

Abstract
Rheumatoid arthritis (RA) is a heterogeneous syndrome of which a subset of patients develops vascular inflammation. The genetic determinants that confer risk for rheumatoid vasculitis are not known, but patients with vascular complications are known to have an expansion of CD4+CD28null T cells, a cell population potentially involved in endothelial damage. CD4+CD28null T cell clones isolated from RA patients with vasculitis were found to express killer cell immunoglobulin–like receptors (KIRs) with the stimulatory KIR2DS2 often present in the absence of opposing inhibitory receptors with related specificities. To test the hypothesis that the KIR2DS2 gene is involved in the development of vasculitis, association studies were performed. The KIR2DS2 gene was significantly enriched among patients with rheumatoid vasculitis compared with normal individuals (odds ratio 5.56, P = 0.001) and patients with RA but no vasculitis (odds ratio 7.96, P = 0.001). Also, the distribution of human histocompatibility leukocyte antigen (HLA)-C, the putative ligand for KIRs, was significantly different in patients with rheumatoid vasculitis in comparison with the control populations. These data suggest that HLA class I–recognizing receptors and HLA class I genes are genetic risk determinants that modulate the pattern of RA expression. Specifically, KIR2DS2 in conjunction with the appropriate HLA-C ligand may have a role in vascular damage by regulating CD4+CD28null T cells.
Keywords: killer cell immunoglobulin–like receptor, T cell, genetic susceptibility, rheumatoid arthritis, vasculitis
Introduction
Rheumatoid arthritis (RA) is a chronic destructive joint disease with crippling potential. Current evidence suggests that susceptibility to RA is under polygenic control with a major contribution of the MHC class II locus 1 2 3 4. Clinically, the disease is heterogeneous in regard to the progression of joint destruction, response to treatment, and involvement of extraarticular organs. It has been proposed that this clinical heterogeneity is genetically determined and that different disease phenotypes are a reflection of different combinations of disease-risk genes 5. In support of this concept, MHC class II polymorphisms correlate with differences in disease pattern and disease aggressiveness. Homozygosity of disease-associated HLA-DRB1*04 alleles predisposes patients with RA to major organ involvement such as in Felty's syndrome and rheumatoid vasculitis 6 7. A role for sex-linked genes has been inferred because disease phenotypes in male and female patients can be distinguished 8. Studies of other possible genetic risk factors that could function as disease modulators are preliminary 1 2 9 10; however, biological markers have been identified that correlate with disease expression and may be gen-etically determined. In particular, a subset of patients with RA has expanded populations of CD4 T cells that lack surface expression of the CD28 molecule 11. CD4+CD28null T cells are uncommon in healthy individuals. The expansion of this cell subset in patients with RA correlates with the clinical phenotype of disease; the highest frequencies are detected in patients with rheumatoid vasculitis 12. Rheumatoid vasculitis is a life-threatening extraarticular complication of the rheumatoid syndrome. A direct role of CD4+CD28null T cells in vascular injury has been suggested because these cells are also a risk factor for inflammation and rupturing of atherosclerotic plaques in acute coronary syndromes 13 14.
CD4+CD28null T cells are functionally distinct from classical CD4 helper T cells. They do not express CD40 ligand, produce large amounts of IFN-γ, and express granzyme B and perforin, giving them the capability to lyse target cells 15 16. Their cytotoxic potential, as well as expression of the NK cell marker CD57, suggests that CD4+CD28null T cells share features with NK cells.
The functional activity of NK cells is tightly regulated by a family of polymorphic receptors that recognize MHC class I molecules 17 18. Interaction of human NK cells with MHC class I molecules is governed by two distinct receptor families, both of which can transmit inhibitory or stimulatory signals. CD94/NKG2 receptors are members of the C-type lectin superfamily and recognize HLA-E molecules 19 20. The various allotypes of HLA-A, B, and C interact with a structurally different set of receptors, the killer cell Ig-like receptors (KIRs), which belong to the Ig superfamily 21 22 23. Different KIR isoforms bind specific MHC class I allotypes; the KIR2D subfamily has specificity for HLA-C–encoded molecules 24. MHC class I–mediated triggering of KIR transduces a dominant inhibitory signal that blocks NK cell cytolytic activity and cytokine production in vitro 25. Ligand binding initiates the phosphorylation of cytoplasmic immunoregulatory tyrosine-based inhibitory motifs (ITIMs) with subsequent recruitment of protein tyrosine phosphatases. The MHC-recognizing receptors of the Ig superfamily also include members that have truncated cytoplasmic domains lacking competent ITIMs. These variants have extracellular domains that are highly homologous to the inhibitory receptors and that also appear to bind MHC class I molecules, but they transmit activation signals 26 27.
The KIR family is a highly polymorphic multigene family of receptors located on chromosome 19q13.4 28 29. Of particular interest, Uhrberg et al. have described considerable variation of KIR expression among Caucasians 30. The authors have detected 18 phenotypes in a population of only 52 individuals. We have, therefore, explored whether MHC class I–recognizing receptors are expressed on CD4+CD28null T cells and whether genetic polymorphisms in the KIR family confer susceptibility to rheumatoid vasculitis.
Materials and Methods
Study Population.
70 unrelated patients fulfilling the 1988 American College of Rheumatology criteria for RA and with no major organ involvement (e.g., Felty's syndrome, interstitial lung disease, or rheumatoid vasculitis), 30 RA patients with rheumatoid vasculitis, and 76 control individuals were enrolled in the study. This study was approved by the Mayo Clinic Internal Review Board and all participants provided written informed consent. All individuals were Caucasians of Western European descent. The diagnosis of rheumatoid vasculitis was based on histomorphology or angiogram. Nonhealing lower extremity skin ulcers without histopathological proof were only accepted as vasculitis if there was no clinically evident atherosclerosis. Periungual nailfold infarcts were not sufficient for the diagnosis of rheumatoid vasculitis.
Flow Cytometry.
Heparinized venous blood was collected and PBMCs were isolated by density gradient centrifugation over Ficoll-Hypaque (Amersham Pharmacia Biotech). Cell surface staining was performed using combinations of the following antibodies: CD4-peridinin chlorophyll protein (PerCP), CD28-FITC, CD158a-PE, CD94-FITC (Becton Dickinson), and CD158b-PE (Beckman Coulter). Samples were analyzed on a FACSCalibur™ flow cytometer (Becton Dickinson) and the frequencies of T cell subsets were calculated using WinMDI software (Joseph Trotter, Scripps Research Institute, La Jolla, CA).
T Cell Cloning and KIR Phenotyping.
CD4+CD28null T cells from three RA patients, two healthy individuals, and one patient with acute coronary syndrome were sorted on a FACS Vantage™ flow cytometer (Becton Dickinson), cloned by limiting dilution cloning, and the T cell clones were maintained as described 22. Donors were selected to express KIR2DS2. Total RNA from T cell clones was extracted using TRIzol reagent (Invitrogen/Life Technologies). cDNA was amplified with the KIR family member–specific primer sets as described by Uhrberg et al. 30 . The nomenclature suggested by E. Long, L. Lanier, and M. Colonna was used as published in Uhrberg et al. 30. To confirm clonality, TCR sequences of selected T cell clones were determined or T cell clones were subcloned and the KIR pattern was reassessed.
KIR and HLA-C Genotyping.
Genomic DNA was isolated using the DNA Isolation Kit for Mammalian Blood (Roche Molecular Biochemicals/Boehringer) or QIAamp Blood Kit (QIAGEN). DNA was amplified with KIR2DS1- and KIR2DS2-specific primers 30 (KIR2DS1, 5′-TCTCCATCAGTCGCATGAA/G-3′, 5′-AGGGCCCAGAGGAAAGTT-3′, and KIR2DS2, 5′-TGCACAGAGAGGGGAAGTA-3′, 5′-CACGCTCTCTCCTGCCAA-3′) in 25 μl reaction volume containing 0.525 U DNA polymerase (Expand High Fidelity PCR System; Roche Molecular Biochemicals/Boehringer), 0.5 mM dNTPs, 0.2 μM sense primer, 0.2 μM antisense primer, and 80–300 ng DNA. PCR was performed in a 9600 thermal cycler (PE Biosystems) using the following conditions: denaturation at 97°C for 10 min followed by 5 cycles of 97°C for 20 s, 60°C for 45 s, 72°C for 90 s, and then 25 cycles of 95°C for 20 s, 58°C for 45 s, and 72°C for 90 s. The amplified products were visualized on ethidium bromide prestained 1% agarose gels.
HLA-C alleles were determined by PCR using the Dynal HLA-C “low resolution” SSP kit (Dynal) according to the manufacturer's instructions.
Costimulation Experiments.
T cell clones that expressed CD158b by FACS® and KIR2DS2 but not KIR2DL2 or KIR2DL3 by PCR were selected. Resting cells (3 × 105/well) were incubated with optimal (1 μg/ml) and suboptimal (50 ng/ml) concentrations of anti-CD3 (Orthoclone OKT3; Ortho Biotech), anti-CD158b (200 ng/ml; GL183; Beckman Coulter), IgG control (200 ng/ml; ICN Biomedicals), or a combination for 30 min on ice. Bound antibodies were cross-linked with rabbit anti–mouse IgG (1 μg/ml; ICN Biomedicals) for 18 h at 37°C in the presence of 1 U/ml recombinant human IL-2 (Proleukin; Chiron Corp.). Total RNA was isolated using TRIzol (Invitrogen/Life Technologies) and analyzed for the transcription of IFN-γ sequences by reverse transcription (RT)-PCR using the following primer set: 5′-ACCGAATAATTAGTCAGCTT-3′ and 5′-ACCTTAAGAAATATTTTAATGC-3′. Transcripts were semiquantified on a Light Cycler (Roche Molecular Diagnostics/Boehringer).
Statistical Analysis.
The frequencies of specific alleles were compared between each of the three study groups using two-by-two tables and either Pearson's Chi-squared statistic or Fisher's exact test (for small counts), and the magnitude of effect was estimated by odds ratios and their 95% confidence intervals. The effects of multiple genes were evaluated simultaneously using logistic regression. All analyses were performed using SAS software.
Results
KIR Expression on CD4 T Cells from Patients with Rheumatoid Vasculitis.
MHC class I–recognizing receptors of the Ig superfamily were detected on CD4 T cells of patients with RA by flow cytometric analysis. As shown in Fig. 1, CD4+CD158b+ cells were found exclusively among CD28null but not CD28+ cells, indicating the presence of KIR2DS2, KIR2DL2, and/or KIR2DL3 on the cell surface. CD4+CD28null T cells were consistently negative for the NK cell molecules, CD16 and CD94. CD158a was infrequently expressed on CD4+CD28null T cells (data not shown).
Figure 1.

CD4+CD28null T cells express MHC class I–recognizing receptors. Peripheral blood CD4 T cells were analyzed by three-color flow cytometry for the expression of MHC class I–recognizing receptors. A representative experiment from a patient with rheumatoid vasculitis is shown. CD158b (encompassing KIR2DS2, KIR2DL2, and KIR2DL3) was exclusively expressed on CD28null but not on CD28+CD4+ T cells. CD158a (KIR2DS1 and KIR2DL1) was infrequently expressed and high expression of CD94/NKG2 dimers was absent (data not shown). Background staining with isotype-matched control antibodies is shown as shaded areas.
The spectrum of KIR-specific antibodies is limited and the available antibodies cannot distinguish between stimulatory and inhibitory receptors. To analyze the array of KIRs on CD4+CD28null T cells, 49 CD4+CD28null T cell clones were generated from three patients with rheumatoid vasculitis. PCR analysis with primer pairs for six inhibitory KIRs and five stimulatory KIRs was used to establish KIR expression patterns. Results are summarized in Fig. 2. Of the activating receptors, expression of KIR2DS2 was prominent and ranged from 58–91% of the clones in the three patients studied. None of the T cell clones expressed transcripts for KIR2DS1, KIR2DS3, or KIR2DS4; between 0–20% of the clones expressed KIR3DS1. The spectrum of inhibitory receptors was broader, and all six isoforms were present in varying frequencies. KIR2DL4 and KIR2DL3 were most frequently detected. KIR2DL1, KIR2DL2, and KIR3DL2 each appeared more frequently in clones from one patient whereas KIR3DL1 was rarely found.
Figure 2.

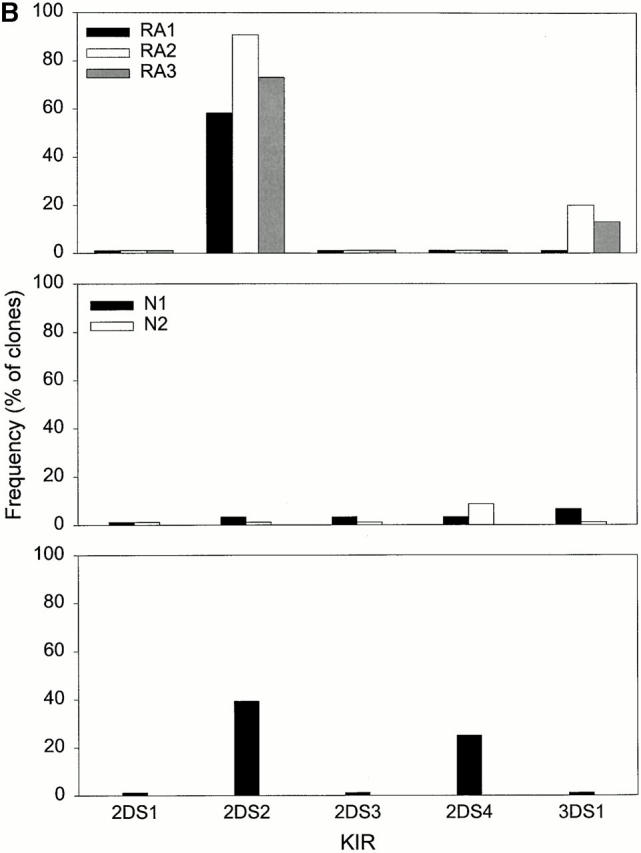
Repertoire of MHC class I–recognizing receptors expressed by CD4 T cells. CD4+CD28null T cell clones established from three patients with rheumatoid vasculitis (top panel) and three controls were characterized for the expression of inhibitory (A) and stimulatory KIR (B) isoforms by RT-PCR. Two controls were healthy individuals (middle panel), one had an acute coronary syndrome (bottom panel). The expression patterns were not random but favored the inhibitory KIR2DL4, KIR2DL2, KIR2DL3, and KIR3DL2. Expression of stimulatory receptors was highly restricted to KIR2DS2 in the patients with RA and the patient with acute coronary syndrome (*not done).
This analysis demonstrated that multiple receptors of the Ig superfamily were simultaneously expressed on CD4+CD28null T cell clones. Although the array of KIR isoforms was broad and several members could appear together, the expression of stimulatory KIR was more restricted with a clear preference for KIR2DS2. To determine whether this pattern is generally seen in CD4+ CD28null T cells or whether it is characteristic for rheumatoid vasculitis, 53 CD4+CD28null T cell clones from two healthy individuals and 28 clones from a patient with an acute coronary syndrome were analyzed (Fig. 2). CD4+CD28null T cells are rarely seen in healthy individuals, and the two control individuals were, therefore, selected to have increased frequencies. However, expansion is regularly observed in acute coronary syndromes to a similar degree as in rheumatoid vasculitis 13 14. Only one KIR2DS2+ T cell clone was found among the 53 clones derived from the two healthy controls. Of the inhibitory receptors, KIR2DL4 and KIR3DL2 remained prominent whereas the frequency of KIR2DL3 expression was slightly lower than in the patients with RA. The pattern in the patient with acute coronary syndrome resembled that in patients with RA in that KIR2DS2 was frequently encountered.
The functional significance of preferential KIR2DS2 expression should depend on the combination of this stimulatory receptor with inhibitory receptors on individual T cell clones. Because KIR2DS2, KIR2DL2, and KIR2DL3 supposedly recognize related antigens, KIR2DS2+ T cell clones were analyzed for the coexpression of KIR2DL2 and KIR2DL3. As shown in Fig. 3, KIR2DS2 was most frequently expressed exclusively (17–54%) or combined with KIR2DL3 (25–36% of the T cell clones) in the RA patients. Coexpression of the three receptor isoforms or combinations with KIR2DL2 were infrequent. These data demonstrate that in patients with rheumatoid vasculitis, KIR2DS2 was frequently unopposed by inhibitory receptors of related specificities on CD4+CD28null T cells. A similar pattern was seen in the T cell clones from the patient with an acute coronary syndrome in which 32% of all clones expressed KIR2DS2 in the absence of KIR2DL3 or KIR2DL2; in contrast, such clones were not found in the healthy donors (Fig. 3).
Figure 3.

Coexpression of MHC class I–recognizing receptors on CD4 T cell clones. KIR2DS2+ T cell clones from Fig. 2 were analyzed for the coexpression of the inhibitory KIR2DL2 and KIR2DL3 receptors, which have a highly homologous extracellular domain to KIR2DS2 and which may recognize the same HLA-C ligands. Approximately one-third of the clones in the patients with rheumatoid vasculitis or acute coronary syndrome (ACS) expressed the stimulatory KIR2DS2 receptor unopposed by any of these two inhibitory receptors whereas such clones were not found in the CD4+CD28null control T cell clones from the healthy individuals.
KIR2DS2 Is Functional As a Stimulatory Receptor on CD4+CD28null T Cells.
KIR2DS2 has a short cytoplasmic domain lacking an immunoreceptor tyrosine-based activation motif (ITAM) motif and appears to lack intrinsic signaling capability. In NK cells, KIR2DS2 is associated with a dimeric adapter molecule, DAP12, that is able to recruit the tyrosine kinases syk and ZAP70 and transmit a positive signal 31 32. To address whether KIR2DS2 is functional on CD4+CD28null T cell clones, clones that expressed KIR2DS2 in the absence of KIR2DL2 and KIR2DL3 were stimulated with a CD158b-specific antibody (GL183). Results of an experiment using IFN-γ production as the readout are shown in Fig. 4. Cross-linking of CD158b in the absence of CD3 stimulation induces the transcription of 23,153 transcripts of IFN-γ mRNA compared with the constitutive expression of 3,941 copies. If CD158b is cross-linked in the presence of suboptimal doses of anti-CD3, transcription of IFN-γ is upregulated from 6,628 copies to 63,300 copies. Enhancement of IFN-γ production by costimulation with GL183 was seen in five additional CD4+CD28null T cell clones that constitutively expressed KIR2DS2 or were transfected with KIR2DS2. Also, cross-linking with GL183 increased T cell proliferation in response to suboptimal anti-CD3 (33, and data not shown).
Figure 4.

Costimulatory function of KIR2DS2. A CD4+CD28null T cell clone that expressed KIR2DS2 but not KIR2DL2 or KIR2DL3 was unstimulated (A) or stimulated with an optimal concentration of anti-CD3 (B), suboptimal concentration of anti-CD3 (C), GL183 alone (D), a suboptimal concentration of anti-CD3 plus control Ig (E), or plus GL183 (F). Cells were harvested after 18 h and transcription of IFN-γ was assessed by RT-PCR. Results are representative of three experiments and are shown in comparison with β-actin transcripts after separation on agarose gels (top panel) or as mean ± standard deviation of IFN-γ transcripts after semiquantification on a Light Cycler.
KIR2DS2 Is a Risk Factor for Rheumatoid Vasculitis.
The preferential expression of KIR2DS2, but not other stimulatory receptors on CD4+CD28null T cells, raised the possibility that KIR2DS2 is a risk factor for developing vascular inflammation in RA. KIR genes are variably present in the Caucasian population with the majority of individuals typing positive for inhibitory KIR. With the exception of KIR2DS4, gene representation of activating receptors is highly variable; 40% of individuals have KIR2DS1 and 40% have KIR2DS2 30. The genomic presence of the KIR2DS1 and KIR2DS2 genes was evaluated in 76 healthy control individuals, 70 patients with RA, and 30 patients with unequivocal rheumatoid vasculitis manifesting as ischemic mononeuritis, ischemic ulcerations, or peripheral gangrene. Results of the genotyping are given in Table . Consistent with previous data, 45–47% of the healthy individuals carried KIR2DS1 or KIR2DS2. Patients with RA, but without major organ involvement, did not differ significantly from the control individuals for either KIR2DS1 (P = 0.563) or KIR2DS2 (P = 0.284). Of the patients with rheumatoid vasculitis, 50% were positive for KIR2DS1, similar to the representation of this gene in control individuals and unselected patients with RA. However, KIR2DS2 was found in 83% of the cohort with rheumatoid vasculitis. The odds ratios for patients with RA vasculitis having KIR2DS2 were 5.56 (P = 0.001, RA vasculitis versus control) and 7.96 (P = 0.001, RA vasculitis versus uncomplicated RA; Table ). In contrast, the odds ratio for unselected RA versus control was 0.70 (P = 0.284). These data indicated that KIR2DS2 did not affect the risk of an individual to develop RA but was strongly associated with the likelihood of vascular complications.
Table 1.
KIR2DS1 and 2DS2 Gene Frequencies Among the Study Cohorts
| Normal (n = 76) | RA (n = 70) | RA Vasculitis (n = 30) | |
|---|---|---|---|
| % | % | % | |
| KIR2DS1 | 45 | 40 | 50 |
| KIR2DS2 | 47 | 39 | 83 |
Table 2.
Odds Ratios for the Association of KIR with Disease Phenotype
| Comparison group | Reference group | Odds ratio (CI) | P | |
|---|---|---|---|---|
| KIR2DS1 | RA | Normal | 0.82 (0.42 − 1.59) | 0.563 |
| KIR2DS2 | RA | Normal | 0.70 (0.36 − 1.35) | 0.284 |
| KIR2DS1 | RA Vasculitis | Normal | 1.24 (0.53 − 2.88) | 0.624 |
| KIR2DS2 | RA Vasculitis | Normal | 5.56 (1.92 − 16.04) | 0.001 |
| KIR2DS1 | RA Vasculitis | RA | 1.50 (0.63 − 3.55) | 0.385 |
| KIR2DS2 | RA Vasculitis | RA | 7.96 (2.72 − 23.31) | 0.001 |
Distribution of HLA-C Alleles in Patients with RA and RA Vasculitis.
Receptors of the KIR2DS subfamily recognize polymorphisms on HLA-C molecules. Therefore, the functional relevance of KIR2DS receptors depends on the HLA-C genotype of the patient. To examine whether patients with rheumatoid vasculitis not only have a skewing in the genomic representation of KIR genes but also of the presumptive ligand, we HLA-C genotyped the three study cohorts (Table ). Compared with ethnically matched controls, patients without RA vasculitis had a decreased frequency of HLA-C*04 and an increased frequency of HLA-C*05. The decreased frequency of HLA-C*04 among patients with RA yielded an odds ratio of 0.39 (P = 0.024), and the enrichment of HLA-C*05 gave an odds ratio of 2.92 (P = 0.024; Table ). Among patients with RA vasculitis, the frequency of HLA-C*04 was even more diminished (odds ratio 0.21 compared with healthy controls, P = 0.033) whereas the frequency of HLA-C*05 alleles was increased fourfold (odds ratio 5.71 compared with healthy controls, P = 0.001). Two additional findings were unique for the patients with vasculitis and not observed in the patients with uncomplicated RA. The representation of HLA-C*03 was almost doubled, with 60% of patients (corresponding to an allele frequency of 33%) typing positive (odds ratio 3.74, P = 0.003). Also, 10–13% of the controls and donors with nonvasculitic RA expressed HLA-C*12 and none of the patients with vasculitis carried that allele; however, this difference was not statistically significant. Although the findings are based on a large number of comparisons, the associations of HLA-C*03 and HLA-C*05 with vasculitis (contrasted with healthy subjects) remain statistically significant, even if corrected for the 15 statistical tests for each of the HLA-C alleles using Bonferroni correction.
Table 3.
HLA-C Allele Frequencies Among the Study Cohorts
| HLA-C | Normal (n = 76) | RA (n = 70) | RA vasculitis (n = 30) |
|---|---|---|---|
| % | % | % | |
| *01 | 5.5 | 5.1 | 8.3 |
| *02 | 3.4 | 5.8 | 6.7 |
| *03 | 17.1 | 19.6 | 33.3 |
| *04 | 13.0 | 5.8 | 3.3 |
| *05 | 4.8 | 11.6 | 18.3 |
| *06 | 6.8 | 8.7 | 8.3 |
| *07 | 32.9 | 30.4 | 18.3 |
| *08 | 3.4 | 3.6 | 1.7 |
| *12 | 6.8 | 5.1 | 0 |
| *13 | 0 | 0 | 0 |
| *14 | 2.1 | 0.7 | 0 |
| *15 | 1.4 | 0 | 0 |
| *16 | 2.1 | 1.4 | 1.7 |
| *17 | 0.7 | 1.4 | 0 |
| *18 | 0 | 0.7 | 0 |
Table 4.
Odds Ratios for the Association of HLA-C with Disease Phenotype
| Comparison group | Reference group | Odds ratio (CI) | P | |
|---|---|---|---|---|
| HLA-C03 | RA | Normal | 1.13 (0.57 − 2.26) | 0.728 |
| HLA-C04 | RA | Normal | 0.39 (0.16 − 0.95) | 0.035 |
| HLA-C05 | RA | Normal | 2.92 (1.12 − 7.60) | 0.024 |
| HLA-C07 | RA | Normal | 0.95 (0.50 − 1.83) | 0.884 |
| HLA-C03 | RA vasculitis | Normal | 3.74 (1.54 − 9.08) | 0.003 |
| HLA-C04 | RA vasculitis | Normal | 0.21 (0.05 − 0.99) | 0.033 |
| HLA-C05 | RA vasculitis | Normal | 5.71 (1.95 − 16.72) | 0.001 |
| HLA-C07 | RA vasculitis | Normal | 0.45 (0.19 − 1.09) | 0.073 |
| HLA-C03 | RA vasculitis | RA | 3.31 (1.36 − 8.08) | 0.007 |
| HLA-C04 | RA vasculitis | RA | 0.55 (0.11 − 2.78) | 0.719 |
| HLA-C05 | RA vasculitis | RA | 1.95 (0.77 − 4.95) | 0.154 |
| HLA-C07 | RA vasculitis | RA | 0.47 (0.19 − 1.15) | 0.096 |
Because HLA-C*03 and KIR2DS2 were both more frequent among the patients with vasculitis than the patients with RA without vasculitis, the independent effect of these two genes was evaluated by modeling their risk with logistic regression. After entering both genes into the model, each demonstrated an independent significant risk associated with vasculitis (odds ratio for HLA-C*03 = 8.6, P < 0.001; odds ratio for KIR2DS2 = 3.69, P = 0.01), suggesting that both contribute a risk to vasculitis, independent of each other. We also evaluated whether there was a significant interaction between these two genes by entering an interaction term into the logistic regression model, but this was found not to be statistically significant (P = 0.483). However, the limited sample size makes it difficult to rule out the possibility of interactions.
Discussion
This study demonstrates that a genetic polymorphism within the family of MHC class I–recognizing receptors is important in vascular complications of RA. KIR genes encode for glycoprotein receptors that bind polymorphic HLA class I ligands and regulate the activation of NK cells and a subset of T cells. The diversity in this gene family is introduced by a minimum of 12 independent loci that are not universally functional. The diversity is further increased by alternative RNA splicing and allelic polymorphisms 17 18 29. This, in conjunction with receptor–ligand interactions regulating NK cells and selected T cells, raised the possibility that this family includes candidate genes that confer susceptibility to developing chronic inflammatory diseases. Previous data have shown that inhibitory KIR genes are expressed in the vast majority of individuals and that the genomic representation of stimulatory receptors is diverse 30. This study focused on KIR2DS2 and was guided by the finding that this gene was preferentially expressed by CD4+CD28null T cell clones isolated from patients with active vasculitis (Fig. 1 and Fig. 2). Data presented demonstrate that the KIR2DS2 gene significantly increases the risk of patients with RA to develop vasculitic complications. Also, the distribution of HLA-C alleles, which represent the presumptive ligand of KIR2DS2, was significantly different in patients with rheumatoid vasculitis, adding further support for the hypothesis that this receptor–ligand pair is involved in the vascular injury occurring in a subset of patients with RA.
The biological significance of KIR receptors in vivo depends on whether these receptors are expressed in individuals that constitutively express the appropriate ligands. KIR2D receptors predominantly recognize HLA-C molecules and are able to distinguish between subtle allelic polymorphisms 34 35. Based on their extracellular domains, the KIR2D receptors have been subsetted into two groups. Receptors recognized by the antibody GL183 (anti-CD158b) include the inhibitory receptors, KIR2DL2 and KIR2DL3, and the stimulatory receptor KIR2DS2. These receptors recognize HLA-C*01, *03, *07, and *08, all of which share a serine at position 77 and an asparagine at position 80 (S77, N80). In contrast, CD158a receptors, including KIR2DL1 and KIR2DS1, are specific for HLA-C alleles that have asparagine at residue 77 and lysine at position 80 (N77, K80).
HLA-C genotyping demonstrated a significant difference in the allelic distribution between patients with RA and ethnically matched healthy controls. HLA-C*05 was enriched and HLA-C*04 was decreased in patients with uncomplicated RA and in patients with rheumatoid vasculitis. In contrast, HLA-C*03 was exclusively enriched in the patients with rheumatoid vasculitis. We excluded linkage disequilibrium with HLA-DRB1*04 as the reason for the enrichment of HLA-C*03 (data not shown). Enrichment of CD158b is consistent with the hypothesis that the KIR2DS2–HLA-C receptor–ligand pair predisposes for rheumatoid vasculitis. The decrease in HLA-C*04 donors in the cohort with rheumatoid vasculitis is consistent with this interpretation. However, the distribution of the remaining HLA-C alleles does not follow the pattern that S77 N80 HLA-C alleles are overrepresented. In particular, the frequency of HLA-C*05, which expresses N77 K80, was increased in patients with RA and also patients with rheumatoid vasculitis. Recent binding studies using soluble KIR receptors and HLA-C molecules have demonstrated that the correlation between KIR specificity and HLA-C polymorphisms is not as strict as initially thought. Winter et al. have shown that the KIR2DL2, a receptor with an extracellular domain very homologous to KIR2DS2, binds to the N77 K80 HLA-C*1503 allele with the same affinity as to HLA-C*01 and HLA-C*03 36. Also, stimulatory receptors, including KIR2DS2, had very low affinity for their presumptive HLA-C ligand, a finding that was attributed to a substitution of tyrosine for phenylalanine at position 45 36 37. We have sequenced KIR2DS2 genes from RA vasculitis patients and have found no allelic polymorphisms, excluding that a sequence variation could influence the specificity profile (data not shown).
Alternatively, peptides bound in the groove of HLA-C alleles could modify the recognition pattern of KIR receptors. This has been elegantly shown for the CD94/NKG2 family of NK receptors that recognize the HLA-E molecule 38. HLA-E binds peptides derived from the signal sequences of certain HLA molecules that have limited sequence polymorphism 39. These polymorphisms influence the affinity of receptor–HLA-E interactions and the functional activity of the NK cell. In analogy, recognition of HLA-C molecules by KIR may also be influenced by the composition of the peptides bound in the MHC groove. Several reports have indicated that amino acid positions 7 and 8 of the peptide are important and could influence the binding of KIR 40 41. It has been argued that variability in the peptides bound is not important for the biological function of inhibitory receptor because most peptides are permissive for recognition. Triggering of the inhibitory KIR would occur even if few nonpermissive peptides are bound 17. However, the concept is attractive for stimulatory KIRs that recognize the dominant HLA-C self-peptide complexes with very low affinity but may bind selected HLA-C peptide complexes with higher affinities.
KIR2DS2 is expressed on NK cells and on some CD4 and CD8 T cells. The association of KIR2DS2 with rheumatoid vasculitis could reflect a functional activity of any of these cell populations that is important in the pathogenetic events inducing vascular damage. However, there is circumstantial evidence to implicate CD4+CD28null T cells. This population of cells is distinctly infrequent in the majority of healthy individuals, but is expanded in a subset of patients with RA 11. Patients with increased frequencies of CD4+CD28null T cells have increased risk of developing extraarticular manifestations, including subcutaneous nodule formation and rheumatoid vasculitis 12. Nodule formation is seen in about 30% of patients with RA and represents a granulomatous reaction after a vascular injury. Rheumatoid vasculitis is a serious condition involving inflammation of mid-size and large arteries. Expanded CD4+CD28null T cells have also been associated with other inflammatory vasculopathies such as Wegener's granulomatosis and acute coronary syndromes 13 14 42. An acute coronary syndrome evolves if inflammation of an atherosclerotic plaque leads to plaque rupture, subsequent thrombosis, and myocardial ischemia 43 44. Patients with increased frequencies of CD4+CD28null T cells and coronary artery disease have an increased risk of developing acute coronary syndromes. In these patients, clonally expanded CD4+CD28null T cells can be demonstrated in the ruptured plaque, suggesting that these cells drive the plaque inflammation that leads to rupture.
CD4+CD28null but not CD4+CD28+ T cells express KIR; it was the preferential expression of the CD158b molecule on these cells and the frequent finding of KIR2DS2+CD4+CD28null T cell clones in rheumatoid vasculitis that led to this study design. The three patients with rheumatoid vasculitis expressed KIR2DS2 on 58–91% of their T cell clones. In contrast, KIR2DS2 expression was very infrequent on CD4 T cell clones from two normal individuals (0 and 3%), although both of them were selected to carry the KIR2DS2 gene and to have an expanded CD4+CD28null T cell compartment comparable to the patients with rheumatoid vasculitis. Of interest, the third selected control patient had an acute coronary event, a disease that has also been correlated with an expansion of CD4+CD28null T cells 13. T cell clones from this patient exhibited a very similar KIR expression pattern as the patients with RA, suggesting that stimulatory KIRs may not only be risk factors for rheumatoid vasculitis but also for other vascular diseases that are associated with increased CD4+CD28null T cell frequencies.
How could KIR2DS2-expressing cells be involved in the disease process? The function of stimulatory KIRs is unclear. In particular, these receptors are usually expressed in the context of several inhibitory receptors. In addition, it has been debated whether these receptors are at all functional in T cells that may lack the necessary adapter molecules. For the CD4+CD28null T cell clones described here, we were able to demonstrate a costimulatory activity of CD158b-specific antibodies to enhance proliferation and cytokine production (33; Fig. 4), confirming earlier reports that KIR2DS2 can function as a costimulatory molecule in response to suboptimal TCR triggering 45 46. Further evidence for a unique role of KIR+CD4+ T cells comes from the observation that in the patients with rheumatoid vasculitis and in the patient with an acute coronary syndrome, CD4+CD28null T cells expressed KIR2DS2 in the absence of opposing inhibitory receptors of the same specificity. Thus, in CD4+CD28null T cells, KIR may be important in the regulation of peripheral tolerance, and the activity of KIR2DS2 may favor the activation of autoreactive T cells. It can easily be envisioned that such a mechanism is important in autoimmune inflammation. However, KIR2DS2 is not a risk factor for the synovial inflammation of RA, suggesting that this putative costimulatory function is not critical for synovitis. The association of KIR2DS2 with vascular complications implicates such a costimulatory activity in the interaction between endothelial cells and immune cells. This may be of particular relevance for CD4+CD28null T cells, which have been shown to be autoreactive and are no longer under the control of the CD28–CD80/CD86 interaction 11.
In summary, we have provided evidence for the model that genetic risk factors determine variations in clinical phenotypes of an autoimmune disease such as RA. Our data are the first to implicate the recently described polymorphisms in MHC class I–recognizing receptors in the pathogenesis of a chronic inflammatory disease. Identifying and understanding risk factors for extraarticular disease in RA is of clinical relevance because of the high morbidity and increased mortality associated with this complication. However, the findings may have implications beyond RA. CD4+CD28null T cells carrying MHC class I–recognizing receptors contribute to vascular inflammation in an array of diseases ranging from Wegener's granulomatosis and rheumatoid vasculitis to life-threatening acute ischemia in coronary artery disease. It remains to be seen whether the ability of CD4+CD28null T cells to inflict endothelial injury and to cause vascular damage is generally linked to genetic polymorphisms of the KIR family and their MHC class I ligands.
Acknowledgments
The authors thank Tammy J. Dahl and James W. Fulbright for manuscript and figure preparation.
Supported in part by grants from the National Institutes of Health (R01 AR42527 and R01 AR41974) and by the Mayo Foundation.
Footnotes
Abbreviations used in this paper: KIR, killer cell Ig–like receptor; RA, rheumatoid arthritis; RT, reverse transcription.
References
- Ollier W., Winchester R. The Germline and somatic genetic basis for rheumatoid arthritis. In: Theofilopoulos A.N., editor. Current Directions in Autoimmunity. S. Karger AG; Basel, Switzerland: 1999. pp. 166–193. [DOI] [PubMed] [Google Scholar]
- Seldin M.F., Amos C.I., Ward R., Gregersen P.K. The genetics revolution and the assault on rheumatoid arthritis. Arthritis Rheum. 1999;42:1071–1079. doi: 10.1002/1529-0131(199906)42:6<1071::AID-ANR1>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Weyand C.M., Goronzy J.J. Pathogenesis of rheumatoid arthritis. Med. Clin. N. Am. 1997;81:29–55. doi: 10.1016/s0025-7125(05)70504-6. [DOI] [PubMed] [Google Scholar]
- Nepom G.T. Major histocompatibility complex-directed susceptibility to rheumatoid arthritis. Adv. Immunol. 1998;68:315–332. doi: 10.1016/s0065-2776(08)60563-5. [DOI] [PubMed] [Google Scholar]
- Weyand C.M., Klimiuk P.A., Goronzy J.J. Heterogeneity of rheumatoid arthritisfrom phenotypes to genotypes. Springer Semin. Immunopathol. 1998;20:5–22. doi: 10.1007/BF00831996. [DOI] [PubMed] [Google Scholar]
- Wordsworth P., Pile K.D., Buckely J.D., Lanchbury J.S., Ollier B., Lathrop M., Bell J.I. HLA heterozygosity contributes to susceptibility to rheumatoid arthritis. Am. J. Hum. Genet. 1992;51:585–591. [PMC free article] [PubMed] [Google Scholar]
- Weyand C.M., Xie C., Goronzy J.J. Homozygosity for the HLA-DRB1 allele selects for extraarticular manifestations in rheumatoid arthritis. J. Clin. Invest. 1992;89:2033–2039. doi: 10.1172/JCI115814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyand C.M., Schmidt D., Wagner U., Goronzy J.J. The influence of sex on the phenotype of rheumatoid arthritis. Arthritis Rheum. 1998;41:817–822. doi: 10.1002/1529-0131(199805)41:5<817::AID-ART7>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Cornelis F., Faure S., Martinez M., Prud'homme J.F., Fritz P., Dib C., Alves H., Barrera P., de Vries N., Balsa A. New susceptibility locus for rheumatoid arthritis suggested by a genome-wide linkage study. Proc. Natl. Acad. Sci. USA. 1998;95:10746–10750. doi: 10.1073/pnas.95.18.10746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelis F. Susceptibility genes in RA. In: Theofilopoulos A.N., editor. Current Directions in Autoimmunity. S. Karger AG; Basel, Switzerland: 2000. pp. 1–16. [DOI] [PubMed] [Google Scholar]
- Schmidt D., Goronzy J.J., Weyand C.M. CD4+CD7−CD28− T cells are expanded in rheumatoid arthritis and are characterized by autoreactivity. J. Clin. Invest. 1996;97:2027–2037. doi: 10.1172/JCI118638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens P.B., Goronzy J.J., Schaid D., Weyand C.M. Expansion of unusual CD4+ T cells in severe rheumatoid arthritis. Arthritis Rheum. 1997;40:1106–1114. doi: 10.1002/art.1780400615. [DOI] [PubMed] [Google Scholar]
- Liuzzo G., Kopecky S.L., Frye R.L., O'Fallon W.M., Maseri A., Goronzy J.J., Weyand C.M. Perturbation of the T-cell repertoire in patients with unstable angina. Circulation. 1999;100:2135–2139. doi: 10.1161/01.cir.100.21.2135. [DOI] [PubMed] [Google Scholar]
- Liuzzo G., Goronzy J.J., Yang H., Kopecky S.L., Holmes D.R., Frye R.L., Weyand C.M. Monoclonal T-cell proliferation and plaque instability in acute coronary syndromes. Circulation. 2000;101:2883–2888. doi: 10.1161/01.cir.101.25.2883. [DOI] [PubMed] [Google Scholar]
- Weyand C.M., Brandes J.C., Schmidt D., Fulbright J.W., Goronzy J.J. Functional properties of CD4+ CD28− T cells in the aging immune system. Mech. Ageing Dev. 1998;102:131–147. doi: 10.1016/s0047-6374(97)00161-9. [DOI] [PubMed] [Google Scholar]
- Namekawa T., Wagner U.G., Goronzy J.J., Weyand C.M. Functional subsets of CD4 T cells in rheumatoid synovitis. Arthritis Rheum. 1998;41:2108–2116. doi: 10.1002/1529-0131(199812)41:12<2108::AID-ART5>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Lanier L.L. NK cell receptors. Annu. Rev. Immunol. 1998;16:359–393. doi: 10.1146/annurev.immunol.16.1.359. [DOI] [PubMed] [Google Scholar]
- Long E.O. Regulation of immune responses through inhibitory receptors. Annu. Rev. Immunol. 1999;17:875–904. doi: 10.1146/annurev.immunol.17.1.875. [DOI] [PubMed] [Google Scholar]
- Borrego F., Ulbrecht M., Weiss E.H., Coligan J.E., Brooks A.G. Recognition of human histocompatibility leukocyte antigen (HLA)-E complexed with HLA class I signal sequence–derived peptides by CD94/NKG2 confers protection from natural killer cell–mediated lysis. J. Exp. Med. 1998;187:813–818. doi: 10.1084/jem.187.5.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee N., Llano M., Carretero M., Ishitani A., Navarro F., Lopez-Botet M., Geraghty D.E. HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc. Natl. Acad. Sci. USA. 1998;95:5199–5204. doi: 10.1073/pnas.95.9.5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna M., Samaridis J. Cloning of immunoglobulin-superfamily members associated with HLA-C and HLA-B recognition by human natural killer cells. Science. 1995;268:405–408. doi: 10.1126/science.7716543. [DOI] [PubMed] [Google Scholar]
- Wagtmann N., Biassoni R., Cantoni C., Verdiani S., Malnati M.S., Vitale M., Bottino C., Moretta L., Moretta A., Long E.O. Molecular clones of the p58 NK cell receptor reveal immunoglobulin-related molecules with diversity in both the extra- and intracellular domains. Immunity. 1995;2:439–449. doi: 10.1016/1074-7613(95)90025-x. [DOI] [PubMed] [Google Scholar]
- D'Andrea A., Chang C., Franz-Bacon K., McClanahan T., Phillips J.H., Lanier L.L. Molecular cloning of NKB1. A natural killer cell receptor for HLA-B allotypes. J. Immunol. 1995;155:2306–2310. [PubMed] [Google Scholar]
- Winter C.C., Long E.O. A single amino acid in the p58 killer cell inhibitory receptor controls the ability of natural killer cells to discriminate between the two groups of HLA-C allotypes. J. Immunol. 1997;158:4026–4028. [PubMed] [Google Scholar]
- Wagtmann N., Rajagopalan S., Winter C.C., Peruzzi M., Long E.O. Killer cell inhibitory receptors specific for HLA-C and HLA-B identified by direct binding and by functional transfer. Immunity. 1995;3:801–809. doi: 10.1016/1074-7613(95)90069-1. [DOI] [PubMed] [Google Scholar]
- Biassoni R., Cantoni C., Falco M., Verdiani S., Bottino C., Vitale M., Conte R., Poggi A., Moretta A., Moretta L. The human leukocyte antigen (HLA)-C-specific “activatory” or “inhibitory” natural killer cell receptors display highly homologous extracellular domains but differ in their transmembrane and intracytoplasmic portions. J. Exp. Med. 1996;183:645–650. doi: 10.1084/jem.183.2.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretta A., Sivori S., Vitale M., Pende D., Morelli L., Augugliaro R., Bottino C., Moretta L. Existence of both inhibitory (p58) and activatory (p50) receptors for HLA-C molecules in human natural killer cells. J. Exp. Med. 1995;182:875–884. doi: 10.1084/jem.182.3.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvakumar A., Steffens U., Palanisamy N., Chaganti R.S., Dupont B. Genomic organization and allelic polymorphism of the human killer cell inhibitory receptor gene KIR103. Tissue Antigens. 1997;49:564–573. doi: 10.1111/j.1399-0039.1997.tb02803.x. [DOI] [PubMed] [Google Scholar]
- Steffens U., Vyas Y., Dupont B., Selvakumar A. Nucleotide and amino acid sequence alignment for human killer cell inhibitory receptors (KIR), 1998. Tissue Antigens. 1998;51:398–413. doi: 10.1111/j.1399-0039.1998.tb02981.x. [DOI] [PubMed] [Google Scholar]
- Uhrberg M., Valiante N.M., Shum B.P., Shilling H.G., Lienert-Weidenbach K., Corliss B., Tyan D., Lanier L.L., Parham P. Human diversity in killer cell inhibitory receptor genes. Immunity. 1997;7:753–763. doi: 10.1016/s1074-7613(00)80394-5. [DOI] [PubMed] [Google Scholar]
- Lanier L.L., Corliss B.C., Wu J., Leong C., Phillips J.H. Immunoreceptor DAP12 bearing a tyrosine-based activation motif is involved in activating NK cells. Nature. 1998;391:703–707. doi: 10.1038/35642. [DOI] [PubMed] [Google Scholar]
- McVicar D.W., Taylor L.S., Gosselin P., Willette-Brown J., Mikhael A.I., Geahlen R.L., Nakamura M.C., Linnemeyer P., Seaman W.E., Anderson S.K. DAP12-mediated signal transduction in natural killer cells. A dominant role for the Syk protein-tyrosine kinase. J. Biol. Chem. 1998;273:32934–32942. doi: 10.1074/jbc.273.49.32934. [DOI] [PubMed] [Google Scholar]
- Namekawa T., Snyder M.R., Yen J.H., Goehring B.E., Leibson P.J., Weyand C.M., Goronzy J.J. Killer cell activating receptors function as costimulatory molecules on CD4+CD28null T cells clonally expanded in rheumatoid arthritis. J. Immunol. 2000;165:1138–1145. doi: 10.4049/jimmunol.165.2.1138. [DOI] [PubMed] [Google Scholar]
- Colonna M., Borsellino G., Falco M., Ferrara G.B., Strominger J.L. HLA-C is the inhibitory ligand that determines dominant resistance to lysis by NK1- and NK2-specific natural killer cells. Proc. Natl. Acad. Sci. USA. 1993;90:12000–12004. doi: 10.1073/pnas.90.24.12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna M., Spies T., Strominger J.L., Ciccone E., Moretta A., Moretta L., Pende D., Viale O. Alloantigen recognition by two human natural killer cell clones is associated with HLA-C or a closely linked gene. Proc. Natl. Acad. Sci. USA. 1992;89:7983–7985. doi: 10.1073/pnas.89.17.7983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter C.C., Gumperz J.E., Parham P., Long E.O., Wagtmann N. Direct binding and functional transfer of NK cell inhibitory receptors reveal novel patterns of HLA-C allotype recognition. J. Immunol. 1998;161:571–577. [PubMed] [Google Scholar]
- Vales-Gomez M., Reyburn H.T., Erskine R.A., Strominger J. Differential binding to HLA-C of p50-activating and p58-inhibitory natural killer cell receptors. Proc. Natl. Acad. Sci. USA. 1998;95:14326–14331. doi: 10.1073/pnas.95.24.14326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vales-Gomez M., Reyburn H.T., Erskine R.A., Lopez-Botet M., Strominger J.L. Kinetics and peptide dependency of the binding of the inhibitory NK receptor CD94/NKG2-A and the activating receptor CD94/NKG2-C to HLA-E. EMBO J. 1999;18:4250–4260. doi: 10.1093/emboj/18.15.4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier L.L. Follow the leaderNK cell receptors for classical and nonclassical MHC class I. Cell. 1998;92:705–707. doi: 10.1016/s0092-8674(00)81398-7. [DOI] [PubMed] [Google Scholar]
- Mandelboim O., Wilson S.B., Vales-Gomez M., Reyburn H.T., Strominger J.L. Self and viral peptides can initiate lysis by autologous natural killer cells. Proc. Natl. Acad. Sci. USA. 1997;94:4604–4609. doi: 10.1073/pnas.94.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peruzzi M., Parker K.C., Long E.O., Malnati M.S. Peptide sequence requirements for the recognition of HLA-B*2705 by specific natural killer cells. J. Immunol. 1996;157:3350–3356. [PubMed] [Google Scholar]
- Moosig F., Csernok E., Wang G., Gross W.L. Costimulatory molecules in Wegener's granulomatosis (WG)lack of expression of CD28 and preferential up-regulation of its ligands B7-1 (CD80) and B7-2 (CD86) on T cells. Clin. Exp. Immunol. 1998;114:113–118. doi: 10.1046/j.1365-2249.1998.00695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross R. Atherosclerosis is an inflammatory disease. Am. Heart J. 1999;138:S419–S420. doi: 10.1016/s0002-8703(99)70266-8. [DOI] [PubMed] [Google Scholar]
- Shah P.K. Plaque disruption and thrombosis. Potential role of inflammation and infection. Cardiol. Clin. 1999;17:271–281. doi: 10.1016/s0733-8651(05)70074-6. [DOI] [PubMed] [Google Scholar]
- Mandelboim O., Davis D.M., Reyburn H.T., Vales-Gomez M., Sheu E.G., Pazmany L., Strominger J.L. Enhancement of class II-restricted T cell responses by costimulatory NK receptors for class I MHC proteins. Science. 1996;274:2097–2100. doi: 10.1126/science.274.5295.2097. [DOI] [PubMed] [Google Scholar]
- Mandelboim O., Kent S., Davis D.M., Wilson S.B., Okazaki T., Jackson R., Hafler D., Strominger J.L. Natural killer activating receptors trigger interferon gamma secretion from T cells and natural killer cells. Proc. Natl. Acad. Sci. USA. 1998;95:3798–3803. doi: 10.1073/pnas.95.7.3798. [DOI] [PMC free article] [PubMed] [Google Scholar]