

Abstract
We sought to understand the relationship between reactive oxygen species (ROS) and the mitochondrial permeability transition (MPT) in cardiac myocytes based on the observation of increased ROS production at sites of spontaneously deenergized mitochondria. We devised a new model enabling incremental ROS accumulation in individual mitochondria in isolated cardiac myocytes via photoactivation of tetramethylrhodamine derivatives, which also served to report the mitochondrial transmembrane potential, ΔΨ. This ROS accumulation reproducibly triggered abrupt (and sometimes reversible) mitochondrial depolarization. This phenomenon was ascribed to MPT induction because (a) bongkrekic acid prevented it and (b) mitochondria became permeable for calcein (∼620 daltons) concurrently with depolarization. These photodynamically produced “triggering” ROS caused the MPT induction, as the ROS scavenger Trolox prevented it. The time required for triggering ROS to induce the MPT was dependent on intrinsic cellular ROS-scavenging redox mechanisms, particularly glutathione. MPT induction caused by triggering ROS coincided with a burst of mitochondrial ROS generation, as measured by dichlorofluorescein fluorescence, which we have termed mitochondrial “ROS-induced ROS release” (RIRR). This MPT induction/RIRR phenomenon in cardiac myocytes often occurred synchronously and reversibly among long chains of adjacent mitochondria demonstrating apparent cooperativity. The observed link between MPT and RIRR could be a fundamental phenomenon in mitochondrial and cell biology.
Keywords: mitochondria, redox, heart, nitric oxide, Ca2+ sparks
Introduction
Beyond a fundamental role in energy metabolism, mitochondria also play key roles in maintenance of cellular redox potential 1, Ca2+ homeostasis 2, and apoptosis 3. A central event in apoptosis is now known to be a phenomenon called the mitochondrial permeability transition (MPT) (for review see references 4 and 5). In addition, mitochondria are both a major source of reactive oxygen species (ROS; reference 6) and a target for their damaging effects 7. Understanding the interplay among these roles and ROS in both normal and pathological conditions has led to renewed interest in mitochondrial function and the MPT.
Observed originally in isolated liver mitochondria 8, the MPT is a proteinaceous pore or megachannel resulting in permeability to ions and solutes up to ∼1,500 daltons and collapse of the mitochondrial membrane potential (ΔΨ) 2 4. Many agents or conditions have been shown to cause pore opening in isolated mitochondria, most notably high Ca2+ loading and various treatments that cause oxidative stress 2. In addition, the MPT is regulated by ΔΨ, H+, Mg2+, adenine nucleotides, NAD(P)H, and the redox state of critical protein thiols 9 10.
Mitochondria and the MPT play major roles in cell death. Agents that block the MPT (cyclosporin A, bongkrekic acid [BA]) block apoptosis 3 11. Various oxidants stimulate whereas antioxidants inhibit apoptosis, suggesting a role for ROS as initiators or downstream mediators of apoptosis 12. In addition, mitochondrial proteins (certain caspases, an apoptosis-inducing factor, and cytochrome c) that are released upon irreversible MPT activation play major roles in apoptosis 3.
A role for the MPT in normal cell function has also been proposed, especially in the context of Ca2+ homeostasis. Physiological stimuli can result in increases in mitochondrial Ca2+ (for review see reference 13), coupling energy demand to ATP production 14 via activation of rate-limiting steps in the respiratory chain 15. As sequential pulses of Ca2+ could summate and produce an overload of mitochondrial Ca2+, the MPT might serve a physiological function to counteract this accumulation by serving as a fast reversible Ca2+ release channel 9. While Hunter and Haworth 8 initially observed MPT reversibility in suspensions of isolated mitochondria, a physiologic role for the MPT in the regulation of mitochondrial Ca2+ homeostasis remains speculative.
Because mitochondria are themselves the major intracellular sources of ROS production, together with the fact that ROS exposure and altered redox state can lead to the MPT, we hypothesized that under certain circumstances this biological system could become self-amplifying and unstable. Thus, we sought to understand how these mechanisms are related and controlled in intact cells. Use of confocal microscopy enabled the recent demonstration of MPT gating in isolated individual mitochondria that was inhibited by catalase 16, supporting causation by ROS. This technique has also been used to show opening of the pore in hepatocytes exposed to MPT inducers 17. We addressed the role of ROS during MPT induction using isolated adult cardiac myocytes, taking advantage of the rigid, lattice-like distribution of mitochondria suitable for high precision confocal line-scan imaging. We present a model of controlled, photoexcitation-induced ROS production within individual mitochondria in these intact cells that triggers MPT induction in a reproducible and frequently reversible fashion. We have also identified a novel phenomenon resulting from this ROS-triggering of the MPT, which we have termed “ROS-induced ROS release” (RIRR).
Materials and Methods
Cardiac Myocyte Isolation.
Single cardiac myocytes were isolated from adult rats (2–4 mo old) by a standard enzymatic technique 18. Cells were suspended in Hepes-buffered solution containing (in millimoles per liter) 137 NaCl, 4.9 KCl, 1.2 MgSO4, 1.2 NaH2PO4, 15 glucose, 20 Hepes, and 1.0 CaCl2, pH 7.4, and stored at room temperature until use.
Confocal Microscopy.
Myocytes were loaded with dyes for >20 min on a microscope stage, incubated in Hepes-buffered solution (same composition as the storage solution) at 23°C, and imaged with a LSM-410 inverted confocal microscope using a Plan-Neofluar 63×/1.4N.A. oil immersion lens (Carl Zeiss, Inc.). Time scans were recorded from mitochondria arrayed along individual myofibrils in a multichannel line-scan mode with excitation at both 488 nm (for 2, 7-dichlorodihydrofluorescein diacetate [DCF], diaminofluorescein diacetate [DAF]-2, and calcein-AM) and 568 nm (for TMRE, TMRM [tetramethylrhodamine, methyl and ethyl ester, respectively], and MitoTracker® Red CMXRos; Molecular Probes, Inc.), collecting simultaneous fluorescence emission at 515–540 nm and >590 nm, respectively. Each image consisted of 512–1,024 line scans obtained at 2–230 Hz, each line comprising 512 pixels spaced at 0.050-μm intervals. The confocal pinhole was set to obtain spatial resolutions of 0.4 μm in the horizontal plane and 1.0 μm in the axial dimension. Some protocols were performed using 351 nm excitation, collecting 400–435 nm fluorescence emission, using a Zeiss c-apo 63×/1.3N.A. water immersion lens. Experiments were carried out at 23°C. Image processing was done using MetaMorph® software (Universal Imaging).
Determination of Glutathione.
Total glutathione (GSH) and glutathione disulfide (GSSG) were measured with the 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB)–GSSG reductase recycling procedure according to Anderson 19 with minor modifications. Glutathione concentrations were calculated in relation to the 5-thio-2-nitrobenzoate (TNB) formation kinetics of identically processed GSH and GSSG standards. For determination of GSSG, the samples were treated with 2-vinylpyridine before addition to the reaction mix. Protein was determined from the centrifuged cell lysate before acid precipitation (BioRad Protein Assay Kit).
Electron Spin Resonance.
Electron paramagnetic resonance (EPR) spectra were recorded in a quartz flat cell with a Bruker ER 300 spectrometer operating at X-band with a TM110 cavity using a modulation frequency of 100 kHz, modulation amplitude of 0.5 G, microwave power of 20 mW, and microwave frequency of 9.785 GHz. Photoactivation of solutions of TMRM (100 μM) in Hepes-buffered medium containing 5-diethoxy-phosphoryl-5-methyl-1-pyrroline-N-oxide (DEPMPO; 10 mM) or 5,5-dimethyl-1-pyrroline-N-oxide (DMPO; 100 mM) inside the EPR cuvette was produced by exposure to light from a 300 W halogen projector bulb passed through a 546 nm center/10 nm FWHM band pass filter (Corion). Temperature of the specimen was maintained at 23°C.
Materials.
TMRE, TMRM, calcein-AM, MitoTracker Red CMXRos, and DCF were purchased from Molecular Probes, Inc.; diethylmaleate (DEM), Nω-nitro-l-arginine methyl ester (L-NAME) and oligomycin were from Sigma-Aldrich; S-nitroso-N-acetylpenicillamine (SNAP), DAF-2, Mn(III)tetrakis (1–methyl-4-pyridyl)porphyrin pentachloride (MnTMPyP), 1,2-bis(o-amino-5-fluorophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetra (acetoxymethyl) ester (BAPTA-AM), Ru360, and 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid (Trolox) were from Calbiochem; and BA was from A.G. Scientific, Inc. All other chemicals were of the purest reagent grade available.
Results
Spontaneous ROS Production at the Sites of Deenergized Mitochondria.
Mitochondria in freshly isolated adult rat cardiac myocytes are normally arrayed in a three-dimensional “lattice” of parallel rows surrounding the contractile myofilaments, as seen in cells loaded with the lipophilic cationic fluorophore TMRM accumulated by mitochondria in proportion to ΔΨ (Fig. 1A and Fig. B). Given the degree of TMRM concentration in mitochondria compared with all other cellular compartments, the extramitochondrial space appears black by contrast. Mitochondrial rows in the axial (or “z”) dimension are sufficiently far apart (Fig. 1 B), so that confocal imaging with 1-μm z-resolution would encompass a single mitochondrial thickness.
Figure 1.


Spatial organization and function of mitochondria in isolated adult rat cardiac myocytes. (A) Confocal plane of a myocyte loaded with TMRM (125 nM; bar = 5 μm). (B) z-section through A (1-μm resolution). Arrows in A and B denote mitochondria lacking TMRM sequestration. (C) Spontaneous ROS production at sites of low mitochondrial membrane potential: red, TMRM (125 nM) fluorescence; green, DCF (10 μM) fluorescence. (D) Exposure to antimycin A (25 μM) produces widespread numbers of depolarized mitochondria (loss of red TMRM fluorescence) together with increased ROS production (increased green DCF fluorescence). (E) Pretreatment with the ROS scavenger Trolox (2 mM) prevents the antimycin A–induced mitochondrial depolarization seen in D (cell labeled with TMRM and DCF as in D).
In TMRM-loaded cells, occasional discrete areas of the mitochondrial lattice lack fluorescence above background despite the (expected) presence of mitochondria (Fig. 1A and Fig. B, arrows), suggesting that ΔΨ has been locally dissipated. Hypothesizing that these deenergized mitochondria exhibit increased ROS production, these black “holes” in TMRM mitochondrial fluorescence were sought in cells dual-loaded with TMRM and the ROS-sensitive dye DCF. Fig. 1 C shows such a cell loaded with both TMRM (red) and DCF (green) in which a discrete area of two or three mitochondria have lost ΔΨ (dark holes in red fluorescence; arrow) in precisely the same region in which increased DCF fluorescence is seen. This suggests that spontaneously deenergized mitochondria could produce significantly elevated levels of ROS, at least transiently. The next series of experiments employs a novel method to study the relationship between mitochondrial ΔΨ and ROS production in live cells.
ROS Triggers the Abrupt Loss of Mitochondrial ΔΨ in Cardiac Myocytes.
In the case of these spontaneously deenergized mitochondria, it was unclear whether local buildup of ROS caused or was the result of the loss of ΔΨ. Increasing endogenous ROS generation using the Complex III inhibitor antimycin A (25 μM) produced widespread numbers of depolarized mitochondria with increased ROS production in cells loaded with both TMRM and DCF (Fig. 1 D). That this effect of antimycin A was prevented by the ROS scavenger Trolox (2 mM) provided further evidence that local ROS accumulation was likely involved in this loss of ΔΨ (Fig. 1 E). As ROS are formed as a byproduct of photoexcitation, we set out to prove the causative role of ROS in this process by devising a laser excitation/confocal fluorescence imaging protocol using mitochondrially sequestered probes to follow ΔΨ and simultaneously cause the incremental photoproduction and accumulation of ROS inside of specific mitochondria (Fig. 2 A). The line-scan (X-T) image in a TMRM-loaded cell, which displays how ΔΨ varies with time in individual mitochondria by inspecting the fluorescence intensity along vertical columns (time axis), shows a series of abrupt transitions (i.e., loss of fluorescence) in each mitochondrion, occurring at variable points during scanning (Fig. 2 A, bottom panel). This pattern of photoexcitation-induced ΔΨ loss in TMRM-loaded cells is characterized by its mean occurrence time, which is typically between 60 and 90 s (Fig. 2 B). Photoexcitation of TMRM causes the production of both ·O2- and ·OH as measured by EPR spectroscopy (Fig. 2 C). Notably, Trolox is a potent scavenger of the ROS produced under these conditions.
Figure 2.


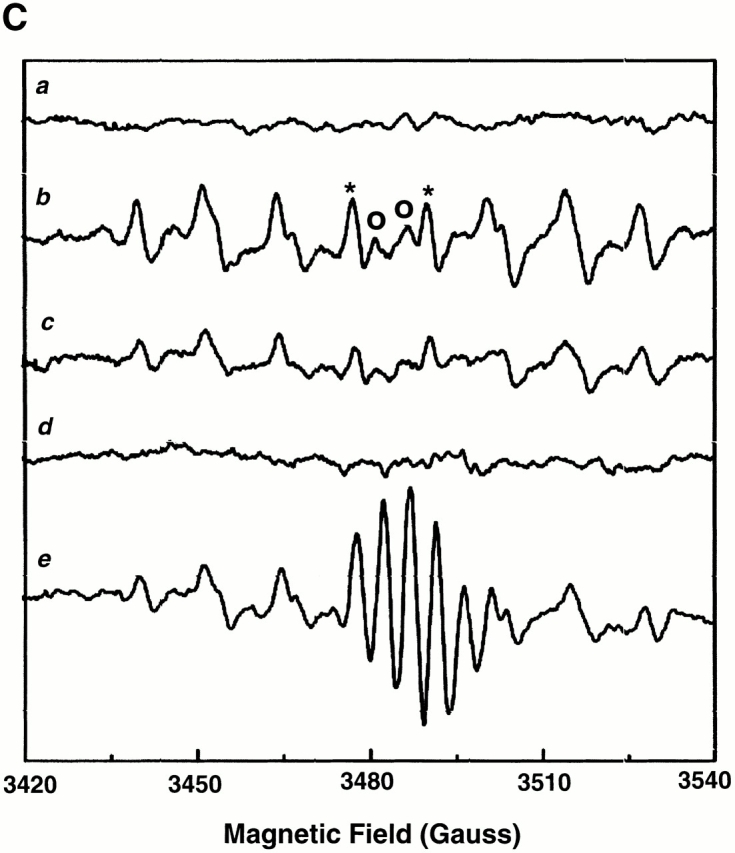
Experimental model: line scanning of mitochondrial arrays and photoexcitation ROS production. (A) Upper panel, confocal image of fluorescence in TMRM-loaded (125 nM) cardiac myocyte. Line drawn on image shows position scanned for experiment in bottom panel. Bottom panel shows 2 Hz line-scan image of TMRM fluorescence along mitochondrial row; time progresses from top (total scan 256 s). The dark regions between vertical columns are junctions between mitochondria. The sudden dissipation of TMRM fluorescence (white-to-black transitions) indicates mitochondrial ΔΨ loss. (B) Frequency distribution of ΔΨ transition times (time to 50% dissipation of ΔΨ) for the mitochondrial ensemble during line-scan imaging at 2 Hz as in A (n = 5 cells). (C) EPR spin-trapping, measured as the formation of DEPMPO/·OH and ·O2 − adducts during photoactivation of TMRM solutions. The system consisted of TMRM (100 μM) and DEPMPO (10 mM) in Hepes-buffered medium, pH 7.4, maintained at 23°C. While no signal was seen without light (a), a prominent spectrum of the DEPMPO/·OH (o symbol labeling central peaks) and ·O2 − (*central peaks) adducts was seen during illumination (b) and abolished partially (50–60%) by catalase (100 U/ml; c) or completely by SOD (1,000 U/ml; d). Trolox (2 mM) quenched these illumination-dependent signals by ∼60 ± 10% and produced a superimposed intense seven-line EPR signal of the Trolox-derived phenoxyl radical (e). Each spectrum is the sum of 24 1-min sequential acquisitions.
ROS-induced ΔΨ Loss Occurs Cooperatively and Reversibly.
The typical pattern of ROS-induced ΔΨ loss occurs synchronously, or “cooperatively,” between mitochondrial pairs along one sarcomere, with occasional higher order synchronization (Fig. 2 A). Infrequently, long-range synchronization between extended mitochondrial groups is observed (>30 mitochondria, over ∼35 μm, < 500 ms; Fig. 3 A). Notably, this phenomenon remains synchronized during repeated episodes of ΔΨ dissipation and, more interestingly, even during the spontaneous and abrupt recovery of TMRM fluorescence.
Figure 3.


(A) Evidence of cooperativity and reversibility of ΔΨ (TMRM) dissipation during 2 Hz line-scan imaging. (B) The abrupt dissipation phase of TMRM fluorescence during line-scan imaging can be described by first-order kinetics. In this example, the dissipation of ΔΨ in a single mitochondrion occurs in step-wise fashion over several seconds. The inset shows all of the segments with negative slope (joined together; after background subtraction), fit by a single exponential model with τ1/2 = 0.54 s.
Clearly, large and abrupt changes in mitochondrial membrane permeability induced by ROS are responsible for these transitions. While these episodes of ΔΨ loss often proceeded to the apparent complete loss of dye fluorescence, many examples were also observed of a step-wise progression by individual mitochondria (Fig. 3 B). This finding raises the question of whether this permeability phenomenon in general was the result of single or multiple conductance states. The inset in Fig. 3 B, using the data from this representative example, shows that each fluorescence segment (joined together) is well fit by a single exponential model. Indeed, an apparent single-state conductance behavior appears to be a general result, because first-order TMRM efflux kinetics described by the same rate constant (1.27 ± 0.12 s−1) during MPT induction was consistently found in all of the cases examined (n = 12) regardless of step-wise or continuous progression.
RIRR in Single Mitochondria.
Observing the spontaneous occurrence of high local ROS production at the sites of deenergized mitochondria (Fig. 1 C), we sought to discover whether the loss of ΔΨ induced by laser scanning would also be accompanied by increased ROS production. In cells dual-labeled with TMRM and DCF (Fig. 4), line-scan imaging induced ΔΨ loss, but in addition, there was an obvious “ROS burst” in each mitochondrion beginning at the moment of ΔΨ loss. ROS production proceeded in two distinct phases: the initial, slow rise due to the accumulation of photoexcitation-related ROS production, i.e., “trigger ROS, ” followed by the ROS burst, occurring simultaneously with ΔΨ dissipation, due to apparent mitochondrial ROS production (Fig. 4 C). We have called this the “ROS-induced ROS release” (RIRR) phenomenon.
Figure 4.
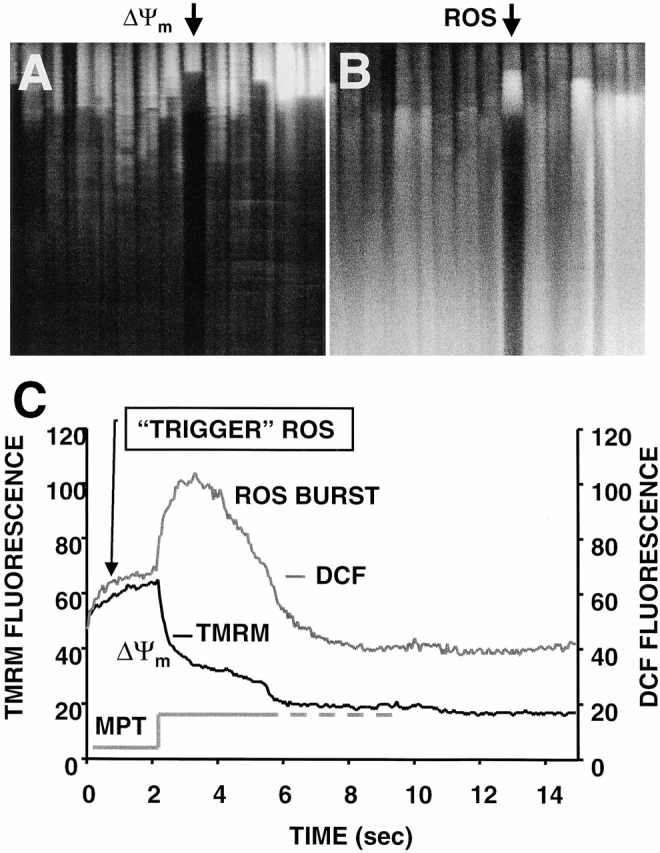
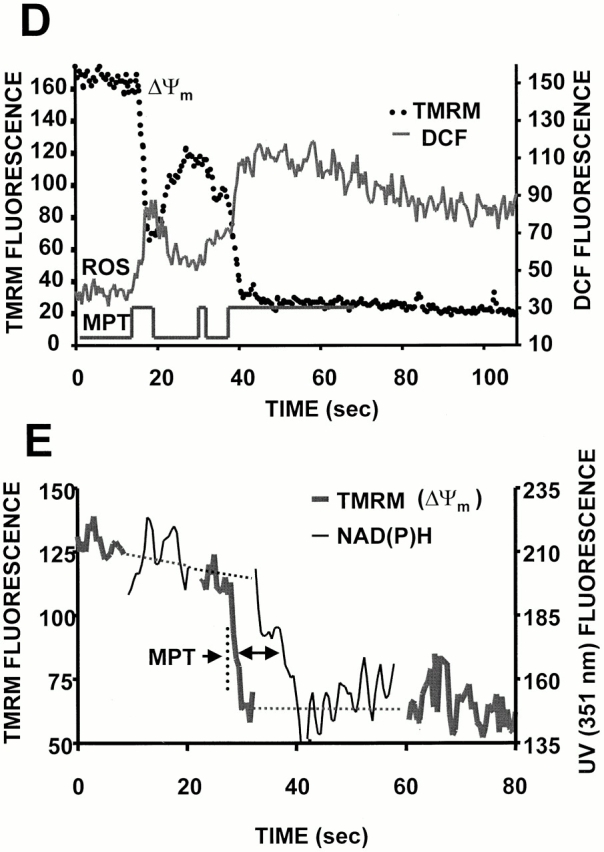
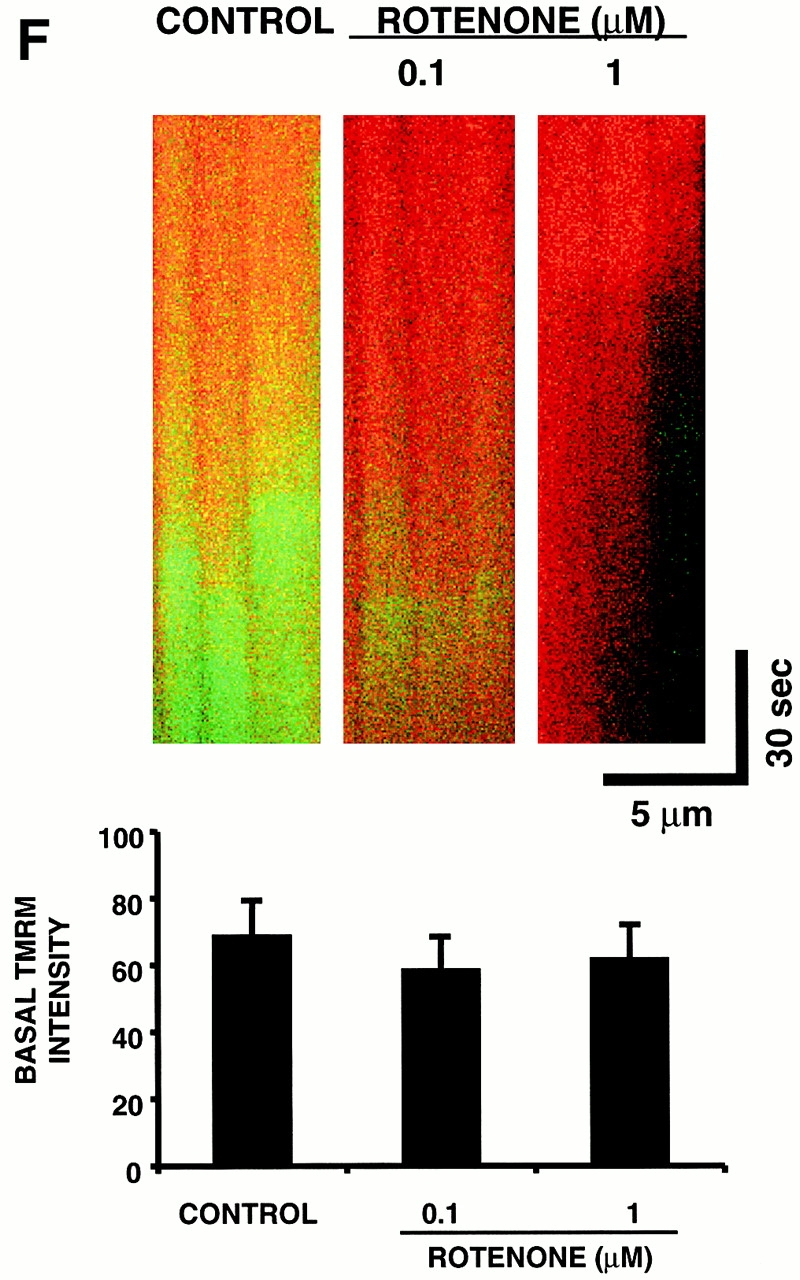
RIRR in single mitochondria. Representative cell that was dual-loaded with 125 nM TMRM (for ΔΨ) and 10 μM DCF (for ROS). (A) Typical pattern of ΔΨ dissipation at 10 Hz line-scan imaging. (B) Generation of ROS, as indicated by the increase in DCF fluorescence (acquired simultaneously with A). (C) Temporal relationship between ΔΨ and ROS production from the mitochondrial pair denoted by arrows in A and B. The trace at the bottom shows the hypothetical opening of the MPT pore. (D) Coordinated flickering of ΔΨ and RIRR in a single mitochondrion at 2 Hz line-scan imaging. (E) Relationship between ΔΨ and NAD(P) redox state during the MPT. ΔΨ and the MPT are assessed by changes in the TMRM (125 nM) fluorescence and the intrinsic autofluorescence excited at 351 nm (index of NAD[P] redox state), respectively, during 2 Hz line-scan imaging. (F) Inhibition of mitochondrial electron transport at Complex I prevents the mitochondrial ROS burst after induction of the MPT. Cell loading with TMRM and DCF and line-scan imaging protocol, as in D, except for the exposure to rotenone (0.1 and 1 μM) as indicated. Representative regions (encompassing groups of about six mitochondria over three sarcomeres) from the respective 2 Hz line-scan protocols are shown from each experimental group (top panel).
The example in Fig. 4 D demonstrates coordinated flickering of ΔΨ (i.e., reversible loss and transient recovery of ΔΨ as in Fig. 3 A) and RIRR in a single mitochondrion. Notably, that the mitochondrial ROS burst phase profile evolves virtually as the mirror image of ΔΨ is not a fluorescence artifact related to some interaction of TMRM and DCF or of their excitation/emission characteristics (i.e., an “inner filter” effect), because RIRR can be demonstrated even in the absence of TMRM by performing laser line scanning using DCF itself as the photosensitizing species (at >10-fold the excitation intensity needed for the ordinary fluorescence measurements, confirming the accompanying ΔΨ loss by finding the dissipation of the 351 nm–excited fluorescence from NAD(P)H, signifying its oxidation; not shown).
Thus, a source of ROS is able to trigger a mitochondrial burst of ROS production. The next step was to prove that the source of the ROS burst involved the diversion of electrons from the electron transport chain (ETC). The redox state of NAD(P)H (indicating the redox state of Complex I) was measured simultaneously with ΔΨ during the usual line-scan imaging protocol (minimizing 351-nm exposure both by maximal attenuation of laser energy together with alternating scans with that for TMRM). The oxidation of NAD(P)H lags seconds behind the ΔΨ loss (Fig. 4 E), which is compatible with the time course of the ROS burst (representing single-electron donation to oxygen) and the cessation of normal flow of respiratory chain electrons. Furthermore, the ROS burst magnitude upon MPT induction is successively decreased with increasing exposure to the Complex I inhibitor, rotenone (0.1 and 1 μM), at concentrations not affecting TMRM sequestration (Fig. 4 F), indicating that the ROS burst derives from the ETC. Neither deferoxamine nor bathophenantroline (each 200 μM) had any effect in these experiments (not shown).
Photodynamic Triggering of RIRR Does Not Depend on a Specific Sensitizing Fluorophore.
Thus far, the photoexcitation of rhodamines or DCF have been shown to initiate the RIRR process. Additional experiments in cells dual-loaded with the chemically distinct, ΔΨ-sensitive dye MitoTracker® Red CMXRos, together with DCF, yield the identical pattern of ΔΨ loss and RIRR compared with that observed with the rhodamines (Fig. 5).
Figure 5.

Triggering the RIRR process is not dependent on a sensitizing fluorophore. Cardiac myocyte dual-loaded with 400 nM MitoTracker Red CMXRos (ΔΨ, red signal) and 10 μM DCF (ROS, green signal); confocal line-scan imaging at 2 Hz.
RIRR Initiation Requires Simultaneous MPT Induction.
Given the abruptness of ΔΨ loss at the onset of RIRR, the most obvious candidate “pore” was the MPT, which allows nonspecific flux of molecules up to ∼1.5 kD across the inner mitochondrial membrane. To test this possibility, cells were loaded with the acetoxymethylester derivative of calcein (a ∼620-dalton inert fluorescent marker), empirically resulting in a predominantly cytoplasmic compartmentalization. Using TMRM photoexcitation to induce RIRR in these cells, we observed that individual mitochondria accumulate calcein (as indicated by the increase of its mitochondria-localized fluorescence) at the moment that ΔΨ loss occurs (Fig. 6 A).
Figure 6.


Demonstration of MPT induction by photoexcited trigger ROS. (A) Cells dual-loaded with TMRM (ΔΨ) and calcein-AM (the latter loaded under conditions that results in a cytosolic distribution initially in excess over that in mitochondria); line-scan imaging at 20 Hz. (B) 10 μM BA partially inhibits the MPT (ΔΨ; a and b) and RIRR (ROS; c and d) versus control. Cells were dual-loaded with TMRM and DCF; line-scan imaging at 2 Hz. (C) 100 μM BA completely inhibits the MPT versus control. Cells are loaded with 125 nM TMRM; line-scan at 2 Hz.
We further confirmed that MPT induction not only occurred at the commencement of RIRR but was also required for its initiation altogether, by using the specific MPT inhibitor, BA 8. BA at low concentration (10 μM) partially inhibits the MPT and RIRR, while at high concentration (100 μM), MPT inhibition was virtually complete (Fig. 6B and Fig. C). TMRM photoexcitation experiments using EPR demonstrated that BA (100 μM) has no significant ROS-scavenging activity (not shown). Exposure to cyclosporin A (ranging from 0.2 to 4 μM), however, had no effect on MPT induction (not shown). Although the MPT has been shown to be mediated by Ca2+ in other models, nevertheless, neither buffering intracellular Ca2+ with BAPTA-AM (15 μM) nor inhibiting the mitochondrial Ca2+ uniporter by Ru360 (6 μM) had any inhibitory effect in these experiments (not shown).
Scavenging Trigger ROS, or Exposure to Exogenous NO, Inhibits the MPT.
To confirm that ROS triggered both MPT and ROS release, experiments were performed in the presence of 2 mM Trolox. Trolox treatment was found to prevent ΔΨ loss during laser line-scan imaging compared with control cells (Fig. 7 A). It is interesting to note that cells pretreated with the membrane-permeant superoxide scavenger Tiron (1–2 mM) or the superoxide dismutase (SOD) mimetics MnTBAP or MnTMPyP (500 μM each) failed to alter the ability to induce the MPT (not shown). Based on the fact that Trolox, but not the ·O2 − scavengers, prevented the MPT and RIRR, it is tempting to speculate that the important ROS species from the standpoint of MPT induction/RIRR is likely to be peroxide rather than ·O2 −.
Figure 7.


Scavenging the ROS trigger or exposure to exogenous NO inhibits the MPT. Cells loaded with 125 nM TMRM and confocal line-scan–imaged at 2 Hz. (A) Mean time to MPT induction in control versus pretreated cells as indicated: Trolox (2 mM); SNAP (100 μM); L-NAME (1 mM). Data represent the average from 8–10 cells in each group. (B) Evidence of endogenous production of NO by mitochondria after MPT induction and inhibition by L-NAME (4 mM). Myocytes were loaded with 125 nM TMRM (red) and 10 mM DAF-2 (green) and line scanned at 100 Hz.
The oxidation of critical thiols, probably on the MPT itself, by ROS may be instrumental in MPT induction. Thus, S-nitrosylation of these critical thiols (i.e., via nitrosative stress) could alter their susceptibility to oxidative stress. In support of this notion, MPT induction was delayed by more that twofold in cells treated with 100 μM SNAP versus control (Fig. 7 A). Since the MPT was apparently stabilized rather than hastened by the SNAP treatment, it would seem that generation of peroxynitrite, a potent inducer of the MPT in isolated mitochondria, was minimal during the trigger stage.
Given the apparently protective role of exogenous NO against MPT induction in cardiac myocytes and the likelihood that endogenous production of NO in these cells could be playing important regulatory roles in excitation–contraction coupling 20 21, we tested whether NO synthase inhibition would affect MPT induction. Pretreatment of cells with 1 mM L-NAME, which was associated with considerably increased variability of the times to MPT induction, nevertheless caused overall shorter times versus control (Fig. 7 A) and abolished spontaneous MPT reversal (not shown). The time course and localization of endogenous NO production in relationship to MPT induction was examined using cells loaded with DAF-2, which develops fluorescence upon reaction with NO. Fig. 7 B shows an increase of mitochondrial fluorescence in cells loaded with DAF-2 after MPT induction (suggesting that NO production has occurred in these areas) but with relatively slower kinetics compared with the ROS burst; the specificity of this phenomenon is demonstrated by the fact of its inhibition by L-NAME.
Altered MPT Characteristics due to Changes in Redox State of Soluble and Protein Thiols.
The data so far suggest that when levels of triggering ROS accumulate past an apparent critical threshold, MPT induction occurs, precipitating the burst phase ROS generated by that particular mitochondrion. Assuming that MPT induction in these experiments resulted from the oxidation of critical protein thiols, and since glutathione is one of the primary redox buffer systems that defends protein thiols against oxidative stress, we examined how changes in this redox buffer induced by DEM exposure would affect MPT induction. Depleting cellular glutathione with 600 μM DEM consistently shortened the MPT induction time by 50–60% (Fig. 8 A). At higher concentrations, DEM is known to eliminate essential thiols 22 forming thioester adducts. Fig. 8 B shows a characteristic, unstable pattern of MPT flickering during scanning in a representative cell exposed to 5 mM DEM, with remarkably repetitive cycles of abrupt ΔΨ loss and recovery that are clearly synchronized across large numbers of mitochondria.
Figure 8.


Altered MPT characteristics after modulation of the redox state of soluble and protein thiols. (A) Comparison of MPT times (during 2 Hz line-scan imaging in TMRM-loaded cells) and cellular glutathione content in control versus 600 μM diethylmaleate-treated cells. (B) Development of apparent unstable MPT pore flickering during 2 Hz line-scan imaging in a representative TMRM-loaded (125 nM) cell exposed to 5 mM diethylmaleate. Arrows indicate movement artifacts.
Induction of Ca2+ Sparks after the MPT RIRR.
This newly described RIRR phenomenon, though only seconds in duration, produces an apparently significant burst of local ROS that could have immediate consequences for the local cellular homeostasis. In particular, since oxidative and nitrosative stress can modulate the spontaneous activity of the ryanodine receptor (see reference 21), we examined the nature of Ca2+ spark activity in the “wake” of MPT induction. As shown in Fig. 9 in a representative cell dual-loaded with TMRM and the Ca2+-sensitive dye fluo-3, there is frequently a period of significantly increased Ca2+ spark frequency at the z-lines (i.e., the site of the T tubule–sarcoplasmic reticulum junction) in the immediate vicinity of the mitochondrion shortly after MPT induction. Ordinarily, the spontaneous background (stochastic) event rate is about two to three sparks per 100 μm·s. However, in proximity of the MPT (defined as within the sarcomere containing the involved mitochondria and within 3 s after MPT occurrence), the event rate approximately doubles to about five to six per 100 μm·s (P < 0.05; Fig. 9, inset). While ordinary background Ca2+ sparks are typically single events, the MPT-associated ones frequently occur as clusters, as seen in Fig. 9.
Figure 9.

Induction of Ca2+ sparks after the MPT. Cell is dual-loaded with 125 nM TMRM (ΔΨ) and fluo-3 (Ca2+) and line-scan imaged at 230 Hz. Representative example showing the dissipation of TMRM fluorescence from a single mitochondrion and a cluster of Ca2+ sparks in the immediate vicinity, within seconds of MPT induction. Inset: comparison of Ca2+ spark rate in proximity of MPT occurrence (i.e., within the sarcomere containing the involved mitochondria and within 3 s after MPT occurrence; n = 90 sparks) versus that at background (n = 150 sparks; P < 0.05).
Discussion
We present a novel method to selectively expose arrays of mitochondria in isolated live adult cardiac myocytes to incremental doses of ROS by photoactivation of mitochondrial dyes while simultaneously recording functional mitochondrial parameters, including ΔΨ, ROS generation, NO production, permeability changes, and redox state. This method of localized excitation causes generation of “triggering” ROS inside individual mitochondria, enabling us to demonstrate the induction of the MPT, and its spontaneous reversibility, in intact cells. The duration of exposure to the triggering ROS necessary to induce the MPT was dependent on the intrinsic ROS-scavenging ability of the cell, as MPT times were dependent upon the cellular glutathione content. We demonstrate that ROS specifically triggers the MPT, because ROS scavengers prevent it.
We term this new mitochondrial phenomenon “ROS-induced ROS release” (RIRR) by analogy to the phenomenon of “Ca2+-induced Ca2+ release” 23. Upon MPT induction, ΔΨ rapidly dissipates, followed by a “burst phase” of ROS generated by that particular mitochondrion and lasting ∼5–10 s (Fig. 4). Occasionally, contiguous mitochondria, in groups of at least 5–10, were observed to undergo synchronous cycles of both induction and reversal of the MPT (Fig. 3). These experiments also show that triggering ROS are not entirely sufficient for the development of burst phase ROS production by mitochondria. If the MPT is prevented with BA and ΔΨ maintained indefinitely, then no burst phase occurs. Thus, it appears that trigger ROS induction of the MPT causes the mitochondrial ROS burst.
Photoinduction of the MPT RIRR process in mitochondria loaded with fluorescent probes is apparently due to the generation of photoproducts, probably originating from singlet oxygen (1O2) and ·O2 −. 1O2 is believed to be the primary ROS resulting from the energy transfer from the excited rhodamine molecule to oxygen 24, and this would be expected to induce lipid peroxidation (alkoxyl and peroxyl radicals). ·O2 − is an effective oxidant itself but can also be transformed by dismutation into H2O2, which, in the presence of Fe2+, forms the strong oxidant ·OH by the Fenton reaction 25. Indeed, we demonstrated by EPR that illumination of TMRM solutions produce ·O2 − and ·OH signals that were suppressed by SOD or catalase, respectively, and scavenged by the α-tocopherol derivative Trolox (Fig. 2 C). Furthermore, Trolox prevention of the MPT RIRR phenomenon argues for initiation by ROS.
As Trolox has a broad antioxidant capacity, we also performed experiments with other ROS scavengers with more selectivity. In contrast to the results with Trolox, the superoxide radical scavenger Tiron as well as the SOD mimetics MnTBAP and MnTMPyP were ineffective. This pointed to the likely importance of peroxide rather than ·O2 −, and possibly the Fenton reaction, in our experiments. Not surprisingly, the impermeable antioxidant catalase (1,500 U/ml), which catalyzes the transformation of H2O2 into water and oxygen, was ineffective. In addition, the iron chelators deferoxamine and bathophenantroline had no effect on the MPT induction, suggesting that Fenton chemistry may not be playing a significant role in induction of MPT RIRR. This suggests that peroxides (and possibly 1O2) form the principal trigger ROS in our experiments. This is supported by the observation that catalase is effective in blocking ΔΨ loss induced by excitation of TMRE-loaded, isolated mitochondria 16. However, it is possible that the lack of effect of deferoxamine might be due to its much higher affinity for Fe3+ compared with Fe2+, which catalyzes the Fenton reaction, while bathophenantroline is known to be poorly cell membrane permeable.
It would be reasonable to assume that these triggering ROS would eventually oxidize some critical protein targets, such as regulatory thiols 26, but that the cellular redox scavengers would tend to prevent this. Thus, it was important to establish that the soluble redox buffer capacity, principally GSH, would govern the characteristic delay between the trigger ROS onset and MPT induction/RIRR. This was confirmed using 0.6 mM DEM, which conjugates to nonprotein thiols 27 and caused both depletion of cellular GSH and the decrease in the time to MPT induction by 60% (Fig. 8 A).
Several independent lines of evidence lead to the conclusion that photoactivation-produced triggering ROS induces the MPT in cardiac mitochondria. First is the abrupt collapse of ΔΨ and its prevention by the ROS scavenger Trolox. Second, an otherwise membrane-impermeant fluorescent molecule, calcein (∼620 daltons), originally present in the cytosol, enters mitochondria concurrently with the loss of ΔΨ (Fig. 6 A). Third, inhibition of the transition by BA (Fig. 6 B), which inhibits the adenine nucleotide translocator (ANT) component of the pore and blocks the MPT 8.
Cyclosporin A (0.2–4 μM) did not prevent the dissipation of ΔΨ in our experiments. Although cyclosporin A is a powerful inhibitor of the MPT in isolated mitochondria, it is known to be ineffective in blocking it in some cell types and under different conditions. For example, in ischemia/reperfusion models with rat heart, the protective effect of cyclosporin A was observed over an extremely narrow concentration range 28. Also, mitochondrial signal peptides 29, butylated hydroxytoluene 30, and thyroxine 31 each induce mitochondrial permeability that is cyclosporin A insensitive.
Certain interesting MPT-related phenomena observed with this model provide mechanistic insights into the nature of the pore and of the communication between mitochondria. Although MPT induction was most typically observed as a synchronous event among mitochondria within a single sarcomere, certain higher levels of coordination were occasionally seen, involving mitochondria in several longitudinally adjacent sarcomeres (observed even beyond the length of 15 sarcomeres; Fig. 3 A). Given the distance and timing of this synchronization, it would seem that the signal coordinating such long-range MPT induction cannot plausibly be conveyed by a simple chemical-diffusive mechanism but rather must be transmitted along some electrical coupling (“cable”) mechanism 32. Recently, the propagation of a mitochondrial “redox signal” was shown to occur both within single cardiomyocytes as well as from one cardiac cell to another, demonstrating both intra- and intercellular mitochondrial communications 33, but this is likely a different mechanism from that observed here.
One of the most important observations about the MPT from our experiments is that it can be spontaneously reversible. As with the pattern of synchronized pore openings, we observed synchronization of spontaneous MPT reversal among large mitochondrial groups (Fig. 3 A). This cyclical pattern of sequential induction and reversal of the MPT results in an apparent “flickering” mode of ΔΨ that eventually becomes irreversible if the trigger does not cease. Reversibility of the MPT induced by photoactivation has also been observed in isolated heart mitochondria 16.
Our data also demonstrate that MPT induction can also occur in apparently halting, step-wise fashion, achieving intermediate stable levels without complete loss of the ΔΨ-tracking dye fluorescence for several seconds at a time (Fig. 3 B). These observations raise the question of whether the MPT pore operates in a single conductance state or if there are multiple possible subconductance states. Studies on isolated mitochondria suggest that there are indeed different conductance states of the MPT 9 10.
Our results in intact cells, however, suggest that the MPT pore most likely exhibits only a single open state and can flicker between the open and closed states. The regular structure of the cardiac myocyte and the confocal imaging conditions we used effectively limit the optical section to that of a single mitochondrion. Therefore, it is unlikely that separate and unrelated events from overlying/underlying planes of mitochondria (i.e., in the axial dimension) are “contaminating” the recording. As all of the ΔΨ fluorescence decay segments during episodes of step-wise MPT progression (Fig. 3 B) can be described by the same first-order kinetic process (Fig. 3 B, inset), and, moreover, that all cells have the same rate constant of TMRM efflux during the MPT, the simplest explanation is that the MPT pore operates with a single open state conductance in the intact rat cardiac myocyte.
DEM at levels an order of magnitude higher than that needed to deplete glutathione (probably reacting with protein thiols [22]) yields a striking result: the MPT induction pattern becomes characteristically unstable with unusual repetitive cycles of abrupt ΔΨ loss and recovery (Fig. 8). This suggests that the trigger ROS may target the same thiol moieties affected by this DEM exposure and possibly develops a certain dynamic “competition” between the formation of disulfide bonds and thioester adducts (of ethyl maleate) at the same critical target(s), each of which causes a protein conformational shift between the open and closed states (respectively) of the pore.
This concept that the competition of reactive species may determine the stability of the regulatory thiol redox state is further supported by the finding that pretreatment of cells with the spontaneous NO donor, SNAP, delayed MPT induction (Fig. 7). NO may interact with protein thiols via S-nitrosylation, a physiologically reversible modification that could alter the susceptibility to the SH/S–S transition, which is known to promote the MPT 4. In this case, NO could be regarded as a stabilizer of thiol groups against harsher oxidizing conditions 21. The overall role of NO in induction of the MPT and in apoptotic cell death is not at all clear, with seemingly contradictory data derived from studies on isolated mitochondria and different cell types 34 35 36. For example, NO has been shown to cause both induction of apoptosis through triggering the MPT 37 38 as well as its inhibition at sites both upstream and downstream of cytochrome c release 39. Furthermore, NO· has been shown to protect cardiomyocytes from oxidative damage induced by organic peroxides 40, whereas NO− is damaging in postischemic myocardial injury 41.
However, for cardiac cells, endogenous NO is likely to achieve micromolar levels in vivo, and, moreover, act in a beneficial manner as an essential modulator of excitation–contraction coupling 20 21 42 without causing apoptosis. The protection from MPT induction in cardiac mitochondria by SNAP demonstrated in our experiments supports this concept. Furthermore, our observation of a slow, L-NAME–sensitive rise of the fluorescence of the NO-sensitive dye, DAF-2, inside mitochondria after induction of the MPT is compatible with a slow generation of NO in (or near) mitochondria after MPT induction (Fig. 7 B). Functionally, pretreatment of cardiac myocytes with L-NAME was associated with considerably increased variability of the times to MPT induction but overall with a tendency to shorter times (Fig. 7 A) and an absence of spontaneous MPT reversal versus control (not shown), suggesting that endogenous NO may contribute to some level of “protective” basal S-nitrosylation.
It would seem that there is a specific mechanistic link between MPT induction and RIRR, because other causes of rapid ΔΨ loss (i.e., those not involving MPT induction) do not result in increased ROS production. In particular, exposure of cells to the mitochondrial uncoupler, carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP), which induces rapid ΔΨ loss via induction of a large membrane proton leak, occurs without any rise of ROS production (not shown). Furthermore, as RIRR can be completely prevented by BA in spite of the prolonged exposure to the ROS trigger in this model, we concluded that MPT induction is obligatory for the ROS burst. It shows that ANT governs RIRR.
The mechanism(s) of the ROS burst accompanying the MPT remains unclear, although under ordinary conditions mitochondrial Complexes I and III are believed to be the major sources of ROS 43 44 45. Separate experiments presented here, (a) examining NAD(P)H oxidation and (b) using rotenone, indicate that the diversion of electrons from the respiratory chain provides the source of the ROS burst (Fig. 4E and Fig. f). Given that trigger ROS cannot cause the ROS burst directly, it is tempting to speculate that occurrence of the MPT itself could change the properties of the phospholipid bilayer surrounding the major mitochondrial enzymes in the inner membrane, thus dramatically affecting their function. Indeed, MPT induction was found to alter the rigidity of the mitochondrial membrane, which could weaken protein–protein interactions necessary for proper function of the respiratory chain 46. If so, the ROS burst might be due to a block of the ETC caused by MPT induction.
The role of mitochondria in maintaining the cellular Ca2+ homeostasis is still under debate. In this connection, Bowser at al. 47 demonstrated that uncouplers or ETC inhibitors caused the progressive elevation of cytosolic [Ca2+] and the frequency of Ca2+ sparks. The latter represents the fundamental Ca2+ release process from the ryanodine receptor of the sarcoplasmic reticulum, which normally is triggered by Ca2+ influx through the adjacent sarcolemmal L-type Ca2+ channels and serves to couple electrical excitation to Ca2+-mediated contraction in cardiac myocytes. In addition to this usual pathway, experiments here demonstrate that the ryanodine receptor also may be activated upon MPT induction (Fig. 9). Although it is possible that mitochondrial Ca2+ may contribute to Ca2+ spark formation 47, we also cannot exclude the possibility that such local Ca2+ release from sarcoplasmic reticulum may be driven by ROS generated in and released by these adjacent mitochondria undergoing MPT induction (a “ROS-induced Ca2+ release” mechanism). Specifically, it has been demonstrated that the oxidation of free thiols on the cardiac ryanodine receptor can lead to its activation 21. A similar process of prooxidant-induced Ca2+ release has been discussed for mitochondria 48. At this point, we are unable to differentiate between the Ca2+ versus ROS induction models driving these Ca2+ sparks, because primary perturbations in mitochondrial Ca2+ efflux cannot readily be made without affecting underlying Ca2+ spark mechanisms, and paradigms involving scavenging ROS would prevent the induction of the MPT in the first place. Regardless of whether Ca2+ or ROS modulate this increase in Ca2+ spark frequency after MPT induction, this phenomenon could induce pathological disturbances in cardiac excitation and rhythm, for example, contributing to postischemic reperfusion arrythmias.
We have identified a novel ROS-triggered phenomenon in mitochondria resulting from MPT induction, called “ROS-induced ROS release” in cardiac myocytes. Given the likely dramatic cellular consequences of ROS production seen in these experiments and its obligatory connection to the MPT, together with the fact that BA prevents both RIRR (this study) and apoptosis 11, it is tempting to speculate that these two phenomena are causally linked. Thus, the specific nature of the RIRR mechanism is probably fundamental, and we propose that it may be related to programmed mitochondrial destruction in cardiac myocytes as well as programmed cell death (apoptosis) in many cell types.
Acknowledgments
We wish to thank Dr. K.W. Fishbein for help solving complex technical problems and Dr. E.G. Lakatta for useful discussions.
This work was supported by the Intramural Research Program, National Institute on Aging (D.B. Zorov, C.R. Filburn, L.-O. Klotz, and S.J. Sollott); National Institutes of Health grants HL38324, HL52315, and HL63744 (J.L. Zweier); and Deutsche Forschungsgemeinschaft grant KL 1245/1-1 (L.-O. Klotz).
Footnotes
Abbreviations used in this paper: BA, bongkrekic acid; EPR, electron paramagnetic resonance; ETC, electron transport chain; MPT, mitochondrial permeability transition; RIRR, ROS-induced ROS release; ROS, reactive oxygen species.
References
- Williamson J.R. Mitochondrial function in heart. Annu. Rev. Physiol. 1979;41:485–506. doi: 10.1146/annurev.ph.41.030179.002413. [DOI] [PubMed] [Google Scholar]
- Gunter T.E., Pfeiffer D.R. Mechanisms by which mitochondria transport calcium. Am. J. Physiol. 1990;258:C755–786. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- Green D.R., Reed J.C. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- Zoratti M., Szabo I. The mitochondrial permeability transition. Biochim. Biophys. Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]
- Boveris A., Oshino N., Chance B. The cellular production of hydrogen peroxide. Biochem. J. 1973;128:617–630. doi: 10.1042/bj1280617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenaz G. Role of mitochondria in oxidative stress and ageing. Biochim. Biophys. Acta. 1998;1366:53–67. doi: 10.1016/s0005-2728(98)00120-0. [DOI] [PubMed] [Google Scholar]
- Hunter D.R., Haworth R.A. The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch. Biochem. Biophys. 1979;195:453–459. doi: 10.1016/0003-9861(79)90371-0. [DOI] [PubMed] [Google Scholar]
- Bernardi P., Petronilli V. The permeability transition pore as a mitochondrial calcium release channela critical appraisal. J. Bioenerg. Biomembr. 1996;28:131–138. doi: 10.1007/BF02110643. [DOI] [PubMed] [Google Scholar]
- Ichas F., Mazat J.P. From calcium signaling to cell deathtwo conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochim. Biophys. Acta. 1998;1366:33–50. doi: 10.1016/s0005-2728(98)00119-4. [DOI] [PubMed] [Google Scholar]
- Zamzami N., Susin S.A., Marchetti P., Hirsch T., Gomez-Monterrey I., Castedo M., Kroemer G. Mitochondrial control of nuclear apoptosis. J. Exp. Med. 1996;183:1533–1544. doi: 10.1084/jem.183.4.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson M.D. Reactive oxygen species and programmed cell death. Trends Biochem. Sci. 1996;21:83–86. [PubMed] [Google Scholar]
- Duchen M.R. Contributions of mitochondria to animal physiologyfrom homeostatic sensor to calcium signalling and cell death. J. Physiol. 1999;516:1–17. doi: 10.1111/j.1469-7793.1999.001aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robb-Gaspers L.D., Burnett P., Rutter G.A., Denton R.M., Rizzuto R., Thomas A.P. Integrating cytosolic calcium signals into mitochondrial metabolic responses. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:4987–5000. doi: 10.1093/emboj/17.17.4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansford R.G. Physiological role of mitochondrial Ca2+ transport. J. Bioenerg. Biomembr. 1994;26:495–508. doi: 10.1007/BF00762734. [DOI] [PubMed] [Google Scholar]
- Huser J., Rechenmacher C.E., Blatter L.A. Imaging the permeability pore transition in single mitochondria. Biophys. J. 1998;74:2129–2137. doi: 10.1016/S0006-3495(98)77920-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieminen A.L., Saylor A.K., Tesfai S.A., Herman B., Lemasters J.J. Contribution of the mitochondrial permeability transition to lethal injury after exposure of hepatocytes to t-butylhydroperoxide. Biochem. J. 1995;307:99–106. doi: 10.1042/bj3070099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capogrossi M.C., Kort A.A., Spurgeon H.A., Lakatta E.G. Single adult rabbit and rat cardiac myocytes retain the Ca2+- and species-dependent systolic and diastolic contractile properties of intact muscle. J. Gen. Physiol. 1986;88:589–613. doi: 10.1085/jgp.88.5.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson M.E. Determination of glutathione and glutathione disulfide in biological samples. Methods Enzymol. 1985;113:548–555. doi: 10.1016/s0076-6879(85)13073-9. [DOI] [PubMed] [Google Scholar]
- Vila-Petroff M.G., Younes A., Egan J., Lakatta E.G., Sollott S.J. Activation of distinct cAMP-dependent and cGMP-dependent pathways by nitric oxide in cardiac myocytes. Circ. Res. 1999;84:1020–1031. doi: 10.1161/01.res.84.9.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L., Eu J.P., Meissner G., Stamler J.S. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- Nishihata T., Caldwell L.J., Sakai K. Inhibitory effect of salicylate on 2,4-dinitrophenol and diethyl maleate in isolated rat intestinal epithelial cells. Biochim. Biophys. Acta. 1988;970:7–18. doi: 10.1016/0167-4889(88)90216-9. [DOI] [PubMed] [Google Scholar]
- Fabiato A., Fabiato F. Contractions induced by a calcium-triggered release of calcium from the sarcoplasmic reticulum of single skinned cardiac cells. J. Physiol. 1975;249:469–495. doi: 10.1113/jphysiol.1975.sp011026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shea C.R., Chen N., Wimberly J., Hasan T. Rhodamine dyes as potential agents for photochemotherapy of cancer in human bladder carcinoma cells. Cancer Res. 1989;49:3961–3965. [PubMed] [Google Scholar]
- Khan A.U., Wilson T. Reactive oxygen species as cellular messengers. Chem. Biol. 1995;2:437–445. doi: 10.1016/1074-5521(95)90259-7. [DOI] [PubMed] [Google Scholar]
- Vercesi A.E., Kowaltowksi A.J., Grijalba M.T., Meinicke A.R., Castilho R.F. The role of reactive oxygen species in mitochondrial permeability transition. Biosci. Rep. 1997;17:43–52. doi: 10.1023/a:1027335217774. [DOI] [PubMed] [Google Scholar]
- Plummer J.L., Smith B.R., Sies H., Bend J.R. Chemical depletion of glutathione in vivo. Methods Enzymol. 1981;77:50–59. doi: 10.1016/s0076-6879(81)77010-1. [DOI] [PubMed] [Google Scholar]
- Griffiths E.J., Halestrap A.P. Protection by cyclosporin A of ischemia/reperfusion-induced damage in isolated rat hearts. J. Mol. Cell. Cardiol. 1993;25:1461–1469. doi: 10.1006/jmcc.1993.1162. [DOI] [PubMed] [Google Scholar]
- Sokolove P.M., Kinnally K.W. A mitochondrial signal peptide from Neurospora crassa increases the permeability of isolated rat liver mitochondria. Arch. Biochem. Biophys. 1996;336:69–76. doi: 10.1006/abbi.1996.0533. [DOI] [PubMed] [Google Scholar]
- Kushnareva Y.E., Campo M.L., Kinnally K.W., Sokolove P.M. Signal presequences increase mitochondrial permeability and open the multiple conductance channel. Arch. Biochem. Biophys. 1999;366:107–115. doi: 10.1006/abbi.1999.1190. [DOI] [PubMed] [Google Scholar]
- Malkevitch N.V., Dedukhova V.I., Simonian R.A., Skulachev V.P., Starkov A.A. Thyroxine induces cyclosporin A-insensitive, Ca2+-dependent reversible permeability transition pore in rat liver mitochondria. FEBS Lett. 1997;412:173–178. doi: 10.1016/s0014-5793(97)00666-2. [DOI] [PubMed] [Google Scholar]
- Amchenkova A.A., Bakeeva L.E., Chentsov Y.S., Skulachev V.P., Zorov D.B. Coupling membranes as energy-transmitting cables. I. Filamentous mitochondria in fibroblasts and mitochondrial clusters in cardiomyocytes. J. Cell Biol. 1988;107:481–495. doi: 10.1083/jcb.107.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romashko D.N., Marban E., O'Rourke B. Subcellular metabolic transients and mitochondrial redox waves in heart cells. Proc. Natl. Acad. Sci. USA. 1998;95:1618–1623. doi: 10.1073/pnas.95.4.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakirev M.Yu, Khramtsov V.V., Zimmer G. Modulation of the mitochondrial permeability transition by nitric oxide. Eur. J. Biochem. 1997;246:710–718. doi: 10.1111/j.1432-1033.1997.00710.x. [DOI] [PubMed] [Google Scholar]
- Wink D.A., Mitchell J.B. Chemical biology of nitric oxideinsights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Rad. Biol. Med. 1998;25:434–456. doi: 10.1016/s0891-5849(98)00092-6. [DOI] [PubMed] [Google Scholar]
- Murphy M.P. Nitric oxide and cell death. Biochim. Biophys. Acta. 1999;1411:401–414. doi: 10.1016/s0005-2728(99)00029-8. [DOI] [PubMed] [Google Scholar]
- Hortelano S., Dallaporta B., Zamzami N., Hirsch T., Susin S.A., Marzo I., Bosca L., Kroemer G. Nitric oxide induces apoptosis via triggering mitochondrial permeability transition. FEBS Lett. 1997;410:373–377. doi: 10.1016/s0014-5793(97)00623-6. [DOI] [PubMed] [Google Scholar]
- Srivastava R.K., Sollott S.J., Khan L., Hansford R., Lakatta E.G., Longo D.L. Bcl-2 and Bcl-X(L) block thapsigargin-induced nitric oxide generation, c-Jun NH(2)-terminal kinase activity, and apoptosis. Mol. Cell. Biol. 1999;19:5659–5674. doi: 10.1128/mcb.19.8.5659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leist M., Single B., Naumann H., Fava E., Simon B., Kuhnle S., Nicotera P. Nitric oxide inhibits execution of apoptosis at two distinct ATP-dependent steps upstream and downstream of mitochondrial cytochrome c release. Biochem. Biophys. Res. Commun. 1999;258:215–221. doi: 10.1006/bbrc.1999.0491. [DOI] [PubMed] [Google Scholar]
- Gorbunov N.V., Tyurina Y.Y., Salama G., Day B.W., Claycamp H.G., Argyros G., Elsayed N.M., Kagan V.E. Nitric oxide protects cardiomyocytes against tert-butyl hydroperoxide-induced formation of alkoxyl and peroxyl radicals and peroxidation of phosphatidylserine. Biochem. Biophys. Res. Commun. 1998;244:647–651. doi: 10.1006/bbrc.1997.7951. [DOI] [PubMed] [Google Scholar]
- Ma X.L., Gao F., Liu G.L., Lopez B.L., Christopher T.A., Fukuto J.M., Wink D.A., Feelisch M. Opposite effects of nitric oxide and nitroxyl on postischemic myocardial injury. Proc. Natl. Acad. Sci. USA. 1999;96:14617–14622. doi: 10.1073/pnas.96.25.14617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinsky D.J., Patton S., Mesaros S., Brovkovych V., Kubaszewski E., Grunfeld S., Malinski T. Mechanical transduction of nitric oxide synthesis in the beating heart. Circ. Res. 1997;81:372–379. doi: 10.1161/01.res.81.3.372. [DOI] [PubMed] [Google Scholar]
- Ricchelli F., Gobbo S., Moreno G., Salet C. Changes of the fluidity of mitochondrial membranes induced by the permeability transition. Biochemistry. 1999;38:9295–9300. doi: 10.1021/bi9900828. [DOI] [PubMed] [Google Scholar]
- Takeshige K., Minakami S. NADH- and NADPH-dependent formation of superoxide anions by bovine heart submitochondrial particles and NADH-ubiquinone reductase preparation. Biochem. J. 1979;180:129–135. doi: 10.1042/bj1800129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nohl H., Jordan W. The mitochondrial site of superoxide formation. Biochem. Biophys. Res. Commun. 1986;138:533–539. doi: 10.1016/s0006-291x(86)80529-0. [DOI] [PubMed] [Google Scholar]
- Korshunov S.S., Korkina O.V., Ruuge E.K., Skulachev V.P., Starkov A.A. Fatty acids as natural uncouplers preventing generation of ·O2 − and H2O2 by mitochondria in the resting state. FEBS Lett. 1998;435:215–218. doi: 10.1016/s0014-5793(98)01073-4. [DOI] [PubMed] [Google Scholar]
- Bowser D.N., Minamikawa T., Nagley P., Williams D.A. Role of mitochondria in calcium regulation of spontaneously contracting cardiac muscle cells. Biophys. J. 1998;75:2004–2014. doi: 10.1016/S0006-3495(98)77642-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter C. Pro-oxidants and mitochondrial Ca2+their relationship to apoptosis and oncogenesis. FEBS Lett. 1993;325:104–107. doi: 10.1016/0014-5793(93)81423-w. [DOI] [PubMed] [Google Scholar]