

Abstract
The enterobacterial pathogen Salmonella induces phagocyte apoptosis in vitro and in vivo. These bacteria use a specialized type III secretion system to export a virulence factor, SipB, which directly activates the host's apoptotic machinery by targeting caspase-1. Caspase-1 is not involved in most apoptotic processes but plays a major role in cytokine maturation. We show that caspase-1–deficient macrophages undergo apoptosis within 4–6 h of infection with invasive bacteria. This process requires SipB, implying that this protein can initiate the apoptotic machinery by regulating components distinct from caspase-1. Invasive Salmonella typhimurium targets caspase-2 simultaneously with, but independently of, caspase-1. Besides caspase-2, the caspase-1–independent pathway involves the activation of caspase-3, -6, and -8 and the release of cytochrome c from mitochondria, none of which occurs during caspase-1–dependent apoptosis. By using caspase-2 knockout macrophages and chemical inhibition, we establish a role for caspase-2 in both caspase-1–dependent and –independent apoptosis. Particularly, activation of caspase-1 during fast Salmonella-induced apoptosis partially relies on caspase-2. The ability of Salmonella to induce caspase-1–independent macrophage apoptosis may play a role in situations in which activation of this protease is either prevented or uncoupled from the induction of apoptosis.
Keywords: monocytes/macrophages, cell death, proteases, natural immunity, bacteria
Introduction
Salmonella species cause a variety of enteric diseases ranging from self-limiting gastroenteritis (mainly due to S. typhimurium) to the more severe systemic typhoid fever (caused by S. typhi). Infections are generally food borne and pose a particularly serious health hazard in regions where the hygienic conditions are inappropriate. After consumption of contaminated food or water, Salmonellae reach the intestine, where they cross the epithelial barrier by invading the specialized M cells of the ileac Peyer's patches. By activating the host signal transduction cascades controlling the actin cytoskeleton, Salmonella induces the formation of membrane ruffles localized at the contact point between bacterium and host cell and is ultimately taken up in large vacuoles 1. By destroying infected M cells, the bacteria gain access to the mesenteric lymph follicles, where they face the host's macrophages. For Salmonella, as well as for many other facultative intracellular pathogens, surviving this encounter is the key to a successful infection. Invasive Salmonella is capable of persisting within the macrophages in spacious vacuoles uncoupled from the normal endocytic route. These vacuoles do not acquire lysosomal markers like cathepsin D or L and may therefore represent a relatively safe intracellular site in which the bacteria can survive and multiply 2.
Besides its ability to survive in infected macrophages, invasive Salmonella induces phagocyte apoptosis in vitro 3 4 5 6. Apoptosis is mediated by a cell-intrinsic suicide program, the activation of which is regulated by different signals originating from both the intracellular and the extracellular milieu. The situation during an in vivo infection is certainly different from experimental setups in vitro in which all of the bacteria are uniformly invasive, and the inoculum is rather overwhelming. A certain amount of phagocyte apoptosis can be detected after infection of mice with Salmonella in vivo 7, but the extent to which apoptosis contributes to the pathogenesis of Salmonella infections is at present unknown. Salmonella species (spp.) share the ability of inducing macrophage apoptosis with Yersinia spp. 8 9 10 and Shigella spp. 11 12 13 14 15, suggesting that this may represent a hallmark of, and perhaps a selective advantage for, the establishment of enterobacterial infections. Both epithelial cell invasion and induction of macrophage apoptosis depend on a functional type III secretion system. Type III protein secretion systems are specialized protein secretion apparatuses capable of translocating bacterial proteins into host cells, and they play a pivotal role in the interaction between a variety of mammalian and plant pathogenic bacteria with their hosts 16. In Salmonella, the type III secretion genes essential for epithelial cell invasion and apoptosis induction are clustered in a region denoted Salmonella pathogenicity island 1 (SPI-1) at centisome 63 of the chromosome 17 18. SipB, a protein encoded by SPI-1, is essential for Salmonella-mediated macrophage apoptosis 3.
Caspases (cystein/aspartic acid proteases) are the effector molecules of the apoptotic program. All enzymes in the family are produced as zymogens and are activated by proteolysis. The so-called “initiator caspases” can activate other family members (the “effector caspases”), establishing a proteolytic cascade. Initiator caspases generally feature a long prodomain containing protein interaction motifs. These motifs mediate the direct or adaptor-mediated oligomerization and activation of the initiator caspases. Effector caspases possess short prodomains, which are cleaved by initiator caspases. Thus activated, the effector caspases process the so-called death substrates, whose cleavage irreversibly commits the cell to apoptosis 19 20.
Despite its evolutionary conservation, the cell death pathway in mammals is complex, and it includes multiple mammalian caspases with apparently similar apoptotic function. A number of caspase genes have been inactivated in the mouse, but the study of caspase-deficient mice has not yielded the expected clues, possibly because of caspase redundancy in most apoptotic processes. In the case of caspase-1, the general conclusion of the study of the mutant mice is that apoptosis is essentially normal in these animals but that they have a severe defect in cytokine production 21 22 23 24 25 26 27 28 29. It was therefore somewhat surprising that both Salmonella- and Shigella-induced macrophage apoptosis require caspase-1. Apoptosis is induced by two structurally similar proteins exported by type III secretion systems, SipB of Salmonella typhimurium and IpaB of Shigella flexneri, by binding to and activating caspase-1 in the host cells 11 30 31. In this study, we show that invasive Salmonella rapidly activates caspase-2 in a caspase-1–independent manner and that caspase-1 is not absolutely required for macrophage apoptosis. Caspase-1–deficient macrophages are killed by invasive Salmonella in a comparatively slow process (4-6 h). SipB is essential for caspase-1–independent macrophage apoptosis, demonstrating that this protein can activate the apoptotic machinery by regulating components distinct from caspase-1. Apoptosis involves the activation of caspase-2, -3, -6, and -8 and the release of cytochrome c from mitochondria. With the exception of caspase-2 activation, these phenomena are not observed during the fast, caspase-1–dependent apoptosis. Caspase-2 plays a role in caspase-1–independent apoptosis and, by contributing to caspase-1 activation, in rapid apoptosis.
Materials and Methods
Bacteria.
Salmonella typhimurium strains SR11 (wild type [wt]), SB111 (invA −; unable to secrete proteins via the type III pathway), and SB169 (sipB −) were grown in 5 ml of Luria-Bertani broth (1% Bacto Tryptone, 0.5% yeast extract, 1% sodium chloride) at 37°C overnight (16–20 h) under agitation. To obtain highly invasive bacteria, overnight cultures were diluted to an OD600 of 0.02 in 50 ml of TYP broth (1.6% Bacto Tryptone, 1.6% yeast extract, 0.5% sodium chloride, 0.25% dipotassium phosphate) and incubated for 5 h under agitation 5.
Cell Culture and Infection.
Bone marrow–derived macrophages from caspase-1– 28 or caspase-2–deficient 32 mice and wt controls were cultured in DMEM supplemented with 10% FCS and 20% L-conditioned medium as a source of CSF-1. Confluent cells (∼5 × 106 cells per 100-mm-diameter tissue culture dish) were cultured for 16–20 h in medium without CSF-1 and then infected with bacterial cultures. In selected experiments, the cells were treated with a caspase-2–specific inhibitor (Z-VDVAD-fmk, 100 μM 33; R&D Systems) for 90 min before infection with Salmonella. A multiplicity of infection (m.o.i.; bacteria per macrophage) of 25 was used. After 30 min of infection, gentamycin was added to the medium (Sigma-Aldrich; 50 μg/ml for 1 h and then 10 μg/ml) to kill extracellular bacteria. All experiments were repeated three to five times.
Cell Lysis and Western Blotting.
Cells from one 100-mm-diameter cell culture dish were washed twice with PBS and lysed in 300 μl of solubilization buffer (10 mM Tris-HCl, pH 7.0; 50 mM sodium chloride; 30 mM sodium pyrophosphate; 1% Triton X-100). Insoluble material was removed by centrifugation (20,000 g, 30 min). For immunoblotting, 30–50 μg of lysates was separated by 10 or 15% SDS-PAGE and transferred onto nitrocellulose membranes. After 1 h blocking in TTBS (10 mM Tris-HCl, pH 8.0; 150 mM sodium chloride; 0.1% Tween 20) supplemented with 5% milk powder, the membranes were probed with the appropriate primary antibodies (actin, caspase-1, caspase-2, and caspase-3 from Santa Cruz Biotechnology, Inc.; caspase-6 and caspase-8 from Chemicon; cytochrome c from BD PharMingen; and cytochrome c oxidase subunit IV [cox-IV] from Molecular Probes) diluted in 1% BSA (fraction V; Sigma-Aldrich) in TTBS before incubation with peroxidase-conjugated secondary antibodies and detection by an enhanced chemiluminescence system (Pierce Chemical Co.).
Subcellular Fractionation.
Cells were scraped into Mito buffer (250 mM sucrose, 20 mM Hepes, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol, 10 μM phenylmethylsulfonyl fluoride, 1× protease inhibitor cocktail [Boehringer Mannheim]). After incubation on ice for 30 min, cells were disrupted at 4°C in a 1-ml syringe fitted with a 25-gauge hypodermic needle (15 strokes). Nuclei were removed by centrifugation at 700 g for 5 min at 4°C. Supernatants were then further centrifuged at 13,000 g for 20 min at 4°C. The resulting supernatants were considered as cytosolic and the pellets as mitochondrial fractions.
Nuclear and Mitochondrial Staining.
3 × 105 macrophages were seeded on a coverslip in a well of a six-well cell culture dish. Mitochondria were stained with Chloromethyl-X-Rosamine (CM-X-ROS, MitoTracker™ Red; Molecular Probes), a potential-sensitive fluorochrome that withstands fixation and permeabilization of cells 34. Before infection, CM-X-ROS was added to the medium (final concentration 50 nM), and macrophages were incubated for 10 min at 37°C before infection with Salmonella. Chromatin condensation in infected macrophages was determined by staining with 4′,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich). In brief, medium was removed and 70 μl of DAPI (0.5 μg/ml; Sigma-Aldrich) was placed on the coverslip. Cells were incubated for 1 min, washed twice with PBS, and fixed with 3% formaldehyde in PBS for 10 min at room temperature. After washing with PBS twice, coverslips were mounted in 20% Mowiol (Sigma-Aldrich) in PBS. Chromatin condensation was assessed in randomly chosen areas of the sample by independent experimenters (300–500 cells per sample).
Electron Microscopy.
Cells were grown on glass coverslips and washed three times before fixation in glutaraldehyde (3% 0.15 M Sorensen's buffer, pH 7.4, for 1 h). Cells were then washed thrice with the same buffer and postfixed in 1% OsO4 in Sorensen's buffer for 1 h. The cells were subsequently dehydrated in ethanol and flat-embedded in epoxy resin (Agar 100). Thin sections (60–80 nm) were mounted on copper grids and contrasted by uranyl acetate and lead citrate. Sections were viewed at 80 kV in a JEM-1210 electron microscope (Jeol Ltd.).
Results
Caspase-1 Is Required for Rapid Salmonella-induced Apoptosis.
Salmonella grown to the transition between the logarithmic and the stationary phase induces macrophage apoptosis with the fastest kinetics reported to date 5. At a m.o.i. of 25, 85% of the cells die within 30 min of infection, showing the morphological hallmarks of apoptosis (shrinkage, chromatin condensation, membrane blebbing; see Fig. 2 B). A functional type III secretion system is essential for the ability of Salmonella to induce apoptosis, and both an invA − and a sipB − mutant are incapable of doing so (Fig. 1 A). SipB of Salmonella 30 and the related protein IpaB of Shigella 15 31 have been reported to bind to caspase-1 and activate it, thereby causing apoptosis. In agreement with this, primary bone marrow–derived macrophages from caspase-1–deficient mice failed to undergo apoptosis within the first 30 min of infection with wt Salmonella (Fig. 1a and Fig. b).
Figure 2.

Ultrastructural features of rapid Salmonella-induced macrophage apoptosis. (A) Uninfected macrophage. (B) wt macrophage after a 20-min infection with invasive Salmonella; both nucleus and cytoplasm are significantly smaller as in uninfected macrophages (compare the bar in A = 2 μm with the bar in B = 1 μm). The nucleus displays electron-dense material and a blistered nuclear envelope. The cytoplasm is in a progressed stage of lysis. It contains structurally intact mitochondria as well as swollen mitochondria with reduced matrix density (red arrowhead). Note also numerous Salmonella (marked by asterisks) associated with the macrophage.
Figure 1.


Caspase-1 is required for rapid macrophage apoptosis. Primary bone marrow–derived macrophages were isolated from wt or caspase-1–deficient mice (casp-1−/−). Cells were stained with CM-X-ROS before infection with Salmonella strains (m.o.i 25). 25 min after infection, cells were stained with DAPI to reveal chromatin condensation and observed under a fluorescent microscope. (A) wt (closed bars) or casp-1−/− macrophages (open bars) were infected with wt Salmonella or with the invasion-defective invA − and sipB − strains. UT, untreated cells. The percentage of cells containing condensed chromatin was determined by microscopical examination of triplicate samples. The SD was <5% in all cases, and it has been omitted. (B) photomicrographs of wt or caspase-1–deficient (caspase-1−/−) macrophages infected with wt Salmonella.
To gain more insight in the mechanism of rapid Salmonella-induced apoptosis, we monitored simultaneously loss of mitochondrial transmembrane potential and chromatin changes during apoptosis. Cells labeled with the potential-sensitive dye CM-X-ROS were infected with invasive Salmonella and stained with DAPI 30 min after infection. The infected cells showed a change in shape, and the CM-X-ROS staining appeared to be more diffused than in the uninfected controls (Fig. 1 B). These changes could also be observed in caspase-1–deficient macrophages, which at this time were not undergoing Salmonella-induced cell death (Fig. 1 A) and were therefore not related to apoptosis. DAPI staining of wt macrophages showed cells with different degrees of chromatin condensation (Fig. 1 B, wt 30 min). In cells with moderate chromatin condensation, the mitochondria retained CM-X-ROS, showing that their inner transmembrane potential was maintained (Fig. 1 B, white arrowhead). Upon completion of apoptosis, the nuclear staining appeared characteristically bright. The mitochondria of these cells released CM-X-ROS, indicating a loss of inner transmembrane potential (Fig. 1 B, yellow arrowheads). This is consistent with the dramatic swelling of some of these organelles observed in transmission electron microscopy of cells in the terminal stage of apoptosis (Fig. 2 B, red arrowhead). Other ultrastructural features of rapid Salmonella-induced macrophage apoptosis were the extreme shrinkage of both nucleus and cytoplasma (compare the bar in Fig. 2 A = 2 μm with that in Fig. 2 B = 1 μm), the blebbing of the cytoplasmic membrane, and the blistered nuclear envelope.
Caspase Activation in the Course of Rapid Salmonella-induced Apoptosis.
To investigate the molecular mechanisms underlying rapid Salmonella-induced apoptosis, quiescent primary bone marrow–derived macrophages were either left untreated or infected with wt or sipB − Salmonella strains. The caspase profile of the cells and the activation state of these enzymes 15 min after infection was assessed in whole cell lysates by immunoblotting with specific antisera. Primary bone marrow–derived macrophages express caspase-1, -2, -3, -6, and -8 (Fig. 3). We monitored the activation of caspase-1 by immunoblotting with an antiserum that recognizes both the zymogen (procaspase-1) and the long subunit of the active enzyme (p20). We confirmed that infection with a wt, but not with a sipB − Salmonella strain, activates this protease (Fig. 3 A). In addition, however, we observed the activation of caspase-2, measured as a decrease in the caspase-2 zymogen. At this early time point, little of the zymogen was cleaved, and intermediate forms (see Fig. 8) could not be detected. Again, the sipB − strain failed to activate caspase-2 (Fig. 3 B). Caspase-3, -6, and -8 were not activated in the course of this rapid apoptosis (Fig. 3C, Fig. D, and Fig. E, respectively). The specific release of cytochrome c from mitochondria was not observed during rapid Salmonella-induced apoptosis (not shown).
Figure 3.

Caspase-1 and –2 are rapidly activated by invasive Salmonella. Primary bone marrow–derived macrophages isolated from wt mice were infected with wt Salmonella or with the invasion-defective sipB − strain. After 15 min of infection, cells were lysed and the activation state of the caspases was analyzed by immunoblotting. (A) caspase-1; (B) caspase-2; (C) caspase-3; (D) caspase-6; (E) caspase-8; (F) actin, used as a loading control.
Figure 8.
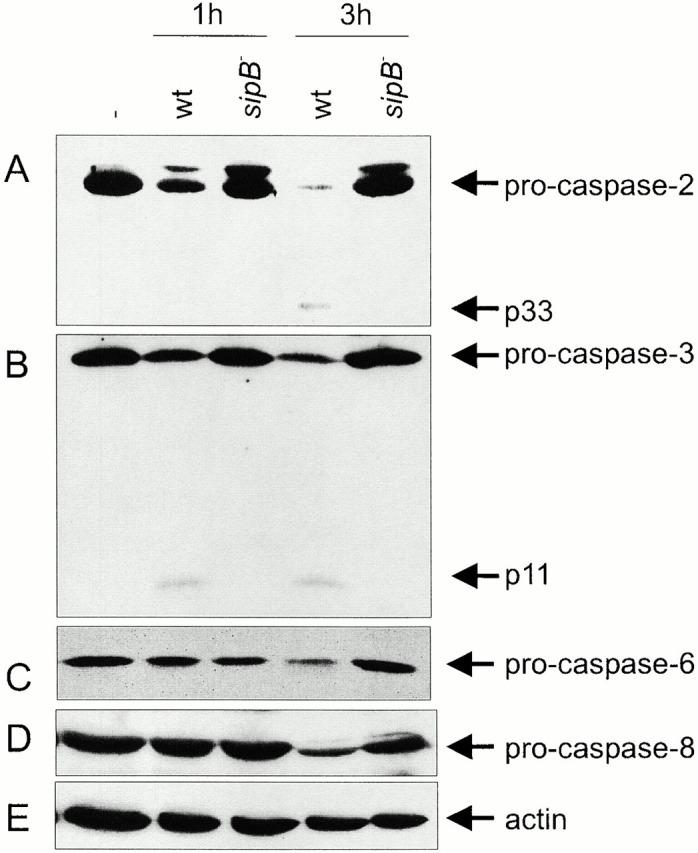
Caspase activation during caspase-1–independent macrophage apoptosis. Primary bone marrow–derived macrophages isolated from caspase-1–deficient mice were infected with wt Salmonella or with the invasion-defective sipB − strain. After 1 and 3 h of infection, cells were lysed, and the activation state of the caspases was analyzed by immunoblotting. (A) caspase-2; (B) caspase-3; (C) caspase-6; (D) caspase-8; (E) actin, used as a loading control. The caspase-2 blot was exposed longer than the immunoblots in Fig. 3 B and Fig. 4 to reveal the progressive cleavage of the zymogen and the appearance of the 33-kD intermediate band.
Caspase-2 Activation by Invasive Salmonella Occurs Independently of Caspase-1.
To assess whether activation of caspase-2 was part of the response initiated by caspase-1, we monitored zymogen cleavage in the early phases of infection in primary bone marrow–derived macrophages from wt and caspase-1–deficient mice. Caspase-2 was rapidly activated in both cases (Fig. 4). As discussed above, only a small fraction of zymogen was cleaved, and intermediate forms were not detected at these early time points. Thus, Salmonella targeted caspase-2 directly, simultaneously with caspase-1.
Figure 4.

Invasive Salmonella activates caspase-2 in a caspase-1–independent manner. Primary bone marrow–derived macrophages from wt or caspase-1–deficient mice (casp-1−/−) were infected with invasive Salmonella (m.o.i 25). 10 and 20 min after infection, cells were lysed and the activation state of caspase-2 was analyzed by immunoblotting. An actin immunoblot is shown as a control for equal loading.
Caspase-1–deficient Macrophages Undergo SipB-dependent, Salmonella-induced Apoptosis.
We next tested whether caspase-1–deficient macrophages underwent apoptosis upon longer infection with the bacteria. Bone marrow–derived, caspase-1–deficient macrophages were either left untreated or were infected with wt or sipB − Salmonella strains. These macrophages succumbed to Salmonella-induced apoptosis after a 4-h infection. As in the case of the rapid apoptosis observed in wt macrophages, induction of cell death requires the SPI-1–encoded type III secretion apparatus, and the invA − and sipB − strains were not cytotoxic (Fig. 5 A). Caspase-1–deficient macrophages showed chromatin condensation after a 4-h infection with Salmonella, but nuclear staining never attained the characteristic compactness and brightness observed in the case of the wt macrophages (compare Fig. 1 B and Fig. 5 B). The mitochondria of these cells, although reduced in number (Fig. 5 B), retained the CM-X-ROS staining, indicating that their transmembrane potential did not dissipate. In infected cells, the mitochondria were organized in a more punctuate pattern. This reorganization was induced by the wt bacteria as well as by the invA − and the sipB − mutant strains and therefore was a consequence of infection rather than of cell death (Fig. 5 B).
Figure 5.


Caspase-1–deficient macrophages are not resistant to apoptosis induced by invasive Salmonella. Primary bone marrow–derived macrophages isolated from wt caspase-1–deficient mice were stained with CM-X-ROS and DAPI and infected with wt Salmonella or with the invasion-defective invA − and sipB − strains (m.o.i 25) as described in the legend to Fig. 1. (A) Caspase-1–deficient macrophages were infected with wt Salmonella or with the invasion-defective invA − and sipB − strains. The percentage of cells containing condensed chromatin was determined as described in the legend to Fig. 1, 25 min (open bars) and 4 h (closed bars) after infection. UT, untreated cells. The SD was <5% in all cases, and it has been omitted. (B) Photomicrographs of caspase-1–deficient macrophages infected with Salmonella.
To characterize the Salmonella-induced death of caspase-1–deficient macrophages ultrastructurally, we performed transmission electron microscopy of cells after a 4-h infection either with wt bacteria or with a sipB − mutant (Fig. 6). The cell infected with wt strain showed a morphology compatible with apoptosis, with cell and nuclear shrinkage and chromatin condensation (Fig. 6 B). Most of the cell was occupied by large vacuoles, and the mitochondria were reduced in number. In contrast to the situation observed in wt macrophages infected with invasive Salmonella (Fig. 2 B), however, these organelles did not show any gross anomalies. Caspase-1–deficient macrophages infected with a sipB − mutant strain were viable, contained fewer vacuoles and some bacteria, and showed no signs of cell damage (Fig. 6 A). Thus, Salmonella is capable to induce caspase-1–independent apoptosis in macrophages, and this requires a functional sipB gene.
Figure 6.

Electron microscopy of wt and caspase-1–deficient macrophages infected with invasive or invasion-deficient Salmonella. (A) Caspase-1–deficient macrophage after a 4-h infection with the sipB − strain; the size ratio between nucleus and cytoplasm shows no significant differences in comparison with uninfected macrophages. Moreover, both the nucleus and the cytoplasm do not show any morphological differences in comparison to control cells (A), and mitochondria do not display any signs of morphological alterations. A number of Salmonella (marked by asterisks) within the section indicate the infection of the cell. (B) Caspase-1–deficient macrophage after a 4-h infection with invasive Salmonella; the nucleus is slightly reduced in size and displays electron-dense material as a result of strong nucleolar segregation. The ratio in size between nucleus and cytoplasm is significantly reduced. The cytoplasm contains giant vacuoles. Absence of vacuoles and a dramatic reduction of cytoplasmic content in a late stage of cell death suggest a release of the vacuolar content into the culture medium (data not shown). Similar to control cells (A), mitochondria appear to be elongated and structurally intact.
Cytochrome c Is Released during Caspase-1–independent, Salmonella-induced Apoptosis.
The mitochondria of infected cells retained their membrane potential even during the late stages of apoptosis, and their morphology was not dramatically altered (Fig. 5 B and Fig. 6 B). However, invasive Salmonella induced progressive release of cytochrome c from the mitochondria of caspase-1–deficient macrophages. The appearance of cytochrome c in the mitochondrial supernatant preceded apoptosis, starting 1 h after infection and accumulating in the later phases (2 and 3 h). In contrast, cox-IV, an integral mitochondrial enzyme, was not present in the mitochondrial supernatant (Fig. 7). This served as a control for the purity of our mitochondrial supernatant and for the specificity of cytochrome c release.
Figure 7.

Cytochrome c (cyt-c) release during caspase-1–independent macrophage apoptosis. Primary bone marrow–derived macrophages isolated from caspase-1–deficient mice were infected with wt Salmonella. The presence of cytochrome c in the mitochondria (mito) or in the mitochondrial supernatant (SN) was assessed by immunoblotting at different times after infection. The blots were reprobed with antisera against cox-IV to control for the purity of our mitochondrial supernatant and for the specificity of cytochrome c release.
Caspase Activation during Caspase-1–independent, Salmonella-induced Apoptosis.
Caspase activation was monitored in the early phases of caspase-1–independent apoptosis (1 h; at this time chromatin condensation has not started and the cells are morphologically indistinguishable from cells infected with the noninvasive strains) as well as in the later phases of the process (3 h; at this time chromatin condensation is evident and the cells are clearly distinguishable from cells infected with noninvasive strains). Progressive caspase-2 activation was readily observed in these cells, as shown by the disappearance of the zymogen (procaspase-2). At later time points, when most of the zymogen was cleaved, the antiserum revealed a 33-kD intermediate form (p33). The apparent discrepancy with the kinetics of procaspase-2 cleavage shown in Fig. 4 stems from different exposure times used. The immunoblot shown in Fig. 4 was exposed for a short time, to visualize the relatively small changes in the amount of procaspase-2 at these early time points. Conversely, the immunoblot in Fig. 8 was exposed longer, to visualize the more dramatic cleavage of procaspase-2 and the appearance of p33 occurring at these later time points. Caspase-2 processing was not observed in cells infected with a sipB − mutant strain (Fig. 8 A). We further investigated whether the other caspases expressed in primary macrophages were activated during caspase-1–independent apoptosis. Caspase-3 activation was assessed using an antiserum that recognizes both the zymogen (procaspase-3) and the short subunit of the active enzyme (p11). Caspase-3 was activated within 1 h of infection and remained active in the late phases of apoptosis (3 h; Fig. 8 B). The activation of caspase-6 and -8 was assessed by immunoblotting with antisera that recognize the zymogens only (procaspase-6 and -8). Processing of caspase-6 and -8 was only evident in the late phases of apoptosis, 3 h after infection with invasive Salmonella. The decrease in the procaspase-6 and -8 bands was specific, as shown by re-probing the blot with an antiserum against actin as a normalization control (Fig. 8C–E). As in the case of caspase-2, caspase-3 -6, and -8 were not activated during infection with the sipB − mutant strain.
Caspase-2 Inhibition Delays Apoptosis in Both wt and Caspase-1–deficient Macrophages.
To gain more insight into the role of caspase-2 in Salmonella-induced apoptosis, we treated primary bone marrow–derived macrophages with a caspase-2 inhibitor before infection with invasive bacteria. The inhibitor-treated cells showed markedly delayed kinetics of apoptosis, although they succumbed to Salmonella 60 min after infection (Fig. 9 B). This result was confirmed by comparing the kinetics of Salmonella-induced apoptosis of primary macrophages from wt or caspase-2–deficient mice (Fig. 9 A). The effect of the inhibitor, however, was more marked than that of gene ablation (compare A and B in Fig. 9).
Figure 9.
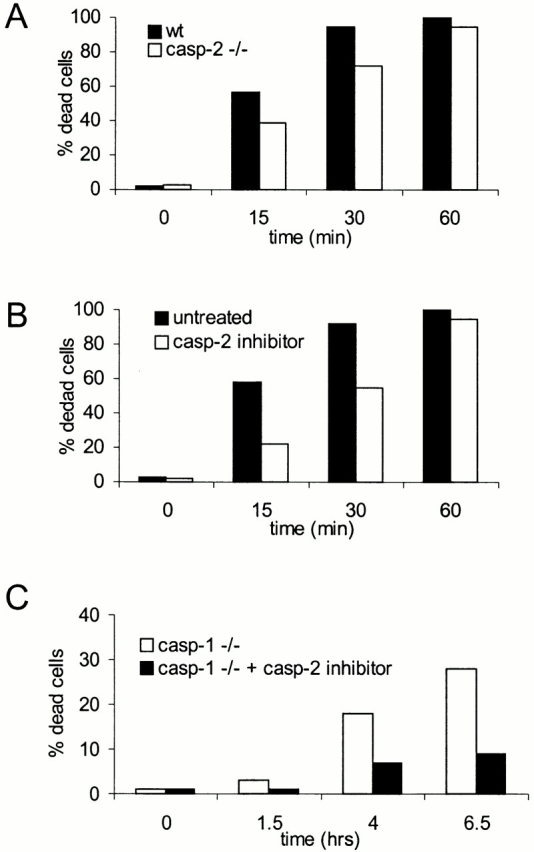
Chemical inhibition or genetic ablation of caspase-2 delays apoptosis induced by invasive Salmonella. Primary bone marrow–derived macrophages isolated from wt, caspase-2–deficient (casp-2−/−), or caspase-1–deficient (casp-1−/−) mice were stained with DAPI and infected with wt Salmonella as described in the legend to Fig. 1. (A) wt (closed bars) and caspase-2–deficient (open bars) macrophages were infected with wt Salmonella. (B) wt macrophages were either left untreated (closed bars) or were treated with a caspase-2 inhibitor (Z-VDVAD-fmk, 100 μM; open bars) for 90 min before infection. (C) Caspase-1–deficient macrophages were either left untreated (open bars) or were treated with a caspase-2 inhibitor as described above. The percentage of cells containing condensed chromatin was determined as described in the legend to Fig. 1, at different points after infection. The SD was <5% in all cases, and it has been omitted.
We next investigated the effect of the caspase-2 inhibitor on caspase-1–independent apoptosis. Treatment of caspase-1–deficient macrophages with the inhibitor resulted in a clear delay in the kinetics of Salmonella-induced apoptosis (Fig. 9 C). As noted above, however, the macrophages did succumb to the infection at later time points (20 h; data not shown).
Caspase-1 Activation by Invasive Salmonella Is Partially Dependent on Caspase-2.
To investigate the role of caspase-2 in caspase-1–dependent apoptosis, we monitored zymogen cleavage in the early phases of infection in primary bone marrow–derived macrophages from wt mice treated with a caspase-2–specific inhibitor. Interestingly, caspase-1 cleavage was clearly impaired in the inhibitor-treated cells (Fig. 10 A). Similar, albeit less pronounced, effects were observed by monitoring caspase-1 cleavage in caspase-2–deficient macrophage (Fig. 10 B). Thus, while Salmonella targeted caspase-2 independently of caspase-1, caspase-2 contributed to caspase-1 activation.
Figure 10.


Caspase-1 activation by invasive Salmonella is partially dependent on caspase-2. (A) Primary bone marrow–derived macrophages from wt mice were treated with a caspase-2 inhibitor (Z-VDVAD-fmk, 100 μM) for 90 min before infection with invasive Salmonella (m.o.i 25). (B) macrophages from wt or caspase-2–deficient mice (casp-2−/−) were infected with invasive Salmonella (m.o.i 25). 15 min after infection, cells were lysed and the activation state of caspase-1 was analyzed by immunoblotting. Actin immunoblots are shown as loading controls.
Discussion
The discovery of the induction of macrophage apoptosis by gram-negative pathogens is one of the most exciting developments in the field of host–parasite relationships. The apoptotic event is one of the fastest reported and is dependent on caspase-1. This finding is particularly intriguing, as caspase-1 is commonly regarded as an enzyme mainly involved in cytokine maturation and release. Here we report a previously unrecognized, caspase-1–independent form of apoptosis induced by invasive Salmonella in macrophages. This apoptotic process is slower than the caspase-1–dependent cell death. It involves both the activation of a caspase cascade comprising caspase-2, -3, -6, and -8 and the release of the apoptogenic cytochrome c from the mitochondria of the infected cells. Caspase-2 possesses a long prodomain, which identifies it as an initiator caspase 35. This protease is activated already 10 min after infection and therefore appears to be at the top of the caspase cascade induced by Salmonella. The second caspase to be activated is caspase-3, followed in the late phase by caspase-6 and -8 (processed 3 h after infection). This order has been derived by comparing the kinetics of caspase degradation using immunoblotting with the respective antibodies and is therefore tentative. Nevertheless, the data are compatible with earlier observations indicating that activation of caspase-2 by various apoptotic stimuli precedes caspase-3 cleavage 36 37 as well as with the ability of caspase-3 to sequentially activate caspase-6 and -8 in vitro 38. In further support of the role of caspase-2 in caspase-1–independent apoptosis, chemical inhibition of caspase-2 significantly delays this process, although it does not prevent it completely. It is therefore still possible that Salmonella may directly target other caspases in addition to caspase-1 and –2.
Specific release of cytochrome c from the mitochondria is a further feature of caspase-1–independent apoptosis. Judging by its kinetics, this event is likely to be a consequence of rather than the reason for the activation of the caspase cascade (Fig. 7). Caspase-3, -6, -7, and -8 can reportedly stimulate cytochrome c release 39. Caspase-3 and -8 may do so by cleaving Bid, a member of the Bcl-2 family, and generating a proapoptotic fragment that induces cytochrome c release 40 41. In Salmonella-infected macrophages, cytochrome c is set free in the absence of loss of mitochondrial membrane potential (Fig. 5 B) or of detectable mitochondrial swelling (Fig. 6 B), as has been reported in the case of Bid-induced release 41 42. It is conceivable that cytochrome c escaping from the mitochondria may feed back on the caspase cascade and accelerate the apoptotic process.
Caspase-2 plays a role in both caspase-1–dependent and –independent apoptosis. Genetic ablation of caspase-2 or its chemical inhibition results in a significant delay in the kinetics of rapid, caspase-1–dependent apoptosis; surprisingly for us, this correlates with a reduced activation of the only other caspase activated at this point, caspase-1. Chemical inhibition of caspase-2 seems to be more effective than genetic ablation. This might be because the inhibitor has unspecific effects other than caspase-2 inhibition. As an alternative explanation, it should be considered that the caspase-2 mice lack both the proapoptotic (caspase-2L) and antiapoptotic (caspase-2S; references 43 and 44) forms of the enzyme. As a result of this, accelerated apoptosis has been described in some cell types derived from these animals 32. The lack of both the pro- and antiapoptotic form of caspase-2 in the knockout mice might be responsible for the different efficacy of the chemical inhibitor and of the genetic modification in inhibiting Salmonella-induced apoptosis and caspase-1 activation. Be that as it may, it is clear that invasive Salmonella targets caspase-1 not only directly via direct SipB binding but also indirectly, via caspase-2 activation.
It is intriguing that both caspase-1–dependent and –independent apoptosis require the function of the type III secretion system encoded for by SPI-1 and, more specifically, the presence of the SipB protein (reference 30 and this study). SipB and its close relative, IpaB from Shigella flexneri, bind caspase-1 directly, but the exact mode of caspase activation has not yet been elucidated. It is tempting to speculate that binding of SipB or IpaB to the protease will cause aggregation and autoactivation of the enzyme (induced proximity model). In this context, it should be noted that caspase-1 and -2, although rather different in sequence, both contain long prodomains featuring highly homologous CARDs (caspase recruitment domains). These domains mediate homophilic binding of caspases to adaptor proteins, leading to caspase aggregation and activation 35. IpaB does not bind to caspase-2 directly 31, and given the similarity between the two bacterial proteins, it is reasonable to predict the same for SipB. This leaves open two main possibilities: (a) SipB may form a complex with a CARD-containing adaptor which in turn will bind to, and cause the activation of, caspase-2; and (b) SipB may function as a translocase for other virulence factors, one of which may be responsible for caspase-2 activation. Clarification of this issue must await further work.
In vitro, caspase-1–dependent apoptosis is a rapid and complete process that kills all exposed macrophages in a remarkably short time. Why, then, does Salmonella initiate a distinct apoptotic process, targeting a different host of apoptogenic proteins? When would this “fallback” apoptosis become relevant? First, there is evidence that, in vivo, caspase activation does not always correlate with apoptosis. Processing of caspases has been observed in nonapoptotic T lymphocytes, and it is in fact required for a number of physiological processes, including cell proliferation 45 46. Interestingly, only a subset of the caspases activated during T cell apoptosis is processed during proliferation. Similarly, active caspase-1 can be found in nonapoptotic, activated mucosal macrophages from patients with inflammatory bowel disease, a relevant example in our context 47. It is conceivable that, in vivo, both caspase-1 and caspase-2 must cooperate to induce efficient apoptosis. Second, it should be taken into account that during infection, the activation state of the macrophages encountered by Salmonella changes dramatically and that activated macrophages are reportedly more resistant than control cells against Salmonella-dependent apoptosis 6. The cells at the site of the infection will be confronted with Salmonella-derived LPS, which has been recently shown to upregulate a novel, endogenous caspase-1–specific inhibitor termed ICEBERG (Dixit, V., personal communication). These cells would then become resistant against caspase-1–dependent apoptosis but would still be killed by the alternative pathway described in this paper.
In addition, macrophages at the site of infection will produce nitric oxide, which will, in turn, decrease the activity of several caspases 48 49, including caspase-1 50. The transcription factors nuclear factor (NF)-κB and activator protein (AP)-1 will be also be activated in these macrophages. Both factors play a major role in the expression of inflammatory cytokines. At the same time, NF-κB 51 52 53 and AP-1 52 53 have been reported to antagonize macrophage apoptosis. If caspase activity in general is lowered by nitric oxide or if the cells become more resistant to apoptosis due to the stimulation of antiapoptotic mechanisms linked to macrophage activation, Salmonella might increase its chances to induce apoptosis by simultaneously targeting caspase-1 and -2.
The occurrence of apoptosis during the early phases of Salmonella infection has been demonstrated experimentally 7. In vivo, the induction of macrophage apoptosis would disable the very cell type that efficiently reduces the bacterial load 54. In addition, the macrophages encountering invasive bacteria would die before being able to produce inflammatory cytokines. Of further advantage for the microbe, apoptosis, in contrast to necrosis, does not lead to massive release of cellular components and therefore does not trigger inflammation. The rapid induction of macrophage apoptosis may be instrumental in establishing/maintaining systemic infection, and if so, it may represent an attractive therapeutic target. However, general caspase inhibitors may interfere with T cell function 45 46, and caspase-1–specific inhibitors might prevent the production of cytokines, which play an important role in the host resistance to infection 55. Understanding the alignment of the apoptotic pathways initiated by Salmonella might prove important for the design of therapeutic protocols that reduce macrophage apoptosis without altering the inflammatory response of the host.
Acknowledgments
We thank Dr. John Mudgett (Merck Sharp & Dohme) for the gift of caspase-1–deficient mice and Dr. Jorge E. Galan (Yale University) for the gift of the sipB − Salmonella strain. We are indebted to Thomas Decker, Vienna Biocenter, and Roberto Testi of the Università di Tor Vergata, Rome for critically reading this manuscript.
This work was supported by grant P13252-MOB of the Austrian Research Fund (to M. Baccarini).
Footnotes
Abbreviations used in this paper: SPI-1, Salmonella pathogenicity island 1; wt, wild type.
K.J. Procyk's present address is Protein Phosphorylation Lab, Imperial Cancer Research Fund, 44 Lincoln's Inn Fields, London WC2A 3PX, UK.
References
- Brumell J.H., Steele-Mortimer O., Finlay B.B. Bacterial invasionforce feeding by Salmonella . Curr. Biol. 1999;9:R277–280. doi: 10.1016/s0960-9822(99)80178-x. [DOI] [PubMed] [Google Scholar]
- Aderem A., Underhill D.M. Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- Chen L.M., Kaniga K., Galan J.E. Salmonella spp. are cytotoxic for cultured macrophages. Mol. Microbiol. 1996;21:1101–1115. doi: 10.1046/j.1365-2958.1996.471410.x. [DOI] [PubMed] [Google Scholar]
- Lindgren S.W., Stojiljkovic I., Heffron F. Macrophage killing is an essential virulence mechanism of Salmonella typhimurium . Proc. Natl. Acad. Sci. USA. 1996;93:4197–4201. doi: 10.1073/pnas.93.9.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg U., Vinatzer U., Berdnik D., von Gabain A., Baccarini M. Growth phase-regulated induction of Salmonella-induced macrophage apoptosis correlates with transient expression of SPI-1 genes. J. Bacteriol. 1999;181:3433–3437. doi: 10.1128/jb.181.11.3433-3437.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monack D.M., Raupach B., Hromockyj A.E., Falkow S. Salmonella typhimurium invasion induces apoptosis in infected macrophages. Proc. Natl. Acad. Sci. USA. 1996;93:9833–9838. doi: 10.1073/pnas.93.18.9833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter-Dahlfors A., Buchan A.M.J., Finlay B.B. Murine salmonellosis studied by confocal microscopySalmonella typhimurium resides intracellularly inside macrophages and exerts a cytotoxic effect on phagocytes in vivo. J. Exp. Med. 1997;186:569–580. doi: 10.1084/jem.186.4.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills S.D., Boland A., Sory M.P., van der Smissen P., Kerbourch C., Finlay B.B., Cornelis G.R. Yersinia enterocolitica induces apoptosis in macrophages by a process requiring functional type III secretion and translocation mechanisms and involving YopP, presumably acting as an effector protein. Proc. Natl. Acad. Sci. USA. 1997;94:12638–12643. doi: 10.1073/pnas.94.23.12638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monack D.M., Mecsas J., Ghori N., Falkow S. Yersinia signals macrophages to undergo apoptosis and YopJ is necessary for this cell death. Proc. Natl. Acad. Sci. USA. 1997;94:10385–10390. doi: 10.1073/pnas.94.19.10385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruckdeschel K., Roggenkamp A., Lafont V., Mangeat P., Heesemann J., Rouot B. Interaction of Yersinia enterocolitica with macrophages leads to macrophage cell death through apoptosis. Infect. Immun. 1997;65:4813–4821. doi: 10.1128/iai.65.11.4813-4821.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Smith M.R., Thirumalai K., Zychlinsky A. A bacterial invasin induces macrophage apoptosis by binding directly to ICE. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:3853–3860. [PMC free article] [PubMed] [Google Scholar]
- Guichon A., Zychlinsky A. Clinical isolates of Shigella species induce apoptosis in macrophages. J. Infect. Dis. 1997;175:470–473. doi: 10.1093/infdis/175.2.470. [DOI] [PubMed] [Google Scholar]
- Zychlinsky A., Prevost M.C., Sansonetti P.J. Shigella flexneri induces apoptosis in infected macrophages. Nature. 1992;358:167–169. doi: 10.1038/358167a0. [DOI] [PubMed] [Google Scholar]
- Zychlinsky A., Perdomo J.J., Sansonetti P.J. Molecular and cellular mechanisms of tissue invasion by Shigella flexneri. Ann. NY Acad. Sci. 1994;730:197–208. doi: 10.1111/j.1749-6632.1994.tb44249.x. [DOI] [PubMed] [Google Scholar]
- Zychlinsky A., Kenny B., Menard R., Prevost M.C., Holland I.B., Sansonetti P.J. IpaB mediates macrophage apoptosis induced by Shigella flexneri . Mol. Microbiol. 1994;11:619–627. doi: 10.1111/j.1365-2958.1994.tb00341.x. [DOI] [PubMed] [Google Scholar]
- Galan J.E., Collmer A. Type III secretion machinesbacterial devices for protein delivery into host cells. Science. 1999;284:1322–1328. doi: 10.1126/science.284.5418.1322. [DOI] [PubMed] [Google Scholar]
- Groisman E.A., Ochman H. How Salmonella became a pathogen. Trends Microbiol. 1997;5:343–349. doi: 10.1016/S0966-842X(97)01099-8. [DOI] [PubMed] [Google Scholar]
- Ochman H., Groisman E.A. Distribution of pathogenicity islands in Salmonella spp. Infect. Immun. 1996;64:5410–5412. doi: 10.1128/iai.64.12.5410-5412.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Los M., Wesselborg S., Schulze-Osthoff K. The role of caspases in development, immunity, and apoptotic signal transductionlessons from knockout mice. Immunity. 1999;10:629–639. doi: 10.1016/s1074-7613(00)80062-x. [DOI] [PubMed] [Google Scholar]
- Thornberry N.A., Lazebnik Y. Caspasesenemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- Fantuzzi G., Ku G., Harding M.W., Livingston D.J., Sipe J.D., Kuida K., Flavell R.A., Dinarello C.A. Response to local inflammation of IL-1 beta-converting enzyme-deficient mice. J Immunol. 1997;158:1818–1824. [PubMed] [Google Scholar]
- Friedlander R.M., Gagliardini V., Hara H., Fink K.B., Li W., MacDonald G., Fishman M.C., Greenberg A.H., Moskowitz M.A., Yuan J. Expression of a dominant negative mutant of interleukin-1 beta converting enzyme in transgenic mice prevents neuronal cell death induced by trophic factor withdrawal and ischemic brain injury. J. Exp. Med. 1997;185:933–940. doi: 10.1084/jem.185.5.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghayur T., Banerjee S., Hugunin M., Butler D., Herzog L., Carter A., Quintal L., Sekut L., Talanian R., Paskind M. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature. 1997;386:619–623. doi: 10.1038/386619a0. [DOI] [PubMed] [Google Scholar]
- Gu Y., Kuida K., Tsutsui H., Ku G., Hsiao K., Fleming M.A., Hayashi N., Higashino K., Okamura H., Nakanishi K. Activation of interferon-gamma inducing factor mediated by interleukin-1beta converting enzyme. Science. 1997;275:206–209. doi: 10.1126/science.275.5297.206. [DOI] [PubMed] [Google Scholar]
- Kuida K., Lippke J.A., Ku G., Harding M.W., Livingston D.J., Su M.S., Flavell R.A. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 1995;267:2000–2003. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- Li P., Allen H., Banerjee S., Franklin S., Herzog L., Johnston C., McDowell J., Paskind M., Rodman L., Salfeld J. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell. 1995;80:401–411. doi: 10.1016/0092-8674(95)90490-5. [DOI] [PubMed] [Google Scholar]
- Norman J., Yang J., Fink G., Carter G., Ku G., Denham W., Livingston D. Severity and mortality of experimental pancreatitis are dependent on interleukin-1 converting enzyme (ICE) J. Interferon Cytokine Res. 1997;17:113–118. doi: 10.1089/jir.1997.17.113. [DOI] [PubMed] [Google Scholar]
- Smith D.J., McGuire M.J., Tocci M.J., Thiele D.L. IL-1 beta convertase (ICE) does not play a requisite role in apoptosis induced in T lymphoblasts by Fas-dependent or Fas-independent CTL effector mechanisms. J. Immunol. 1997;158:163–170. [PubMed] [Google Scholar]
- Wong W.W. ICE family proteases in inflammation and apoptosis. Agents Actions Suppl. 1998;49:5–13. doi: 10.1007/978-3-0348-8857-8_2. [DOI] [PubMed] [Google Scholar]
- Hersh D., Monack D.M., Smith M.R., Ghori N., Falkow S., Zychlinsky A. The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc. Natl. Acad. Sci. USA. 1999;96:2396–2401. doi: 10.1073/pnas.96.5.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilbi H., Moss J.E., Hersh D., Chen Y., Arondel J., Banerjee S., Flavell R.A., Yuan J., Sansonetti P.J., Zychlinsky A. Shigella-induced apoptosis is dependent on caspase-1 which binds to IpaB. J. Biol. Chem. 1998;273:32895–32900. doi: 10.1074/jbc.273.49.32895. [DOI] [PubMed] [Google Scholar]
- Bergeron L., Perez G.I., Macdonald G., Shi L., Sun Y., Jurisicova A., Varmuza S., Latham K.E., Flaws J.A., Salter J.C. Defects in regulation of apoptosis in caspase-2-deficient mice. Genes Dev. 1998;12:1304–1314. doi: 10.1101/gad.12.9.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talanian R.V., Quinlan C., Trautz S., Hackett M.C., Mankovich J.A., Banach D., Ghayur T., Brady K.D., Wong W.W. Substrate specificities of caspase family proteases. J. Biol. Chem. 1997;272:9677–9682. doi: 10.1074/jbc.272.15.9677. [DOI] [PubMed] [Google Scholar]
- Macho A., Decaudin D., Castedo M., Hirsch T., Susin S.A., Zamzami N., Kroemer G. Chloromethyl-X-Rosamine is an aldehyde-fixable potential-sensitive fluorochrome for the detection of early apoptosis. Cytometry. 1996;25:333–340. doi: 10.1002/(SICI)1097-0320(19961201)25:4<333::AID-CYTO4>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Colussi P.A., Harvey N.L., Shearwin-Whyatt L.M., Kumar S. Conversion of procaspase-3 to an autoactivating caspase by fusion to the caspase-2 prodomain. J. Biol. Chem. 1998;273:26566–26570. doi: 10.1074/jbc.273.41.26566. [DOI] [PubMed] [Google Scholar]
- Harvey N.L., Butt A.J., Kumar S. Functional activation of Nedd2/ICH-1 (caspase-2) is an early process in apoptosis. J. Biol. Chem. 1997;272:13134–13139. doi: 10.1074/jbc.272.20.13134. [DOI] [PubMed] [Google Scholar]
- Li H., Bergeron L., Cryns V., Pasternack M.S., Zhu H., Shi L., Greenberg A., Yuan J. Activation of caspase-2 in apoptosis. J. Biol. Chem. 1997;272:21010–21017. doi: 10.1074/jbc.272.34.21010. [DOI] [PubMed] [Google Scholar]
- Slee E.A., Harte M.T., Kluck R.M., Wolf B.B., Casiano C.A., Newmeyer D.D., Wang H.G., Reed J.C., Nicholson D.W., Alnemri E.S. Ordering the cytochrome c–initiated caspase cascadehierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9–dependent manner. J. Cell Biol. 1999;144:281–292. doi: 10.1083/jcb.144.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossy-Wetzel E., Green D.R. Caspases induce cytochrome c release from mitochondria by activating cytosolic factors. J. Biol. Chem. 1999;274:17484–17490. doi: 10.1074/jbc.274.25.17484. [DOI] [PubMed] [Google Scholar]
- Li H., Zhu H., Xu C.J., Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- Luo X., Budihardjo I., Zou H., Slaughter C., Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- Shimizu S., Tsujimoto Y. Proapoptotic BH3-only bcl-2 family members induce cytochrome c release, but not mitochondrial membrane potential loss, and do not directly modulate voltage-dependent anion channel activity. Proc. Natl. Acad. Sci. USA. 2000;97:577–582. doi: 10.1073/pnas.97.2.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Miura M., Bergeron L., Zhu H., Yuan J. Ich-1, an Ice/ced-3-related gene, encodes both positive and negative regulators of programmed cell death. Cell. 1994;78:739–750. doi: 10.1016/s0092-8674(94)90422-7. [DOI] [PubMed] [Google Scholar]
- Kumar S., Kinoshita M., Noda M. Origin, expression and possible functions of the two alternatively spliced forms of the mouse Nedd2 mRNA. Cell Death Differ. 1997;4:378–387. [Google Scholar]
- Kennedy N.J., Kataoka T., Tschopp J., Budd R.C. Caspase activation is required for T cell proliferation. J. Exp. Med. 1999;190:1891–1896. doi: 10.1084/jem.190.12.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam A., Cohen L.Y., Aouad S., Sekaly R.P. Early activation of caspases during T lymphocyte stimulation results in selective substrate cleavage in nonapoptotic cells. J. Exp. Med. 1999;190:1879–1890. doi: 10.1084/jem.190.12.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlindon M.E., Galvin A., McKaig B., Gray T., Sewell H.F., Mahida Y.R. Investigation of the expression of IL-1beta converting enzyme and apoptosis in normal and inflammatory bowel disease (IBD) mucosal macrophages. Clin. Exp. Immunol. 1999;116:251–257. doi: 10.1046/j.1365-2249.1999.00884.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Billiar T.R., Talanian R.V., Kim Y.M. Nitric oxide reversibly inhibits seven members of the caspase family via S-nitrosylation. Biochem. Biophys. Res. Commun. 1997;240:419–424. doi: 10.1006/bbrc.1997.7672. [DOI] [PubMed] [Google Scholar]
- Kim Y.M., Talanian R.V., Billiar T.R. Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J. Biol. Chem. 1997;272:31138–31148. doi: 10.1074/jbc.272.49.31138. [DOI] [PubMed] [Google Scholar]
- Kim Y.M., Talanian R.V., Li J., Billiar T.R. Nitric oxide prevents IL-1beta and IFN-gamma-inducing factor (IL-18) release from macrophages by inhibiting caspase-1 (IL-1beta-converting enzyme) J. Immunol. 1998;161:4122–4128. [PubMed] [Google Scholar]
- Kitamura M. NF-kappaB-mediated self defense of macrophages faced with bacteria. Eur. J. Immunol. 1999;29:1647–1655. doi: 10.1002/(SICI)1521-4141(199905)29:05<1647::AID-IMMU1647>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- von Knethen A., Callsen D., Brune B. Superoxide attenuates macrophage apoptosis by NF-kappa B and AP-1 activation that promotes cyclooxygenase-2 expression. J. Immunol. 1999;163:2858–2866. [PubMed] [Google Scholar]
- von Knethen A., Callsen D., Brune B. NF-kappaB and AP-1 activation by nitric oxide attenuated apoptotic cell death in RAW 264.7 macrophages. Mol. Biol. Cell. 1999;10:361–372. doi: 10.1091/mbc.10.2.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenstock E., Jann K. Natural resistance of mice to Salmonella typhimuriumbactericidal activity and chemiluminescence response of murine peritoneal macrophages. J. Gen. Microbiol. 1981;125:173–183. doi: 10.1099/00221287-125-1-173. [DOI] [PubMed] [Google Scholar]
- Mastroeni P., Clare S., Khan S., Harrison J.A., Hormaeche C.E., Okamura H., Kurimoto M., Dougan G. Interleukin 18 contributes to host resistance and gamma interferon production in mice infected with virulent Salmonella typhimurium . Infect. Immun. 1999;67:478–483. doi: 10.1128/iai.67.2.478-483.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]