

Abstract
Plasma von Willebrand factor (vWF) is a multimeric protein that mediates adhesion of platelets to sites of vascular injury. Only the very large vWF multimers are effective in promoting platelet adhesion in flowing blood. A protein disulfide bond reductase in plasma reduces the average multimer size of vWF secreted by endothelial cells. This activity has been isolated from human endothelial cell conditioned medium and shown to be the trimeric glycoprotein, thrombospondin-1 (TSP-1). Incubation of purified TSP-1 with vWF resulted in formation of thiol-dependent complexes of TSP-1 and vWF, generation of new thiols in vWF, and reduction in the average multimer size of vWF. The ratio of the concentrations of TSP-1 and vWF in plasma reflected with average multimer size of vWF. The higher the plasma TSP-1/vWF molar ratio, the smaller the average vWF multimer size. In addition, administration of TSP-1 to mice resulted in reduction in the average multimer size of plasma vWF. Interaction of TSP-1 with vWF is mediated by TSP-1 type 1 properdin domains and the vWF A3 domain. These results indicate that TSP-1 regulates the multimeric size and therefore hemostatic activity of vWF.
Keywords: endothelial cell, reductase, disulfide, thrombosis, hemostasis
Introduction
Binding of platelets to von Willebrand factor (vWF) in the subendothelium of a damaged blood vessel is the initial step in formation of a hemostatic plug 1. vWF is also the carrier for procoagulant factor VIII protecting it from inactivation by activated protein C and factor Xa in the circulating blood. vWF is synthesized by vascular endothelial cells and megakaryocytes and circulates in blood as a series of multimers containing a variable number of ∼500 kD homodimers 2. The largest vWF multimers have a molecular mass of ∼20,000 kD and are comparable in length to the diameter of a medium platelet (2 μM).
vWF dimers are assembled from pairs of ∼250 kD polypeptide subunits in the endoplasmic reticulum via di-sulfide bridges between cysteine residues located in the COOH-terminal regions. Intersubunit disulfide bonds involve one or three of the Cys residues at positions 2,008, 2,010, and 2,048 3. Subsequently, multimers are formed by interdimeric disulfide linking of NH2-terminal domains in a parallel orientation 4. Interdimeric disulfide bonds involve Cys379 and one or more of the Cys residues at positions 459, 462, and 464 5.
Only the large multimeric forms of vWF are hemostatically active 6. This relates to affinity of vWF for its ligands. Binding of multimeric vWF to collagen occurs with ∼100-fold higher affinity than binding of monomeric vWF fragments. Similarly, large vWF multimers bind to activated platelets with up to 10-fold higher affinity than small multimers, or with ∼100-fold higher affinity than monomeric fragments (for a review, see reference 6). Large multimers of vWF have considerably higher ristocetin cofactor activity per unit antigen than small multimers 7, and the unusually large vWF multimers secreted by endothelial cells have been shown to be more effective than the largest plasma forms in inducing platelet aggregation under conditions of high fluid shear 8. Some thrombotic disorders are characterized by altered vWF multimer size. Thrombotic thrombocytopenic purpura (TTP) is usually associated with unusually large vWF multimers in the blood which are thought to precipitate intravascular platelet clumping 9. Conversely, lower than average multimer size characterizes the bleeding diathesis of type II von Willebrand disease.
vWF secreted by cultured endothelial cells into the medium in the presence of plasma cryosupernatant is of smaller average size than the vWF deposited into the subendothelial matrix 10. Frangos et al. 10 proposed that a substance in plasma cryosupernatant is responsible for reducing the size of the large vWF multimeric forms secreted into the vascular lumen to the somewhat smaller vWF multimers ordinarily found in plasma. They found that the vWF depolymerizing activity was inactivated by thiol blocking agents and there was no evidence for proteolysis of the depolymerized vWF. This prompted Phillips et al. 11 to suggest that the activity was a type of limited disulfide bond reductase.
We recently reported that the conditioned medium of cultured macrovascular and microvascular endothelial cells contained an activity which reduced the average multimer size of plasma or purified vWF 12. The reducing activity was ablated by pretreatment with heat or thiol blocking agents, but not by a range of specific proteinase inhibitors. Reduction in vWF multimer size was associated with formation of new thiols in vWF and there was no evidence for additional proteolytic processing of vWF. These findings suggested that the disulfide bonds that link the vWF multimers were being reduced by a vWF reductase secreted by endothelial cells. This reductase has been isolated and shown to be the trimeric glycoprotein, thrombospondin-1 (TSP-1).
Materials and Methods
Proteins and Reagents.
Leupeptin, phenylmethylsulfonyl fluoride, soybean trypsin inhibitor, and D-Phe-Pro-Arg-chloromethyl ketone were from Calbiochem, and N-ethylmaleimide (NEM), reduced glutathione (GSH), and EDTA were from Sigma-Aldrich. 3-(N-maleimidylpropionyl)biocytin (MPB) was from Molecular Probes. vWF, TSP-1, and scrambled peptides were synthesized by Auspep. The peptide purity was >95%. Purified vWF from human plasma was a gift from Dr. M. Berndt (Baker Medical Research Institute, Melbourne, Australia) and was purified according to Booth et al. 13. TSP-1 was purified from human platelet concentrates as described previously 14 with some modifications 15. Buffers containing either 0.1 mM or 2 mM CaCl2 were used throughout the chromatographic purification of TSP-1. TSP-1 concentration was calculated using an absorption coefficient for a 1% solution at 280 nm of 10.9. The murine anti–TSP-1 monoclonal antibody hybridoma cell line, HB8432 16, was obtained from American Type Culture Collection. Antibody was produced in ascites and purified using Protein G-Sepharose (Amersham Pharmacia Biotech). All other reagents were of analytical grade.
Normal and TTP Plasmas.
Blood was collected by atraumatic venipucture into citrate. Plasma was prepared by centrifugation at 2,000 g for 20 min at 4°C and stored at −20°C.
TTP1.
Plasma was obtained during therapeutic plasmapheresis of a 73-yr-old female with TTP. For 2 wk before admission she had been treated with “triple therapy” for Helicobacter pylorii. On admission she had severe diarrhea, was confused, and was in cardiac and renal failure (serum creatinine was 621 mol/liter [60–111]). The hemoglobin was 99 g/liter (120–153), white cell count 8.4 × 109 per liter 4 5 6 7 8 9 10 11, and platelet count 23 × 109 per liter (150–400). She responded to repeated plasmapheresis and dialysis and was discharged well.
TTP2.
A 2-yr-old girl was admitted to hospital with a fever and evidence of a petechial rash. Her platelet count and hemoglobin was 15 × 109 per liter and 62 g/liter, respectively. The blood film was polychromatic with the occasional nucleated red blood cell and a moderate number of schistocytes. The reticulocyte count was raised and the direct antiglobulin test was negative. Her renal function was normal. A diagnosis of a microangiopathic hemolytic anemia/probable TTP was made and plasma therapy commenced. After the infusion of 200 ml of plasma her platelet count rose progressively to normal over 72 h with gradual resolution of her hemolytic anemia. She has been readmitted with thrombocytopenia and a microangiopathic hemolytic anemia on numerous occasions, usually with an accompanying febrile illness. Periods of remission have ranged from 1–9 mo. Increments in her platelet count are usually noted within 48 h of the infusion of a single unit of plasma. She has not received glucocorticoids and her glucose-6-phosphate dehydrogenase assay was normal.
TTP3.
A 29-yr-old man was admitted to hospital with a history of fever, confusion, malaise, abdominal pain, oral mucosal bleeding, melena, and renal impairment. On admission he was severely thrombocytopenic and anemic with a platelet count of 2 × 109 per liter and hemoglobin of 56 g/liter. A presumptive diagnosis of acalculous cholecystitis and immune thrombocytopenia with associated bleeding was made and the patient commenced on antibiotic therapy and intravenous Ig infusion. The plasma fibrinogen level and D-Dimer assay were normal. He failed to respond to treatment and over the next few days his blood film demonstrated increasing numbers of fragmented erythrocytes. He was commenced on plasma exchange with cryosupernatant replacement. He responded well with a rapid resolution of his hemolytic anemia and fever with a slower but progressive improvement of his thrombocytopenia and renal impairment.
Assays for vWF Multimer Size.
TTP plasma was incubated with 20 mM Hepes, 0.14 M NaCl, 1 mM CaCl2, 1 mM MgCl2, pH 7.4 buffer, conditioned media of HMEC-1 cells, or the Hepes buffer containing TSP-1 for 1 or 24 h at 37°C. Volumes and concentrations of reactants are indicated in the figure legends. Aliquots of the reactions were diluted 10-fold in the Hepes buffer and assayed for collagen binding affinity and vWF antigen as described by Favaloro et al. 17. On some occasions, aliquots of the reactions were resolved on 1% agarose gel electrophoresis 18, transferred to polyvinylidine difluoride (PVDF) membrane (Dupont/NEN Life Science Products), blotted with 2 μg/ml of horseradish peroxidase–conjugated anti-vWF polyclonal antibodies (Dako), and visualized using chemiluminescence (Dupont/NEN Life Science Products). Murine plasma vWF was assayed in the same way except that anti–rat vWF polyclonal antibodies (Cedarlane) were used.
Purification of vWF Reductase.
Conditioned medium of HMEC-1 cells was prepared using Nunc Cell Factories. HMEC-1 (∼80,000 cells/cm2 of cell factory area) were seeded into cell factories in MCDB-131 medium (GIBCO BRL) containing 10 ng/ml epidermal growth factor (GIBCO BRL), 1 μg/ml hydrocortisone (Sigma-Aldrich), and 10% fetal calf serum (GIBCO BRL). When the cells were at ∼80% confluence they were washed twice with phosphate-buffered saline (Sigma-Aldrich) and incubated with serum-free MCDB-131 medium at 37°C, 5% CO2 for 30 h. The conditioned medium was collected, centrifuged at 1200 g for 10 min and passed through a 0.2-μm Millipore filter to remove detached cells and cellular debris, and stored at −20°C. 30 liters of conditioned medium was concentrated to 350 ml using a Amicon spiral-wound concentrator with a 10-kD cutoff membrane. The proteinase inhibitors, leupeptin (10 μM), phenylmethylsulfonyl fluoride (1 mM), and soybean trypsin inhibitor (10 μg/ml) were added to the concentrated medium to minimize proteolytic degradation of the vWF reductase. The concentrated medium was applied to a 150-ml column of heparin-Sepharose (2.5 × 30 cm; Amersham Pharmacia Biotech) equilibrated with 20 mM Hepes, 1 mM CaCl2, 1 mM MgCl2, and 0.02% NaN3, pH 7.4 buffer. The column was washed with 3 bed volumes of the Hepes buffer at a flow rate of 0.5 ml/min to elute unbound proteins and developed with a 2.2-liter linear NaCl gradient from 0 to 1 M in the Hepes buffer. vWF reductase activity eluted at ∼0.3 M NaCl (∼700 ml). The fractions containing vWF reductase activity (∼45 ml) were concentrated to 5 ml, dialyzed against 20 mM Hepes, 0.05 M NaCl, 1 mM CaCl2, 1 mM MgCl2, 0.02% NaN3, pH 7.4 buffer, and applied to a 210-ml column of Sephacryl S-300 HR (1.5 × 120 cm; Amersham Pharmacia Biotech) at a flow rate of 0.5 ml/min. The vWF reductase activity eluted at ∼85 ml.
Electrophoresis and Western Blotting.
Samples were resolved on 4–15% SDS-PAGE 19. On one occasion, proteins were reduced with 20 mM dithiothreitol and the cysteines alkylated with 40 mM iodoacetamide before SDS-PAGE. On another occasion, samples were resolved on SDS-PAGE, transferred to PVDF membrane, and blotted with the HB8432 monoclonal antibody (used at 2 μg/ml). The murine antibody was blotted with rabbit anti–mouse peroxidase–conjugated antibodies (Dako; used at 1:2,000 dilution) and detected by chemiluminescence (DuPont/NEN Life Science Products).
Assay for Formation of New Thiols in vWF.
The biotin-linked maleimide, MPB, was used to measure reduction of vWF disulfide bond(s) by TSP-1. The protocol was essentially as described by Xie et al. 12. In brief, purified human vWF (2 μg/ml) was incubated with 20 mM Hepes, 0.14 M NaCl, 1 mM CaCl2, 1 mM MgCl2, pH 7.4 buffer, HMEC-1–conditioned medium, or the Hepes buffer containing purified human TSP-1 or peptides for 60 min at 37°C. Free thiol(s) formed in vWF by reduction of disulfide bond(s) were labeled with MPB (100 μM) for 10 min at 37°C, and the unreacted MPB was quenched with GSH (200 μM) for 10 min at 37°C. The MPB-labeled vWF was incubated in microtiter plate wells coated with anti–human vWF polyclonal antibodies and the biotin label was detected using StreptABComplex/HRP (Dako).
Assay for TSP-1.
The HB8432 monoclonal antibody (100 μl of 5 μg/ml in 15 mM Na2CO3, 35 mM NaHCO3, 0.02% NaN3, pH 9.6 buffer) was adsorbed to Nunc PolySorp 96-well plates overnight at 4°C in a humid environment. Wells were washed once with 20 mM Hepes, 0.14 M NaCl, pH 7.4 buffer (HBS) containing 0.05% Tween 20 (HBS/Tween), nonspecific binding sites blocked by adding 200 μl of 2% BSA in HBS and incubating for 90 min at 37°C, and then washed two times with HBS/Tween. Normal plasma was depleted of TSP-1 by immunoaffinity chromatography on HB8432-Sepharose CL-4B matrix and then spiked with known amounts of purified platelet TSP-1. The spiked plasmas and normal or patient plasmas were diluted in HBS/Tween and 100 μl aliquots added to antibody-coated wells and incubated for 30 min at room temperature with orbital shaking. Plasmas were assayed in triplicate. Wells were washed three times with HBS/Tween and 100 μl of 5 μg/ml of biotin-labeled HB8432 antibody added and incubated for 30 min at room temperature with orbital shaking. Labeled HB8432 antibody was prepared using the Biotin-XX Protein Labeling Kit from Molecular Probes. Detection of bound TSP-1 with the same monoclonal antibody used to coat the wells was possible because of the homotrimeric structure of TSP-1. Wells were washed three times with HBS/Tween, and 100 μl of 1:100 dilution of StreptABComplex/HRP (Dako) in HBS/Tween was added and incubated for 30 min at room temperature with orbital shaking. Wells were washed three times with HBS/Tween and the peroxidase detected as described previously 17. Control wells contained TSP-1–depleted plasma. Absorbances were a linear function of TSP-1 concentration over the range 1–100 ng/ml.
Assay of Binding of TSP-1 to vWF.
Purified human vWF (100 μl of 5 μg/ml in 0.1 M NaHCO3, pH 8.3 buffer) were adsorbed to Nunc PolySorp 96-well plates overnight at 4°C in a humid environment. Wells were washed once with HBS, nonspecific binding sites blocked by adding 200 μl of 2% BSA in HBS and incubating for 90 min at 37°C, and then washed two times with HBS. Coated wells were incubated with TSP-1 (0 to 10 μg/ml) in 20 mM Hepes, 0.14 M NaCl, 1 mM CaCl2, 1 mM MgCl2, pH 7.4 buffer containing 0, 5, or 20 mM NEM for 30 min at room temperature with orbital shaking. On one occasion, wells not coated with vWF but blocked with BSA were incubated with 10 μg/ml TSP-1. The wells were washed four times with Hepes buffer containing 1 M NaCl and 100 μl of 10 μg/ml anti-TSP1 monoclonal antibody was added and incubated for 30 min at room temperature with orbital shaking. The wells were washed three times with HBS and 100 μl of 1:1,000 dilution of rabbit anti–mouse peroxidase-conjugated antibodies was added and incubated for 30 min at room temperature with orbital shaking. Wells were washed three times with HBS and the peroxidase detected as described previously 17.
Results
Reduction in the Average Multimer Size of vWF by HMEC-1 Cells.
Incubation of TTP1 patient plasma with the conditioned medium of the human dermal microvascular endothelial cell line, HMEC-1 20, resulted in decrease in the average multimer size of vWF (Fig. 1 A). Specifically, the very large multimers were lost (see bracket in Fig. 1 A). There was negligible endogenous vWF in the HMEC-1–conditioned medium (not shown). The loss of the large vWF multimers upon incubation with HMEC-1–conditioned medium was reflected in decrease in affinity of the vWF for collagen (Fig. 1 B). Affinity of vWF for collagen is an accurate and sensitive measure of average vWF multimer size 17 21. Collagen binding was expressed relative to the vWF antigen level which takes into account any variation in the total vWF in the assays.
Figure 1.

Reduction in the average multimer size of vWF by conditioned medium from HMEC-1 cells. (A) TTP1 patient plasma (10 μl) was incubated with Hepes-buffered saline containing 1 mM CaCl2 and MgCl2 (TTP, lane 1) or the conditioned media of HMEC-1 cells (+ECcm, lane 2) (90 μl) for 1 h at 37°C and aliquots of the reaction (10 μl) were resolved on 1% agarose gel electrophoresis. The vWF was transferred to PVDF membrane and Western blotted using peroxidase-conjugated anti-vWF polyclonal antibodies. The bracket highlights the change in the proportion of large vWF multimers in the population. (B) Aliquots of the reactions described in panel A were analyzed for vWF antigen levels and collagen binding affinity. The results are expressed as the ratio of the collagen binding activity and vWF antigen level. The bars and errors are the mean and SD of triplicate determinations.
Purification of vWF Reductase.
The reducing activity secreted by endothelial cells was associated with a protein with an anionic pI that binds heparin and contains reactive thiol(s) 12. 30 liters of conditioned medium from HMEC-1 cells was collected (910 mg), concentrated, and applied to heparin-Sepharose. The bound proteins were resolved with a linear NaCl gradient. vWF reductase activity eluted at ∼0.3 M NaCl (Fig. 2 A). The peak of activity was pooled (∼3 mg), concentrated, and gel filtered on Sephacryl S-300 HR. The vWF reductase activity resolved in the leading peak (∼0.1 mg; Fig. 2 B). The enzyme had a molecular mass of ∼500 kD on SDS-PAGE which reduced to ∼170 kD after reduction and alkylation of the protein (Fig. 2 C). This subunit structure was very similar to that of TSP-1, which is a homotrimer of ∼170 kD subunits that is secreted by endothelial cells 22 and functions in cell–cell and cell–matrix interactions 23. The HMEC-1 protein was recognized by an anti–TSP-1 monoclonal antibody in Western blot (Fig. 2 D), and immunoprecipitation of TSP-1 from HMEC-1–conditioned medium accounted for all the vWF reductase activity in the medium (Fig. 2 E).
Figure 2.

Purification of vWF reductase. (A) 30 liters of conditioned medium from the human dermal microvascular endothelial cell line, HMEC-1, was collected, concentrated, and applied to heparin-Sepharose. The bound proteins were resolved with a linear NaCl gradient. vWF reductase activity eluted at ∼0.3 M NaCl. (B) The peak of activity from the heparin-Sepharose column was pooled, concentrated, and gel filtered on Sephacryl S-300 HR. The vWF reductase activity resolved in the leading peak. (C) A sample (30 μl) of the pooled activity from the Sephacryl S-300 HR column was resolved on 4–15% SDS-PAGE under nonreducing (lane 1) or reducing (lane 2) conditions and silver stained. The vWF reductase had a molecular mass of ∼500 kD which reduced to ∼170 kD after reduction and alkylation of the protein. The positions of M r markers are shown at left. (D) Purified human platelet TSP-1 (50 ng, lane 1) or a sample (30 μl) of the pooled activity from the Sephacryl S-300 HR column was resolved on 4–15% SDS-PAGE under nonreducing conditions, transferred to PVDF membrane, and blotted with the anti–TSP-1 monoclonal antibody, HB8432. The positions of M r markers are shown at left. (E) HMEC-1–conditioned medium (1 ml) was incubated alone or with an anti–TSP-1 or control anti-vWF monoclonal antibody (10 μg/ml) and Protein G-Sepharose beads (50 μl of packed beads) for 1 h at 4°C on a rotating wheel. The Sepharose beads were pelleted by centrifugation and the supernatant was assayed for vWF reductase activity. TTP1 patient plasma (10 μl) was incubated with the HMEC-1–conditioned medium (+ECcm) supernatants (90 μl) for 1 h at 37°C and aliquots of the reaction were analyzed for vWF antigen levels and collagen binding affinity. The results are expressed as the ratio of the collagen binding activity and vWF antigen level. The bars and errors are the mean and SD of triplicate determinations.
Reduction in the Average Multimer Size of vWF by TSP-1 In Vitro.
TSP-1 is a major component of platelet α-granules which is secreted upon platelet activation and aggregation. TSP-1 was purified to homogeneity from pooled outdated human platelet concentrates and tested for vWF reductase activity. Each subunit of TSP-1 contains a free thiol that can reside on any 1 of 12 different cysteines in the COOH-terminal Ca2+-binding repeats and globular domain of TSP-1 24. TSP-1 can be isolated in two different disulfide-bonded conformational states by varying the Ca2+ ion concentration used in the purification buffers. TSP-1 purified in buffers containing 0.1 mM Ca2+ is a homogeneous TSP-1 population in that the free thiol is at Cys974 25, whereas TSP-1 purified in buffers containing 2 mM Ca2+ is probably a heterogeneous population of molecules in which the free thiol resides on 1 of any 12 Cys 24.
Both forms of platelet TSP-1 reduced the average multimer size of vWF in buffer (not shown) or in plasma (Fig. 3 A, data for the 0.1 mM Ca2+ form of TSP-1 is shown). In particular, the very large multimers were lost (see bracket in Fig. 3 A). Decrease in vWF multimer size was associated with decrease in affinity of vWF for collagen (Fig. 3 B). TSP-1–mediated reduction in vWF multimer size was concentration and time dependent (Fig. 3 B). It is noteworthy that the weight ratio of TSP-1 to vWF, and not the absolute concentration of TSP-1, influenced the extent of reduction in vWF multimer size (Fig. 3 B). The effect of TSP-1 on the collagen binding of vWF shown in Fig. 3 B was matched by the decrease in vWF multimer size measured by agarose gel electrophoresis (not shown). Three different preparations of platelet TSP-1 had the same vWF reducing activity (not shown), and addition of TSP-1 to TTP2 and TTP3 patient plasmas also reduced the average multimer size of the plasma vWF (not shown). The vWF reductase activity in endothelial cell–conditioned medium is inhibited by EDTA 12. In accordance with this observation, chelation of Ca2+ with EDTA ablated the vWF reductase activity of TSP-1 (not shown).
Figure 3.
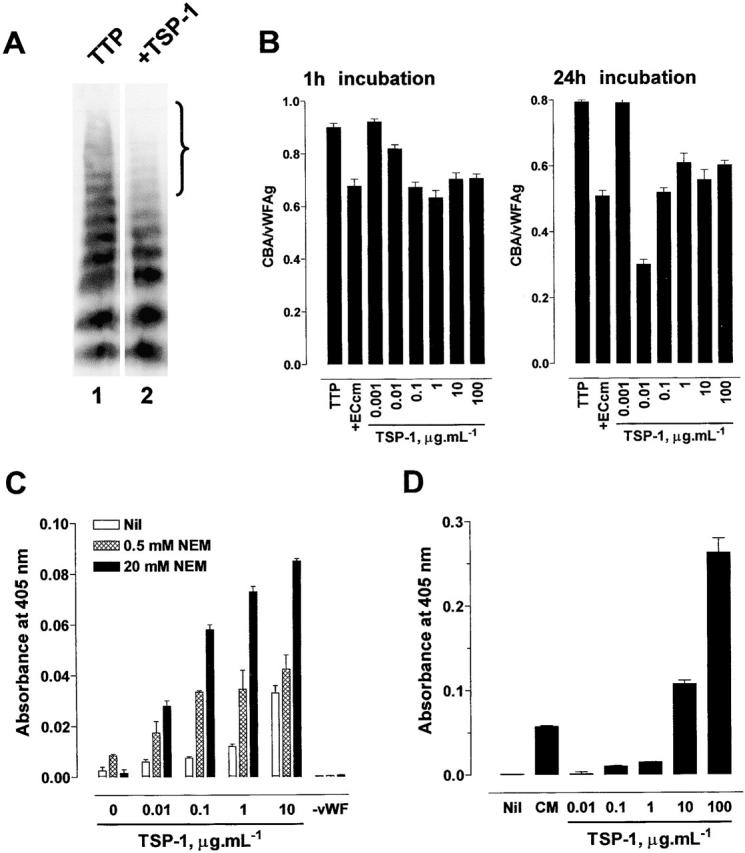
Reduction in the average multimer size of vWF by TSP-1 in vitro. (A) TTP1 patient plasma (10 μl) was incubated with Hepes-buffered saline containing 1 mM CaCl2 (TTP, lane 1) or purified platelet TSP-1 (1 μg/ml, lane 2) in the Hepes/CaCl2 buffer (90 μl) for 1 h at 37°C and aliquots of the reaction (10 μl) were resolved on 1% agarose gel electrophoresis. The vWF was transferred to PVDF membrane and Western blotted using peroxidase-conjugated anti-vWF polyclonal antibodies. The bracket highlights the change in the proportion of large vWF multimers in the population. (B) TTP1 patient plasma (10 μl) was incubated with HMEC-1–conditioned medium (+ECcm) or purified platelet TSP-1 (0.001 to 100 μg/ml) in Hepes-buffered saline containing 1 mM CaCl2 (90 μl) for 1 or 24 h at 37°C and aliquots of the reaction were analyzed for vWF antigen levels and collagen binding affinity. The results are expressed as the ratio of the collagen binding activity and vWF antigen level. The bars and errors are the mean and SD of triplicate determinations. (C) Interaction of TSP-1 with vWF. Microtiter plate wells coated with purified human vWF and blocked with BSA were incubated with purified human TSP-1 (0 to 10 μg/ml) in Hepes-buffered saline containing 1 mM CaCl2 and no (white bars), 5 mM (hatched bars), or 20 mM (black bars) NEM for 30 min at room temperature. On one occasion, wells not coated with vWF but blocked with BSA were incubated with 10 μg/ml TSP-1. The wells were washed with the Hepes buffer containing 1 M NaCl to minimize noncovalent interactions and the bound TSP-1 was meas-ured using the anti-TSP1 monoclonal antibody, HB8432, and peroxidase-conjugated secondary antibody. The bars and errors are the mean and SD of triplicate determinations. (D) Generation of new thiols in vWF by TSP-1. Purified human vWF (2 μg/ml) was incubated with Hepes-buffered saline containing 1 mM CaCl2 (Nil), HMEC-1–conditioned medium (CM), or purified human TSP-1 (0.01 to 100 μg/ml) in the Hepes/CaCl2 buffer for 60 min at 37°C. The reactions were labeled with MPB (100 μM) and the unreacted MPB was quenched with GSH (200 μM). Aliquots of the reactions were incubated in microtitre plate wells coated with anti-vWF polyclonal antibodies and the adsorbed vWF was incubated with streptavidin peroxidase to measure the incorporated MPB. The bars and errors are the mean and SD of triplicate determinations.
The first step in reduction of a disulfide bond is nucleophilic attack on the substrate disulfide bond by a reductant thiol which results in formation of a disulfide-linked complex between the substrate and the reductant. Release of the reductant from the complex requires nucleophilic attack on the disulfide linkage by another thiol, usually of the reductant. We reasoned that NEM might trap intermediate covalent complexes of TSP-1 and vWF by blocking the TSP-1 thiol responsible for separating TSP-1 and vWF. Maleimides react rapidly and specifically with cysteine thiols at neutral pH. This prediction was tested by measuring formation of salt-resistant complexes between TSP-1 and vWF and the effect of the thiol-blocking reagent, NEM, on complex formation. Microtiter plate wells coated with vWF were incubated with TSP-1 in the absence or presence of NEM and then washed with high salt buffer to minimize noncovalent interactions between TSP-1 and vWF. Up to eightfold more TSP-1 bound to vWF in the presence of NEM (Fig. 3 C). Complex formation increased with increasing NEM concentration. There was no binding of TSP-1 to wells not coated with vWF.
Reduction of disulfide bonds in vWF was anticipated to result in a net increase in free thiols in the vWF population 12. The biotin-linked maleimide, MPB, was used to measure the formation of new thiols in vWF upon incubation with TSP-1. vWF was incubated with TSP-1 and free thiol(s) formed in vWF were labeled with MPB and the unreacted MPB was quenched with GSH. Aliquots of the reactions were incubated in microtiter plate wells coated with anti-vWF polyclonal antibodies and the adsorbed vWF was incubated with streptavidin peroxidase to measure the incorporated MPB. There was negligible existing free thiols in the purified vWF 12. Incubation of vWF with either HMEC-1–conditioned medium or purified TSP-1 resulted in incorporation of MPB into vWF and the incorporation increased with increasing TSP-1 concentration (Fig. 3 D). Chelation of Ca2+ in the reactions with EDTA ablated TSP-1–facilitated formation of free thiols in vWF (not shown).
Correlation of vWF Multimer Size with the Concentration of TSP-1 in Plasma.
The results of Fig. 3 indicated that the extent of reduction in vWF multimer size was a function of the ratio of the concentrations of TSP1 and vWF. It was anticipated, therefore, that the molar ratio of TSP-1/vWF would be higher in plasmas from healthy subjects than in plasmas from patients with TTP. This ratio was measured in 11 healthy subject plasmas and 3 TTP patient plasmas. A sensitive enzyme-linked immunosorbent assay for plasma TSP-1 was developed using the anti–TSP-1 monoclonal antibody, HB8432 16, that recognizes the central stalk-like region of TSP-1 (unpublished data). TSP-1 concentrations as low as 1 ng/ml were measured using the assay.
The average multimer size of the vWF in the healthy subject plasmas (CBA/vWFAg of 1.01 ± 0.04 [mean ± SD, n = 11]) was significantly lower than that in the TTP patient plasmas (CBA/vWFAg of 1.10 ± 0.01 [mean ± SD of triplicate determinations], 1.37 ± 0.04, and 1.10 ± 0.02 for TTP1, TTP2, and TTP3 plasmas). The average concentration of TSP-1 in the normal plasmas was 81 ± 16 ng/ml (mean ± 2 SD, n = 11), which is in good agreement with other estimates. For instance, Hao et al. 26 reported a mean plasma TSP-1 concentration in 24 healthy adults of 64 ± 18 ng/ml. The concentration of TSP-1 in the TTP plasmas was 39 ± 7 ng/ml (mean ± 2 SD of triplicate determinations), 24 ± 1 ng/ml, and 45 ± 1 ng/ml for TTP1, TTP2, and TTP3, respectively, which is significantly lower than the concentration in normal plasma.
The TSP-1/vWFAg ratio in the normal plasmas was 286 ± 54 ng/ml/OD (mean ± SD, n = 11). The TSP-1/vWFAg ratio in the TTP plasmas was 169 ± 33 ng/ml/OD (mean ± SD of triplicate determinations), 74 ± 5 ng/ml/OD and 123 ± 4 ng/ml/OD for TTP1, TTP2, and TTP3, respectively, which is significantly lower than the ratio in normal plasma. In addition, there was an inverse relationship between the average vWF multimer size (CBA/vWFAg) and the TSP-1/vWFAg ratio in the normal and TTP patient plasmas.
Characterization of the Binding Sites on TSP-1 and vWF.
CD36 is a membrane receptor for TSP-1 on endothelial cells, platelets, monocytes, and several tumor cell lines. TSP-1 binds to amino acid residues 87 to 99 within a single disulfide loop of CD36 27. Examination of the vWF amino acid sequence revealed a region of the A3 domain (dalgfavrylt, residues 1,008–1,018) with high homology to the TSP-1 binding sequence of CD36 (gpytyrvrFla, residues 89–99). 7 of 11 amino acids are the same or very similar.
The vWF A3 domain peptide (residues 1,008–1,018) or a scrambled peptide was synthesized and tested for inhibition of TSP-1 reduction of vWF. Platelet TSP-1 was preincubated with the peptide and the reaction was started by addition of vWF. The vWF was labeled with MPB and detected with streptavidin peroxidase as described in Fig. 3 D. The A3 domain peptide was a potent inhibitor of reduction of vWF by TSP-1 with half-maximal effect at ∼0.01 μM (Fig. 4 A). The scrambled peptide had no effect on vWF reduction up to 10 μM.
Figure 4.
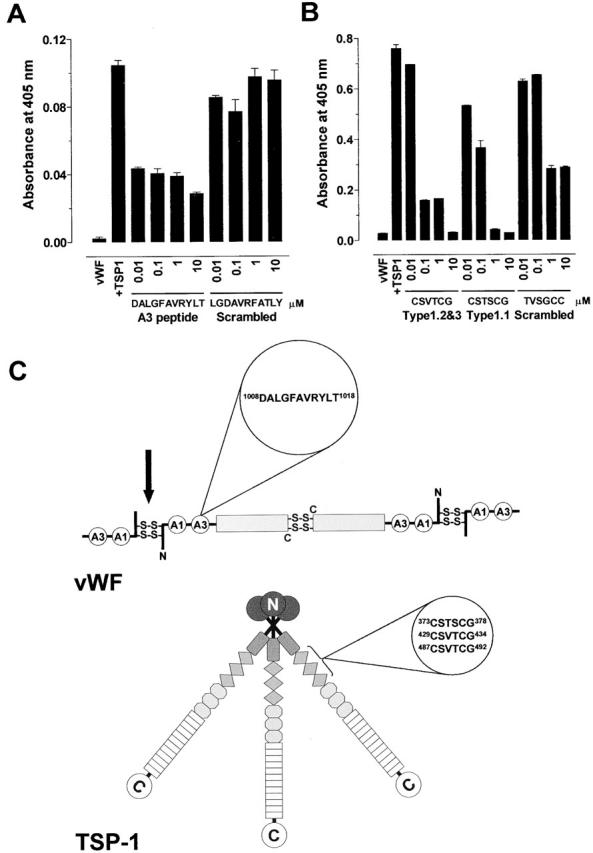
Characterization of the binding sites on TSP-1 and vWF. (A) Platelet TSP-1 (10 μg/ml) was incubated with Hepes-buffered saline containing 1 mM CaCl2 and vWF A3 domain peptide DALGFAVRYLT or scrambled peptide LGDAVRFATLY (0.01 to 100 μM) for 10 min at 37°C. The reaction was started by addition of purified vWF (2 μg/ml) and incubated for 60 min at 37°C. The vWF was labeled with MPB and detected with streptavidin peroxidase. The bars and errors are the mean and SD of triplicate determinations. (B) Purified vWF (2 μg/ml) was incubated with Hepes-buffered saline containing 1 mM CaCl2 and TSP-1 type 1 peptides CSVTCG, CSTSCG, or scrambled peptide TVSGCC (0.01 to 100 μM) for 10 min at 37°C. The reaction was started by addition of platelet TSP-1 (10 μg/ml) and incubated for 60 min at 37°C. The vWF was labeled with MPB and detected with streptavidin peroxidase. The bars and errors are the mean and SD of triplicate determinations. (C) Cartoon of the quaternary structures of vWF and TSP-1 and position of the amino acid sequences that mediate their interaction. Dimers of vWF are assembled from subunits via disulfide bridges between cysteine residues located in the COOH-terminal regions. Multimers are formed from dimers by interdimeric disulfide linking of NH2-terminal domains in a parallel orientation. The A1 and A3 domains of the vWF subunit are indicated. We propose that TSP-1 reduces the interdimeric disulfide bonds (black arrow). The TSP-1 subunit contains a unique heparin-binding domain at the NH2 terminus, followed by a connecting region that link three subunits via interchain disulfide bonds, a procollagen-like module, three properdin-like or type 1 modules, three epidermal growth factor–like or type 2 modules, 12 unique calcium-binding loops (or seven type 3 repeats), and a unique COOH-terminal globular domain.
TSP-1 binds to CD36 via the TSP-1 type 1 properdin domains. The CSTSCG sequence in the first TSP-1 type 1 repeat and the CSVTCG sequence in the second and third type 1 repeats are responsible for the interaction with CD36 28 29. The TSP-1 type 1 domain peptides or a scrambled peptide was synthesized and tested for inhibition of TSP-1 reduction of vWF. vWF was preincubated with the peptide and the reaction was started by addition of TSP-1. The vWF was labeled with MPB and detected with streptavidin peroxidase as described in Fig. 3 D. The type 1 domain peptides inhibited reduction of vWF by TSP-1 with half-maximal effect at ≤ 0.1 μM (Fig. 4 B). The scrambled peptide had no effect at 0.1 μM and caused ∼50% inhibition at 10 μM.
Reduction in the Average Multimer Size of vWF by TSP-1 In Vivo.
The ability of TSP-1 to reduce the average multimer size of vWF in vivo was examined by administering TSP-1 to mice via intraperitoneal injection and measuring the consequence for the average multimer size of plasma vWF. Balb c mice (7 to 9 wk of age) were administered either buffer vehicle (n = 4) or 1 mg/kg purified human platelet TSP-1 (n = 4) via 0.1 ml intraperitoneal injections. There were two male and two female mice in each group. Blood was collected by cardiac puncture into EDTA 24 h later and the plasma was analyzed in triplicate for vWF antigen levels and collagen binding affinity. The average multimer size of the plasma vWF was significantly lower (P < 0.05) in mice treated with TSP-1 (CBA/vWFAg = 0.83 ± 0.05) than with buffer vehicle (CBA/vWFAg = 1.65 ± 0.32). The concentration of human TSP-1 in the plasma of the treated mice 24 h after injection was measured using the TSP-1 assay and found to be ∼1 μg/ml.
Discussion
TSP-1 reduced the average multimer size of vWF in vitro and in vivo. Reduction in vWF multimer size was associated with formation of thiol-dependent complexes of TSP-1 and vWF and generation of new thiols in vWF.
The extent of reduction of vWF by TSP-1 was dependent on the molar ratio of TSP-1 and vWF and time of incubation and not simply on the absolute concentration of TSP-1. In experiments with purified TSP-1 and ∼1 μg/ml plasma vWF, for example, the optimal TSP-1 concentration for reduction in 1 h was ∼0.1 μg/ml compared with ∼0.01 μg/ml in 24 h. The weight concentrations of vWF and TSP-1 in normal plasma are ∼10 μg/ml 30 and ∼0.1 μg/ml (our results and reference 26), respectively. Assuming a weight-average M r for vWF of ∼2 × 106 31 and M r for TSP-1 of 0.5 × 106, the molar concentrations of vWF and TSP-1 in normal plasma are ∼5 nM and ∼0.2 nM, respectively. TSP-1 targets the very large vWF multimers in plasma, which represent ≤10% of the total vWF population (see Fig. 3 A). The moles of TSP-1 and moles of vWF it reacts with in normal plasma, therefore, is roughly equal.
Our findings predicted that the higher the plasma TSP-1/vWF molar ratio the smaller the average vWF multimer size. We anticipated, therefore, that the molar ratio of TSP-1/vWF would be lower in plasmas from patients with TTP than in plasmas from healthy subjects. This was the case in the plasmas of the three patients with TTP examined in this study. The concentration of TSP-1 in the TTP patient plasmas was ≤55% of the average concentration in plasmas from healthy subjects. Similarly, the TSP-1/vWF ratio in the patient plasmas was ≤59% of the average ratio in normal plasma. Moreover, there was an inverse relationship between the average vWF multimer size and the TSP-1/vWFAg ratio of the plasmas.
These observations define a disulfide bond reductase/isomerase activity of TSP-1. Reductase/isomerase active sites are characterized by closely spaced cysteines in the consensus sequence, CGXC 32. The cysteine thiols cycle between the reduced dithiol and oxidized disulfide bond in coordination with a dithiol or disulfide of a protein substrate which can result in reduction, formation, or interchange of disulfide bonds in the protein substrate. There is a CGAC sequence in the first type 2 repeat of TSP-1 but there is no evidence that these two cysteines are redox active 24. In contrast, a complex intramolecular disulfide interchange operates in the COOH-terminal Ca2+-binding repeats and globular domain of TSP-1 24. These disulfide exchange events are likely to be involved in reduction of vWF by TSP-1 because of the requirement for Ca2+ ions for TSP-1 reductase activity. Different disulfide-bonded forms of TSP-1 have different Arg-Gly-Asp (RGD)-dependent cell adhesive activity 25, inhibit neutrophil enzymes with markedly differing potencies 33 34, bind Ca2+ with different stoichiometries 35, and bind platelet-derived growth factor with very different affinities 15.
Interaction of TSP-1 with vWF is mediated by TSP-1 type 1 properdin domains and the vWF A3 domain. We propose that binding of the TSP-1 type 1 domain(s) to the vWF A3 domain(s) enables engagement of the TSP-1 C-termini with the vWF interdimer disulfide bonds. A model for the mechanism by which TSP-1 reduces vWF multimer size is shown in Fig. 5. TSP-1 preferentially reduced the large vWF multimers. We suggest that the multimeric size of vWF will be balanced by the avidity of TSP-1 for vWF, which will decrease as vWF multimer size decreases, and the potential for reformation of intersubunit disulfide bonds between vWF molecules, which will increase as the number of smaller vWF molecules increase 12. Frangos et al. 10 showed that small vWF multimers can react to form larger multimers in plasma.
Figure 5.

Model for reduction of vWF by TSP-1. TSP-1 contains three unpaired cysteines per molecule, presumably one per subunit (references 24 and 25). These thiols are only partially accessible to alkylation by NEM and inaccessible to alkylation by iodoacetamide in the Ca2+-replete form of TSP-1 (reference 24). We suggest that interaction of vWF with TSP-1 results in a conformational change in TSP-1 which triggers the reduction of vWF intersubunit disulfide bonds. Chelation of Ca2+ with EDTA inhibits reductase activity. Nucleophilic attack by a TSP-1 thiol on a vWF intersubunit disulfide bond results in reduction of the disulfide bond with formation of an intermediate disulfide-linked complex between TSP-1 and vWF. Nucleophilic attack by a second TSP-1 thiol results in release of vWF and formation of an intramolecular disulfide bond in TSP-1. Reduction of one disulfide bond in vWF is represented but two, three, or four intersubunit disulfide bonds may need to be reduced to change multimeric size (reference 5). Each step in the model is potentially reversible. The model predicts the formation of intermediate covalent complexes between TSP-1 and vWF, which may be trapped with thiol-blocking agents such as NEM (see Fig. 3 B), and a net increase in free cysteine thiols in the vWF population (see Fig. 3 C).
The average multimer size of vWF can be reduced by cleavage of the Tyr842-Met843 peptide bond by a plasma metalloproteinase. Deficiency of this enzyme is associated with abnormally large vWF multimers in the blood of patients with chronic relapsing TTP 36 37. The relationship between TSP-1 and the plasma metalloproteinase for control of vWF multimer size is unknown but they are clearly separate activities 12. It is possible that TSP-1 and the vWF cleaving metalloproteinase act in concert to control of the average multimer size of plasma vWF 9. It is noteworthy that vWF binds to the type 1 domains of TSP-1, which are the same domains that bind CD36. Anti-CD36 antibodies have been identified in patients with TTP 38. It may be that perturbation of the TSP-1–CD36 interaction in TTP contributes to the abnormally large vWF multimers associated with this disease.
The prevalence of large vWF multimers in the plasma vWF population determines vWF's hemostatic activity 6. Our findings imply that TSP-1 is involved in normal control of vWF hemostatic activity, and perturbation of secretion or function of TSP-1 or polymorphism in the TSP-1 gene may contribute to thrombotic and vascular disorders.
Acknowledgments
The authors thank Dr. M.C. Berndt for the purified vWF and Drs. J. Pimanda, J. Paxton, and N. Wickham for TTP patient plasma and clinical details.
This work was supported by the National Health and Medical Research Council of Australia, the National Heart Foundation of Australia, and the NSW Health Department.
Footnotes
Abbreviations used in this paper: GSH, reduced glutathione; MPB, 3-(N-maleimidylpropionyl)biocytin; NEM, N-ethylmaleimide; PVDF, polyvinylidine difluoride; TSP-1, thrombospondin-1; TTP, thrombotic thrombocytopenic purpura; vWF, von Willebrand factor.
References
- Sadler J.E. Biochemistry and genetics of von Willebrand factor. Ann. Rev. Biochem. 1998;67:395–424. doi: 10.1146/annurev.biochem.67.1.395. [DOI] [PubMed] [Google Scholar]
- Counts R.B., Paskell S.L., Elgee S.K. Disulfide bonds and the quaternary structure of factor VIII/von Willebrand factor. J. Clin. Invest. 1978;62:702–709. doi: 10.1172/JCI109178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsumi A., Tuley E.A., Bodó I., Sadler J.E. Localization of disulfide bonds in the cysteine knot domain of human von Willebrand factor. J. Biol. Chem. 2000;275:25585–25594. doi: 10.1074/jbc.M002654200. [DOI] [PubMed] [Google Scholar]
- Wagner D.D., Lawrence S.O., Ohlsson-Wilhelm B.M., Fay P.J., Marder V.J. Topology and order of formation of interchain disulfide bonds in von Willebrand factor. Blood. 1987;69:27–32. [PubMed] [Google Scholar]
- Dong Z., Thoma R.S., Crimmins D.L., McCourt D.W., Tuley E.A., Sadler J.E. Disulfide bonds required to assemble functional von Willebrand factor multimers. J. Biol. Chem. 1994;269:6753–6758. [PubMed] [Google Scholar]
- Furlan M. von Willebrand factormolecular size and functional activity. Ann. Hematol. 1996;72:341–348. doi: 10.1007/s002770050184. [DOI] [PubMed] [Google Scholar]
- Furlan M., Perret B.A., Beck E.A. Studies on factor VIII-related protein. III. Size distribution and carbohydrate content of human and bovine factor VIII. Biochim. Biophys. Acta. 1979;579:325–333. doi: 10.1016/0005-2795(79)90060-6. [DOI] [PubMed] [Google Scholar]
- Moake J.L., Turner N.A., Stathopoulos N.A., Nolasco L.H., Hellums J.D. Involvement of large plasma von Willebrand factor (vWF) multimers and unusually large vWF forms derived from endothelial cells in shear stress-induced platelet aggregation. J. Clin. Invest. 1989;78:1456–1461. doi: 10.1172/JCI112736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moake J.L. Studies on the pathophysiology of thrombotic thrombocytopenic purpura. Sem. Hematol. 1997;34:83–89. [PubMed] [Google Scholar]
- Frangos J.A., Moake J.L., Nolasco L., Phillips M.D., McIntire L.V. Cryosupernatant regulates accumulation of unusually large vWF multimers from endothelial cells. Am. J. Physiol. 1989;256:H1635–H1644. doi: 10.1152/ajpheart.1989.256.6.H1635. [DOI] [PubMed] [Google Scholar]
- Phillips M.D., Moake J.L., Nolasca L., Garcia R. Plasma von Willebrand factor processing activity functions like a disulfide bond reductasereversible decrease of multimer size. Thromb. Haemostasis. 1993;69:1199. [Google Scholar]
- Xie L., Chesterman C.N., Hogg P.J. Reduction of von Willebrand factor by endothelial cells. Thromb. Haemostasis. 2000;84:506–513. [PubMed] [Google Scholar]
- Booth W.J., Furby F.H., Berndt M.C., Castaldi P.A. Factor VIII/von Willebrand factor has potent lectin activity. Biochem. Biophys. Res. Commun. 1984;118:495–501. doi: 10.1016/0006-291x(84)91330-5. [DOI] [PubMed] [Google Scholar]
- Murphy-Ullrich J.E., Mosher D.F. Localization of thrombospondin in clots formed in situ. Blood. 1985;66:1098–1104. [PubMed] [Google Scholar]
- Hogg P.J., Hotchkiss K.A., Jiménez B.M., Stathakis P., Chesterman C.N. Interaction of platelet-derived growth factor with thrombospondin 1dependence on the disulfide-bond arrangement in thrombospondin 1. Biochem. J. 1997;326:709–716. doi: 10.1042/bj3260709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe E.A., Ruggiero J.T., Leung L.L., Doyle M.J., McKeown-Longo P.J., Mosher D.F. Cultured fibroblasts synthesize and secrete thrombospondin and incorporate it into extracellular matrix. Proc. Natl. Acad. Sci. USA. 1983;80:998–1002. doi: 10.1073/pnas.80.4.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favaloro E.J., Grispo L., Exner T., Koutts J. Development of a simple collagen based ELISA assay aids in the diagnosis of, and permits sensitive discrimination between type I and type II, von Willebrand's disease. Blood Coagul. Fibrinolysis. 1991;2:285–291. doi: 10.1097/00001721-199104000-00011. [DOI] [PubMed] [Google Scholar]
- Ruggeri Z.M., Zimmerman T.S. Variant von Willebrand's diseasecharacterization of two subtypes by analysis of multimer composition of factor VIII/von Willebrand factor in plasma and platelets. J. Clin. Invest. 1980;65:1318–1325. doi: 10.1172/JCI109795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli U.K. Cleavage of structural proteins during assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Ades E.W., Candal F.J., Swerlick R.A., George V.G., Summers S., Bosse D.C., Lawley T.J. HMEC-1establishment of an immortalized human microvascular endothelial cell line. J. Invest. Dermatol. 1992;99:683–690. doi: 10.1111/1523-1747.ep12613748. [DOI] [PubMed] [Google Scholar]
- Siekmann J., Turecek P.L., Schwarz H.P. The determination of von Willebrand factor activity by collagen binding assay. Haemophilia. 1998;4:15–24. doi: 10.1046/j.1365-2516.1998.0040s3015.x. [DOI] [PubMed] [Google Scholar]
- Mosher D.F., Doyle M.J., Jaffe E.A. Synthesis and secretion of thrombospondin by cultured human endothelial cells. J. Cell Biol. 1982;93:343–348. doi: 10.1083/jcb.93.2.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawler J. The functions of thrombospondin-1 and -2. Curr. Opin. Cell Biol. 2000;12:634–640. doi: 10.1016/s0955-0674(00)00143-5. [DOI] [PubMed] [Google Scholar]
- Speziale M.V., Detwiler T.C. Free thiols of platelet thrombospondin. J. Biol. Chem. 1990;265:17859–17867. [PubMed] [Google Scholar]
- Sun X., Skorstengaard K., Mosher D.F. Disulfides modulate RGD-inhibitable cell adhesive activity of thrombospondin. J. Cell Biol. 1992;118:693–701. doi: 10.1083/jcb.118.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao K.-J., Klein P.A. A monoclonal antibody-based enzyme-linked immunosorbent assay for quantitation of plasma thrombospondin. Am. J. Clin. Pathol. 1986;86:317–323. doi: 10.1093/ajcp/86.3.317. [DOI] [PubMed] [Google Scholar]
- Asch A.S., Liu I., Briccetti F.M., Barnwell J.W., Kwakye-Berko F., Dokun A., Goldberger J., Pernambuco M. Analysis of CD36 binding domainsligand specificity controlled by dephosphorylation of an ectodomain. Science. 1993;262:1436–1440. doi: 10.1126/science.7504322. [DOI] [PubMed] [Google Scholar]
- Tuszynski G.P., Rothman V.L., Deutch A.H., Hamilton B.K., Eyal J. Biological activities of peptides and peptide analogues derived from common sequences present in thrombospondin, properdin, and malarial proteins. J. Cell Biol. 1992;116:209–217. doi: 10.1083/jcb.116.1.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asch A.S., Silbiger S., Heimer E., Nachman R.L. Thrombospondin sequence motif (CSVTCG) is responsible for CD36 binding. Biochem. Biophys. Res. Commun. 1992;182:1208–1217. doi: 10.1016/0006-291x(92)91860-s. [DOI] [PubMed] [Google Scholar]
- Borchiellini A., Fijnvandraat K., ten Cate J.W., Pajkrt D., van Deventer S.J., Pasterkamp G., Meijer-Huizinga F., Zwart-Huinink L., Voorberg J., van Mourik J.A. Quantitative analysis of von Willebrand factor propeptide release in vivoeffect of experimental endotoxemia and administration of 1-deamino-8-D-arginine vasopressin in humans. Blood. 1996;88:2951–2958. [PubMed] [Google Scholar]
- Berndt M.C., Du X., Booth W.J. Ristocetin-dependent reconstitution of binding of von Willebrand Factor to purified human platelet membrane glycoprotein Ib-IX complex. Biochemistry. 1988;27:633–640. doi: 10.1021/bi00402a021. [DOI] [PubMed] [Google Scholar]
- Ferrari D.M., Soling H.D. The protein disulfide-isomerase familyunravelling a string of folds. Biochem. J. 1999;339:1–10. [PMC free article] [PubMed] [Google Scholar]
- Hogg P.J. Thrombospondin 1 as an enzyme inhibitor. Thromb. Haemostasis. 1994;72:787–792. [PubMed] [Google Scholar]
- Hotchkiss K.A., Chesterman C.N., Hogg P.J. Catalysis of disulfide isomerization in thrombospondin 1 by protein disulfide isomerase. Biochemistry. 1996;35:9761–9767. doi: 10.1021/bi9603938. [DOI] [PubMed] [Google Scholar]
- Misenheimer T.M., Mosher D.F. Calcium ion binding to thrombospondin 1. J. Biol. Chem. 1995;270:1729–1733. doi: 10.1074/jbc.270.4.1729. [DOI] [PubMed] [Google Scholar]
- Furlan M., Robles R., Solenthaler M., Wassmer M., Sandoz P., Lämmle B. Deficient activity of von Willebrand factor-cleaving protease in chronic relapsing thrombotic thrombocytopenic purpura. Blood. 1997;89:3097–3103. [PubMed] [Google Scholar]
- Furlan M., Robles R., Galbusera M., Remuzzi G., Kyrle P.A., Brenner B., Krause M., Scharrer I., Aumann V., Mittler U., Solenthaler M., Lammle B. von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndrome. N. Engl. J. Med. 1998;339:1578–1584. doi: 10.1056/NEJM199811263392202. [DOI] [PubMed] [Google Scholar]
- Tandon N.N., Rock G., Jamieson G.A. Anti-CD36 antibodies in thrombotic thrombocytopenic purpura. Br. J. Haematol. 1994;88:816–825. doi: 10.1111/j.1365-2141.1994.tb05122.x. [DOI] [PubMed] [Google Scholar]