

Abstract
One mechanism regulating the ability of different subsets of T helper (Th) cells to respond to cytokines is the differential expression of cytokine receptors. For example, Th2 cells express both chains of the interferon γ receptor (IFN-γR), whereas Th1 cells do not express the second chain of the IFN-γR (IFN-γR2) and are therefore unresponsive to IFN-γ. To determine whether the regulation of IFN-γR2 expression, and therefore IFN-γ responsiveness, is important for the differentiation of naive CD4+ T cells into Th1 cells or for Th1 effector function, we generated mice in which transgenic (TG) expression of IFN-γR2 is controlled by the CD2 promoter and enhancer. CD4+ T cells from IFN-γR2 TG mice exhibit impaired Th1 polarization potential in vitro. TG mice also display several defects in Th1-dependent immunity in vivo, including attenuated delayed-type hypersensitivity responses and decreased antigen-specific IFN-γ production. In addition, TG mice mount impaired Th1 responses against Leishmania major, as manifested by increased parasitemia and more severe lesions than their wild-type littermates. Together, these data suggest that the sustained expression of IFN-γR2 inhibits Th1 differentiation and function. Therefore, the acquisition of an IFN-γ–unresponsive phenotype in Th1 cells plays a crucial role in the development and function of these cells.
Keywords: T helper type 1 cells; interferon type II; interferon receptors; hypersensitivity, delayed; cytokines
Introduction
CD4+ T cells, or Th cells, are key regulators of a vast array of immune responses. The regulatory and effector functions of these cells are mediated, in part, by the cytokines they produce. Mature, effector Th cells can be subdivided into two functionally distinct subsets based on their cytokine secretion profiles. Th1 cells secrete IFN-γ, TNF-β, and IL-2, mediate immune responses against intracellular pathogens, and are associated with pathological processes such as organ-specific autoimmune diseases 1. Conversely, Th2 cells produce IL-4, IL-5, IL-6, and IL-13, mediate immune responses against extracellular pathogens, and are associated with allergic immune responses 1 2 3. It has been established that, after activation through their TCR–CD3 complex, naive precursor Th cells can give rise to either one of these two distinct mature Th populations. The process of Th subset phenotype acquisition has been shown to be affected by a host of factors, including strength of stimulus (antigen dose and level of costimulation), route of immunization, and by the cytokines present in the immediate microenvironment of the cells 4 5 6. Cytokines can either positively or negatively regulate the development of Th subsets. IL-12 and IFN-γ are associated with Th1 development, whereas IL-4 is a Th2-polarizing stimulus. Moreover, IL-4 and IL-10 inhibit Th1 development, whereas IFN-γ is thought to suppress polarization of cells towards the Th2 pathway 7 8 9. More recent work has focused on understanding the mechanism by which these cytokines regulate Th subset development, as well as the molecular differences between the Th subsets that result from this process.
During their development, Th1 and Th2 cells acquire differential responsiveness to several classes of extracellular ligands including cytokines 10 11 12. We and others have shown that responsiveness to IFN-γ is regulated during Th cell development 13 14. Unlike precursor Th and Th2 cells, which express a functional receptor for IFN-γ on their cell surface, Th1 cells do not express the second chain of IFN-γR (IFN-γR2; previously termed AF-1 or IFN-γRβ), and are therefore unable to respond to IFN-γ. Consequently, IFN-γ induces signal transducer and activator of transcription (Stat) 1 activation and IFN-γ–dependent gene expression in Th2 cells, but not in Th1 cells, suggesting that the regulation of IFN-γR2 expression participates in some way in Th cell differentiation or function 13. However, the role of the regulation of IFN-γ responsiveness during mature Th cell phenotype acquisition is unknown.
To examine the importance of IFN-γR2 regulation during Th cell differentiation, we generated transgenic (TG) mice in which IFN-γR2 cDNA expression is controlled by the human CD2 promoter and enhancer, resulting in constitutive expression of IFN-γR2 in all T cells 15. Analysis of IFN-γR2 TG mice reveals that the transgene causes defects in both Th1 differentiation and Th1-dependent immunity. Naive TG Th cells fail to develop into Th1 cells under Th1-polarizing conditions in vitro. Furthermore, in vivo and ex vivo Th1-dependent immune responses such as T cell memory, class switching to IgG2a, and resistance to infection with Leishmania major are severely impaired in TG mice. Our results demonstrate that the loss of IFN-γ responsiveness is not only associated with, but is required for, Th1 phenotype acquisition and for the development of normal Th1 effector function.
Materials and Methods
Generation of IFN-γR2 TG Mice.
IFN-γR2 cDNA 16 was subcloned into the human CD2 promoter/enhancer cassette 15 and was injected into fertilized oocytes (C57BL/6 × CBA/J) that were reimplanted into pseudopregnant C57BL/6 × CBA/J F1 mice (The Jackson Laboratory). Litters were screened for founders by Southern blot of SacI-digested genomic DNA probed with IFN-γR2 cDNA. Subsequent litters were screened by PCR using the following primers: 5′-GCACGTGGTTAAGCTCTCG (located in the CD2 promoter) and 5′-TGTCTCTGTGATGTCCGTACA. The mice used for all experiments were bred into the C57BL/6 genetic background for at least four generations. All animal experiments conformed to Columbia University's Institutional Animal Care and Use Protocols.
Ribonuclease Protection Assay.
Ribonuclease protection assay was performed as described previously 17. In brief, a SacI-ClaI fragment, diagrammed in Fig. 1 A, from the CD2-IFN-γR2 construct was subcloned into the pBluescript II SK+ phagemid (Stratagene). Radiolabeled antisense mRNA probe was synthesized using an RNA transcription kit (Stratagene). Radiolabled probe (105 cpm) was hybridized with 30 μg of tissue RNA in 80% deionized formamide at 42°C overnight. Samples were then digested with RNases A and T1 and run on an 8 M urea-6% acrylamide gel.
Figure 1.

The generation of IFN-γR2 TG mice, transgene expression, and IFN-γ responsiveness. (A) A diagram of the CD2 promoter IFN-γR2-CD2 enhancer construct used to generate IFN-γR2 TG mice. The template used to synthesize the antisense mRNA probe used in RNase protection assays is diagrammed above the TG construct. UTR, untranslated region. (B) The expression of the endogenous (End.) and TG alleles of IFN-γR2 in different organs was detected by RNase protection. (C) IFN-γR2 gene expression in Th1 and Th2 clones was detected by reverse transcription PCR and visualized by autoradiography. T, transgenic; E, endogenous. (D) The detection by electrophoretic mobility shift assays of activated STAT complexes by Th1 and Th2 clones in response to treatment with IL-4 or IFN-γ. All experiments were repeated three times with similar results.
Cell Culture.
Cells were grown in complete RPMI containing 10 μg/ml penicillin-streptomycin, 2 mM l-glutamine, 0.1 mM nonessential amino acids, 0.1 mM β-mercaptoethanol, 1 mM sodium pyruvate, 10 mM Hepes, and 10% fetal bovine serum.
Cytokines, ELISA, Abs, Antigens, and Adjuvants.
KLH, DNP-KLH, CFA, and IFA were purchased from Calbiochem. Abs (mAbs) were purchased from BD PharMingen. ELISA was performed according to the BD PharMingen protocol and recommended reagents. Recombinant human IL-2 was provided by the National Cancer Institute Biological Research Branch (Frederick, MD). Recombinant murine IFN-γ and IL-12 were purchased from Genzyme. Recombinant murine IL-4 was a gift of Dr. Satwant Nurula of Schering-Plough Corp. (Kenilworth, NJ).
CD4+ T Cell Purification.
CD4+ T cells were purified by negative selection as described previously 18. In brief, single-cell suspensions from lymph nodes or spleens containing no red blood cells were first incubated with rat anti–mouse mAbs against B cells (anti-B220/CD45R), monocytes (anti-CD11b), and CD8+ T cells (anti-CD8a/Ly-2) at 20 μg/ml each, washed, and then incubated with anti–rat IgG Dynabeads (Dynal). Ab-coated cells were removed using a magnetic concentrator (Dynal). Naive Th lymphocytes (CD4+Mel-14hi) were purified by cell sorting using a FACStar™ flow cytometer (Becton Dickinson).
In Vitro Th1 and Th2 Polarization and Proliferation.
In vitro Th cell differentiation has been described previously 18. In brief, 5 × 105 naive CD4+ T cells were cultured for 7 d on anti-CD3–coated plates (10 μg/ml) in complete RPMI in the presence of IL-2 (20 U/ml). Th2 cultures were supplemented with IL-4 (10 ng/ml) plus anti–IFN-γ mAb (XMG1.2, 15 μg/ml). Th1 cultures were supplemented with anti–IL-4 mAb (11B11, 10 μg/ml) and with either IFN-γ (10 ng/ml) or IL-12 (10 U/ml). Cells (5 × 105 live cells per well) were then restimulated on anti-CD3–coated plates in RPMI and IL-2. 48 h after restimulation, culture supernatants were assayed for IFN-γ and IL-4 by ELISA. Polarized cultures were then pulsed for 6 h with [3H]thymidine (1 μCi/well).
Cell Division Cycle Profile Analysis Using Carboxyfluorescein Diacetate Succinimidyl Ester.
CD4+ splenocytes (purified as described above) were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE; Molecular Probes) in a modification of a previously described technique 19. In brief, cells were washed with serum-free RPMI. Cells (107 cells/ml) were then labeled with 10 μM CFSE in serum-free RPMI at 37°C for 10 min, and CFSE was then neutralized with complete RPMI. CFSE-labeled CD4+ T cells were cultured on anti-CD3 mAb plus anti-CD28 mAb–coated plates and polarized as described above. Initial CFSE labeling efficiency and the fluorescein intensities at the end of each experiment were detected using a FACScan™ flow cytometer (Becton Dickinson). Histogram overlays and peak distribution analyses were performed using CELLQuest™ software (Becton Dickinson). All plots represent live, activated CD4+ cells.
Generation and Characterization of KLH-specific Th1 and Th2 Clones.
Mouse immunization was performed as described previously 20. In brief, mice were injected with 100 μl DNP-KLH (1 mg/ml in CFA) in the hind footpad. 7 d later, CD4+ T cells were purified from inguinal lymph nodes and stimulated with antigen (KLH, 100 μg/ml) plus irradiated C57BL/6 splenocytes as APCs 21. KLH-specific Th1 and Th2 clones were generated by limiting dilution T cell cloning 22. Th1 and Th2 clones were defined based on their production of IFN-γ and IL-4, respectively.
IFN-γR2 expression in Th clones was detected by semiquantitative reverse transcription PCR in the presence of [32P]dCTP using total RNA as a template 23 24. Endogenous IFN-γR2 was amplified from cDNA using the primer pair 5′-GCGTCCACCCCGCGGTCCCGG and 5′-GTCTCTGTGATGTCCGTACA. TG IFN-γR2 was amplified using the primer pair 5′-GCACGTGGTTAAGCTCTCG and 5′-TGTCTCTGTGATGTCCGTACA.
7 d after antigenic stimulation, KLH-specific Th clones were treated with either IL-4 or IFN-γ. Whole cell protein extracts were then prepared and assayed for activated Stat complexes by electromobility shift assay using a βGAS probe, as described previously 18.
CD4+ T cells were isolated from KLH-immunized mice as described above. 105 cells/well were cultured with 7 × 105 APCs and KLH. 48 h after stimulation, culture supernatants were analyzed for cytokines by ELISA. At 72 h, these cultures were pulsed with [3H]thymidine as described above.
Responses to Listeria monocytogenes.
L. monocytogenes was provided by D. Hirsh (Columbia University, New York, NY) and was passaged, cultured, and titered as described previously 25. Heat-killed L. monocytogenes (HKLM) was prepared as described previously 25. 4–8-wk-old mice were infected by intraperitoneal injection with a sublethal dose of L. monocytogenes (2 × 106 CFU). In vivo and in vitro assays were performed 4 wk after infection.
Delayed-type hypersensitivity (DTH) was performed as described previously 25. In brief, sensitized mice were injected with HKLM (107 CFU) in PBS in the left hind footpad. The contralateral footpad was injected with vehicle control. Footpad swelling was measured 48 h after injection, and is represented as the difference between the average of triplicate measurements of thickness in the HKLM- and in the vehicle-injected footpads.
In vitro assays of Th cell function were performed as follows: CD4+ T cells, purified from the spleens of infected mice, were stimulated at 3 × 105 cells/well with either anti-CD3 mAbs and IL-2 (as described above), or with irradiated APCs (7 × 105 cells/well) and HKLM (107 CFU/well). Culture supernatants were removed 48 h after stimulation and assayed for cytokine content by ELISA. Proliferation rates of cells in each well were determined after an 18-h pulse with [3H]thymidine.
Antigen-specific Ab Production.
Immunization and antiserum production were performed using a published protocol 26. In brief, three to five mice per genotype were injected intraperitoneally with DNP-KLH (as above). Mice received serial booster immunizations (as described above except DNP-KLN was emulsified in IFA) every 30 d. 10 d after each injection, sera were obtained from immunized mice. KLH-specific Ab (IgG2a and IgG1) levels in these sera were detected by ELISA, as described 27.
Cytokine Production from In Vivo–primed CD4+ T Cells.
Mice were immunized with DNP-KLH, and CD4+ T cells were purified from local lymph nodes as described elsewhere. 105 CD4+ T cells were stimulated with 7 × 105 APCs and KLH 21. 48 h later, culture supernatants were harvested and assayed for IL-4 and IFN-γ by ELISA.
L. major Infection.
TG mice (fifth backcross to C57BL/6) and controls were infected with 1.5 × 107 stationary phase L. major promastigotes as described previously 28. In vitro Leishmania-specific IFN-γ production and in vivo parasite loads were determined as described previously 28.
Results
IFN-γR2 TG Mice.
To generate mice in which IFN-γR2 expression is not regulated during Th cell differentiation, the gene encoding the IFN-γR2 was placed under the control of the human CD2 promoter and enhancer (Fig. 1 A). Two different lines of TG mice, TG1 and TG2, were generated. The IFN-γR2 transgene is specifically expressed in the thymus and spleen, but not in the liver or kidney, as shown by ribonuclease protection (Fig. 1 B, and the riboprobe construct in Fig. 1 A). The expression of the transgene is higher in the thymus than in the spleen in both lines of IFN-γR2 TG mice (Fig. 1 B). It is likely that the multiple TG mRNA species protected in this assay reflects the existence of more than one transcriptional initiation site in the CD2 promoter 15.
IFN-γR2 TG mice appear normal and thrive in both conventional and barrier animal facilities. Gross examination of organs from TG mice reveals no apparent abnormalities. Moreover, flow cytometric analysis of thymocytes (CD4 versus CD8, and CD3 versus CD25), splenocytes (CD4 versus CD8 versus B220, and B220 versus IgD versus IgM), and bone marrow cells (B220 versus IgM) demonstrates that IFN-γR2 TG mice have normal lymphocyte populations in these organs (data not shown).
Altered IFN-γR2 Expression and Dysregulated IFN-γ Signaling in IFN-γR2 TG Th Clones.
KLH-specific Th1 (producing IFN-γ but not IL-4) and Th2 (producing IL-4 but not IFN-γ) clones from DNP-KLH–immunized wild-type (WT) and TG mice were generated, and the expression of IFN-γR2 in these clones was determined. Consistent with previous reports 13 14, expression of endogenous IFN-γR2 was detected in Th2 but not Th1 clones derived from WT mice (Fig. 1 C). In contrast, both Th1 and Th2 clones generated from the two lines of TG mice express IFN-γR2 from the endogenous and TG loci (Fig. 1 C). This suggests that expression of the transgene affects the expression of the endogenous IFN-γR2 locus.
Because expression of IFN-γR2 is obligatory for the transduction of the IFN-γ signal, the altered pattern of IFN-γR2 gene expression seen in the TG Th1 clones suggests that these cells may have different IFN-γ signaling properties from WT Th1 cells. As expected, after IFN-γ treatment, activated Stat1 complexes were detected in WT Th2 but not in WT Th1 clones (Fig. 1 D). However, both Th1 and Th2 clones derived from TG mice were able to activate Stat1 in response to IFN-γ. Interestingly, Stat1 is constitutively active in TG Th1 clones (Fig. 1 D). The presence of Stat1 in the STAT–DNA complexes was confirmed by supershift with anti-Stat1 mAb (data not shown). Activation of STATs in response to IL-4 did not differ between the Th1 and Th2 clones derived from TG and WT mice (Fig. 1 D). Detection of activated Stat1 in response to IFN-γ treatment in TG CD4+ T cell extracts confirms that both IFN-γR1 and IFN-γR2 are expressed on the surface of these cells 29. These data indicate that the IFN-γR2 transgene is expressed in TG Th cells and endows TG Th1 cells with the ability to activate Stat1 in response to IFN-γ.
Defective In Vitro Th1 Polarization in IFN-γR2 TG Mice.
Expression of the IFN-γR2 transgene prevents the loss of responsiveness to IFN-γ in Th1 cells. To examine whether the transgene affects the acquisition of effector Th phenotypes, naive T lymphocytes (CD4+Mel-14hi) from TG1, TG2, and WT mice were used to reconstitute Th differentiation in vitro. Th1-polarized CD4+ T cells derived from WT mice produced significantly higher amounts of IFN-γ than Th1-polarized CD4+ T cells isolated from either of the two IFN-γR2 TG lines (Fig. 2 A). Similar results were obtained regardless of whether Th1 polarization was induced by IFN-γ or IL-12. In the absence of polarizing stimuli, TG Th cultures produced lower amounts of IFN-γ but similar amounts of IL-4 as WT cultures (data not shown). Furthermore, the levels of IL-4 produced by the Th2-polarized populations did not differ significantly among CD4+ T cells isolated from WT mice and the two TG lines (Fig. 2 B). These results indicate that naive IFN-γR2 TG CD4+ cells have a reduced Th1 developmental potential compared with their WT counterparts. Moreover, despite their Th1 defect, TG CD4+ cells do not have an enhanced Th2 potential in this cell culture system.
Figure 2.
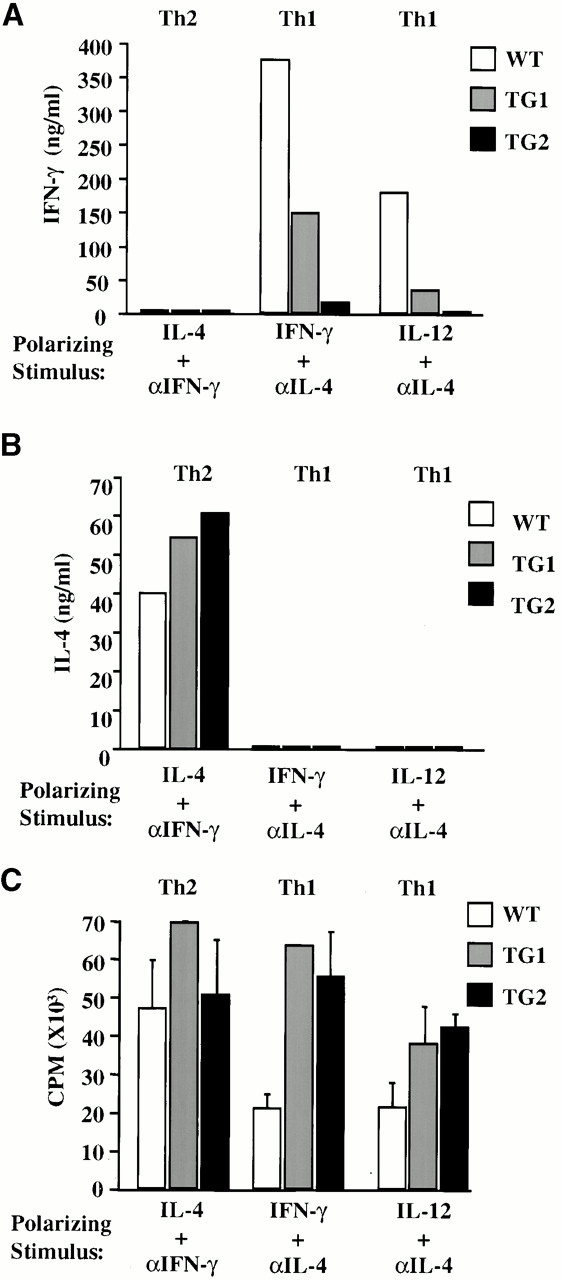
IFN-γ signaling disrupts Th1 differentiation in vitro, but does not affect the proliferation of polarized cells. CD4+Mel-14hi T cells were purified from spleens, stimulated with plate-bound anti-CD3 mAbs, and polarized in vitro for 7 d with IL-4 and anti–IFN-γ mAb (Th2) or anti–IL-4 and either IFN-γ or IL-12 (Th1). (A) IFN-γ and (B) IL-4 levels in culture supernatants were determined by ELISA 48 h after restimulation. (C) Proliferation rates of in vitro–differentiated cells were determined after a 6-h pulse with [3H]thymidine. These experiments were performed three times yielding similar results.
IFN-γ Signaling Does Not Affect Th Cell Proliferation.
A potential explanation for the impaired in vitro polarization seen in TG Th cells may be that the transgene causes an inherent proliferative defect in these cells. The retention of IFN-γ responsiveness imparted by the transgene could potentially render TG Th1 cells susceptible to the antiproliferative effects of autocrine, paracrine, or exogenous IFN-γ 30. To explore this possibility, we assayed the proliferation rates of in vitro–differentiated cells upon restimulation. Newly differentiated TG Th1 and Th2 cell lines proliferated at least as well as their WT counterparts upon restimulation with anti-CD3 mAbs in the presence of IL-2 (Fig. 2 C).
The experiments above did not examine the proliferative capacity of these cells early in the process of polarization towards the Th1 and Th2 phenotypes. It may be that it was not possible to detect significant differences in proliferation between polarized WT and TG Th cells because before restimulation, cell numbers were equalized among wells, thereby masking potential differences in proliferation rates under polarizing conditions. To examine this possibility, we tested whether Th1-polarizing conditions reduce the mitotic potential of TG Th cells by tracking the division of individual cells in our in vitro Th differentiation system 19 31. Purified CD4+ splenocytes from WT and TG mice were labeled with CFSE. Labeled cells were stimulated on anti-CD3–coated plates under several different polarizing conditions. 4 d after stimulation, the distribution of the CFSE label in each culture was analyzed by flow cytometry. A pattern of discrete generations of activated Th1-polarized cells results from the sequential halving of the CFSE label (Fig. 3 A). We detected no appreciable differences in peak distribution among Th1-polarized WT, TG1, and TG2 cultures (Fig. 3 B). Therefore, TG and WT Th cells undergo an equivalent number of cell division cycles when cultured under Th1-polarizing conditions. Furthermore, WT and TG CD4+ T cell cultures had a similar cell division profile regardless of the polarizing stimulus (data not shown). These data demonstrate that TG and WT Th cells proliferate at equivalent rates both during and after polarization towards Th1 or Th2 phenotypes. The defects in in vitro Th1 differentiation imparted by the IFN-γR2 transgene do not appear to be caused by altered proliferation of TG Th cells.
Figure 3.

IFN-γ signaling does not affect the mitotic profile of Th1-polarized cultures. (A) CFSE-labeled, purified CD4+ splenocytes from WT, TG1, and TG2 mice (top, middle, and bottom, respectively) were cultivated on anti-CD3 plus anti-CD28–coated plates and Th1 polarized with anti–IL-4 mAbs and IL-12. 4 d after the initiation of the culture, cells were analyzed by flow cytometry for CFSE fluorescence gating on live CD4+ cells. The sequential dilution of intracellular fluorescein, depicted as peak progression from right to left, represents progressive cell division. The CFSE label intensity at the initiation of each culture is represented as a dashed histogram overlaid at the right of each panel. (B) A chart numerically representing the percentages of cells in each peak of the histograms in A. Results are representative of three independent experiments.
Impaired Th1 Function and Th1 Memory Responses in IFN-γR2 TG Mice.
Naive Th cells overexpressing IFN-γR2 appeared to have a reduced ability to acquire a Th1 phenotype in vitro, suggesting that in vivo Th1-mediated immunity in IFN-γR2 TG mice may be impaired as well. To address this, several Th1-mediated immune responses were examined. DTH is a local, Th1-dependent inflammatory response elicited by subcutaneous administration of an antigen to a sensitized animal. DTH responses to L. monocytogenes were examined by administration of HKLM to the footpads of previously infected WT and TG mice. Listeria-sensitized WT mice responded to antigenic rechallenge with a robust swelling of the footpad (Fig. 4 A). In contrast, sensitized TG mice and naive animals responded poorly to antigenic rechallenge, suggesting that TG mice fail to develop normal Th1-dependent memory responses.
Figure 4.

IFN-γ signaling disrupts Th1-dependent memory responses in vivo. (A) DTH responses to L. monocytogenes. Naive and sensitized (previously infected) mice were injected subcutaneously with HKLM in the hind footpad. Swelling was measured 48 h later and represented as the difference in thickness between the HKLM-injected and control (contralateral, PBS-injected) footpads. Each bar represents one to four mice. (B–D) Cytokine secretion and proliferation of Th cells in response to antigenic rechallenge. Splenic CD4+ T cells from naive and sensitized mice were stimulated with APCs in the presence or absence of HKLM. 48 h later, (C) IFN-γ and (D) IL-2 levels in culture supernatants were quantified by ELISA. (B) Proliferation rates of these cultures were determined by measuring their [3H]thymidine incorporation in the presence of IL-2. All experiments were repeated at least three times with similar results.
To determine whether the DTH defect observed in TG mice resides within the CD4+ T cell compartment, antigen-specific memory Th cell responses were examined in vitro. CD4+ T cells purified from the spleens of mice sensitized with L. monocytogenes were tested for their ability to proliferate and secrete cytokines in response to in vitro antigenic rechallenge. Whereas WT Th cells secreted appreciable amounts of IFN-γ and IL-2, TG Th cells secreted negligible amounts of these Th1 cytokines in response to stimulation with HKLM presented by WT APCs (Fig. 4C and Fig. D). No IL-4 was detected in either WT or TG Th cultures (data not shown). However, these TG Th cells proliferated similarly to WT Th cells in response to rechallenge with listerial antigens (Fig. 4 B). Therefore, it is likely that TG mice are unable to mount a DTH response to HKLM because, although antigen-specific TG CD4+ T cells develop in response to infection with L. monocytogenes, these cells do not appear to be Th1 and are therefore unable to orchestrate cellular immune responses.
An alternative explanation for the defective DTH response in the TG mice may be that Th1 cells do not develop in these mice because of impaired or altered antigen presentation. To test this possibility, we first examined the function of APCs from TG mice. There was no difference between the ability of WT and TG APCs to elicit cytokine secretion and proliferative responses from normal Th cells in this assay (Tau, G.Z., and P.B. Rothman, unpublished observations). In addition, stimulating TG cells with WT APCs did not ameliorate the observed dysfunction in Th1 cytokine production by IFN-γR2 TG Th cells (Fig. 4C and Fig. D). These data support the conclusion that, in IFN-γR2 TG mice, the defect in Th1-dependent immunity is intrinsic to the CD4+ T cell compartment.
IFN-γ Signaling Abrogates In Vivo Th1-dependent Ab Production.
Protein antigens can induce a mixed Th1/Th2 cellular response when used as immunogens. Th1 and Th2 cytokines induce distinct Ig heavy chain isotype class switching events in B cells, leading to production of IgG2a and IgG1, respectively 32. The reduced ability of TG Th cells to produce IFN-γ suggests that IFN-γR2 TG mice may produce an altered pattern of Ig isotypes in response to immunization with protein antigen. To address this possibility, WT and TG mice were immunized with KLH, and the levels of different KLH-specific Ig isotypes were measured after the primary immunization, the first booster, and the second booster. As expected, KLH-specific IgG1 and IgG2a mAbs were undetectable after the primary immunization and the first booster with KLH in either WT or TG mice (Fig. 5C and Fig. D). After the second booster, both TG and WT controls produced significant levels of KLH-specific IgG1, suggesting that the Th2 component of the anti-KLH immune response is normal in TG mice (Fig. 5 C). On the other hand, although WT animals produced significant amounts of antigen-specific IgG2a, KLH-specific Abs of this Ig isotype were not detected in TG animals (Fig. 5 D). This observation supports earlier data demonstrating impaired Th1 immunity but normal Th2 responses in IFN-γR2 TG mice.
Figure 5.

KLH-specific responses. (A and B) IFN-γ production and proliferation of in vivo–primed CD4+ T cells. 7 d after in vivo immunization with KLH, purified CD4+ T cells were stimulated in vitro with APCs in the presence or absence of KLH. (A) IFN-γ content of the culture supernatants was determined 48 h after stimulation, and (B) the proliferation rates of these cultures were determined 72 h after stimulation after a 6-h pulse with [3H]thymidine. (C and D) KLH-specific Ig production. Mice were serially immunized with KLH. After each immunization, sera were analyzed for KLH-specific (C) IgG1 and (D) IgG2a content by ELISA. These experiments were repeated twice with similar results.
It is probable that the impaired in vivo production of IgG2a by B cells is secondary to a T cell defect. CD4+ cells isolated from KLH-immunized TG mice failed to produce IFN-γ but proliferated normally in response to stimulation with this antigen, suggesting that non–IFN-γ–secreting KLH-specific Th cells do develop in TG mice (Fig. 5A and Fig. B). To examine B cell function, IgM+ B cells were purified from WT and TG mice and stimulated with LPS and either IFN-γ or IL-4 33 34. WT and TG B cells produced similar levels of IgG2a or IgG1 in vitro (data not shown). Therefore, there does not appear to be an intrinsic defect in the ability of TG B cells to undergo Ig heavy chain class switching. Together, these data demonstrate that the altered Ab production in the TG mice results from aberrant Th1 cell development.
Impaired Th1 Response to L. major Infection in IFN-γR2 TG Mice.
A healing response to infection with L. major, an intracellular protozoan, requires robust Th1 immunity 35. Since it appears that IFN-γR2 TG mice have impaired Th1 responses, it is possible that these mice may be susceptible to infection with L. major. While monitoring the course of footpad infection with this pathogen, it was found that TG mice develop larger, more persistent lesions than their WT littermates (Fig. 6 A). This correlates with a 10-fold greater parasitemia in TG mice, indicating that these mice are unable to elaborate normal parasite control mechanisms (Fig. 6 C). CD4+ T cells isolated from infected WT mice exhibited robust proliferative responses as well as IFN-γ and IL-2 production after stimulation with L. major antigens (Fig. 6 B, and data not shown). On the other hand, TG CD4+ T cells produced negligible amounts of IFN-γ and IL-2, but proliferated normally in response to the identical stimulus (Fig. 6 B, and data not shown). These data suggest that although TG mice are capable of developing antigen-specific Th cells in response to infection with L. major, these cells do not have a Th1 phenotype.
Figure 6.
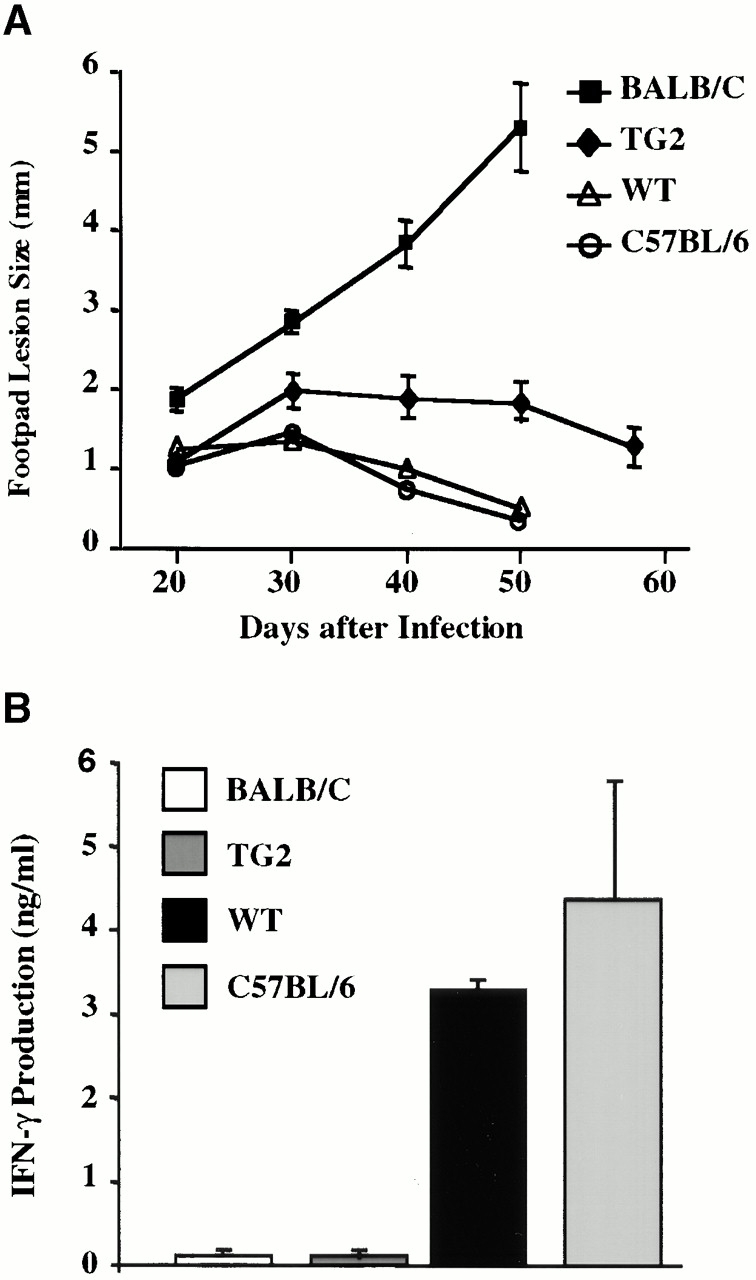

Responses to chronic L. major infection. (A) The course of L. major infection. Lesion size was determined at 7–10-d intervals by calculating the average difference in thickness between the infected and uninfected footpads of at least five mice per group. (B) L. major–specific IFN-γ production. T cells were isolated from lesion-draining popliteal lymph nodes and stimulated in vitro with APCs and leishmanial antigens. At 48 h, the IFN-γ content of the culture supernatants was determined by ELISA. (C) Parasite load. The number of parasites in cultures of cell suspensions from popliteal lymph nodes was determined by limiting dilution.
Resistance to L. major infection is mouse strain dependent 35. C57BL/6 (the genetic background of the TG mice) mice mount healing responses, whereas BALB/c mice succumb to infection with this pathogen. It is believed that the exquisite sensitivity of BALB/c mice to this pathogen results from their natural propensity towards Th2 responses, as manifested by greater IL-4 and IgE production and lower IFN-γ and IL-12 levels relative to those detected in C57BL/6 mice 35 36. Although IFN-γR2 TG mice are more susceptible to infection with L. major than WT mice, they do not develop the progressive, nonhealing lesions seen in BALB/c mice. Moreover, unlike in BALB/c mice, infected TG T cells do not produce elevated amounts of IL-4 in response to L. major antigens, nor are higher IgE levels detected in the serum of infected TG mice (data not shown). These observations are consistent with our previous data, but are uncharacteristic of other systems in which Th1 responses are impaired 35. This unique Th subset imbalance, characterized by impaired Th1 parameters in the absence of increased Th2 parameters, is less severe than that reported for BALB/c mice, but nevertheless correlates well with the intermediate susceptibility phenotype of TG mice (Fig. 6).
Discussion
Prior studies have demonstrated that cytokine and chemokine responsiveness is regulated during Th cell differentiation. However, the significance of many of these regulatory processes is not well understood. We sought to determine the importance of regulating IFN-γ responsiveness for Th subset development and for effector Th function by generating mice whose Th cells are unable to regulate expression of IFN-γR2. Expression of the IFN-γR2 transgene imparts IFN-γ responsiveness to Th1 cells, which do not normally respond to this cytokine.
IFN-γR2 TG mice are unable to mount efficient Th1-dependent in vivo immune responses such as DTH and healing after infection with L. major. In addition, these mice exhibit diminished ex vivo IFN-γ production in response to immunization with L. monocytogenes and DNP-KLH. Decreased IFN-γ levels are likely responsible for the lower amounts of KLH-specific IgG2a that are produced by the TG mice after immunization. These data were corroborated in experiments recapitulating Th cell differentiation in vitro. Here, naive TG Th cells were found to lack the potential to become Th1 cells in an in vitro culture system that specifically examines the intrinsic polarization potential of Th cells since it is devoid of other cell types. This suggests that the primary immunological defect seen in the TG mice resides within the CD4+ T cell compartment. Together, these data indicate that the ectopic expression of IFN-γR2 profoundly impairs Th1 cell development and Th1-type immune responses in these mice.
Our data suggest that defects in other, non-T cell compartments are not responsible for the Th1 defect observed in TG mice. B cells function normally in these mice. Also, TG macrophages do not express the transgene and normally upregulate expression of MHC class II molecules in response to infection with L. monocytogenes. (Tau, G.Z., and P.B. Rothman, unpublished observations). There were no detectable differences between WT and TG mice in their ability to survive primary listeriosis, indicating that listerial control mechanisms of the innate immune system are intact (Tau, G.Z., and P.B. Rothman, unpublished observations).
There are several different mechanisms by which the IFN-γR2 transgene may affect Th1-dependent immune responses. One possibility is that the IFN-γ responsiveness of Th1 cells imparted by the transgene makes these cells susceptible to the antiproliferative and antimetabolic effects of IFN-γ. Several studies have suggested that IFN-γ may, in fact, exert its Th1-promoting effects indirectly, by restricting the outgrowth of Th2 cells or by antagonizing the Th2 developmental pathway rather than by promoting Th1 phenotype acquisition 30 37. As TG Th1 cells would gain the ability to secrete IFN-γ during their differentiation, they would, in effect, inhibit their own proliferation or development, resulting in an initially normal but ultimately abortive commitment to the Th1 pathway. The observation that, during Th1 polarization, the mitotic program of activated TG CD4+ cells is no different than that of their WT counterparts speaks against the proliferative disadvantage hypothesis. Also, we detected no increased propensity towards Th2 responses in TG mice, supporting the abortive commitment hypothesis. Furthermore, IFN-γ appeared to have no antiproliferative effect on TG Th1 cell clones and on TG Th cells during polarization (Tau, G.Z., and P.B. Rothman, unpublished observations). However, it is possible that in vitro–generated, and in vivo–occurring TG CD4+ IFN-γ producers have become selectively refractory to the growth-inhibitory effects of this cytokine despite the fact that certain IFN-γ–dependent signaling molecules, such as Stat1, are activated in these cells in response to IFN-γ treatment.
We demonstrated that Stat1 is constitutively active in TG but not in WT Th1 clones. Consequently, a vast array of Stat1-regulated genes may be expressed in TG but not in WT Th1 cells. In support of this, we have shown that the endogenous IFN-γR2 gene itself is expressed in TG Th1 cells but not in WT Th1 cells (Fig. 1 C). This could potentially affect a host of cellular programs in TG mice, including the Th1 differentiation process and Th1 effector functions. Therefore, it is possible that signaling downstream of IFN-γ or the expression of IFN-γ–dependent genes creates intracellular conditions that are incompatible with a Th1 effector phenotype. Consequently, the TG “Th1” cells that do develop may be unable to mediate Th1 functions other than IFN-γ production and may, in fact, not be true Th1 cells.
It is interesting that Th1 cell defects are observed in both IFN-γR2–deficient and IFN-γR2–overexpressing mice. The mechanisms underlying these defects are likely to be different. It appears that the lack of efficient Th1 cell generation in IFN-γR2–deficient mice results, at least in part, from decreased levels of the IL-12R 18. Therefore, IFN-γ signaling may be important for the initiation of Th1 cell differentiation. In contrast, the data herein demonstrate that sustained IFN-γ signaling is deleterious for Th1 cell development and suggest that the temporal regulation of IFN-γR2 expression is essential in Th1 cell differentiation.
Unlike other TG or knockout mouse models examining Th1 cell development and function, IFN-γR2 TG mice exhibit intermediate susceptibility to infection with L. major 38 39 40. Perhaps the block in Th1 development in TG mice occurs after commitment to the Th1 pathway, whereas in other mouse models (in which Th1 function is more severely compromised) the Th1 pathway is blocked earlier and naive Th cells are effectively unable to begin differentiation along the Th1 pathway, and therefore default to the Th2 pathway. Regardless, the Th1 block caused by the IFN-γR2 transgene appears to be substantial enough to give rise to significant alterations in this and other cell-mediated immune responses. Further analysis of differential gene expression in Th1 cells expressing IFN-γR2 may provide insight into the molecular events that define Th1 cell differentiation, and those events that can disrupt this developmental process.
Acknowledgments
We wish to acknowledge Yulin Wang for his significant contribution in generating the data presented in the figures. We also wish to thank Smita Mauze for her technical assistance, Matt Adlam for oocyte injection, and Martijn Nawijn for his critical reading of the manuscript.
The work at Columbia University was supported by American Chemical Society grant IM783 and National Institutes of Health grant PO1AI39675. The DNAX Research Institute of Molecular and Cellular Biology is supported by the Schering-Plough Corp.
Footnotes
Abbreviations used in this paper: CFSE, carboxyfluorescein diacetate succinimidyl ester; DTH, delayed-type hypersensitivity; HKLM, heat-killed Listeria monocytogenes; STAT, signal transducer and activator of transcription; TG, transgenic; WT, wild-type.
References
- Mosmann T.R., Coffman R.L. TH1 and TH2 cellsdifferent patterns of lymphokine secretion lead to different functional properties. Annu. Rev. Immunol. 1989;7:145–173. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- Paul W.E., Seder R.A. Lymphocyte responses and cytokines. Cell. 1994;76:241–251. doi: 10.1016/0092-8674(94)90332-8. [DOI] [PubMed] [Google Scholar]
- Abbas A.K., Murphy K.M., Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383:787–793. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- Constant S., Pfeiffer C., Woodard A., Pasqualini T., Bottomly K. Extent of T cell receptor ligation can determine the functional differentiation of naive CD4+ T cells. J. Exp. Med. 1995;182:1591–1596. doi: 10.1084/jem.182.5.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constant S.L., Bottomly K. Induction of Th1 and Th2 CD4+ T cell responsesthe alternative approaches. Annu. Rev. Immunol. 1997;15:297–322. doi: 10.1146/annurev.immunol.15.1.297. [DOI] [PubMed] [Google Scholar]
- O'Garra A. Cytokines induce the development of functionally heterogeneous T helper cell subsets. Immunity. 1998;8:275–283. doi: 10.1016/s1074-7613(00)80533-6. [DOI] [PubMed] [Google Scholar]
- Fitch F.W., McKisic M.D., Lancki D.W., Gajewski T.F. Differential regulation of murine T lymphocyte subsets. Annu. Rev. Immunol. 1993;11:29–48. doi: 10.1146/annurev.iy.11.040193.000333. [DOI] [PubMed] [Google Scholar]
- Moore K.W., O'Garra A., de Waal Malefyt R., Vieira P., Mosmann T.R. Interleukin-10. Annu. Rev. Immunol. 1993;11:165–190. doi: 10.1146/annurev.iy.11.040193.001121. [DOI] [PubMed] [Google Scholar]
- Sher A., Coffman R.L. Regulation of immunity to parasites by T cells and T cell-derived cytokines. Annu. Rev. Immunol. 1992;10:385–409. doi: 10.1146/annurev.iy.10.040192.002125. [DOI] [PubMed] [Google Scholar]
- Austrup F., Vestweber D., Borges E., Lohning M., Brauer R., Herz U., Renz H., Hallmann R., Scheffold A., Radbruch A., Hamann A. P- and E-selectin mediate recruitment of T-helper-1 but not T-helper-2 cells into inflamed tissues. Nature. 1997;385:81–83. doi: 10.1038/385081a0. [DOI] [PubMed] [Google Scholar]
- Sallusto F., Lanzavecchia A., Mackay C.R. Chemokines and chemokine receptors in T-cell priming and Th1/Th2-mediated responses. Immunol. Today. 1998;19:568–574. doi: 10.1016/s0167-5699(98)01346-2. [DOI] [PubMed] [Google Scholar]
- Szabo S.J., Jacobson N.G., Dighe A.S., Gubler U., Murphy D.M. Developmental commitment to the Th2 lineage by extinction of IL-12 signaling. Immunity. 1995;2:665–675. doi: 10.1016/1074-7613(95)90011-x. [DOI] [PubMed] [Google Scholar]
- Pernis A., Gupta S., Gollob K.J., Garfein E., Coffman R.L., Schindler C., Rothman P. Lack of interferon γ receptor β chain and the prevention of interferon γ signaling in TH1 cells. Science. 1995;269:245–247. doi: 10.1126/science.7618088. [DOI] [PubMed] [Google Scholar]
- Bach E.A., Szabo S.J., Dighe A.S., Ashkenazi A., Aguet M., Murphy K.M., Schreiber R.D. Ligand-induced autoregulation of IFN-γ receptor β chain expression in T helper cell subsets. Science. 1995;270:1215–1218. doi: 10.1126/science.270.5239.1215. [DOI] [PubMed] [Google Scholar]
- Lang G., Wotton D., Owen M.J., Sewell W.A., Brown M.H., Mason D.Y., Crumpton M.J., Kioussis D. The structure of the human CD2 gene and its expression in transgenic mice. EMBO (Eur. Mol. Biol. Organ.) J. 1988;7:1675–1682. doi: 10.1002/j.1460-2075.1988.tb02995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmi S., Bohni R., Stark G., Di Marco F., Aguet M. A novel member of the interferon receptor family complements functionality of the murine interferon γ receptor in human cells. Cell. 1994;76:803–810. doi: 10.1016/0092-8674(94)90355-7. [DOI] [PubMed] [Google Scholar]
- Gilman M. Ribonuclease protection assay Ausubel F.M., Brent R., Kingston R.E., Moore D.D., Seidman J.G., Smith J.A., Struhl K. Current Protocols in Molecular Biology. Vol. 1 1989. 4 John Wiley & Sons, Inc; New York: 7.1–4.7.8. [Google Scholar]
- Lu B., Ebensperger C., Dembic A., Wang Y., Kvatyuk M., Lu T., Coffman R.L., Pestka S., Rothman P. Targeted disruption of the interferon γ receptor 2 gene results in severe immune defects in mice. Proc. Natl. Acad. Sci. USA. 1998;95:8233–8238. doi: 10.1073/pnas.95.14.8233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons A.B., Parish C.R. Determination of lymphocyte division by flow cytometry. J. Immunol. Methods. 1994;171:131–137. doi: 10.1016/0022-1759(94)90236-4. [DOI] [PubMed] [Google Scholar]
- Fitch F.W., Gajewski T.F. Production of T cell clones Coligan J.E., Kruisbeek A.M., Margulies D.H., Shevach E.M., Strober W. Current Protocols in Immunology. Vol. 1 1997. 3 John Wiley & Sons, Inc; New York: 13.1–3.13.11. [Google Scholar]
- Kruisbeek A.M. Proliferative assays for T cell function Coligan J.E., Kruisbeek A.M., Margulies D.H., Shevach E.M., Strober W. Current Protocols in Immunology. Vol. 1 1991. 3 John Wiley & Sons, Inc; New York: 12.1–3.12.14. [Google Scholar]
- Berkowitz N., Braunstein N.S. T cell responses specific for subregions of allogeneic MHC molecules. J. Immunol. 1992;148:309–317. [PubMed] [Google Scholar]
- Chomczynski P. Single-step RNA isolation from cultured cells or tissue Ausubel E.M., Brent R., Kingston R.E., Moore D.D., Seidman J.G., Struhl K. Current Protocols in Molecular Biology. Vol. 1 1988. 4 Massachusetts General Hospital and Harvard Medical School; Boston: 2.4–4.2.8. [Google Scholar]
- Murray L.J., Lee R., Martens C. In vivo cytokine gene expression in T cell subsets of the autoimmune MRL/Mp-lpr/lpr mouse. Euro. J. Immunol. 1990;20:163–170. doi: 10.1002/eji.1830200124. [DOI] [PubMed] [Google Scholar]
- Irikura V.M., Hirsch E., Hirsh D. Effects of interleukin-1 receptor antagonist overexpression on infection by Listeria monocytogenes . Infect. Immun. 1990;67:1901–1909. doi: 10.1128/iai.67.4.1901-1909.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper H.M. Production of antibodies Coligan J.E., Kruisbeek A.M., Margulies D.H., Shevach E.M., Strober W. Current Protocols in Immunology. Vol. 1 1995. 2 John Wiley & Sons, Inc; New York: 4.1–2.4.9. [Google Scholar]
- Germann T., Bongartz M., Dlugonska H., Hess H., Schmitt E., Kolbe L., Kolsch E., Podlaski F.J., Gately M.K., Rude E. Interleukin-12 profoundly up-regulates the synthesis of antigen-specific complement-fixing IgG2a, IgG2b and IgG3 antibody subclasses in vivo . Eur. J. Immunol. 1995;25:823–829. doi: 10.1002/eji.1830250329. [DOI] [PubMed] [Google Scholar]
- von der Weid T., Beebe A.M., Roopenian D.C., Coffman R.L. Early production of IL-4 and induction of Th2 responses in the lymph node originate from an MHC class-1-independent CD4+ NK1.1 T cell population. J. Immunol. 1996;157:4421–4427. [PubMed] [Google Scholar]
- Bach E.A., Aguet M., Schreiber R.D. The IFNγ receptora paradigm for cytokine receptor signaling. Annu. Rev. Immunol. 1997;15:563–591. doi: 10.1146/annurev.immunol.15.1.563. [DOI] [PubMed] [Google Scholar]
- Gajewski T.F., Fitch F.W. Anti-proliferative effect of IFN-γ in immune regulation. I. IFN-γ inhibits the proliferation of Th2 but not Th1 murine helper T lymphocyte clones. J. Immunol. 1988;140:4245–4252. [PubMed] [Google Scholar]
- Wells A.D., Gudmundsdottir H., Turka L.A. Following the fate of individual T cells throughout activation and clonal expansion. Signals from T cell receptor and CD28 differentially regulate the induction and duration of a proliferative response. J. Clin. Invest. 1997;100:3173–3183. doi: 10.1172/JCI119873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffman R.L., Lebman D.A., Rothman P. Mechanism and regulation of immunoglobulin isotype switching. Adv. Immunol. 1993;54:229–270. doi: 10.1016/s0065-2776(08)60536-2. [DOI] [PubMed] [Google Scholar]
- Isakson P.C., Pure E., Vitetta E.S., Krammer P.H. T cell–derived B cell differentiation factor(s). Effect on the isotype switch of murine B cells. J. Exp. Med. 1982;155:734–748. doi: 10.1084/jem.155.3.734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapper C.M., Paul W.E. Interferon-gamma and B cell stimulatory factor-1 reciprocally regulate Ig isotype production. Science. 1987;236:944–947. doi: 10.1126/science.3107127. [DOI] [PubMed] [Google Scholar]
- Reiner S.L., Locksley R.M. The regulation of immunity to Leishmania major . Annu. Rev. Immunol. 1995;13:151–177. doi: 10.1146/annurev.iy.13.040195.001055. [DOI] [PubMed] [Google Scholar]
- Heinzel F.P., Sadick M.D., Holaday B.J., Coffman R.L. Reciprocal expression of interferon γ or interleukin 4 during the resolution or progression of murine leishmanaisisevidence of expansion of distinct helper T cell subsets. J. Exp. Med. 1989;169:59–72. doi: 10.1084/jem.169.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski T.F., Joyce J., Fitch F.W. Antiproliferative effect of IFN-γ in immune regulation. III. Differential selection of Th1 and Th2 murine helper T lymphocyte clones using recombinant IL-2 and recombinant IFN-γ. J. Immunol. 1989;143:15–22. [PubMed] [Google Scholar]
- Wang Z.E., Reiner S.L., Zheng S., Dalton D.K., Locksley R.M. CD4+ effector cells default to the Th2 pathway in interferon gamma–deficient mice infected with Leishmania major . J. Exp. Med. 1994;179:1367–1371. doi: 10.1084/jem.179.4.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattner F., Magram J., Ferrante J., Launois P., Di Padova K., Behin R., Gately M.K., Louis J.A., Alber G. Genetically resistant mice lacking interleukin-12 are susceptible to infection with Leishmania major and mount a polarized Th2 cell response. Eur. J. Immunol. 1996;26:1553–1559. doi: 10.1002/eji.1830260722. [DOI] [PubMed] [Google Scholar]
- Swihart K., Fruth U., Messmer N., Hug K., Behin R., Huang S., Del Giudice G., Aguet M., Louis J.A. Mice from a genetically resistant background lacking the interferon gamma receptor are susceptible to infection with Leishmania major but mount a polarized T helper cell 1–type CD4+ T cell response. J. Exp. Med. 1995;181:961–971. doi: 10.1084/jem.181.3.961. [DOI] [PMC free article] [PubMed] [Google Scholar]