

Abstract
Immature CD4+CD8+ thymocytes rearrange their T cell receptor (TCR)-α gene locus to generate clonotypic α/β TCR, after which a few cells expressing selectable TCR are signaled to further differentiate into mature T cells. Because of requirements for self-tolerance, immature CD4+CD8+ thymocytes are programmed to die in the thymus in response to a variety of stimuli that do not induce death of mature T cells. We now demonstrate that, in contrast to all previously described stimuli, immature CD4+CD8+ thymocytes are selectively more resistant than mature T cells to apoptotic death induced by DNA intercalating agents. Importantly, we demonstrate that DNA intercalating agents induce double-stranded DNA breaks in both immature thymocytes and mature T cells, but immature thymocytes tolerate these DNA breaks, whereas mature T cells are signaled to die by an Atm-dependent but p53-independent death mechanism. Thus, our results indicate that absence of an Atm-dependent but p53-independent pathway allows immature thymocytes to survive double-stranded DNA breaks. It is likely that the unique ability of immature thymocytes to survive DNA-damaging intercalating agents reflects their tolerance of double-stranded DNA breaks that occur normally during antigen receptor gene rearrangements.
Keywords: DNA damage, apoptosis, thymic development, actinomycin D, rearrangement
Introduction
Within the thymus, immature thymocytes undergo an ordered process of TCR gene rearrangements to build the antigen-specific TCR complex 1. Surface expression of the α/β TCR first occurs at the CD4+CD8+ (double-positive [DP]) stage 2, and it is at this stage that cells are screened for the specificity of the TCR they express 3. Only 3–5% of immature DP thymocytes are selected to mature into long-lived T cells 4, with the remainder dying either of neglect or in response to active negative selection 3. It is thought that immature thymocytes are programmed to die of neglect unless actively rescued by TCR-mediated positive selection 5. Indeed, immature DP thymocytes are induced to die by many different stimuli, including those that induce mature T cells to grow and proliferate 6. The paradoxical death response of immature thymocytes to stimuli that activate mature T cells is thought to reflect requirements for self-tolerance during selection of the T cell repertoire in developing thymocytes 7. Death of immature thymocytes during development is mediated by apoptosis and can be blocked by inhibitors of protein synthesis such as cycloheximide (CHX) and inhibitors of transcription such as actinomycin D (ActD; reference 6).
Consequently, we were surprised to discover in this study that ActD and other DNA intercalating agents induced death of mature T cells but not immature thymocytes, despite inducing DNA breaks in both cell types. We show that death of mature T cells induced by DNA intercalating agents is due to an apoptotic mechanism mediated by an Atm-dependent but p53-independent response to DNA damage that is not activated in immature thymocytes. Thus, DNA-damaging intercalating agents are the first identified death stimuli to which immature thymocytes are resistant relative to their mature progeny. It is likely that the resistance of immature thymocytes to DNA-damaging intercalating agents reflects a necessary tolerance to DNA breaks that occur normally during TCR gene rearrangements.
Materials and Methods
Mice.
Normal C57BL/6 (B6) mice were obtained from the Frederick Cancer Research and Development Center. B6Smn.C3H-gld (B6.gld) mice deficient in Fas ligand were obtained from The Jackson Laboratory. The creation of Atm-deficient mice (allele designation Atmins5790neo) has been previously described 8. Progeny of heterozygous matings were genotyped by PCR 9. p53 −/− mice were obtained from Taconic Laboratories, and p53 genotyping was performed by PCR as described 10. Bcl-2–transgenic mice expressing human Bcl-2 under the control of the proximal lck promoter in T lineage cells have been described 11. All mice were housed in a specific pathogen–free facility and used at 4–12 wk of age.
Cell Isolations and Culture.
Single-cell suspensions of thymocytes or splenocytes from mice were cultured overnight (12–16 h) at a density of 2 × 106 cells/ml in cell culture medium containing 10% FCS (Hyclone), after which they were counted in a hemocytometer. CD4+CD8+ (DP) thymocytes were isolated by panning on anti-CD8 coated plates as described 12 and were routinely 96–99% pure. ActD obtained from Sigma-Aldrich was used at concentrations ranging from 3 to 30 μg/ml. CHX (Sigma-Aldrich) was used at 10 μg/ml. ZVAD-FMK and ZFA-FMK were obtained from Enzyme Systems. The intercalating agent dihydroethidium (DhE) was obtained from Molecular Probes. The transcription inhibitor 5, 6-dichloro-1-β-D-ribofuranosylbenzimidazole (DRB) was obtained from Sigma-Aldrich. To calculate recovery of a cell population, we multiplied the frequency of that given cell population determined by flow cytometry with the overall cell recovery. Data were normalized to the number of cells recovered when cells were cultured in medium alone, in which case recoveries were from 75 to 90%.
Flow Cytometric Analysis.
Single-cell suspensions of thymocytes or splenocytes were assessed by three-color flow cytometry using anti-CD8 CY5 (CT-CD8a; Caltag); anti-CD4–PE (GK1.5; Becton Dickinson), and anti–Qa-2–biotin (clone 1-1-2; PharMingen), followed by streptavidin conjugated to fluorescein (Caltag). In some experiments, cells were stained with either anti–Qa-2–biotin for thymocytes or anti–TCR-β–biotin (clone H57-597; PharMingen) for splenocytes, followed by streptavidin conjugated to CY5 (Caltag). Cells were then stained with annexin V–FITC and propidium iodide (PI; PharMingen).
Immunofluorescence.
Approximately 3 × 106 lymphocytes were collected, washed twice with PBS, suspended in 250 μl, and transferred onto a 24-well plate containing coverslips coated with 150 μg/ml poly-l-lysine. Cells were spun at 1,100 rpm for 10 min to attach cells onto the coverslip, fixed with methanol at −20°C for 20 min, washed three times with PBS, blocked with 2.5% goat serum + 1% BSA at room temperature for 60 min at 4°C, incubated with γ-H2AX primary antibody 13 14 for 1 h, washed, and incubated with Alexa 488–conjugated goat anti–rabbit secondary antibody. Nuclear counterstaining was performed by incubating slides with 1:500 Topro-3 (Molecular Probes) for 25 min. Slides were then mounted with Mowiol and examined using confocal microscopy.
Results
Selective Resistance of Immature Thymocyte Subpopulations to Death Mediated by ActD.
To examine the effect of ActD on thymocytes, single-cell suspensions of thymocytes were cultured overnight in 10 μg/ml of ActD, following which cells were harvested, counted, and analyzed by flow cytometry for surface expression of CD4, CD8, and the maturation marker Qa-2. Overall cell recoveries were minimally affected by treatment with ActD (Fig. 1). Flow cytometric analysis, however, revealed selective loss of specific thymocyte populations. Surface expression of Qa-2 defines populations of thymocytes that are mature and have achieved functional competence 15 16. Immature CD4+ CD8+ (DP) thymocytes are uniformly Qa-2−, whereas mature single-positive (SP) T cells in extrathymic peripheral tissues are uniformly Qa-2+. Expression of Qa-2 by SP thymocytes is acquired relatively late during intrathymic maturation, such that only 30–40% of CD4 SP thymocytes are Qa-2+, whereas 85–90% of CD8 SP thymocytes are Qa-2+. Surprisingly, we found that sensitivity to ActD treatment correlated with expression of Qa-2 in that all Qa-2+ thymocyte populations were depleted by treatment with ActD (Fig. 1 A and cell recoveries in Fig. 1 B). In contrast, immature Qa-2− thymocytes, which includes DP and immature CD4 SP thymocytes, were selectively resistant to ActD treatment.
Figure 1.


Differential resistance of immature thymocytes and mature T cells to ActD-induced death. (A) Qa-2+ mature thymocytes are selectively susceptible to ActD-induced death. 2 × 106 thymocytes were cultured overnight, either in medium alone or in medium containing 10 μg/ml of ActD. Contour plots depicting surface expression of CD4 versus CD8, and Qa-2 expression on gated CD4 SP, CD8 SP, or DP thymic subpopulations is shown. Death of Qa-2+ cells occurs 8–16 h after initiation of culture. (B) Cell recoveries of ActD-treated T cell subpopulations. To compare different populations on the same graph, the data are normalized such that recoveries in medium alone (open bars) are represented at 100%, with the absolute number of cells in each subpopulation recovered indicated within the bar. Recoveries in ActD (filled bars) are represented as a percentage of cells recovered when cultured in medium alone. Error bars represent 1 SEM. DP Qa-2+ thymocytes and splenic T cells as well as Qa-2− CD8 SP thymocytes do not constitute defined populations and so are labeled “Not defined.” Recoveries of total, DP, CD4 SP, or CD8 SP populations are shown among Qa-2− thymocytes, Qa-2+ thymocytes, and splenic T cells (all Qa-2+).
As mature SP thymocytes were sensitive to ActD, we asked whether splenic T cells that are uniformly mature and Qa-2+ 15 would be similarly sensitive to ActD treatment. We treated single-cell suspensions of splenocytes with ActD in overnight cultures (Fig. 1 B). As was the case with mature Qa-2+ thymocytes, CD4 SP and CD8 SP Qa-2+ splenic T cells were uniformly sensitive to ActD. Thus, immature Qa-2− thymocytes are resistant to death induced by ActD whereas mature Qa-2+ T cells are sensitive to ActD-induced death.
Because the cellular pattern of death induced by ActD (mature T cells >> immature thymocytes) was opposite to that observed in normal lymphocytes with all other stimuli 6, we wished to characterize the death induced by ActD further. Death in a variety of systems results from cleavage of preformed protease procaspases to active caspases 17. As is evident in Fig. 2 A, addition of the caspase inhibitor ZVAD-FMK to cultures prevented death induced by ActD, whereas addition of the control peptide ZFA-FMK had no effect. Thus, the death induced by Act-D is caspase dependent. This death was prevented by overexpression of the antiapoptotic protein Bcl-2, as CD8 SP thymocytes from Bcl-2–transgenic mice were completely protected from the effects of ActD in overnight culture (Fig. 2 B, top). ActD-induced death was not dependent on Fas–Fas ligand interactions, as CD8 SP thymocytes from B6.gld mice were also sensitive to ActD (Fig. 2 B, bottom). The death of mature Qa-2+ T cells induced by ActD is the result of apoptosis, as further revealed by annexin V+ staining of viable cells while they are still PI− (Fig. 2 C). Thus, the death induced by ActD in mature T cell subpopulations operates by a Bcl-2–regulated apoptotic pathway that results in caspase activation and that is independent of Fas–Fas ligand interactions.
Figure 2.



Death of Qa-2+ T cells induced by ActD is apoptosis mediated by caspases. (A) ActD-induced death requires activation of caspases. Single-cell suspensions of B6 thymocytes were cultured overnight in medium (open bars), ActD alone, with the caspase inhibitor ZVAD in the presence of ActD, or with control ZFA in the presence of ActD (all filled bars). Cell recoveries of Qa-2+ CD8 SP thymocytes are shown normalized to recovery in medium alone. (B) Death of cells treated with ActD is prevented by Bcl-2 and does not require Fas–Fas ligand interaction. Single-cell suspensions of thymocytes from B6 control mice, mice expressing a Bcl-2 transgene in T lineage cells, or B6.gld mice were cultured overnight in medium alone (open bar) or ActD (filled bars). Recoveries of representative CD8 SP cells (Qa-2+) are shown normalized to recoveries of cells cultured in medium alone. (C) Death of cells treated with ActD results from apoptosis. Single-cell suspensions of thymocytes or splenocytes were cultured overnight in medium alone (dashed line) or ActD (solid line) and then stained with annexin V, PI, and either Qa-2 or TCR-β. Annexin V staining on PI− cells is shown for Qa-2− thymocytes, Qa-2+ thymocytes, and TCR-β+ splenic T cells and reveals the translocation of phosphatidylserine from the inner to the outer leaflet of the membrane before the loss of membrane integrity. Percentages of annexin V+ cells in cultures with ActD are indicated, after subtraction of the percentage of annexin V+ cells in cultures with medium alone. expt., experiment.
ActD is a transcriptional inhibitor that functions by intercalating into DNA 18 and as such eventually inhibits protein synthesis. To elucidate the mechanism of ActD-induced death, we first treated thymocytes with the protein synthesis inhibitor CHX to ask whether ActD-induced death was due to its inhibition of protein synthesis. In fact this was not the case, as inhibition of protein synthesis with CHX neither induced Qa-2+ thymocyte death nor protected these cells from death induced by ActD (Fig. 3 A). We next examined whether transcriptional inhibition was the basis for ActD-induced death of Qa-2+ thymocytes by assessing the effect of DRB, a transcriptional inhibitor that does not intercalate into DNA but instead directly inhibits RNA polymerase II 19. DRB was unable to induce death of Qa-2+ thymocyte subpopulations (Fig. 3 B), even though the concentration of DRB used was sufficient to prevent CD4 and CD8 transcription (not shown). Finally, we considered that ActD-induced death of mature Qa-2+ T cells resulted from its intercalation into DNA by assessing the effect of a different intercalating agent, DhE (a derivative of ethidium bromide that is able to enter living cells). Importantly, DhE did induce death of thymocyte subpopulations in a pattern identical to that induced by ActD (Fig. 3 C). Mature Qa-2+ cells were selectively lost in cell cultures treated with DhE, whereas Qa-2− cells again remained resistant. Thus, mature T cells but not immature thymocytes are susceptible to death induced by DNA intercalating agents.
Figure 3.



Mechanism of ActD-induced death. (A) Inhibition of protein synthesis is not sufficient to induce cell death. Single-cell suspensions of thymocytes from B6 mice were cultured overnight in medium alone (open bar), ActD, or CHX at 10 μg/ml. Some cells were pretreated with CHX for 6 h before addition of ActD (CHX + ActD). Cell recoveries of CD8 SP Qa-2+ cells are shown and are normalized to the recovery in medium alone. The concentration of CHX used was sufficient to prevent CD4 and CD8 protein synthesis (not shown). (B) Transcriptional inhibition is not sufficient to induce cell death. Single-cell suspensions of thymocytes from B6 mice were cultured overnight in medium alone (open bar), ActD, or graded doses of the transcriptional inhibitor DRB (all filled bars). Cell recoveries of CD8 SP Qa-2+ cells are shown and are normalized to the recovery in medium alone. (C) The DNA intercalating agent DhE also induces death of Qa-2+ thymocytes. Single-cell suspensions of B6 thymocytes were cultured in medium alone (open bars), ActD, or in graded doses of the intercalating agent DhE that is able to enter live cells (all filled bars). Cell recoveries of Qa-2+ cells are shown normalized to the recovery of cells cultured in medium alone. Recoveries of Qa-2− thymocytes is shown in the left panel and those of Qa-2+ thymocytes in the right panel.
A Pathway of DNA Damage Response in Mature but Not Immature T Cells.
The ability of DNA intercalating agents to induce cells to die suggested that such death might be a cellular response to DNA damage. We therefore wished to examine whether DNA intercalating agents such as ActD inflicted DNA damage in treated T cells. However, the process of apoptosis itself results in oligonucleosomal fragmentation of DNA 6 after caspase activation. We therefore examined LNT cells from Bcl-2–transgenic mice, as Bcl-2–transgenic cells do not fragment DNA in response to apoptotic signals that otherwise induce death in nontransgenic T cells 17. To examine whether DNA intercalating agents were indeed inducing DNA damage, we examined phosphorylation of histone H2AX on serine residue 139 (Fig. 4). Serine phosphorylation of histone H2AX on position 139 is known to result from a wide variety of DNA-damaging insults that produce DNA double-stranded breaks (dsbs) and is an early and specific indicator of DNA damage 13 14. We therefore compared the level of serine phosphorylation of H2AX (γ-H2AX) in ActD-treated LN mature T cells (95% T cells) by immunostaining. Untreated LNT cells showed essentially no γ-H2AX in overnight culture (Fig. 4). Upon addition of ActD, multiple foci of γ-H2AX were visible in LNT cells. Thus, DNA intercalating agents such as ActD induce DNA damage in mature T cells.
Figure 4.
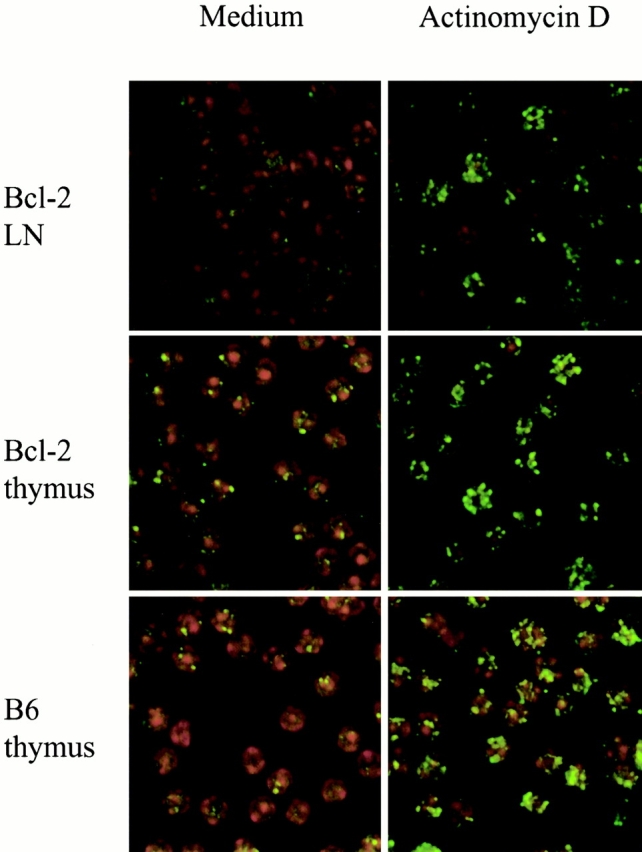
ActD induces dsbs as indicated by phosphorylation of the oligonucleosomal histone H2AX on serine 139 (γ-H2AX). LNT cells from mice transgenic for Bcl-2 were cultured either in medium alone or in ActD at 10 μg/ml. Thymocytes from Bcl-2–transgenic mice or B6 mice were also cultured in medium or ActD at 10 μg/ml. The level of γ-H2AX within cultured cells was then visualized by immunostaining (green fluorescence). Nuclear counterstaining was performed with Topro-3 (red fluorescence).
To determine if ActD also induced DNA damage in immature thymocytes, we next examined γ-H2AX in untreated and ActD-treated immature thymocyte populations (Fig. 4). Interestingly, we found that immature thymocytes, unlike mature LNT cells, had a few γ-H2AX foci even in the absence of ActD, indicative of dsbs occurring during TCR gene rearrangements, a point that will be addressed in further experiments elsewhere. Importantly, the number of γ-H2AX foci increased dramatically in immature thymocytes after ActD treatment, indicating that ActD induced DNA damage in both immature thymocytes and mature T cells. We conclude that immature thymocytes and mature T cells do not differ in the DNA damage inflicted by ActD but do differ in their response to this type of DNA damage.
To gain further insight into the mechanism of death induced by DNA intercalating agents, we assayed mutant strains of mice engineered to lack molecules important for the cellular response to DNA damage. We compared mice deficient in either p53 10 or Atm 8 with wild-type mice (Fig. 5). Cells from p53 −/− mice were indistinguishable from wild-type cells in that Qa-2− thymocytes were resistant to death induced by ActD, whereas Qa-2+ T cells in both thymus and spleen were sensitive to death induced by ActD in a dose-dependent manner. In contrast, Atm −/− cells were relatively resistant to death induced by ActD. Indeed, Qa-2+ cells from Atm −/− mice were resistant to doses of ActD that resulted in the death of the majority of Qa-2+ cells from wild-type and p53 −/− mice. These data confirm that the mechanism of death induced by ActD is due to a cellular response to DNA damage and demonstrate that death of Qa-2+ mature T cells is due to an Atm-dependent but p53-independent pathway.
Figure 5.

Death induced by ActD in Qa-2+ T cells is Atm dependent but not p53 dependent. Whole thymocytes or splenocytes from B6 mice (□), p53 −/− mice (⋄), and Atm −/− mice (○) were cultured in medium or ActD at the indicated concentrations. Cell recoveries of DP Qa-2− immature thymocytes (left panel), CD8 SP Qa-2+ mature thymocytes (center panel), and CD8 SP splenocytes (right panel) cultured in ActD are shown normalized to recovery in medium alone (0 μg/ml ActD on abscissa).
Discussion
We have demonstrated a novel pathway of death that results from DNA damage induced by DNA intercalating agents in quiescent mature T cells. This death induces phosphatidylserine translocation from the inner to the outer leaflet of the plasma membrane before loss of membrane integrity, results from caspase activation, and can be regulated by Bcl-2, and so we conclude that death is due to cellular apoptosis. In mature T cells, apoptosis results from an Atm-dependent signaling pathway that does not require the tumor suppressor gene p53. In contrast to mature T cells, immature thymocytes are uniquely resistant to apoptosis induced by this Atm-dependent signaling pathway even though their DNA is also damaged, likely reflecting the necessity for immature thymocytes to tolerate dsbs that occur normally during rearrangement of clonotypic TCRs 20.
Immature thymocytes and mature T cells have been previously shown to respond very differently to the same stimuli (for review see references 5 and 6). Indeed, immature DP thymocytes die in response to a wide variety of stimuli that do not result in death of mature T cell populations 6. We were therefore surprised to discover that immature thymocytes were resistant to death that was induced in mature T cells by DNA intercalating agents, as we knew of no other stimulus resulting in this pattern of cell death in normal lymphocytes. Immature DP thymocytes represent a unique stage of T cell differentiation at which cells rearrange and express their TCR-α locus to generate the clonotypic α/β TCR 2. It is known that rearrangement at the TCR-α locus results in recombination activating gene–mediated DNA breaks 20, and sequential rearrangements at the TCR-α locus may impose a particularly heavy burden of DNA breaks on this cell population 21. It is likely that developmentally regulated alterations in the cellular response to DNA dsbs are required to prevent apoptosis of immature thymocytes undergoing receptor rearrangements and that these specific alterations are responsible for the unique ability of immature thymocytes to tolerate DNA breaks induced by DNA intercalating agents.
Whereas immature DP thymocytes do not die in response to ActD-induced DNA damage, DP thymocytes do die in response to DNA damage induced by gamma irradiation. The altered cellular pattern of cell death to DNA damage induced by ActD versus gamma irradiation reflect differing molecular responses to the DNA damage inflicted by these two different stimuli. Death of quiescent lymphocytes from DNA damage induced by gamma irradiation requires p53, with the role of ATM still unresolved 22 23. In contrast, our study demonstrates that death from DNA damage induced by ActD is Atm dependent but p53 independent. These differing molecular responses to DNA damage induced by ActD versus gamma irradiation likely reflect differences in the type of DNA damage that these two different stimuli inflict. Gamma irradiation damages DNA by producing free radicals, whereas DNA intercalating agents alter DNA sites that may then be cleaved by enzymes such as topoisomerases, resulting in dsbs 24. Importantly, this study demonstrates that the Atm-dependent but p53-independent apoptotic death pathway that is activated by DNA intercalating agents in quiescent mature T cells is not activated in immature thymocytes. Even though the Atm gene itself is expressed in immature thymocytes 25, we suspect that upstream activators or downstream substrates of Atm are more likely to be modulated in immature thymocytes to inhibit apoptosis than the Atm gene itself. Unfortunately, little is yet known about the mechanism by which Atm is activated in response to DNA damage 26, but it is clear that Atm regulates the activity of a growing list of substrates other than p53, including c-abl, Brca-1 and 2, and nuclear factor κB 27 28 29. In this regard, a p53-independent pathway of apoptosis in response to DNA damage that requires IFN regulatory factor 1 has been previously demonstrated in lymphoblasts, although not in quiescent T cells 30 31.
In conclusion, our results indicate that downregulation of an Atm-dependent, p53-independent death pathway allows immature thymocytes undergoing antigen receptor gene rearrangements to tolerate dsbs.
Acknowledgments
We thank William M. Bonner for antibody to γ-H2AX, Richard J. Hodes and Lisa Petiniot for their comments on this manuscript, and Henry Chen and Tilmann M. Brotz for technical assistance.
References
- Kisielow P., von Boehmer H. Development and selection of T cellsfacts and puzzles. Adv. Immunol. 1995;58:87–209. doi: 10.1016/s0065-2776(08)60620-3. [DOI] [PubMed] [Google Scholar]
- von Boehmer H., Fehling H.J. Structure and function of the pre-T cell receptor. Annu. Rev. Immunol. 1997;15:433–452. doi: 10.1146/annurev.immunol.15.1.433. [DOI] [PubMed] [Google Scholar]
- Marrack P., Kappler J. Positive selection of thymocytes bearing alpha beta T cell receptors. Curr. Opin. Immunol. 1997;9:250–255. doi: 10.1016/s0952-7915(97)80144-6. [DOI] [PubMed] [Google Scholar]
- Egerton M., Scollay R., Shortman K. Kinetics of mature T-cell development in the thymus. Proc. Natl. Acad. Sci. USA. 1990;87:2579–2582. doi: 10.1073/pnas.87.7.2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J.J. Programmed cell death in the immune system. Adv. Immunol. 1991;50:55–85. doi: 10.1016/s0065-2776(08)60822-6. [DOI] [PubMed] [Google Scholar]
- Cohen J.J. Apoptosis. Immunol. Today. 1993;14:126–130. doi: 10.1016/0167-5699(93)90214-6. [DOI] [PubMed] [Google Scholar]
- Smith C.A., Williams G.T., Kingston R., Jenkinson E.J., Owen J.J. Antibodies to CD3/T-cell receptor complex induce death by apoptosis in immature T cells in thymic cultures. Nature. 1989;337:181–184. doi: 10.1038/337181a0. [DOI] [PubMed] [Google Scholar]
- Barlow C., Hirotsune S., Paylor R., Liyanage M., Eckhaus M., Collins F., Shiloh Y., Crawley J.N., Ried T., Tagle D. Atm-deficient micea paradigm of ataxia telangiectasia. Cell. 1996;86:159–171. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Liao M.J., Yin C., Barlow C., Wynshaw-Boris A., van Dyke T. Atm is dispensable for p53 apoptosis and tumor suppression triggered by cell cycle dysfunction. Mol. Cell. Biol. 1999;19:3095–3102. doi: 10.1128/mcb.19.4.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timme T.L., Thompson T.C. Rapid allelotype analysis of p53 knockout mice Biotechniques. 17 1994. 460, 462–463 [PubMed] [Google Scholar]
- Linette G.P., Grusby M.J., Hedrick S.M., Hansen T.H., Glimcher L.H., Korsmeyer S.J. Bcl-2 is upregulated at the CD4+ CD8+ stage during positive selection and promotes thymocyte differentiation at several control points. Immunity. 1994;1:197–205. doi: 10.1016/1074-7613(94)90098-1. [DOI] [PubMed] [Google Scholar]
- Nakayama T., June C.H., Munitz T.I., Sheard M., McCarthy S.A., Sharrow S.O., Samelson L.E., Singer A. Inhibition of T cell receptor expression and function in immature CD4+CD8+ cells by CD4. Science. 1990;249:1558–1561. doi: 10.1126/science.2120773. [DOI] [PubMed] [Google Scholar]
- Rogakou E.P., Boon C., Redon C., Bonner W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou E.P., Pilch D.R., Orr A.H., Ivanova V.S., Bonner W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- Vernachio J., Li M., Donnenberg A.D., Soloski M.J. Qa-2 expression in the adult murine thymus. A unique marker for a mature thymic subset. J. Immunol. 1989;142:48–56. [PubMed] [Google Scholar]
- Ramsdell F., Jenkins M., Dinh Q., Fowlkes B.J. The majority of CD4+8− thymocytes are functionally immature. J. Immunol. 1991;147:1779–1785. [PubMed] [Google Scholar]
- Rathmell J.C., Thompson C.B. The central effectors of cell death in the immune system. Annu. Rev. Immunol. 1999;17:781–828. doi: 10.1146/annurev.immunol.17.1.781. [DOI] [PubMed] [Google Scholar]
- Muller W., Crothers D.M. Studies of the binding of actinomycin and related compounds to DNA. J. Mol. Biol. 1968;35:251–290. doi: 10.1016/s0022-2836(68)80024-5. [DOI] [PubMed] [Google Scholar]
- Zandomeni R., Mittleman B., Bunick D., Ackerman S., Weinmann R. Mechanism of action of dichloro-beta-d-ribofuranosylbenzimidazoleeffect on in vitro transcription. Proc. Natl. Acad. Sci. USA. 1982;79:3167–3170. doi: 10.1073/pnas.79.10.3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsden D.A., van Gent D.C., Gellert M. Specificity in V(D)J recombinationnew lessons from biochemistry and genetics. Curr. Opin. Immunol. 1997;9:114–120. doi: 10.1016/s0952-7915(97)80167-7. [DOI] [PubMed] [Google Scholar]
- Petrie H.T., Livak F., Schatz D.G., Strasser A., Crispe I.N., Shortman K. Multiple rearrangements in T cell receptor alpha chain genes maximize the production of useful thymocytes. J. Exp. Med. 1993;178:615–622. doi: 10.1084/jem.178.2.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Baltimore D. Dual roles of ATM in the cellular response to radiation and in cell growth control. Genes Dev. 1996;10:2401–2410. doi: 10.1101/gad.10.19.2401. [DOI] [PubMed] [Google Scholar]
- Elson A., Wang Y., Daugherty C.J., Morton C.C., Zhou F., Campos-Torres J., Leder P. Pleiotropic defects in ataxia-telangiectasia protein-deficient mice. Proc. Natl. Acad. Sci. USA. 1996;93:13084–13089. doi: 10.1073/pnas.93.23.13084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross W.E., Bradley M.O. DNA double-stranded breaks in mammalian cells after exposure to intercalating agents. Biochim. Biophys. Acta. 1981;654:129–134. doi: 10.1016/0005-2787(81)90145-3. [DOI] [PubMed] [Google Scholar]
- Chen G., Lee E. The product of the ATM gene is a 370-kDa nuclear phosphoprotein. J. Biol. Chem. 1996;271:33693–33697. doi: 10.1074/jbc.271.52.33693. [DOI] [PubMed] [Google Scholar]
- Canman C.E., Lim D.S., Cimprich K.A., Taya Y., Tamai K., Sakaguchi K., Appella E., Kastan M.B., Siliciano J.D. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- Brown K.D., Barlow C., Wynshaw-Boris A. Multiple ATM-dependent pathwaysan explanation for pleiotropy. Am. J. Hum. Genet. 1999;64:46–60. doi: 10.1086/302223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim D.S., Kim S.T., Xu B., Maser R.S., Lin J., Petrini J.H., Kastan M.B. ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature. 2000;404:613–617. doi: 10.1038/35007091. [DOI] [PubMed] [Google Scholar]
- Hirao A., Kong Y.Y., Matsuoka S., Wakeham A., Ruland J., Yoshida H., Liu D., Elledge S.J., Mak T.W. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science. 2000;287:1824–1827. doi: 10.1126/science.287.5459.1824. [DOI] [PubMed] [Google Scholar]
- Strasser A., Harris A.W., Jacks T., Cory S. DNA damage can induce apoptosis in proliferating lymphoid cells via p53-independent mechanisms inhibitable by Bcl-2. Cell. 1994;79:329–339. doi: 10.1016/0092-8674(94)90201-1. [DOI] [PubMed] [Google Scholar]
- Tamura T., Ishihara M., Lamphier M.S., Tanaka N., Oishi I., Aizawa S., Matsuyama T., Mak T.W., Taki S., Taniguchi T. An IRF-1-dependent pathway of DNA damage-induced apoptosis in mitogen-activated T lymphocytes. Nature. 1995;376:596–599. doi: 10.1038/376596a0. [DOI] [PubMed] [Google Scholar]