and mandatory interaction with GTP/ras 13. PKC follows a different paradigm that involves binding of a combination of different lipid second messengers and, depending on isoform, also Ca2+ 14. In addition to phosphorylation of select amino acids, the conversion, like cRaf, of cytoplasmic to membrane protein takes place.
Oxidative activation has been identified as an alternative pathway. Both cRaf and several PKC isoforms have been found to respond rapidly to reactive oxygen species (ROS) in vivo and in vitro 15 16. This milestone discovery is apt to lead to an understanding of how changes in the redox microenvironment are sensed by the signaling apparatus and converted to biochemical action. The chain of molecular events leading to enzyme activation is unclear. Konishi et al. reported that tyrosine phosphorylation in the catalytic domain of PKC was a prerequisite for activation by H2O2 16. The question of whether oxidative activation is by direct action and, if so, which structures in the regulatory and/or catalytic domains of PKC and Raf are modified by redox reaction remains unanswered. The presence of conserved cysteine-rich, zinc-stabilized structures in the regulatory domains of cRaf and PKC 17 18 19 invites the hypothesis that the initial attack is on the sulfhydryl groups. By coincidence, our laboratory has found that the zinc finger (ZiF) domains serve as high affinity binding sites for retinoids, as reported below. Furthermore, we demonstrate that retinol serves as cofactor to facilitate the redox activation of both Raf and PKC kinases. Because retinol binds to the cysteine-rich domain, the focus of attention is on this structural element. The ZiF domains of PKC and Raf not only function as critical regulatory structures in the classical hormone receptor–mediated pathways, but they also appear of similar importance in the redox activation pathway. Although the actual chemical and structural modifications of the mammalian ZiF domains remain to be solved, a fitting precedent exists in the prokaryotic chaperone, heat shock protein (Hsp)33, where a related ZiF domain has been demonstrated to function as a redox switch 20—in the absence of retinoids. Oxidizing conditions have been demonstrated to cause cysteine residues of the bacterial ZiF domain to form disulfide bonds and consequently to activate the chaperone function. This process was reversible by reduction with glutathione.
Our work extends the range of actions of retinoids from the well-known effects in gene transcription 8 9 and vision 21, mediated by retinoic acids and retinaldehyde, respectively, to regulation of signal transduction. As stated above, the broadening of the field has long been predicted since the immune deficiency and male sterility resulting from experimental vitamin A deficiency in laboratory animals are never completely reversed by retinoic acid alone. A third branch of vitamin A action is exemplified by the redox regulation of serine/threonine kinases by retinol and its hydroxylated metabolites, 14HRR and DHR 5 6. Since the capacity to take up vitamin A and convert it to 14HRR goes beyond immune cells, and includes essentially all nucleated cells, and since all cells utilize PKC and cRaf in central signal pathways, this new paradigm is likely to play a fundamental role in signal transduction.
Materials and Methods
Immunological Reagents
Rabbit antibody to the cRaf C20-terminal peptide was purchased from Santa Cruz Biotechnology, Inc. Antibodies to phosphorylated and nonphosphorylated extracellular signal–regulated kinase (ERK)1 and 2 were from New England Biolabs, Inc. Anti-Flag mAb M2 was from Eastman Kodak Co., and agarose-conjugated M2 was from Sigma-Aldrich. mAb to PKCα was from Transduction Laboratories.
Retinoids
All-trans isomers of anhydroretinol (AR), 14HRR, and DHR were synthesized as described previously 6 7 22. Trans-retinol was purchased from Sigma-Aldrich and purified by high pressure liquid chromatography on a C18 column. 3H-retinol was purchased from DuPont/NEN Life Science Products.
Glutathione S-Transferase Fusion Proteins: Vector Construction, Protein Expression, and Purification
The following cDNA fragments were used, encoding cysteine-rich domains of human cRaf-1 with different lengths, corresponding to amino acids 136–186 (devoid of tryptophan [trp]), 136–195 with trp at position 187), and 4–256; the Drosophila PKC C1A domain (amino acids 40–101) and Drosophila C1B (amino acids 105–261 plus a trp residue at position 262); kinase suppressor of ras (KSR; amino acids 333–378); and Vav (amino acids 464–518, with an added trp residue at position 519). The cDNA fragments were generated by PCR 23 24. They were cloned into the BamH1-Sma1 or the BamH1-EcoR1 sites of pGEX2T (Amersham Pharmacia Biotech). The glutathione S-transferase (Gst) fusion proteins were expressed in the BL21/DE3 strain of Escherichia coli (Novagen). The growth conditions were as follows: the bacteria were initially grown at 37°C to an optical density at 600 nm of 0.5, then transferred to room temperature. When the optical density reached 0.7–0.8, protein synthesis was induced by isopropyl-thio-β-d-galactopyranoside, and the cells were harvested 2 h later. Bacteria were lysed by passing twice through a French press, and the proteins were purified by affinity chromatography on the glutathione-agarose matrix (Sigma-Aldrich) according to a standard protocol. Purity by Coomassie blue staining of SDS polyacrylamide gels was >90%.
Construction of Flag-cRaf ZiF.
The cRaf cys domain was cloned by PCR using the primers 5′-GAT TTC CTG GAT AAT TCC CTC ACA ACA CAC and 3′-ACC AAT AGT GGA ATT GGA TCC CAA TAA CAG TTG TCT. The product was inserted into the EcoR1-BamH1 sites of the Sigma-Aldrich vector pFlag-CMV-2. Fidelity was confirmed by sequencing.
Binding Assays.
Gst fusion proteins were dissolved in PBS. The latter was purged of oxygen by sparging with helium for 15 min. Fluorescence excitation and emission spectra of protein–retinoid complexes, as well as quantitative fluorescence measurements with retinoid titrations, were carried out in a JASCO Spectrofluorimeter (model FP777). Protein concentrations were at 250 nM, and retinoids were added to proteins from 25-μM stock solutions in methanol. All measurements were repeated two to four times. Binding constants were calculated by nonlinear curve fitting according to the theorem by Norris et al. 25.
In Vivo Binding Assay.
COS-7 cells were transfected by the calcium phosphate method with DNA encoding Flag-tagged full length cRaf or the Raf-ZiF domain fragment. Transfected and control cultures were starved of retinoids for 2 d, then incubated with 5 nM 3H-retinol for 30 min. To assess binding specificity, labeled cells were preincubated with indicated concentrations of cold all-trans retinol. Because at 1 μM and above retinol forms micelles, these concentrations cannot be evaluated in binding competition assays. Extracts were prepared by repeated freeze–thawing, and cRAf proteins were isolated by immunoprecipitation. Immunoprecipitates were washed extensively with saline containing 0.5 M NaCl and 0.5% bovine albumin, and bound radioactivity was measured. Results were expressed as differentials of immunoprecipitates between transfected and nontransfected cultures.
Cell Culture Experiments
Retinol Starvation and Reconstitution.
NIH 3T3 cells were grown in 60-mm dishes in MEM high glucose medium supplemented with 7% fetal bovine serum (FBS). Upon reaching half confluence, the medium was replaced by MEM supplemented with 2 μM BSA, 2 μM linoleic acid, and 70 nM transferrin and culturing was continued for 4–5 d. To replenish retinoids, sets of cultures received 2 μM all-trans retinol 12 h before assaying, as specified in the experiments.
Stimulation of 3T3 Cells by Reactive Oxygen.
To distinguish cRaf effects from concomitant PKC effects, cultures of 3T3 cells, depleted of retinol or repleted with retinol, were blocked by culturing in 25 nM staurosporine for 40 min before activation by ROS. Next, H2O2 was added at concentrations between 300 and 10 μM, and culturing was continued for specified periods of time. For harvesting, cells were washed with ice-cold PBS and snap-frozen in 75 μl extraction buffer (buffer A: 50 mM Tris-HCl, 100 mM NaCl, 1 mM Na3VO4, 100 g/ml PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin). To obtain whole cell lysates, 1% NP-40 was added, and the cells were thawed, incubated on ice for 30 min, and centrifuged for 30 min at 32,500 g. Protein concentrations were determined by the Bio-Rad Protein Assay kit (Bio-Rad Laboratories).
Western Blots.
Equal quantities of total cell extracts, cytosolic proteins, or residual proteins were separated on 7.5% SDS-PAGE gels, transferred to polyvinylidene difluoride membranes, immunostained by standard methods, and detected on x-ray films by enzyme-coupled chemiluminescence (New England Nuclear Life Science Products). The results of Western blots were quantified densitometrically by PhosphorImager® (Molecular Dynamics).
Activation by UVB.
COS cells transfected with the pCMV-Raf-FLAG vector, donated by Dr. R. Davis (University of Massachusetts, Worcester, MA), were grown in serum-free DME without phenol red for 2 d. Cells treated with retinoids and/or oxygen scavengers as specified in the experiments were irradiated with UVB at 400 μw/cm2. Cultures were incubated at 37°C for the indicated time periods and harvested (see below).
Enzyme Assays.
Endogenous cRaf/mitogen-activated protein kinase (MAPK) activity was detected by Western blot with the phospho-ERK–specific antibody kit as described by the manufacturer (New England Biolabs, Inc.). To ensure equal loading of cRaf, the transfer membrane was cut in two, and the segment containing proteins >65 kD was immunostained for cRaf protein and the segment <65 kD was stained for phospho-ERK. The latter was then stripped and reprobed with antibody specific for nonphosphorylated ERK. Films were scanned in the PhosphorImager®, and the results of MAPK activity were corrected for the amounts of ERK protein present in each sample.
PKC Kinase Assay.
Detergent lysates of 3T3 cells depleted of retinol, and repleted for 12 h with 2 μM retinol as described above and activated by H2O2 without blocking with staurosporine, were immunoprecipitated with PKCα antibody (Signal Transduction Laboratories). Kinase activity was determined in the washed immunoprecipitates with H1 histone as substrate, as described by Konishi et al. 16.
Raf Immunoprecipitation/Kinase Assay.
COS-7 cells were transfected by the calcium phosphate method in 60-mm dishes. Cells were lysed using 100 μl lysis buffer: 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 2mM EDTA, 1mM EGTA, 1% Triton X-100, 25 μg/ml each leupeptin and aprotinin, 1 mM PMSF, 1 mM Na3VO4, 30 mM NaF, and 30 mM β-glycerophosphate. The lysates were precleared with 30 μl of a 50:50 protein G–agarose slurry, and the Flag-Raf protein was precipitated using 30 μl of anti-Flag M2 affinity gel (Sigma-Aldrich). The immunoprecipitates were washed four times with lysis buffer containing 0.5 M NaCl, and twice with kinase buffer (35 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 0.5 mM EGTA, and 1 mM Na3VO4. The kinase reaction was performed in 20 μl kinase buffer using 200 ng of kinase-disabled His-MEK(K97M) (provided by Dr. Kolesnick, Memorial Sloan-Kettering Cancer Center, New York, NY) as substrate, 60 μM ATP, and 10 μCi [γ-32P]ATP (6,000 Ci/mmol). The reaction was carried out for 20 min at 30°C, and terminated by the addition of 10 μl 5× Laemmli buffer.
Results
The human cRaf-1 fragments spanning amino acids 4–256, or 136–195, comprising the 50–amino acid ZiF domain 18 HNFARKTFLKLAFCDICQKFLLNGFRCQTCGYKFHEHCSTKVPTMC, corresponding to the consensus sequence HX12CX2CXnCX2CX4HX2CX7C (H = histidine, C = cysteine, X = any amino acid [including a natural trp residue at position 187]), were fused to the Gst gene and expressed in E. coli. Expressed proteins, referred to below as Gst-Raf ZiF, were purified by affinity chromatography to >90% purity and tested for the ability to bind retinol. Studies of the interaction of retinol with proteins is often confounded by the hydrophobic nature of this ligand, which renders standard binding assays unsuitable for quantitative measurements. In contrast, fluorescence-based methodologies allow monitoring of the binding of retinol in protein binding sites without the need for physical separation of bound and free species. Thus, four fluorescence-based methods were used to investigate the characteristics of the interactions of retinol with the ZiF domains of cRaf and PKC: quenching of intrinsic protein (trp) fluorescence; resonance energy transfer of intrinsic protein fluorescence (FRET); enhancement of retinol fluorescence; and expression of vibronic fine structure.
Quenching of the Intrinsic Fluorescence of the Protein upon Ligand Binding.
Because the absorption spectrum of retinol significantly overlaps with the emission spectrum of tryptophan and tyrosine residues, binding of retinol is often accompanied by quenching of the intrinsic protein fluorescence 26. The data in Fig. 1 show that binding of a stoichiometric amount of retinol to the ZiF domain of cRaf indeed resulted in a decrease of the protein fluorescence, and this was accompanied by FRET as evidenced by a small, but distinctive, retinol fluorescence with the characteristic emission maximum at 466 nm. To determine the binding affinity, retinol was added in increments, and the protein fluorescence emission was measured after each addition. Plotting the fluorescence intensity versus retinol concentration yielded, after correction for inner filtering, clear evidence for saturable binding with a calculated affinity of 22 ± 3.5 nM (Fig. 2). Titrations of retinol were also evaluated by FRET (Fig. 3). Using Gst-Raf Zif136–195, protein-saturable binding was observed and an affinity constant of 25.8 nM was computed. A truncated protein, Gst-Raf Zif136–186, which lacked the trp residue at position 187 crucial for protein fluorescence, did not elicit a FRET signal, although by fluorescence enhancement (see below) this protein bound retinol with similar affinity.
Figure 1.
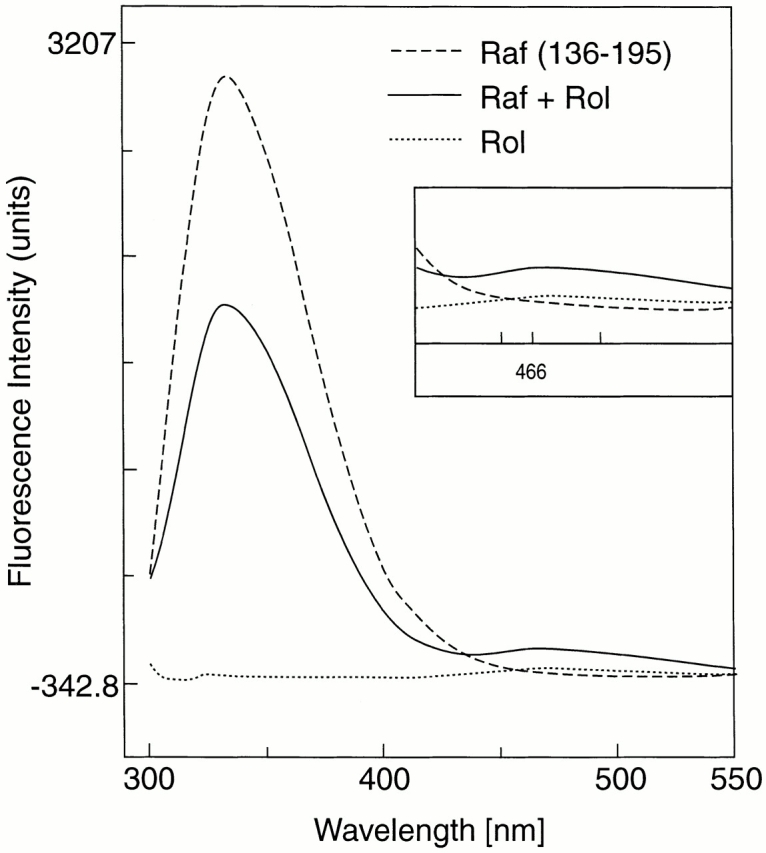
Quenching of protein fluorescence and resonance energy transfer. Fluorescence emission spectra of 250-nM solution of Gst fusion protein comprising amino acids 136–195 of human cRaf-1, excited at 280 nm in the presence (solid line) or absence (dashed line) of 250 nM all-trans retinol (Rol) and spectrum of retinol in buffer (dotted line). A fluorescence peak at 466 nm, although small, indicated energy transfer (inset). The naturally occurring trp residue at position 187 may not be optimally placed for efficient energy transfer.
Figure 2.

Determination of binding constant by fluorescence quench method. The 500-nM solution of Gst-Raf ZiF136–195 fusion protein in PBS was titrated with all-trans retinol added in 25-nM increments (⋄). Fluorescence intensity values (excitation at 280 nm; emission at 330 nm) corrected for inner filtering were plotted versus retinol concentration. Affinity constants were computed according to Norris et al. (reference 25). Shown are the vehicle control (□), a titration of retinol in tryptophanamide solution (•), and Gst protein (▵).
Figure 3.
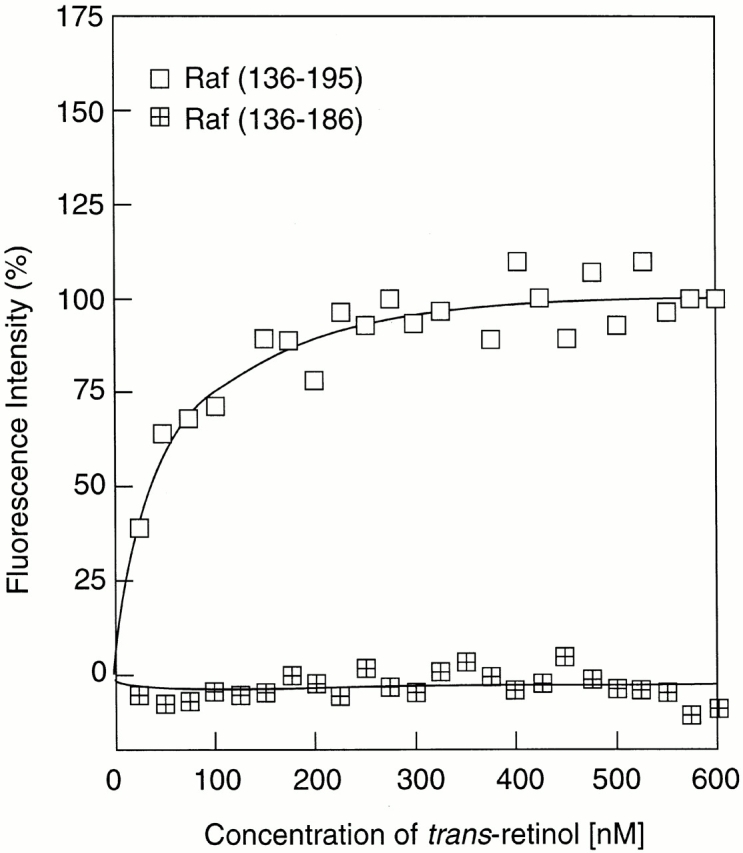
Determination of binding constant by fluorescence resonance energy transfer. All-trans retinol was added in 25-nM increments to a 500-nM solution of the Gst fusion protein comprising the cRaf fragment 136–195, containing a natural trp residue at position 187 (open squares), or to a truncated fragment 136–186 (crossed squares).The protein was excited at 280 nm, and the emission of retinol monitored at λ max466 nm. The binding constant of the former protein was calculated according to Norris et al. (reference 25), whereas the latter did not elicit FRET.
The binding of retinoid was dependent on the conformation of the protein, as shown for Gst-Raf Zif4–256, since denaturation by guanidine hydrochloride abolished binding (14HRR and retinol behaved similarly). Renaturation of the protein restored saturable binding (Fig. 4).
Figure 4.

Dependence of retinoid binding on protein conformation. The 1-μM solution of Gst-Raf4–256 was titrated with 14HRR and fluorescence quenching was monitored (excitation at 280 nm; emission at 330 nm). The native protein (▪) showed saturable binding, whereas binding was lost after denaturation with guanidine hydrochloride (•). Binding was restored after renaturation (□).
cRaf is not unique in its ability to bind retinol, as a similar result was obtained with Gst fusion proteins comprising the first ZiF domain, C1A, of PKCα, or the C1B ZiF domains of PKCδ, or both human and mouse KSR, but not that of human Vav (data not shown). With the Drosophila and yeast PKC C1A ZiF Gst fusion proteins, both quenching and FRET were pronounced, especially when retinol was added in threefold molar excess (data not shown). Titrations of retinol yielded saturable binding at nanomolar affinity with stoichiometry consistent with a single binding site. The affinity constants based on a minimum of four independent titrations are summarized in Table .
Table 1.
Binding Constants of Raf and PKC Cys Domain for Retinol
| ZiF domain | Amino acids | Binding constant | |
|---|---|---|---|
| nM ± SE | |||
| Human cRaf-1 | C1 | 4–271 | 21.3 ± 3.0 |
| Human cRaf-1 | C1 | 136–195 | 22.0 ± 3.5 |
| Human PKCα | C1A | 37–87 | 20.7 ± 4 |
| Human PKCα | C1B | w100–151 | >500 |
| Human PKCδ | C1A | 159–209 | 95.2 ± 1.3 |
| Human PKCδ | C1B | 231–280 | 67.3 ± 1.3 |
| Human PKCμ | C1A | 34–290 | 18.1 ± 8 |
| Human PKCμ | C1B | w263–328 | 17.3 ± 4 |
| Human PKCζ | C1 | 123–182 | 34.1 ± 6.7 |
| Drosophila PKC | C1A | 42–102 | 18.5 ± 3.7 |
| Drosophila PKC | C1B | 107–161w | >500 |
| S. cerevisiae PKC | C1A | w415–461 | 17.9 ± 5.1 |
| S. cerevisiae PKC | C1B | 476–531 | >500 |
| Human Vav | C1 | 464–518w | >500 |
Summary of binding constants. Recombinant Gst fusion proteins encompassing the indicated ZiF domains of cRaf and various PKC isoforms were obtained at >90% purity. Where no endogenous one existed, a trp residue (w) was engineered at the position indicated. Binding constants and SE covering at least four independent measurements were calculated from fluorescence quench curves of tryptophan emission as shown in Fig. 2. Values obtained by fluorescence enhancement were in close agreement with values obtained by the trp quench method.
Not All ZiF Domains Bind Retinol.
By the criterium of fluorescence quenching, the C1B domains of human PKCα, Drosophila and yeast PKC, and the Vav ZiF domain, as mentioned, do not bind retinol with appreciable affinity, indicating specificity on the part of the protein. To further test for binding specificity, several unrelated signal transduction proteins were investigated, including ras, Hsp90, 14-3-3, immunophilin p50, and the pleckstrin homology domain of PKCμ 27, all expressed as Gst fusion proteins, as well as Gst protein itself. None of these showed quenching of the protein emission spectrum, suggesting, but not proving, that retinol does not interact with these proteins (Fig. 2, and data not shown).
Fluorescence Enhancement Assays.
Binding to the Raf ZiF fusion proteins was confirmed independently by enhancement of the retinol fluorescence emission spectrum excited at λmax 325 nm. Increased quantum yield results from movement of the ligand, retinol, from polar solvent to nonpolar environment of the binding pocket. This is shown qualitatively for PKCα C1A and PKCδ C1B (Fig. 5), as well as quantitatively for Gst-Raf ZiF136–195 and the trp-minus Raf ZiF136–186 fragment (Fig. 6). Using titrations of retinol, binding was found to be saturable. The calculated binding constant was comparable to that obtained from protein quenching experiments. Fluorescence enhancement measurements were extended to the mammalian, Drosophila, and yeast PKC ZiF domains, yielding very similar results to the results of quench assays (see Table ).
Figure 5.
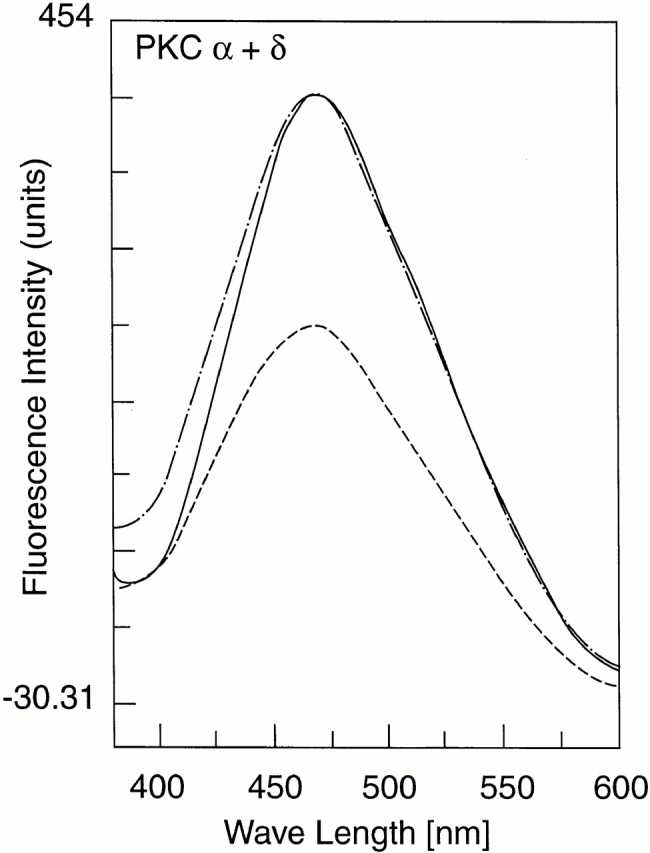
Enhancement of fluorescence emission of bound retinol. The fluorescence emission spectrum (excitation at 325 nm) of a 250-nM all-trans retinol solution is shown in PBS (dashed line), and in stoichiometric mixtures with Gst PKCα C1A (solid line) or Gst PKCδ C1B fusion proteins (broken line).
Figure 6.

Determination of binding constants by fluorescence enhancement. The Gst-Raf136–195 (□) and Gst-Raf136–186 (•) fusion protein solutions (250 nM) was titrated with all trans-retinol added in 25-nM increments. Retinol was excited at 325 nm and the fluorescence intensity was monitored at 466 nm. Binding constants were computed according to Norris et al. (reference 25).
Vibronic Fine Structure.
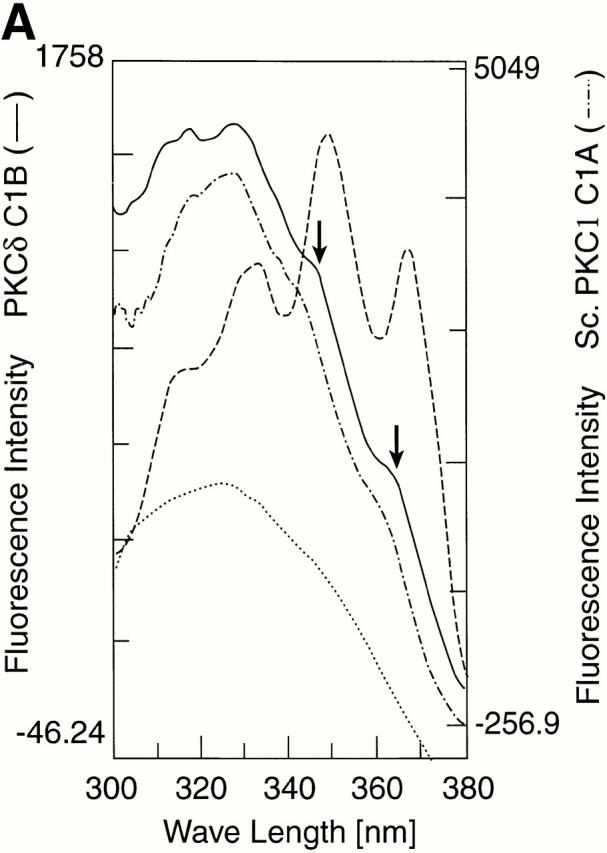
Ligation of retinol by proteins has been shown to impose a planar conformation on the ligand, resulting in a red shift in the retinol fluorescence excitation spectrum and expression of vibronic fine structure. It is known, for example, that the completely enclosed, barrel-shaped binding pocket of cellular retinol binding protein induces a profound red shift and vibronic fine structure (28; Fig. 7). Binding of retinol to Saccharomyces cerevisiae PKC1 or the PKCδ C1B ZiF (Fig. 7 A) as well as the mammalian ZiFs, PKCα C1A and cRaf, and the Drosophila PKC C1A ZiF (data not shown) produced distinct red shift and partial fine structure although the magnitude of the effect was smaller than the one observed with the cellular retinol binding protein (CRBP)–retinol complex. The distinct shoulders in the excitation spectrum elicited by bound retinol occurred at 348 and 365 nm (Fig. 7 A, arrows) and hence coincided with the reported peaks of retinol–CRBP as well as the naturally planar retinoid, 14HRR. Mixing retinol with the fusion proteins comprising the Drosophila C1B or PKCα C1B ZiF domain did not result in enhanced fluorescence emission of retinol, red shift, or vibronic fine structure (data not shown), confirming binding selectivity on the part of different ZiF domains. As will be reported elsewhere in detail HRR, retinoic acid, and AR also bound cRaf-1 ZiF fusion protein with similar affinity as retinol.
Figure 7.
Red shift and vibronic fine structure of bound retinol. (A) Retinol was ligated to equimolar solutions of ZiF domains of human PKCδ C1B (solid line), S. cerevisiae (Sc.) PKC1 C1A (broken line), or CRBP (dashed line), or dissolved in Tris buffer (dotted line). In the fluorescence excitation spectra shown, monitored at the retinol λmax 466 nm, the red shift is pronounced and vibronic fine structure developed partially as three distinct shoulders at wavelengths 330, 348, and 365 nm (arrows) after ligation to the human or yeast PKC ZiF domain. Vibronic fine structure is maximal in the retinol–CRBP complex (reference 28). (B) Retinol binds to cRaf in vivo. COS cells expressing Flag-tagged cRaf (n = 4) or cRaf ZiF fragment (n = 2) were loaded with the indicated concentrations of unlabeled retinol, followed by 3H-retinol. Values for radioactivity in immunoprecipitates were corrected for radioactivity in control immunoprecipitates from nontransfected cells.

Binding of retinol to full length cRaf and to the ZiF domain, expressed as Flag-tagged proteins in COS cells, was evaluated in vivo by use of 3H-labeled retinol. Fig. 7 B demonstrates that immunoprecipitates of transfected COS cells bound retinol. Binding was specific, as free cold retinol was able to displace radioactive retinol in a dose-dependent manner.
Retinol as Cofactor in the Alternate Redox Pathway of Activation.
Having established that retinol binds the regulatory domain of cRaf-1, it was of interest to inquire into the underlying purpose. An exhaustive search of activation of any kinase cascade by retinol alone was unfruitful. Similarly, despite our earlier definition of retinol as growth factor 4 and the demonstration that retinol cooperates with platelet-derived growth factor 29, attempts to modulate the activation of the Raf/MAPK pathway by receptor-mediated stimulation in a variety of cells by retinol, or its active metabolites, 14HRR and AR, produced inconclusive results. The failure of AR to show any effect in signaling on its own was intriguing, since AR has been shown to diminish the survival of a variety of cells types 7, to cause depolymerization of actin filaments in fibroblasts 30, and even to lead to activation of programmed cell death by a mechanism involving conventional kinases 10. Because retinoids did not appear to intersect with conventional phosphorylation signal cascades, attention was focused on the alternate redox activation pathway that recently has been shown to control both PKC and cRaf/MAPK 15 16 31. Both have been shown to become catalytically active under oxidizing conditions.
To test whether retinol could influence the redox activation of cRaf, Jurkat cells grown under retinol-free conditions were replenished with different concentrations of retinol, activated by peroxide 15 min later, and analyzed for endogenous MAPK activity using phospho-ERK measurements on Western blots 32. A distinct dose-dependent enhancement of MAPK activation by retinol was evident (Fig. 8 A). To determine dose responses of peroxide on cRaf, 3T3 fibroblasts were first depleted of endogenous retinoids by extended culturing (4–5 d) in serum-free medium. One set of cultures was subsequently replenished with a physiological amount of retinol at 2 μM, added 12 h before the experiment. Because PKC binds retinol, is activated by H2O2, and can activate the downstream cRaf kinase, this pathway was blocked by brief culture with staurosporine. Different doses of H2O2 were added, and the cells were harvested, lysed, and analyzed by Western blot (Fig. 8 B), with subsequent quantitation by densitometry (Fig. 8 C). In the representative experiment shown in Fig. 8 B as well as four others, ERK1 and 2 phosphorylation was significantly higher in retinol-containing compared with retinol-free cultures over the entire dose range (P < 0.002). The shift in the H2O2 dose–response indicated that retinol increased the sensitivity to redox activation. Activation by 5% FBS showed no differences between retinol-sufficient and retinol-deficient cultures. To test whether the kinetics of activation were changed by retinol, three sets of cultures were compared with each other: 3T3 cells grown continuously in 2 μM retinol (Fig. 9 A, top), 3T3 cells depleted of retinol then reconstituted with retinol 12 h before the experiment, and retinol-deficient 3T3 cells. The results of activation by 300 μM H2O2 (Fig. 9 A) and their quantitation by imaging of phosphorylated p42, corrected for the amount of nonphosphorylated p42 detected on the same membrane, are shown in Fig. 9 B. Measurements of phosphorylated p44 yielded similar results (data not shown). These data indicate that MAPK activities peaked around the same time (10–15 min), but were two- to threefold higher in retinol-repleted cultures compared with retinol-deficient ones.
Figure 8.
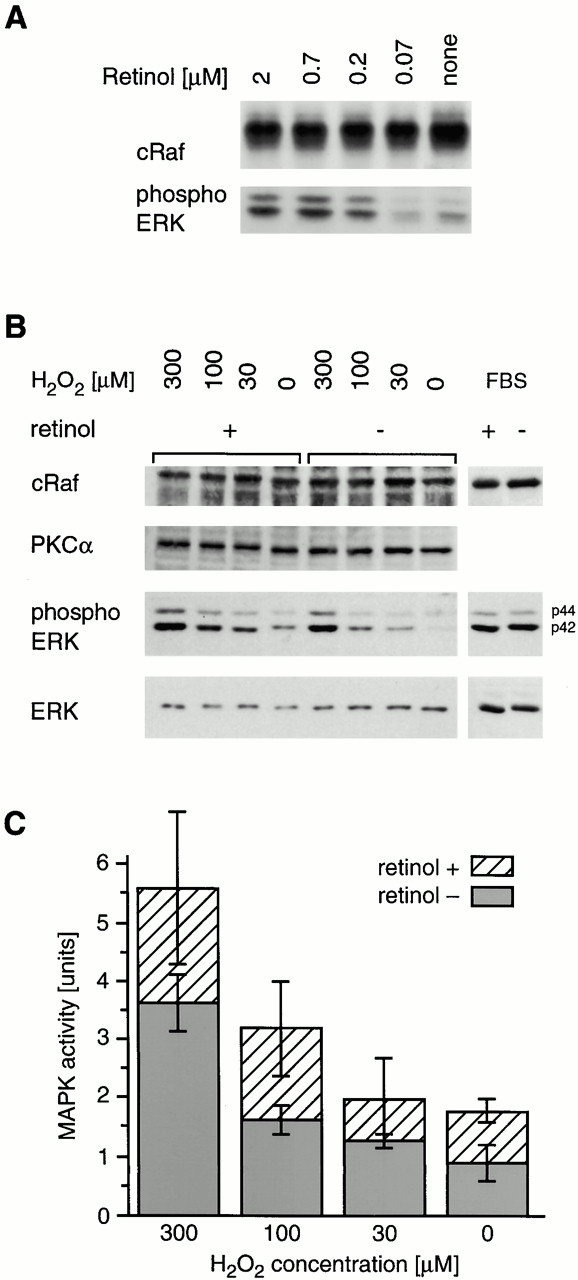
Retinol enhances the oxidative activation of cRaf/MAPK. (A) Jurkat cells grown in serum-free medium were activated for 10 min with 100 μM H2O2 in the presence of different concentrations of retinol. 3T3 fibroblasts were starved of retinol for 4 d, and part of the cultures was reconstituted with 2 μM retinol overnight. Cells were then blocked with 25 nM staurosporine and activated for 15 min with H2O2. (B) Western blot showing phosphorylation of endogenous ERK1 and 2 in retinol-starved and retinol-reconstituted cultures. (C) Quantitation of the amounts of phosphorylated ERK2 of 3T3 fibroblasts treated as in A. Shown are the averages of four replicate retinol-sufficient cultures (hatched bars) or retinol-deficient cultures (gray bars) for each H2O2 concentration, corrected for amounts of total ERK proteins.
Figure 9.
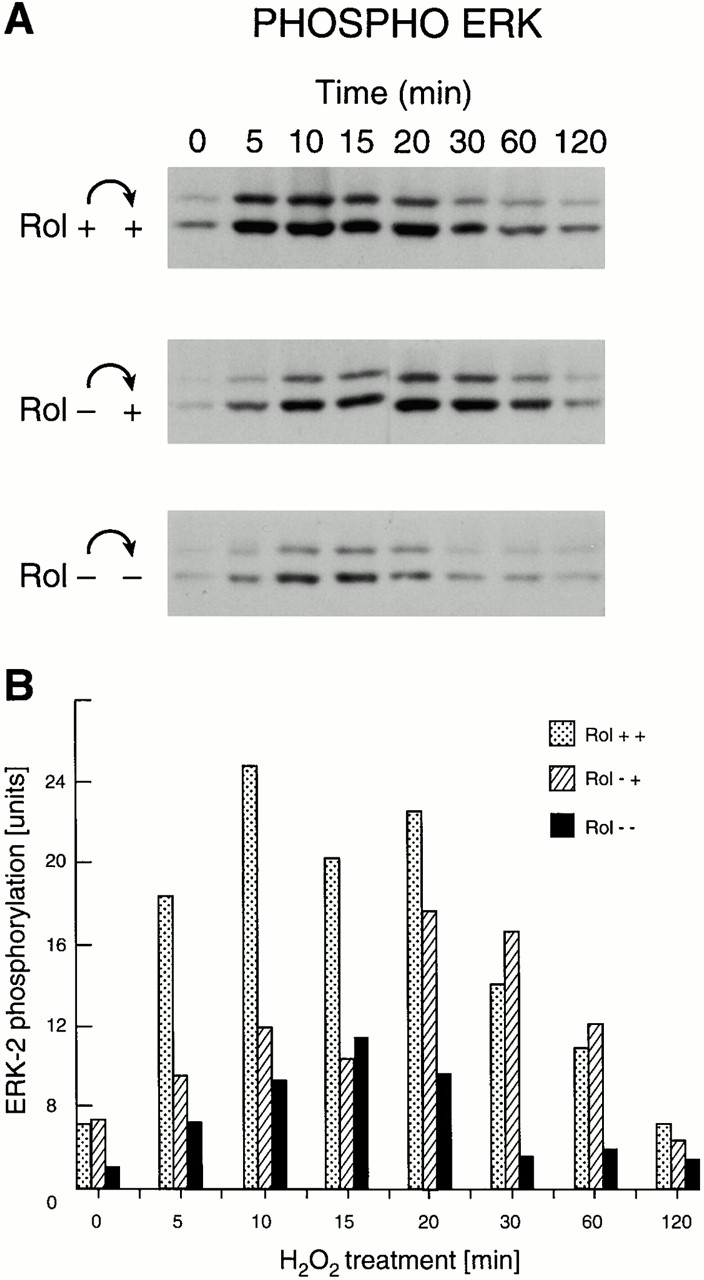
Kinetics of oxidative activation of endogenous cRaf/MAPK. (A) Western blot showing activation by 100 μM H2O2 of endogenous ERK1 and 2 in 3T3 fibroblast cultures, passaged continuously in retinol (Rol)-sufficient medium (top), or starved for 4 d then reconstituted with retinol (middle), or kept in retinol-deficient medium without reconstitution (bottom). (B) Quantitation by imaging of the amounts of ERK2 phosphorylation corrected for amounts of nonphosphorylated ERK2.
As PKCα bound retinol with an affinity comparable to that of cRaf and is known to undergo activation by the redox pathway 15 16 33, it was of interest whether retinol acted as a costimulator in this circumstance as well. 3T3 cells were rendered retinol deficient and subsequently reconstituted with 2 μM retinol as described above. Cells were activated for 10 min with different doses of H2O2, and cytoplasmic extracts were assayed for PKC kinase activity by standard immunoprecipitation/kinase assay 16. The results of Fig. 10 show a marked increase in PKC activity when retinol was present compared with its absence. Retinol caused no intrinsic PKC activation on its own. The pooled imaging data from 6 independent experiments yielded a 20-fold increase of PKCα kinase activity at maximum stimulation (200 μM H2O2) over unstimulated cells in the presence of retinol, compared with a 7-fold increase in the absence of retinol, for an overall 3-fold retinol enhancement effect (P < 0.001). The optimum dose, determined at 1 μM (data not shown), was similar to that showing enhancement of the MAPK pathway. Activation of PKC by 150 nM phorbol ester was not significantly enhanced by retinol.
Figure 10.

Retinol as cofactor in the oxidative activation of PKCα. Normalized results of immunoprecipitation/kinase assays of six independent titrations of H2O2 with retinol (Rol)-depleted (white bars) and retinol-reconstituted (black bars) 3T3 fibroblasts. Except for unstimulated cells, the evaluation by Student's t test showed significantly higher PKC kinase activity in retinol-reconstituted cultures for each H2O2 concentration compared with retinol-deficient cultures (P < 0.001).
To address the question whether endogenous ROS was augmented by retinol, COS cells transfected with Flag-tagged cRaf were irradiated with UVB, known to generate intracellular singlet oxygen and other ROS 34 35 36 37. Testing kinase activity in immunoprecipitated Flag-Raf revealed a modest activation in the absence of retinol and a substantial (2.2 ± 0.4–fold; P < 0.003) augmentation when cells were replenished with 2 μM retinol (see Fig. 11 A, representing one of three experiments). Addition of oxygen scavengers, sodium azide (100 mM) and N-acetylcysteine (1 mM) abolished the Raf activation. By contrast, addition of deuterated water known to stabilize singlet oxygen 37 produced distinct, though moderate, enhancement of Raf activity (Fig. 11 B).
Figure 11.
(A) Activation of Raf kinase activity by endogenously generated ROS. Cos-7 cells were activated by UVB in the presence or absence of retinol (Rol). Raf kinase activity was determined in immunoprecipitates with anti-Flag antibody. Raf kinase activity was transient, reaching a maximum at 10 min. (B) Neutralization of ROS by sodium azide and N-acetylcysteine (L-NAC) resulted in diminished kinase response at the 10-min time point, whereas addition of deuterated water enhanced the kinase response. MEK, MAPK.


Discussion
The significance of our findings is threefold: first, the unprecedented binding of vitamin A to the cysteine-rich region of the regulatory domains of serine/threonine kinases; second, the demonstrated prooxidant effect of retinol; third, the strong correlation between presence of retinol, its binding to PKC and Raf, and the enhancement of redox activation of catalytic capacity. On that score, the ZiF domain of serine/threonine kinases qualifies as receptor of retinoids. Our discovery is apt to broaden the field of retinol physiology, which up to now has been dominated by retinoic acid as transactivator of nuclear receptors 8 9. The newly discovered cytoplasmic retinoid receptors, i.e., the ZiF domains of serine/threonine kinases, are ubiquitously expressed and moreover conserved in evolution from yeast to humans. That the capacity to metabolize retinol to 14HRR and DHR is equally shared by all mammalian and invertebrate cell lines tested (our unpublished results) implies coevolution of this receptor–ligand system over millions of years. If so, the binding of retinoids to the Zif domain must serve a basic function.
Using Gst fusion proteins containing the ZiF domains of cRaf and four mammalian, one insect, and one yeast PKC 38 isoforms, binding at an affinity in the range of tens of nanomoles was established by four fluorimetric assays. Quenching of the intrinsic protein fluorescence, fluorescent resonance energy transfer, enhancement of retinoid fluorescence, and acquisition of partial fine structure all furnished concordant, and hence conclusive, evidence for binding (Fig. 1 Fig. 2 Fig. 3 Fig. 4 Fig. 5 Fig. 6 Fig. 7). Nonspecific interactions of retinol with a variety of hydrophobic proteins, some (like 14-3-3 [12], Hsp90, and immunophilins) part of the kinase signaling complexes 11, were not in evidence. Moreover, not every ZiF domain bound retinol. The C1B domain of mammalian α isoform, Drosophila and yeast PKC, and the C1A domain of PKCδ bound either with low affinity or not at all, despite sequence homologies to those that bound 23, indicating that retinol binding was protein selective. Moreover, the capacity to bind retinoids was vested in the tertiary structure, since denaturation of cRaf-1 by guanidine hydrochloride abolished the binding capacity and renaturation restored it completely (Fig. 4). As small lipid-like molecules, retinoids can freely pass through membranes, have free access to all compartments, and can rapidly partition to the sites of highest affinity 39, just as diacylglycerides are presumed to do, unless such sites are occupied by other ligands. By testing in in vitro assays for the potential binding interference by known lipid ligands of cRaf 40 41 and PKC ZiF domains 17 18 42, the retinol binding site was found to be distinct from the binding sites for acidic lipids (i.e., phosphatidylserine), diacylglycerol, and phorbol ester (our unpublished results). It was therefore a reasonable assumption that under culture conditions in media containing an excess of retinol (2 μM), all available sites on PKC and cRaf were occupied by retinol or the metabolites, 14HRR and DHR, our test cells producing no other known retinol metabolites. Attempts to demonstrate binding of retinol in vivo to cRaf were successful although the use of detergents was not permissible owing to the lipid nature of retinoids. Therefore, only crude purification of the cRaf complex by immunoprecipitation was achieved, and the exact in vivo binding site in the complex remained unknown. On the other hand, the Raf-ZiF fragment bound radioactive retinol in vivo (Fig. 7 B). Displacement of bound by free retinol indicated specificity.
The function of the class of hydroxylated retinoids in vivo is best described as survival factor 4 29. Virtually all cultured cells tested, whether animal or insect in origin, have the ability to synthesize 14HRR and DHR (5 6; our unpublished results). Many of these are profoundly affected by vitamin A deprivation, although adaptive mechanisms must exist whereby cells, and indeed whole animals, can overcome vitamin A deficiency. The question of how hydroxylated retinoids support cell survival is an issue too complex to address in this report. We have instead focused on the cRaf and PKCα kinases in order to learn which of the several known activation pathways may be affected by bound retinol. Extensive experimentation had convinced us that retinoids themselves caused neither cytosol-to-membrane translocation nor catalytic activation of the cRaf/MAPK complex or the PKCα kinase (Fig. 10). Similarly, the classical receptor-mediated activation pathway of cRaf/MAPK proved to function largely independently of retinol. This is shown in the control experiment of Fig. 8, where stimulation by 5% FBS produced equal MAPK activity in retinol-deficient and retinol-sufficient 3T3 cells, and this result is echoed by the lack of enhancement of PKC activity stimulated by PMA (Fig. 10).
During the past few years, it has become increasingly clear that oxidation presents an alternative for activation of both cRaf and PKC kinases. This “oxidative stress pathway” has its roots in numerous cell biological observations that oxidizing agents can cause a broad spectrum of reactions, including lymphocyte proliferation 43 and apoptosis. The molecular dissection of this phenomenon has led to the realization that a variety of well-known transcriptional regulators, notably nuclear factor κB, AP1, and SP1, and their upstream signaling cascades respond to oxidative stress 44 45 46. PKC in a variety of cells and MAPK in cardiac myocytes 47 stand out as targets for reactive oxygen. While oxidative stress has helped to reveal its existence, the redox pathway may more aptly signify a general regulation circuit powered by the endogenous redox system of cells and operative at all times.
PKC has been reported to become readily activated by H2O2 in vivo, and there is suggestive evidence that oxidative activation can also be accomplished with purified PKC, although uncontrolled oxidation most often leads to destruction 15 33. If the former observation is verified, the initial chemical reaction is likely to be with the protein itself. It has also been reported that oxidative activation of PKC is accompanied by phosphorylation of distinct tyrosines in the catalytic domain, implying the recruitment of an upstream kinase 16. This finding still begs the question where the initial chemical modification may take place. Taking into account that the regulatory domains of the PKC and cRaf kinases contain cysteine-rich domains 41 48 49 50, chemically highly susceptible to oxidation, that this domain contains the retinol binding site, and furthermore that it is crucial for kinase regulation, the likely target of oxidation may actually lie in this structure. Although our cell experiments clearly establish the connection between retinol, its binding to the ZiF domain, and enzyme activation by an oxidative step, the chemical proof remains to be established whether sulfhydryl groups become oxidized. A precedent for this mechanism, however, has been recently described for the redox regulation of the bacterial chaperone, Hsp33 20. This enzyme contains a closely related ZiF structure, which underwent selective oxidation affecting cysteine residues critical for the coordination of zinc. As this process was reversible by reduction, the authors suggested that this structure served as a redox switch to turn on the chaperone under oxidizing conditions and off under reducing conditions.
How might retinoids come into play? The important clue is probably the presence of the system of conjugated double bonds characteristic of all natural retinoids. This polyene system is responsible for the pro- and antioxidant properties of the molecule. In other words, the electron transfer capacity of retinoids is rooted in the π electrons of the polyene system. In nature, this unique capacity is exploited in photosynthetic systems for straight electron transfer from carotene to chlorophyll P680, and in vision to capture the energy of a photon-excited π electron for isomerization of 11-cis- to trans-retinal 51. It is conceivable that the polyene of retinol interacts with the microenvironment of the protein and exerts a catalyst-like effect. Redox-sensitive structures (foremost cysteines) of the ZiF domains might be tagged for preferred oxidation. Our cell biological data that show heightened sensitivity to oxidative kinase activation (Fig. 8 Fig. 9 Fig. 10 Fig. 11) are consistent with this view.
Peroxide is but one pharmacologic agent that induces oxidative activation of Raf and PKC. Internally generated oxygen radicals had the same consequence, as demonstrated by UV irradiation. The presence of free radicals, including singlet oxygen, was required for kinase activation, since radical scavengers inhibited kinase activation, whereas deuterated water, a known stabilizer of singlet oxygen 37, enhanced kinase activation (Fig. 11). Whether by pharmacologic means or by endogenously created oxidizing conditions, the presence of retinol consistently enhanced the kinase activity, implying that retinol functioned as a cofactor in the redox activation pathway. As PKC and Raf are receptors of retinoids and are independently activated by a redox step, chances are that the retinol influence on the protein is a direct one.
Acknowledgments
The authors are indebted to Dr. F. Derguni for gifts of retinoids and valuable discussions. We also wish to acknowledge the gifts of PKCμ cDNA from Dr. K. Pfizenmaier and Dr. F.J. Johannes, University of Stuttgart, Stuttgart, Germany; the Gst PKCδ C1B expression vector from Dr. P. Blumberg, National Institutes of Health, Bethesda, MD; PKCα cDNA from Dr. T. Powell, Memorial Sloan-Kettering Cancer Center, New York, NY; the PKCζ cDNA from Dr. Y. Nishizuka and Dr. U. Kikkawa, Kobe University, Kobe, Japan; the yeast PKC1 cDNA from Dr. J. Thorner, University of California at Berkeley, Berkeley, CA; and the Drosophila PKC cDNA from Dr. A. Rosenthal, Genentech, Inc., South San Francisco, CA. Dr. Robert Glazer, George Washington University, Washington, DC, Dr. Roger Davis, University of Massachusetts, Worcester, MA, and Dr. Ulf Rapp, University of Würzburg, Würzburg, Germany, kindly provided PKC cDNAs and expression plasmids of Flag-cRaf and cRaf, respectively.
This work was supported by grants from the National Cancer Institute (CA49933, and CA69718 to C. Swenson) and the Focused Giving Program of Johnson & Johnson, Inc.
Footnotes
Abbreviations used in this paper: AR, anhydroretinol; CRBP, cellular retinol binding protein; ERK, extracellular signal–regulated kinase; FBS, fetal bovine serum; FRET, resonance energy transfer of intrinsic protein fluorescence; Gst, glutathione S-transferase; Hsp, heat shock protein; MAPK, mitogen-activated protein kinase; PKC, protein kinase C; ROS, reactive oxygen species; trp, tryptophan; ZiF, zinc finger.
References
- Underwood B.A. Vitamin A in human nutrition. In: Sporn M.B., Roberts A.B., Goodman D.S., editors. The Retinoids. Raven Press; New York: 1994. pp. 211–227. [Google Scholar]
- Wolbach S.B., Howe P.R. Tissue changes following deprivation of fat-soluble A vitamin. J. Exp. Med. 1925;42:753–763. doi: 10.1084/jem.42.6.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross A.C., Hammerling U. Retinoids and the immune system. In: Sporn M.B., Roberts A.B., Goodman D.S., editors. The Retinoids. Raven Press; New York: 1994. pp. 524–544. [Google Scholar]
- Garbe A., Buck J., Hammerling U. Retinoids are important cofactors in T cell activation. J. Exp. Med. 1992;176:109–117. doi: 10.1084/jem.176.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck J., Derguini F., Levi E., Nakanishi K., Hammerling U. Intracellular signaling by l4-hydroxy-retro-retinol. Science. 1991;254:1654–1656. doi: 10.1126/science.1749937. [DOI] [PubMed] [Google Scholar]
- Derguini F., Nakanishi K., Hämmerling U., Chua R., Eppinger T., Levy E., Buck J. 13,14-Dihydroxy-retinol, a new bioactive retinol metabolite. J. Biol. Chem. 1995;270:18875–18880. doi: 10.1074/jbc.270.32.18875. [DOI] [PubMed] [Google Scholar]
- Buck J., Grun F., Kimura S., Noy N., Derguini F., Hammerling U. Anhydroretinola naturally occurring inhibitor of lymphocyte physiology. J. Exp. Med. 1993;178:675–680. doi: 10.1084/jem.178.2.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans R.M. The steroid and thyroid hormone receptor superfamily. Science. 1988;240:889–895. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green S., Chambon P. Nuclear receptors enhance our understanding of transcription regulation. Trends Genet. 1988;4:309–314. doi: 10.1016/0168-9525(88)90108-4. [DOI] [PubMed] [Google Scholar]
- O'Connell M.J., Chua R., Hoyos B., Buck J., Chen Y.Q., Derguini F., Hammerling U. Retro-retinoids in regulated cell growth and death. J. Exp. Med. 1996;184:549–555. doi: 10.1084/jem.184.2.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stancato L.F., Chow Y.-H., Hutchison K.A., Perdew G.H., Jove R., Pratt W.B. Raf exists in a native heterocomplex with hsp90 and p50 that can be reconstituted in a cell-free system. J. Biol. Chem. 1993;268:21711–21716. [PubMed] [Google Scholar]
- Fu H., Xia K., Pallas D.C., Cui C., Conroy K., Narsinham R.P., Mammon H., Collier R.J., Roberts T.M. Interaction of the protein kinase Raf-1 with 14-3-3 proteins. Science. 1994;266:126–129. doi: 10.1126/science.7939632. [DOI] [PubMed] [Google Scholar]
- Morrison D.K., Kaplan D.R., Rapp U.R., Roberts T.M. Signal transduction from membrane to cytoplasmgrowth factors and membrane bound oncogene products increase Raf-1 phosphorylation and associated protein kinase activity. Proc. Natl. Acad. Sci. USA. 1988;85:8855–8859. doi: 10.1073/pnas.85.23.8855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizuka Y. Intracellular signalling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- Gopalakrishna R., Anderson W.B. Ca2+- and phospholipid-independent activation of protein kinase C by selective oxidative modification of the regulatory domain. Proc. Natl. Acad. Sci. USA. 1989;86:6758–6762. doi: 10.1073/pnas.86.17.6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi H., Tanaka M., Takemura Y., Matsuzaki H., Ono Y., Kikkawa U., Nishizuka Y. Activation of protein kinase C by tyrosine phosphorylation in response to H2O2 . Proc. Natl. Acad. Sci. USA. 1997;94:11233–11237. doi: 10.1073/pnas.94.21.11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns D.J., Bell R.M. Protein kinase C contains two phorbol ester binding domains. J. Biol. Chem. 1991;266:18330–18338. [PubMed] [Google Scholar]
- Kazanietz M.G., Bustelo X.R., Barbacid M., Kolch W., Mishak H., Wong G., Pettit G.R., Bruns J.D., Blumberg P.M. Zinc finger domains and phorbol ester pharmacophore. J. Biol. Chem. 1994;269:11590–11594. [PubMed] [Google Scholar]
- Mott H.R., Carpenter J.W., Zhong S., Ghosh S., Bell R.M., Campbell S.L. The solution structure of the Raf-1 cysteine rich domaina novel Ras and phospholipid binding site. Proc. Natl. Acad. Sci. USA. 1996;93:8312–8317. doi: 10.1073/pnas.93.16.8312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakob U., Muse M., Eser M., Bardwell J.C.A. Chaperone activity with a redox switch. Cell. 1999;96:341–352. doi: 10.1016/s0092-8674(00)80547-4. [DOI] [PubMed] [Google Scholar]
- Wald G. Molecular basis for visual excitation. Science. 1968;162:230–232. doi: 10.1126/science.162.3850.230. [DOI] [PubMed] [Google Scholar]
- Derguini F., Nakanishi K., Hammerling U., Buck J. Intracellular signalling activity of synthetic (14R-), (14S-) and (14RS-)-14-hydroxy-4,14-retro-retinol. Biochemistry. 1994;33:623–628. doi: 10.1021/bi00169a001. [DOI] [PubMed] [Google Scholar]
- Hurley J.H., Newton A.C., Parker P., Blumberg P., Nishizuka Y. Taxonomy and function of C1 protein kinase C homology domains. Protein Sci. 1997;6:477–480. doi: 10.1002/pro.5560060228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G., Kazanietz M.G., Blumberg P.M., Hurley J.H. Crystal structure of the Cys2 activator-binding domain of protein kinase C delta in complex with phorbol ester. Cell. 1995;81:917–924. doi: 10.1016/0092-8674(95)90011-x. [DOI] [PubMed] [Google Scholar]
- Norris A.W., Cheng L., Giguere V., Rosenberger M., Li E. Measurement of subnanomolar retinoic acid binding affinities for cellular retinoic acid binding proteins by fluorometric titration. Biochim. Biophys. Acta. 1994;1209:10–18. doi: 10.1016/0167-4838(94)90130-9. [DOI] [PubMed] [Google Scholar]
- Cogan U., Kopelman M., Mokaday S., Shinitzky M. Binding affinities of retinol and related compounds to retinol binding proteins. Eur. J. Biochem. 1976;65:71–78. doi: 10.1111/j.1432-1033.1976.tb10390.x. [DOI] [PubMed] [Google Scholar]
- Prestle J., Pfizenmaier K., Brenner J., Johannes F.J. Protein kinase C mu is located at the Golgi compartment. J. Cell Biol. 1996;124:1401–1410. doi: 10.1083/jcb.134.6.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong D., Chytil F. Purification of cellular retinol and retinoic acid binding proteins from rat tissue. Methods Enzymol. 1980;67:288–296. doi: 10.1016/s0076-6879(80)67036-0. [DOI] [PubMed] [Google Scholar]
- Chen Y., Derguini F., Buck J. Vitamin A in serum is a survival factor for fibroblasts. Proc. Natl. Acad. Sci. USA. 1997;94:10205–10208. doi: 10.1073/pnas.94.19.10205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korichneva I., Hammerling U. F-actin as a functional target for retro-retinoidsa potential role in anhydroretinol-triggered cell death. J. Cell Sci. 1999;112:2521–2528. doi: 10.1242/jcs.112.15.2521. [DOI] [PubMed] [Google Scholar]
- Goldstone S.D., Hunt N.H. Redox regulation of the mitogen-activated protein kinase pathway during lymphocyte activation. Biochim. Biophys. Acta. 1997;1355:353–360. doi: 10.1016/s0167-4889(96)00150-4. [DOI] [PubMed] [Google Scholar]
- Sturgill T.W., Ray L.B., Erikson E., Maller J.L. Insulin-stimulated MAP-2 kinase phosphorylates and activates ribosomal protein S6 kinase II. Nature. 1988;334:715–718. doi: 10.1038/334715a0. [DOI] [PubMed] [Google Scholar]
- Gundimeda U., Hara S.K., Anderson W.B., Gopalakrishna R. Retinoids inhibit the oxidative modification of protein kinase C induced by oxidant tumor promoters. Arch. Biochem. Biophys. 1993;300:526–530. doi: 10.1006/abbi.1993.1072. [DOI] [PubMed] [Google Scholar]
- Yasui H., Sakurai H. Chemiluminescent detection and imaging of reactive oxygen species in live mouse skin exposed to UVA. Biochem. Biophys. Res. Commun. 2000;269:131–136. doi: 10.1006/bbrc.2000.2254. [DOI] [PubMed] [Google Scholar]
- Chan W.H., Yu J.S. Inhibition of UV irradiation induced oxidative stress and apoptotic biochemical changes in human epidermal carcinoma A431 cells by genistein. J. Cell. Biochem. 2000;78:73–84. doi: 10.1002/(sici)1097-4644(20000701)78:1<73::aid-jcb7>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Kasid U., Suy S., Dent P., Ray S., Whiteside T.L., Sturgill T.W. Activation of Raf by ionizing radiation. Nature. 1996;382:813–816. doi: 10.1038/382813a0. [DOI] [PubMed] [Google Scholar]
- Scharffetter-Kochanek K., Wlaschek M., Brenneisen P., Schauen M., Blaudschun R., Wenk J. UV-induced reactive oxygen species in photocarcinogenesis and photoaging. Biol. Chem. 1997;378:1247–1257. [PubMed] [Google Scholar]
- Levin D.E., Fields F.O., Kunisawa R., Bishop J.M., Thorner J. A candidate protein kinase C gene, PGC1, is required for the S. cerevisiae cell cycle. Cell. 1990;62:213–224. doi: 10.1016/0092-8674(90)90360-q. [DOI] [PubMed] [Google Scholar]
- Noy N., Xu Z.-J. Thermodynamic parameters of the binding of retinol to binding proteins and to membranes. Biochemistry. 1990;29:3888–3892. doi: 10.1021/bi00468a014. [DOI] [PubMed] [Google Scholar]
- Improta-Brears T., Ghosh S., Bell R.M. Mutational analysis of Raf cysteine rich domainrequirement for a cluster of basic aminoacids for interaction with phosphatidylserine. Mol. Cell. Biochem. 1999;198:171–178. doi: 10.1023/a:1006981411691. [DOI] [PubMed] [Google Scholar]
- Daub M., Jockel J., Quack T., Weber C.K., Schmitz F., Rapp U.R., Wittinghofer A., Block C. The RafC1 cysteine-rich domain contains multiple distinct regulatory epitopes which control ras-dependent Raf activation. Mol. Cell. Biol. 1998;18:6698–6710. doi: 10.1128/mcb.18.11.6698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marom M., Parish C.A., Giner J.L., Rando R.R. Minimal structural requirements for diglyceride-site directed activators of protein kinase C. Tetrahedron. 1997;53:10041–10050. [Google Scholar]
- Novogrodsky A., Stenzel K.H., Rubin A.L. Stimulation of human peripheral blood lymphocytes by periodate, galactose oxidase, soy bean agglutinin and peanut agglutinindifferent effects of adherent cells. J. Immunol. 1977;118:852–857. [PubMed] [Google Scholar]
- Baeuerle P.A. Pro-inflammatory signalinglast pieces in the NF-kappaB puzzle? Curr. Biol. 1998;8:R19–R22. doi: 10.1016/s0960-9822(98)70010-7. [DOI] [PubMed] [Google Scholar]
- Los M., Droege W., Stricker K., Baeuerle P.A., Schulze-Osthoff K. Hydrogen peroxide as a potent activator of T lymphocyte functions. Eur. J. Immunol. 1995;25:159–165. doi: 10.1002/eji.1830250127. [DOI] [PubMed] [Google Scholar]
- Schreck R., Rieber P., Baeuerle P. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO (Eur. Mol. Biol. Organ.) J. 1991;10:2247–2252. doi: 10.1002/j.1460-2075.1991.tb07761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seko Y., Tobe K., Takahashi N., Kaburagi Y., Kadowaki T., Yazaki Y. Hypoxia and hypoxia/regeneration activate src family tyrosine kinases and p21 ras in cultured rat cardiac myocytes. Biochem. Biophys. Res. Commun. 1996;226:530–535. doi: 10.1006/bbrc.1996.1389. [DOI] [PubMed] [Google Scholar]
- Ono Y., Fuji T., Igarashi K., Kono T., Tanaka C., Kikkawa U., Nishizuka Y. Phorbol ester binding to protein kinase C requires a cysteine-rich zinc-finger-like sequence. Proc. Natl. Acad. Sci. USA. 1989;86:4868–4871. doi: 10.1073/pnas.86.13.4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow Y.-H., Pumiglia K., Jun T.H., Dent P., Sturgill T.W. Functional mapping of the N-terminal regulatory domain in the human Raf-1 protein kinase. J. Biol. Chem. 1995;270:14100–14106. doi: 10.1074/jbc.270.23.14100. [DOI] [PubMed] [Google Scholar]
- Ghosh S., Xie W.Q., Quest A.F.G., Mabrouk G.M., Strum J.C., Bell R.M. The cysteine rich region of Raf-1 kinase contains zinc, translocates to liposomes, and is adjacent to a segment that binds GTP-Ras. J. Biol. Chem. 1994;269:10000–10007. [PubMed] [Google Scholar]
- Shichi H. Biochemistry of Vision 1983. Academic Press; New York: pp. 275 [Google Scholar]