

Abstract
Monocyte recruitment to the central nervous system (CNS) is a necessary step in the development of pathologic inflammatory lesions in experimental autoimmune encephalomyelitis (EAE), a murine model of multiple sclerosis. Monocyte chemoattractant protein (MCP)-1, a potent agonist for directed monocyte migration, has been implicated in the pathogenesis of EAE. Here we report that deficiency in CC chemokine receptor (CCR)2, the receptor for MCP-1, confers resistance to EAE induced with a peptide derived from myelin oligodendrocyte glycoprotein peptide 35–55 (MOGp35–55). CCR2−/− mice immunized with MOGp35–55 failed to develop mononuclear cell inflammatory infiltrates in the CNS and failed to increase CNS levels of the chemokines RANTES (regulated on activation, normal T cell expressed and secreted), MCP-1, and interferon (IFN)-inducible protein 10 (IP-10) as well the chemokine receptors CCR1, CCR2, and CCR5. Additionally, T cells from CCR2−/− immunized mice showed decreased antigen-induced proliferation and production of IFN-γ compared with wild-type immunized controls, suggesting that CCR2 enhances the T helper cell type 1 immune response in EAE. These data indicate that CCR2 plays a necessary and nonredundant role in the pathogenesis of EAE.
Keywords: receptors, chemokine; chemokines; macrophages; encephalomyelitis; cytokines
Introduction
Chemokines are chemotactic cytokines that share important structural features and the ability to attract leukocytes. The regulated interactions of chemokines with their respective cell surface receptors mediate the recruitment of specific leukocyte subpopulations to sites of inflammation. The two major families of chemokines are distinguished by whether the first two of the four cysteine residues are separated by one amino acid (CXC, α-chemokines) or occur adjacent to each other (CC, β-chemokines). Whereas CXC chemokines are chemotactic mainly for neutrophils and lymphocytes, CC chemokines preferentially attract monocytes, eosinophils, basophils, and lymphocytes with variable selectivity. The CC chemokine family includes the monocyte chemoattractant protein (MCP)-eotaxin subfamily, containing MCP-1, -2, -3, -4, and -5 as well as eotaxin, all of which share ∼60% identity 1.
MCP-1 is a potent in vivo monocyte chemoattractant secreted by a variety of cell types in response to proinflammatory stimuli 2. Chemotactic and activating functions of MCP-1 are directed at monocytes, activated T cells, NK cells, and basophils. MCP-1 binds solely to CC chemokine receptor (CCR)2, a seven-transmembrane–spanning protein that is functionally linked to downstream signaling pathways through heterotrimeric G proteins 3. CCR2 is expressed on peripheral blood monocytes and activated T cells 4 and serves as the receptor for five ligands, MCP-1, -2, -3, -4, and –5 3.
MCP-1 and CCR2 have been implicated in a number of inflammatory diseases, including multiple sclerosis (MS; references 5 6 7), a chronic inflammatory disease of the central nervous system (CNS), and experimental autoimmune encephalomyelitis (EAE), the animal model for MS 8 9 10 11 12 13. EAE is mediated by activated neuroantigen-specific CD4+ Th1 cells and is characterized histologically by mononuclear cell infiltrates consisting of monocytes and Ag-specific and nonspecific CD4+ and CD8+ T cells 14. Monocyte recruitment to the CNS is a necessary step for the development of inflammatory lesions in EAE 15, and MCP-1 mRNA is upregulated during the acute phase of disease induced by immunization with myelin oligodendrocyte glycoprotein peptide 35–55 (MOGp35–55) or other encephalitogenic Ags 9 10. Vaccination of mice with a naked MCP-1 DNA, resulting in anti–MCP-1 Abs, inhibited the generation EAE 16. In addition, CCR2 signaling appears to promote Th1 immune responses in vivo 17 18 19. Taken together, these data indicate that MCP-1 and CCR2 might play an important role in the initiation and progression of EAE.
To test the hypothesis that CCR2 signaling is required for the pathogenesis of EAE, we induced disease in CCR2−/− and CCR2+/+ animals with the encephalitogenic peptide MOGp35–55. We found that mice lacking CCR2 are resistant to EAE, fail to develop mononuclear cell infiltrates, and display decreased Ag-specific proinflammatory responses in the secondary lymphoid organs. These results suggest an essential nonredundant in vivo function for CCR2 in the pathogenesis of EAE.
Materials and Methods
Mice and EAE Induction.
CCR2−/− mice were generated by homologous recombination as previously described 17. CCR2−/− and CCR2+/+ (WT) littermate controls were generated from matings between heterozygous (CCR2+/−) mice (50/50 Sv129 × C57BL/6) and raised in identical specific pathogen–free conditions. To induce EAE, CCR2−/− and WT littermates between 12 and 16 wk of age were immunized in the footpad with a total of 100 μg of MOGp35–55 (MGH Peptide Synthesis Facility, Charlestown, MA) emulsified in CFA (Difco Labs.); mice also received two intraperitoneal injections of 200 ng of Pertussis toxin (List Biological Laboratories) on days 0 and 2 after immunization. Animals were scored for signs of EAE using the following criteria: 1, tail paralysis; 2, hind limb weakness; 3, hind limb paralysis; 4, hind limb plus forelimb paralysis; 5, moribund or dead.
Cytokine Production and Proliferation.
Spleens from mice in each group were collected on days 8, 11, 20, 23, and 26 after disease induction; popliteal LNs draining the immunization site were collected on day 9. The organs were dispersed into a single-cell suspension, washed, and cultured at 4 × 106 cells/ml in serum-free X-Vivo 20 medium (Biowhittaker) with 100 μg/ml MOGp35–55. Supernatants were collected at 24 h for IL-2 and IL-4, 48 h for IFN-γ and IL-10, 72 h for TGF-β and IL-6, and 96 h for TNF-α. Quantitative ELISA was performed for interleukins and TNF-α using paired mAbs and protocol supplied by the manufacturer (PharMingen). Active and latent (with acid activation) TGF-β levels were determined by ELISA (Promega). For proliferation, cells were cultured in triplicate at 2 × 106 cells/ml with various doses of MOGp35–55, anti-CD3 mAb (PharMingen), or Con A (Fisher Scientific). [3H]Thymidine was added in the last 16 h of a 96-h stimulation with Ag or in the last 16 h of a 48-h stimulation with anti-CD3 mAb or Con A. Plates were harvested and counts read using a beta counter. Values used represent the mean value of three wells.
CNS Histology.
Brains and spinal cords from mice in each group were collected on days 8, 11, 15, 20, 23, and 26 after disease induction and fixed in 10% formalin in PBS. Paraffin-embedded tissues were sectioned 5 μm thick at 60-μm intervals and stained with hematoxylin and eosin as well as the myelin-specific stain Luxol fast blue. Slides were analyzed in a blinded fashion for lesion activity and type of infiltrating cells.
RNase Protection Assay.
Brains from mice in each group were collected on days 8, 11, 15, 20, 23, and 26 after disease induction and snap frozen in liquid nitrogen. Total RNA was prepared from frozen brain by standard guanidium isothiocyanate CsCl2 gradient centrifugation. The RNA probes used to detect chemokine and chemokine receptor mRNAs as well as the GADPH control for RNA loading was generated by in vitro transcription (PharMingen). Samples of total RNA (5 μg) were hybridized with 3 × 105 cpm/μl of α-[32P]UTP (3,000 Ci/mmol; NEN Life Science Products) labeled probe at 56°C overnight. RNase A and RNase Ti (PharMingen) digestion were carried at 37°C for 45 min. The digested products were separated on a 5% acrylamide–8 M urea gel, which was dried and film exposed (Eastman Kodak Co.) for 5–16 h.
Phagocyte Migration to the LN.
Mice were injected subcutaneously in the footpad with 100 μl of PBS/CFA 1:1 emulsion containing 1 μl of FluoroSpheres of 0.5-μm diameter (Molecular Probes). Popliteal LNs were collected 10 d after immunization, specimens were frozen in TissueTek medium (Sakura), and 5-μm frozen sections were made at 60-μm intervals. Sections were examined for fluorescence using a UV microscope.
Results and Discussion
CCR2− /− Mice Are Resistant to Induction of EAE.
To test the contribution of CCR2 signaling in the initiation and progression of EAE, CCR2−/− and CCR2+/+ mice were immunized with the encephalitogenic peptide MOGp35–55 and scored for signs of disease (Fig. 1). MOGp35–55 is an effective encephalitogen in the susceptible strain C57/BL6 bearing the H-2b MHC haplotype 20. As mice used in these experiments were littermates generated by crossing C57BL6/Sv129 parents heterozygous at the CCR2 locus, the offspring expressed H-2b and would be expected to remain susceptible to MOGp35–55-induced EAE. As expected, wild-type offspring developed clinical EAE, with a mode incidence of 80%. In contrast, their CCR2-deficient littermates completely failed to express signs of clinical disease in 24 of 26 CCR2-deficient mice analyzed. The two CCR2-deficient mice that developed disease had significantly delayed onset and decreased maximal disease severity compared with the wild-type littermates (data not shown).
Figure 1.

Incidence of disease in CCR2−/− and wild-type (WT) mice immunized with MOGp35–55. Groups of 5–10 mice were immunized with MOGp35–55 in CFA and received two intraperitoneal injections of Pertussis toxin. Results are expressed as mean disease score. This experiment is representative of n = 3.
Paucity of Inflammatory Lesions in CCR2−/− Mice Immunized with MOGp35–55.
To explore the basis of resistance to disease in CCR2-deficient mice, we examined the CNS tissues from mice in both groups. On histological examination (n = 6 mice per group), there were numerous meningeal and CNS parenchymal inflammatory/demyelinating lesions exclusively in the wild-type mice that also showed signs of clinical EAE (Fig. 2). These lesions consisted mainly of lymphocytic and monocytic infiltrates with few granulocytes. There were also varying degrees of optic and perioptic neuritis (data not shown). In contrast, CCR2-deficient littermates failed to develop CNS inflammation, which correlated with the absence of clinical EAE in these mice (Fig. 2).
Figure 2.

Histology of CNS tissue from MOGp35–55-immunized mice. Hematoxylin and eosin–stained, formalin-fixed cerebellum (A and B) and spinal cord (C and D) from CCR2+/+ (A and C) and CCR2−/− mice (B and D) after immunization with MOGp35–55. Representative (n = 6 mice per group) perivascular inflammatory lesions (indicated by arrows) composed of mononuclear cells are shown in CCR2+/+ brain and spinal cord sections. Note the absence of lesions and inflammatory cells in the sections from the brain and spinal cord of CCR2−/− mice. Original magnification 240×.
CNS Chemokine and Chemokine Receptor Expression Is Not Detected in Immunized CCR2−/− Mice.
To address the mechanism underlying the inability of CCR2−/− mice to produce active lesions, we examined the CNS chemokine profile from CCR2−/− and CCR2+/+ mice during the development of EAE. RNase Protection assays performed on brains from wild-type mice immunized with MOG35–55 revealed the upregulation of RANTES (regulated on activation, normal T cell expressed and secreted), IFN-inducible protein 10 (IP-10), and MCP-1. Increased levels of these chemokines were detected on day 11, peaked on days 20–23, and declined by day 26. In contrast, chemokine expression was not induced in the CNS of CCR2−/− immunized mice at all time points examined (Fig. 3 A). These results support the notion that CCR2-bearing cells are needed to produce an active lesion characterized by the production of proinflammatory chemokines that amplify the local inflammatory response.
Figure 3.

Chemokine and chemokine receptor expression in brains of MOGp35–55-immunized mice. RNase Protection analysis on 5 μg of total RNA isolated from brains of CCR2+/+ and CCR2−/− animals at the indicated times (in days) after immunization. Unprotected probe for each chemokine (A) and each chemokine receptor (B), which run slightly higher than protected RNA species, is indicated in the first and/or last lane.
In accordance with an increase in RANTES and MCP-1 expression in the brains of wild-type immunized mice, we found that the expression of their receptors was also upregulated. CCR1 and CCR5 (RANTES) and CCR2 (MCP-1) mRNA levels increased coincident with the increase of their ligands in wild-type mice (Fig. 3 B). In contrast, no increase in chemokine receptor expression was detected in CCR2-deficient immunized mice. Low levels of CCR5 mRNA were seen in CCR2-deficient and in wild-type mice before the onset of clinical disease. These results demonstrate that the upregulation of CCR1, CCR2, and CCR5 in the brains of mice with EAE requires the activity of CCR2-expressing cells.
Decreased MOGp35–55-specific T Cell Responses in CCR2−/− Mice.
As CCR2-deficient mice are resistant to developing inflammatory lesions and clinical signs of EAE, we asked whether this is associated with alterations in the MOGp35–55-specific immune response. To address this question, we analyzed the phenotype of T cells generated by immunization with MOGp35–55 in CCR2-deficient and wild-type mice (Fig. 4). Draining LN cells from wild-type mice showed robust proliferative responses to Ag, with stimulation indices approximately two times higher than in LN cells from the CCR2-deficient animals (Fig. 4). IFN-γ, a proinflammatory Th1 cytokine known to be involved in the pathogenesis of EAE, was also decreased in CCR2-deficient mice compared with wild-type mice (Fig. 4). Both groups secreted similar levels of IL-6 (Fig. 4). Levels of IL-2, TNF-α, IL-4, and IL-10 as well as active TGF-β were undetectable in culture supernatants (data not shown).
Figure 4.
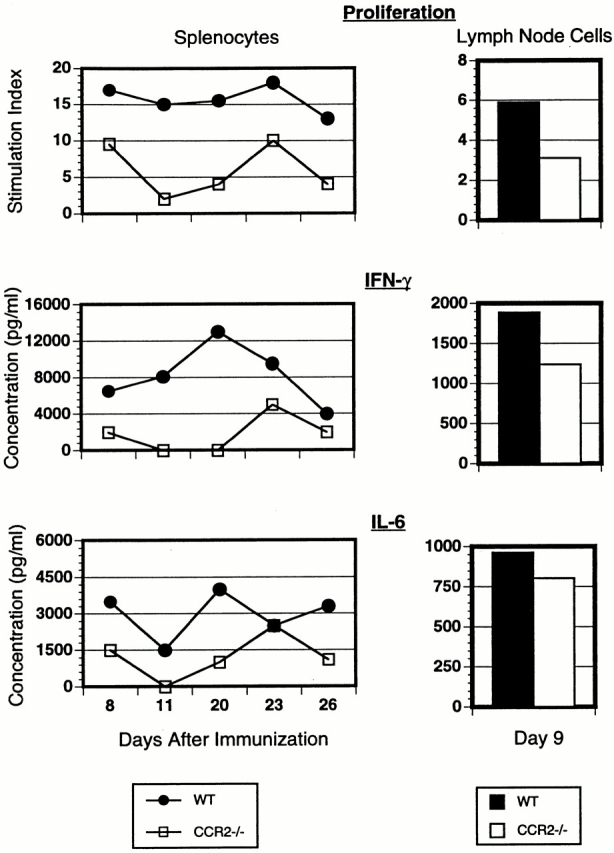
Ag-specific proliferative responses and cytokine production by spleen and LN cells from CCR2+/+ and CCR2−/− mice. Cells were isolated on days 8, 11, 20, 23, and 26 (spleen) and day 9 (LN; pooled from three mice per group) after disease induction and stimulated in vitro with MOGp35–55. Proliferative responses are shown as stimulation indices for each group responding to Ag concentration of 100 μg/ml. Cytokine concentration was measured from supernatants collected after 48 h (IFN-γ) or 72 h (IL-6) of in vitro culture with peptide (100 μg/ml).
We also examined the phenotype of MOGp35–55-specific T cells found in the spleen at different points in the course of disease. At days 8, 11, 20, 23, and 26 after disease induction, splenocytes from CCR2-deficient mice also proliferated less robustly to Ag and secreted lower levels of IFN-γ and IL-6 than splenocytes from wild-type controls, paralleling the finding of decreased Th1 expression in draining LNs. In contrast, when cells were stimulated with nonspecific stimuli such as Con A or the anti-CD3 mAb, splenocytes from CCR2-deficient mice showed proliferation that was equal to that of cells from wild-type littermates (data not shown). This suggests that the decreased effector responses were Ag specific and did not result from a generalized defect in T cell activation or function. Taken together, these observations indicate that resistance to EAE in CCR2-deficient mice is associated with a decreased Ag-specific Th1 effector response.
As T cell differentiation and activation in vivo involves effective Ag presentation, we also tested whether the observed differences in Ag-specific T cell responses in vitro could result from impaired Ag presentation in the LN. Dendritic cells are potent immunostimulatory cells found in the T cell areas of the LN. Recent studies concerning the origin of these cells suggest that monocytes recruited to an inflammatory focus acquire Ag by phagocytosis and then rapidly leave the tissue as they migrate into the LN and differentiate into dendritic cells 21. Therefore, we asked whether deficiency in CCR2, a key monocyte chemoattractant receptor, impairs the accumulation of phagocytes in the LN. In preliminary experiments, we found that CCR2−/− mice did not show decreased phagocyte migration to the LN 10 d after they were immunized with fluorescent beads in CFA (data not shown). This indicates that monocyte recruitment to the site of immunization and the LN are not overtly impaired in the CCR2−/− mice and most likely do not account for the observed decreased T cell responses.
Our observations provide functional evidence that CCR2 signaling is essential in the inflammatory cascade that culminates in clinical disease. These findings suggest that MCP-1 or other CCR2 agonists, induced in the CNS by activated neuroantigen-specific T cells, are necessary for the recruitment of peripheral blood monocytes into the brain and spinal cord. These monocytes serve to amplify the initial local T cell–mediated response in the CNS into an active lesion where macrophages become sufficiently numerous and activated to digest the myelin sheath, leading to demyelination, axonal dysfunction, and clinical manifestations of disease. Our results implicating CCR2 as an important receptor mediating monocyte recruitment in vivo are consistent with similar findings in other inflammatory disease models characterized by macrophage tissue accumulation 17 19 22 23 24.
In addition, our data suggests that CCR2 signaling enhances Ag-specific effector Th1 responses in vivo. Previous in vivo studies have also found decreased Th1 immune responses in CCR2−/− mice 17 18 19, although the mechanism for this observation has not been completely elucidated. MCP-1 has been shown to directly costimulate T cell activation in vitro, suggesting that CCR2 signaling may directly augment T cell proliferation and cytokine secretion 25 26. Our data further the notion that CCR2 signaling plays a role in Th1 immune responses in vivo.
In summary, we demonstrate that CCR2 is required for the pathogenesis of an organ-specific T cell and macrophage-mediated inflammatory disease, EAE, a model of MS. Accordingly, the existence of MCP-1 and CCR2 gene mutations in the human population may be important determinants in the initiation and/or progression of autoimmune diseases, and CCR2 antagonism might represent a potential therapeutic approach for such conditions.
Acknowledgments
We would like to thank Ray Sobel for expert analysis of histopathology and Anthony Slavin for help with this work.
This work was supported by National Institutes of Health grants KO8-DA00522 (to R.S. Klein), R01-AI43458 (to H.L. Weiner), and RO1-CA69212 and RO1-AI4699 (to A.D. Luster) and a Charles E. Culpeper Medical Scientist Award to A.D. Luster.
References
- Luster A.D. Chemokineschemotactic cytokines that mediate inflammation. N. Engl. J. Med. 1998;388:436–445. doi: 10.1056/NEJM199802123380706. [DOI] [PubMed] [Google Scholar]
- Gu L., Tseng S.C., Rollins B.J. Monocyte chemoattractant protein-1. Chem. Immunol. 1999;72:7–29. doi: 10.1159/000058723. [DOI] [PubMed] [Google Scholar]
- Charo I.F. CCR2from cloning to the creation of knockout mice. Chem. Immunol. 1999;72:30–41. doi: 10.1159/000058724. [DOI] [PubMed] [Google Scholar]
- Frade J.M., Mellado M., del Real G., Gutierrez-Ramos J.C., Lind P., Martinez A.C. Characterization of the CCR2 chemokine receptorfunctional CCR2 receptor expression in B cells. J. Immunol. 1997;159:5576–5584. [PubMed] [Google Scholar]
- Simpson J.E., Newcombe J., Cuzner M.L., Woodroofe M.N. Expression of monocyte chemoattractant protein-1 and other beta-chemokines by resident glia and inflammatory cells in multiple sclerosis lesions. J. Neuroimmunol. 1998;84:238–249. doi: 10.1016/s0165-5728(97)00208-7. [DOI] [PubMed] [Google Scholar]
- Van Der Voorn P., Tekstra J., Beelen R.H., Tensen C.P., Van Der Valk P., De Groot C.J. Expression of MCP-1 by reactive astrocytes in demyelinating multiple sclerosis lesions. Am. J. Pathol. 1999;154:45–51. doi: 10.1016/S0002-9440(10)65249-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balashov K., Rottman J., Weiner H., Hancock W. CCR5+ and CXCR3+ T cells are increased in multiple sclerosis and their ligands MIP-1 and IP-10 are expressed in demyelinating brain lesions. Proc. Natl. Acad. Sci. USA. 1999;96:6873–6878. doi: 10.1073/pnas.96.12.6873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulkower K., Brosnan C.F., Aquino D.A., Cammer W., Kulshrestha S., Guida M.P., Rapoport D.A., Berman J.W. Expression of CSF-1, c-fms, and MCP-1 in the central nervous system of rats with experimental allergic encephalomyelitis. J. Immunol. 1993;150:2525–2533. [PubMed] [Google Scholar]
- Kennedy K.J., Strieter R.M., Kunkel S.L., Lukacs N.W., Karpus W.J. Acute and relapsing experimental autoimmune encephalomyelitis are regulated by differential expression of the CC chemokines macrophage inflammatory protein-1alpha and monocyte chemotactic protein-1. J. Neuroimmunol. 1998;92:98–108. doi: 10.1016/s0165-5728(98)00187-8. [DOI] [PubMed] [Google Scholar]
- Juedes A.E., Hjelmstrom P., Bergman C.M., Neild A.L., Ruddle N.H. Kinetics and cellular origin of cytokines in the central nervous systeminsight into mechanisms of myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis. J. Immunol. 2000;164:419–426. doi: 10.4049/jimmunol.164.1.419. [DOI] [PubMed] [Google Scholar]
- Godiska R., Chantry D., Dietsch G.N., Gray P.W. Chemokine expression in murine experimental allergic encephalomyelitis. J. Neuroimmunol. 1995;58:167–176. doi: 10.1016/0165-5728(95)00008-p. [DOI] [PubMed] [Google Scholar]
- Glabinski A.R., Tani M., Strieter R.M., Tuohy V.K., Ransohoff R.M. Synchronous synthesis of alpha- and beta-chemokines by cells of diverse lineage in the central nervous system of mice with relapses of chronic experimental autoimmune encephalomyelitis. Am. J. Pathol. 1997;150:617–630. [PMC free article] [PubMed] [Google Scholar]
- Jiang Y., Salafranca M.N., Adhikari S., Xia Y., Feng L., Sonntag M.K., deFiebre C.M., Pennell N.A., Streit W.J., Harrison J.K. Chemokine receptor expression in cultured glia and rat experimental allergic encephalomyelitis. J. Neuroimmunol. 1998;86:1–12. doi: 10.1016/s0165-5728(98)00005-8. [DOI] [PubMed] [Google Scholar]
- Karpus W.J., Ransohoff R.M. Chemokine regulation of experimental autoimmune encephalomyelitistemporal and spatial expression patterns govern disease pathogenesis. J. Immunol. 1998;161:2667–2671. [PubMed] [Google Scholar]
- Tran E.H., Hoekstra K., van Rooijen N., Dijkstra C.D., Owens T. Immune invasion of the central nervous system parenchyma and experimental allergic encephalomyelitis, but not leukocyte extravasation from blood, are prevented in macrophage-depleted mice. J. Immunol. 1998;161:3767–3775. [PubMed] [Google Scholar]
- Youssef S., Wildbaum G., Maor G., Lanir N., Gour-Lavie A., Grabie N., Karin N. Long-lasting protective immunity to experimental autoimmune encephalomyelitis following vaccination with naked DNA encoding C-C chemokines. J. Immunol. 1998;161:3870–3879. [PubMed] [Google Scholar]
- Boring L., Gosling J., Chensue S., Kunkel S., Farese R.J., Broxmeyer H., Charo I. Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J. Clin. Invest. 1997;100:2552–2561. doi: 10.1172/JCI119798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warmington K.S., Boring L., Ruth J.H., Sonstein J., Hogaboam C.M., Curtis J.L., Kunkel S.L., Charo I.R., Chensue S.W. Effect of C-C chemokine receptor 2 (CCR2) knockout on type-2 (schistosomal antigen-elicited) pulmonary granuloma formationanalysis of cellular recruitment and cytokine responses. Am. J. Pathol. 1999;154:1407–1416. doi: 10.1016/S0002-9440(10)65394-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynor T.R., Kuziel W.A., Toews G.B., Huffnagle G.B. CCR2 expression determines T1 versus T2 polarization during pulmonary Cryptococcus neoformans infection. J. Immunol. 2000;164:2021–2027. doi: 10.4049/jimmunol.164.4.2021. [DOI] [PubMed] [Google Scholar]
- Wekerle H., Kojima K., Lannes-Vieira J., Lassmann H., Linington C. Animal models. Ann. Neurol. 1994;36:S47–53. doi: 10.1002/ana.410360714. [DOI] [PubMed] [Google Scholar]
- Randolph G.J., Inaba K., Robbiani D.F., Steinman R.M., Muller W.A. Differentiation of phagocytic monocytes into lymph node dendritic cells in vivo. Immunity. 1999;11:753–761. doi: 10.1016/s1074-7613(00)80149-1. [DOI] [PubMed] [Google Scholar]
- Kurihara T., Warr G., Loy J., Bravo R. Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J. Exp. Med. 1997;186:1757–1762. doi: 10.1084/jem.186.10.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuziel W.A., Morgan S.J., Dawson T.C., Griffin S., Smithies O., Ley K., Maeda N. Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc. Natl. Acad. Sci. USA. 1997;94:12053–12058. doi: 10.1073/pnas.94.22.12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boring L., Gosling J., Cleary M., Charo I.F. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–897. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- Taub D.D., Turcovski-Corrales S.M., Key M.L., Longo D.L., Murphy W.J. Chemokines and T lymphocyte activationI. Beta chemokines costimulate human T lymphocyte activation in vitro. J. Immunol. 1996;156:2095–2103. [PubMed] [Google Scholar]
- Taub D.D., Ortaldo J.R., Turcovski-Corrales S.M., Key M.L., Longo D.L., Murphy W.J. Beta chemokines costimulate lymphocyte cytolysis, proliferation, and lymphokine production. J. Leukoc. Biol. 1996;59:81–89. doi: 10.1002/jlb.59.1.81. [DOI] [PubMed] [Google Scholar]