

Abstract
Epidermal Langerhans cells (LCs) play a key role in immune defense mechanisms and in numerous immunological disorders. In this report, we show that percutaneous infection of C57BL/6 mice with the helminth parasite Schistosoma mansoni leads to the activation of LCs but, surprisingly, to their retention in the epidermis. Moreover, using an experimental model of LC migration induced by tumor necrosis factor (TNF)-α, we show that parasites transiently impair the departure of LCs from the epidermis and their subsequent accumulation as dendritic cells in the draining lymph nodes. The inhibitory effect is mediated by soluble lipophilic factors released by the parasites and not by host-derived antiinflammatory cytokines, such as interleukin-10. We find that prostaglandin (PG)D2, but not the other major eicosanoids produced by the parasites, specifically impedes the TNF-α–triggered migration of LCs through the adenylate cyclase–coupled PGD2 receptor (DP receptor). Moreover, the potent DP receptor antagonist BW A868C restores LC migration in infected mice. Finally, in a model of contact allergen-induced LC migration, we show that activation of the DP receptor not only inhibits LC emigration but also dramatically reduces the contact hypersensitivity responses after challenge. Taken together, we propose that the inhibition of LC migration could represent an additional stratagem for the schistosomes to escape the host immune system and that PGD2 may play a key role in the control of cutaneous immune responses.
Keywords: dendritic cells, migration, Schistosoma, eicosanoids, cAMP
Introduction
Dendritic cells (DCs) are professional APCs that initiate primary immune responses in lymphoid tissues 1. Among them, epidermal Langerhans cells (LCs) play a key role in the establishment of cutaneous immunity. Under normal, noninflammatory conditions, LCs reside in the epidermis anchored to neighboring keratinocytes (KCs) via homotypic E-cadherin interactions 2. In this environment, they display an immature phenotype characterized by high antigen uptake and processing abilities and poor T cell stimulatory function. However, in response to stimulation occurring during infection or topical application of allergens, LCs activate, and a proportion of them migrates via afferent lymphatics to regional LNs where they accumulate as immunostimulatory DCs 1 3. Similarly, emigration of LCs from the epidermis may be initiated via an antigen-independent manner, for instance by skin irritants, ultraviolet irradiation, or microbial CpG motifs 4 5. During their migration, LCs undergo a complex process of maturation, becoming less effective in capturing and processing antigens but more specialized in stimulating naive T lymphocytes. This latter property is partly mediated by an increased expression of MHC class I and class II and costimulatory molecules including intracellular adhesion molecule (ICAM)-1 (CD54), CD40, B7-1 (CD80), and B7-2 (CD86) 6.
The molecular mechanisms that govern LC migration have been the purpose of extensive research in the past few years. Accumulating evidence suggests that the synthesis of inflammatory cytokines, particularly TNF-α and IL-1β, is one of the first events in the multistep cascade leading to LC departure from the epidermis 7 8. These cytokines are respectively produced by KCs and LCs in response to skin-penetrating pathogens or to contact allergens, and affect the interactions between KCs and LCs by diminishing the expression of E-cadherin 9 and by stimulating actin-dependent movements of LCs 10. Other adhesion molecules such as ICAM-1, very late antigen (VLA)-6 (CD49f), CD40, and CD44 also play a role in the migratory properties of LCs. In addition, the importance of the seven transmembrane–spanning G protein–coupled receptor family in driving LC motility has also been reported 11 12 13 14. Among them, the CC chemokine receptor (CCR)7 is sharply upregulated during LC maturation and is crucial to attracting LCs into the LNs 11 12. On the other hand, LC emigration is associated with a rapid decrease in the expression of receptors for inflammatory chemokines such as CCR1 13.
Mechanisms controlling the emigration of epidermal LCs after activation have been reported. In this phenomenon, the antiinflammatory cytokine IL-1ra has been shown to block the binding of IL-1β to its receptor 15. Similarly, IL-4 and IL-10 may act as negative regulators of LC migration. Takayama et al. 16 recently showed that IL-4 interferes with the TNF-α–induced mobilization of LCs by downregulating the expression of TNFR-II on LCs. In a model of contact allergen-induced LC migration, Wang et al. 17 suggested that IL-10 impedes LC emigration, at least in part, by downregulating the synthesis of IL-1β and TNF-α by epidermal cells. Although other components may also be involved 18 19, these studies suggest that LC motility is tightly controlled by the homeostatic balance between pro- and antiinflammatory cytokines produced early in the skin, and that this balance quantitatively and qualitatively affects the resulting adaptive immune response 20. During cutaneous infections, skin-penetrating pathogens may directly or indirectly influence such events. For instance, the intracellular parasite Leishmania major favors the rapid production of inflammatory cytokines in the skin and provokes LC migration to the skin-draining LNs (SLNs; reference 21).
In this report, we have analyzed the effects of the helminth parasite Schistosoma mansoni, the causative agent of schistosomiasis, on the activation state and migratory abilities of LCs. Indeed recent demonstrations that schistosomes interfere with some inflammatory pathways in host cells 22 23 24 and that certain pathogens, particularly viruses and intracellular parasites, can profoundly alter the functions of DCs 25 26 27 28 29, prompted us to initiate this study. Schistosoma mansoni has a complex migratory route within its vertebrate host that is initiated by the penetration of the larvae (termed cercariae) through the skin. In the cutaneous environment, transformation of cercaria into schistosomulum is accompanied by the release of a wide range of proteases and fatty acid derivatives which facilitate parasite migration through the skin 30. Additionally, parasite larvae closely interact with cutaneous immunocompetent cells, while remaining in the skin for 3 to 4 d. The nature and immunological consequences of these interactions have not yet been fully studied. Here, we show that, after murine infection, schistosomula activate LCs but, surprisingly, impede their migration to the SLNs. This inhibitory effect, which also occurs in a TNF-α–induced model of LC migration, is mediated by excreted/secreted (ES) lipophilic factors produced by parasite larvae, particularly by PGD2. We speculate that schistosomes may utilize this stratagem to limit and/or orientate the host immune response. We also propose a new function for PGD2 in skin homeostasis and in the regulation of the cutaneous immune response.
Materials and Methods
Reagents and Abs.
All reagents were purchased from Sigma-Aldrich unless otherwise notified. PGD2, PGE2, PGF2α, 5-hydroxyeicosatetranoic acid (HETE), 15-HETE, leukotriene (LT)B4, LTC4, and BW245C were from Cayman Chemical. BW A868C was donated by Dr. S. Lister (Glaxo Wellcome, Greenford, UK). The anti-I-Ad/I-Ed mAbs (clone M5/114, rat IgG2b) and the anti-DEC-205 (NLDC-145, rat IgG2a) were provided by Drs. A. Ager (National Institute for Medical Research, London, UK) and D. Sacks (National Institutes of Health, Bethesda, MD), respectively. The FITC-conjugated anti-CD80 (hamster IgG), anti-CD86 (rat IgG2a), and biotin-conjugated anti-CD11c (hamster IgG) mAbs were purchased from BD PharMingen. The following were used as secondary Abs: biotin-conjugated anti–rat and anti-FITC peroxidase-conjugated (Boehringer). The biotinylated reagents were detected using ABC complex horseradish peroxidase (HRP; Dako). The neutralizing anti–IL-10 mAb (clone JES052A5, rat IgG1) was from R&D Systems and the isotype control mAb from Caltag Laboratories.
Cell Lines.
The LC line XS52 has been established from mouse epidermis and presents the phenotypic and functional features of LC 31. XS52 was cultured in RPMI containing 10% (vol/vol) heat-inactivated FCS in the presence of 2 ng/ml GM-CSF (Biosource International) and 10% (vol/vol) NS47 fibroblast supernatant as described 32. Mouse Pam212 KCs were cultured in Eagle's MEM complemented with 10% FCS and 0.05 mM CaCl2 31.
Mice, Parasites and Infection Protocols.
Young adult wild-type (WT) and IL-10–deficient (knockout [KO]) C57BL/6 mice (6- to 8-wk old) were purchased from Iffa-Credo. The S. mansoni (Puerto Rican strain) life cycle was maintained in Biomphalaria glabrata snails as the intermediate host and the hamster Mesocricetus auratus as the definitive host. Skin schistosomula and schistosomula ES products (SESP; the supernatant of a 4-h culture containing 103 parasites/ml) were prepared as described 23 24. The methanol/chloroform-extracted fraction from the SESP (termed the lipophilic fraction) was obtained by a modified Folch extraction protocol 24. The organic phase was dried under a stream of nitrogen and resuspended in DMSO (for biological studies) or methanol (for HPLC analysis) (50 μl/50 ml parasite culture). For S. mansoni infection, mice were anesthetized with pentobarbital (30 mg/kg; Sanofi) and exposed to 250 cercariae by immersion of the ears for 25 min.
Identification and Quantification of Eicosanoids.
Eicosanoids recovered from the schistosomula culture medium were extracted as described above and analyzed by HPLC on a 3.9 × 150 mm Novapack C-18 reverse phase column (Waters). Elution was carried out at a rate of 0.5 ml/min with acetonitrile/water (40:60 vol/vol) plus 0.01% (vol/vol) trifluoroacetic acid. Peak elution was monitored at 195 nm for PGs, 230 nm for conjugated dienes, and 270 nm for LTs. Identification and quantification of various HPLC peaks were performed by injecting known quantities of eicosanoid standards. Enzyme immunoassay (EIA) was also used to quantify PGF2α, PGE2, PGD2, LTB4, and LTC4 directly from the parasite culture supernatant with kits provided by Cayman. Results in Table represent the concentration of individual eicosanoid detected per ml of culture (103 parasites/ml).
Table 2.
Schistosomula Eicosanoid Production as Determined by HPLC and/or EIA
| Eicosanoid | Retention time | Wavelength | Concentration | Concentration | ||
|---|---|---|---|---|---|---|
| min | nm | pg/ml | μm | pg/ml | μm | |
| PGF2α | 4.32 | 195 | 2,359 | 6.53 | 1,841 | 5.09 |
| PGE2 | 5.68 | 195 | 2,273 | 6.43 | 2,589 | 7.32 |
| PGD2 | 6.57 | 195 | 691 | 1.96 | 752 | 2.13 |
| 15-HETE | 98 | 230 | 263 | 0.82 | nd | nd |
| 5-HETE | 150 | 230 | 218 | 0.68 | nd | nd |
| LTB4 | 26 | 270 | nd | nd | 18 | 0.05 |
| LTC4 | 66 | 270 | nd | nd | 14 | 0.04 |
Shown is one representative experiment out of four. nd, not determined.
Cytokine and Ab Administration.
Recombinant murine TNF-α (specific activity ≥ 5 × 107 U/mg) (R&D Systems) was reconstituted in sterile PBS containing 0.1% (wt/vol) BSA as a carrier protein. Mice were intradermally injected with 50 ng TNF-α (30 μl) into both ear pinnae with 27 3/4-gauge stainless steel needles. Epidermal sheets were analyzed 1 h after injection, a time previously shown to be optimal for TNF-α–induced LC emigration 7. Treatment of mice with Abs was as follows: 1 h before infection, C57BL/6 mice were injected intradermally with 40 μg of neutralizing anti–IL-10 or isotype-matched control mAb diluted in sterile PBS (final volume: 30 μl).
Preparation and Analysis of Epidermal Sheets.
The epidermis was separated from the dermis by means of ammoniumthiocyanate as described previously 33. Epidermal sheets were fixed in paraformaldehyde (PFA; 2% in PBS) for 10 min at room temperature, and washed three times with PBS. For immunohistochemical staining, sheets were placed for 15 min in 3% H2O2 to inhibit endogenous peroxidase, washed three times with PBS, and incubated for 30 min with PBS plus 1% (wt/vol) blocking reagent (Boehringer). Epidermal sheets were then incubated with primary Abs for 90 min and washed in PBS before adding either biotinylated conjugated goat anti–rat Ig or peroxidase-conjugated anti-FITC for an additional 30 min. In the final step, sheets were developed with 3-amino 9-ethyl carbazol, washed three times in PBS, and mounted onto glass slides in Immumount (Shandon) for immunohistochemical analysis. LCs were enumerated by counting MHC class II–positive cells. Epidermal sheets were prepared from each experimental group and for each sheet 10 random fields were examined. Cell frequency was converted to LC/mm2 and results were expressed as mean ± SD. The statistical significance of differences between experimental groups was calculated using the Student's t test.
Skin Explant Assay.
After treatment, ears were rinsed in 70% ethanol and split into dorsal and ventral halves with forceps 34. Four sheets were floated, dermal side down, on 4 ml RPMI supplemented with 25 mM Hepes, 10% FCS, and gentamycin (50 μg/ml). After 24 h incubation at 37°C in a 5% CO2 incubator, epidermal sheets were prepared and LCs were enumerated as described above.
Immunochemical Analysis of SLNs.
SLNs were removed 18 h after TNF-α treatment and fixed in a formaldehyde-free zinc fixative (ImmunoHistoFix; Interstiles sprl) for 7 d at 4°C. After dehydration in graded alcohol baths, embedding was performed by three successive immersions in ImmunoHistoWax (Interstiles sprl) at 37°C. Sections of 5 μm thickness were dewaxed in acetone for 5 min and immunostained with anti-CD11c Ab as described above. For immunohistochemical analysis, sections were counterstained with hematoxylin and mounted in Immumount.
Induction and Elicitation of Contact Hypersensitivity Responses.
Mice were sensitized by painting 10 μl of a 0.5% solution of FITC prepared in acetone/dibutylphtalate (1:1, vol/vol; vehicle) on the total surface of the left ear. 30 μl of BW245 (100 nM) or DMSO (as a control) was injected intradermally 15 min before and 5 h after sensitization. Contact hypersensitivity (CHS) was elicited 5 d after the sensitization by painting the dorsal and ventral surface of the right ear with 10 μl of 0.5% FITC 35. Ear thickness was measured using an engineers' micrometer (Mitutoyo) 24 h after challenge. Results are expressed as ear swelling, which was calculated by subtracting the thickness of the ear before the challenge from the thickness of the ear after the challenge. In experiments where elicitations were not required, mice were killed 18 h (for the determination of epidermal LC density) or 24 h after sensitization. To determinate the number of migrating FITC-positive DCs in the SLNs, single cell suspensions were prepared from auricular LNs and DCs were enriched by centrifugation on a 14.5% (wt/vol) metrizamide gradient. DCs were then stained with the biotin-conjugated anti-CD11c mAb followed by phycoerythrin-streptavidin. The percentage of CD11c +FITC+ LN cells was determined on a FACSCalibur™ flow cytometer (Becton Dickinson). Data were analyzed using CELLQuest™ software.
mRNA Extraction and Reverse Transcription PCR Amplification.
Ears from mice were excised, the epidermis were separated from the dermis, and total RNA was isolated using TRIzol reagent (Life Technologies). RNA from resting XS52 and Pam212 cells were isolated as described above. cDNA was synthesized from 1 μg of total RNA with random hexamer primers and Superscript reverse transcriptase (Life Technologies) using standard procedures. PCR amplifications were performed with the primer pairs indicated in Table . Amplified products were subjected to 1% agarose gel electrophoresis and visualized by ethidium bromide staining.
Table 1.
Sequences of Primers Used for PCR Amplification of cDNA, Product Sizes, and PCR Cycle Numbers
| Gene | Primer | Sequence | Size (bp) | Cycle |
|---|---|---|---|---|
| β-actin | 5′ | 5′-GTCGGGCGCCCCAGGCACCA | 539 | 28 |
| 3′ | 5′-CTCCTTAATGTCACGCACGATTTC | |||
| TNF-α | 5′ | 5′-AACCACCAAGTGGAGGAGCAGC | 312 | 36 |
| 3′ | 5′-TGACCTCAGCGCTGAGTTGGTCC | |||
| IL-1β | 5′ | 5′-TGAAGGGCTGCTTCCAAACCTTTGACC | 322 | 36 |
| 3′ | 5′-TGTCCATTGAGGTGGAGAGCTTTCAGC | |||
| IL-1ra | 5′ | 5′-CCTGCAAGATGCAAGCCTTCAGG | 353 | 35 |
| 3′ | 5′-CAGCCTCTAGTGTTGTGCAGAGG | |||
| IL-4 | 5′ | 5′-GAATGTACCAGGAGCCATATC | 384 | 38 |
| 3′ | 5′-CTCAGTACTACGAGTAATCCA | |||
| TNFR-II | 5′ | 5′-GTAGGCCTTGAGCAGCAGCACCT | 312 | 35 |
| 3′ | 5′-GTGTCTCTGTAGTCTCACACGG | |||
| IL-10 | 5′ | 5′-TCCTTAATGCAGGACTTTAAGGGTTAC | 246 | 38 |
| 3′ | 5′-GACACCTTGGTCTTGGAGCTTATTAAA | |||
| DP receptor | 5′ | 5′-GAAGTTCGTGCAGTACTGTCCAG | 435 | 35 |
| 3′ | 5′-TCCACTATGGAAATCACAGACAG |
Results
S. mansoni Induces LC Activation In Vivo.
The distribution of epidermal LCs was visualized by immunohistochemical staining with anti-MHC class II molecule Abs in noninfected or S. mansoni–infected skins (Fig. 1A and Fig. B). In a kinetic study (1 to 120 h), the morphology of epidermal LCs dramatically differed in infected sheets compared with controls at all time points examined. In infected skins, LCs markedly increased in size and exhibited a more dendritic morphology with typical interdigiting cellular processes. Moreover, the MHC class II staining on LCs from infected epidermis was more intense. Immunolabeling with the LC-specific Ab NLDC-145 confirmed the activated phenotype of LCs in infected skins (not shown). We next analyzed other surface markers known to be expressed by mature LCs. Although we could barely detect CD86-positive cells in control epidermis (Fig. 1 C), the expression of CD86 on LCs was strongly upregulated in infected skins, 12 h (Fig. 1 D) to 120 h (not shown) after parasite penetration. Interestingly, most of the CD86-positive cells were located in the vicinity of the parasite or around its “ghost.” In contrast, we were unable to detect any CD80-positive cells either in noninfected or infected skins whatever time after infection (not shown). Taken together, our data suggest that, early after S. mansoni infection, LCs display clear signs of activation, exhibiting a more dendritic appearance and expressing higher amounts of MHC class II and CD86 molecules.
Figure 1.

Immunohistochemical staining of murine epidermal sheets after transcutaneous infection by S. mansoni. Epidermal sheets were prepared either from noninfected or from S. mansoni–infected mice and LCs were stained for MHC class II (A and B) or for CD86 (C and D). The arrow indicates the “ghost” of parasite. The isotype control mAb did not reveal any reactivity (not shown). Original magnification: ×400.
S. mansoni Induces Retention of Activated LCs in the Epidermis In Vivo and Ex Vivo.
As after activation, LCs normally migrate from the epidermal site of antigen capture to the SLNs, we attempted to study the migratory behavior of LCs after S. mansoni penetration. To this end, the frequency of MHC class II–positive cells was determined in the epidermis at different times after infection (1 to 120 h). As shown in Fig. 2 A, the density of MHC class II–positive epidermal cells in naive and noninfected mice ranged between 450 to 470 LCs/mm2. Surprisingly, in comparison to control epidermis, the number of LCs/mm2 remaining in the epidermis was not reduced 1 to 120 h after S. mansoni infection. To confirm this data, we used a complementary approach based on the spontaneous migration of LCs from skin explants cultured in vitro for 24 h (34; Fig. 2 B). In noninfected mice, compared with a freshly isolated epidermis, the number of LC/mm2 remaining in the epidermis after 24 h culture dramatically decreased to 188 ± 22 (vs. 452 ± 32, 58% reduction). In contrast, in infected animals, the density of LCs remained constant 6 to 48 h after infection and decreased significantly to 225 ± 11 (50% reduction) 120 h after infection, a period that coincides with the departure of the parasites from the skin. Together, both in vivo and ex vivo approaches suggest that parasite infection causes the activation of epidermal LCs but prevents their migration from the epidermis.
Figure 2.

Effect of S. mansoni infection on the migration of epidermal LCs in vivo (A) and ex vivo (B). (A) Epidermal sheets were prepared at different times after the infection (1 to 120 h) and the number of LC/mm2 was determined after anti-MHC class II staining. Controls included epidermal sheets from naive mice and from mice exposed to water without parasites (Non-infected). Results are expressed as means ± SD and are representative of four independent experiments (n = 7). (B) Skin explants were obtained from ears of noninfected or S. mansoni–infected mice (6, 24, 48, and 120 h). The number of LC/mm2 was determined in the epidermis from the explants after 24 h of culture and compared with the epidermis from fresh skin (naive). Results are expressed as means ± SD and are representative of three independent experiments (n = 4). Significant differences are designated by * (P < 0.001).
S. mansoni Inhibits the TNF-α–induced Migration of LCs.
We next investigated whether S. mansoni infection could alter LC migration in a system known to promote a strong LC departure to the SLNs 7. For this purpose, mice were injected into ear pinnae with TNF-α and the capacity of LCs to emigrate from the skin was then assessed 1 h after injection. As shown in Fig. 3 A, in noninfected mice, TNF-α caused ∼54% reduction in LC frequency compared with control mice (carrier). By contrast, infection by S. mansoni inhibits the TNF-α–induced LC migration 6 and 24 h after infection. Interestingly, the migratory ability of LCs was restored in mice infected 120 h before TNF-α treatment. Taken together, these results show that schistosomula transiently interfere with TNF-α to inhibit LC migration from the epidermis to the SLNs. To further confirm this, we visualized the accumulation of DCs in the SLNs isolated from TNF-α–treated mice previously infected (24 h before TNF-α treatment) or not with S. mansoni. As observed in Fig. 3 B, in noninfected mice, TNF-α administration resulted in increased number of CD11c-positive cells in the T cell areas of the SLNs whereas few CD11c-positive cells were detected in the SLNs from TNF-α–treated S. mansoni–infected mice.
Figure 3.


Effect of S. mansoni infection on the TNF-α–induced LC migration in vivo. (A) 6, 24, and 120 h after infection, mice (four mice/time point) were intradermally injected with 30 μl of PBS/BSA (carrier) containing or not containing 50 ng TNF-α into both ear pinnae. Ears were removed 1 h later, epidermal sheets were prepared, and the number of LC/mm2 was determined by immunohistochemistry. In TNF-α–treated S. mansoni-infected mice, we noted that LCs remained interdigitated among surrounding KCs and still expressed E-cadherin (not shown). The experiment shown is representative of five experiments (n = 8) and values are means ± SD. (B) Detection of CD11c-expressing cells in SLNs from TNF-α–treated mice previously (24 h), or not, infected with S. mansoni (original magnification: ×200).
IL-10 Is Not Sufficient to Inhibit LC Migration in S. mansoni–infected Mice.
To determine the mechanism by which S. mansoni inhibits LC migration, we first investigated by reverse transcription (RT)-PCR the presence of mRNAs for cytokines known to control LC mobility at various times after infection (1 to 120 h). As shown in Fig. 4 A, compared with noninfected mice (0 h), we observed a rapid and sustained increase in TNF-α and IL-1β mRNA levels in the epidermis of infected mice suggesting that the signals required for LC departure may be present. We then tested the hypothesis that the observed inhibitory effect could be associated with the expression of antiinflammatory cytokines. Interestingly, we detected a marked up-regulation of IL-10 mRNA, particularly between 6 and 24 h after infection. In contrast, we found that infection did not significantly affect the basal level of IL-1ra mRNA expression. In infected mice, the level of IL-4 mRNA increased progressively between 1 to 120 h after infection and this change was accompanied by a gradual decrease in mRNA levels of TNFR-II, but not by a total disappearance of the signal, in infected mice. Altogether, based on recent findings 17, our data suggest that IL-10 might be involved in the control of LC migration after S. mansoni infection. To test this hypothesis, IL-10 KO mice were infected and the density of LCs on epidermal sheets was assessed by immunohistochemistry 24 h after infection. As shown in Fig. 4 B, the number of LC/mm2 was identical in the epidermis of noninfected and infected IL-10–deficient mice, whereas TNF-α dramatically depleted the population of LCs by >60%. It is worth mentioning that in IL-10 KO mice, LCs exhibited an activated phenotype after parasite infection. Similar results were obtained in WT mice by using anti–IL-10 neutralizing mAbs injected intradermally before infection (Fig. 4 B). These data indicate that the inhibition of LC migration in S. mansoni–infected skin probably involve other factors than antiinflammatory host-derived cytokines.
Figure 4.

(A) RT-PCR analysis of mRNAs specific for pro- and antiinflammatory cytokines in the epidermis of S. mansoni–infected mice. Epidermal sheets were prepared from noninfected (0 h) or infected (1, 6, 24, and 120 h) mice, total RNA extracted, and RT-PCR was carried out using the primers shown in Table . Representative results of three independent experiments are shown. (B) Role of IL-10 in the inhibition of LC migration. IL-10 KO or WT mice were infected (or not) and 24 h after infection, epidermal sheets were prepared and the number of LCs/mm2 determined by immunohistochemistry. Before infection, WT mice were treated with neutralizing anti–IL-10 or isotype-matched mAbs (IgG1). As a positive control, TNF-α was intradermally injected 1 h before the analysis. Significant differences are designated by * (P < 0.001).
ES Lipophilic Substances from Schistosomula Inhibit the TNF-α–induced LC Migration In Vivo.
We then investigated the possibility that factors released by parasites themselves may directly affect LC migration. To this end, SESP were intradermally injected into the ear pinnae and the density of LCs remaining in the epidermis was determined 1 h after TNF-α injection. As seen in Fig. 5 A, SESP had a strong inhibitory effect on the TNF-α–induced LC mobility. We have previously demonstrated that the SESP contain bioactive lipophilic compounds able to activate host cells 24. We therefore tested the effect of the lipophilic fraction from the SESP, obtained by a modified Folch extraction protocol, on the TNF-α–induced LC departure. Compared with the control (DMSO), we found that the lipophilic fraction dose dependently abrogates the departure of LCs from the epidermis (Fig. 5 B). Previous studies revealed that parasite larvae secrete various arachidonic acid–derived eicosanoids and that PGD2, PGE2, 5-HETE, 15-HETE, LTB4, and LTC4 are the major compounds 30 36. Using different chromatographic systems, such as thin-layer chromatography (not shown) and HPLC, we confirmed these data except for LTs (detected in low amounts in the lipophilic fraction) and PGF2α (present in a detectable level) (Fig. 6). As represented in Table , quantification by HPLC and/or EIA revealed that, in our culture conditions, the parasite culture supernatant contains micromolar concentrations of PGF2α, PGE2, PGD2, 5-HETE, 15-HETE, and nanomolar concentrations of LTB4 and LTC4.
Figure 5.
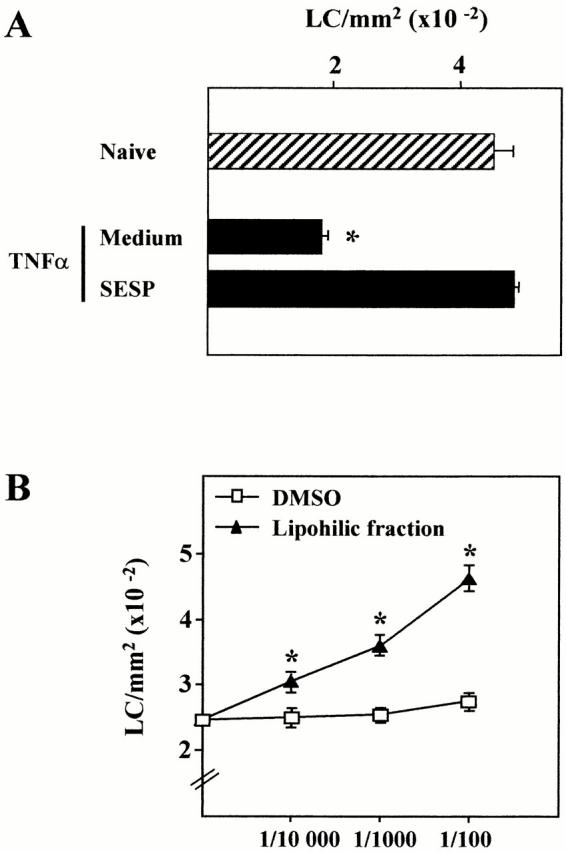
Effect of SESP on the TNF-α–induced LC migration in vivo. (A) The supernatant of a 4-h culture of schistosomula (SESP) or (B) increasing amounts of the lipophilic fraction from the SESP (diluted in DMSO) were intradermally injected to mice. After 20 min, mice were treated with 50 ng of TNF-α and the epidermal sheets were analyzed 1 h after for the determination of LC density. These data are representative of three experiments (n = 4). Significant differences are designated by * (P < 0.001).
Figure 6.

HPLC analysis of S. mansoni schistosomula eicosanoid production. 1 μl of the lipophilic fraction was injected, fractions were collected every 30 s, and monitored using a densitometer (wavelength: 195, 230, and 270 nm). The elution position of external standards are indicated. Note that the scales of the arbitrary values are different in each panel.
PGD2 Specifically Inhibits the TNF-α–induced LC Migration through a cAMP-dependent Pathway.
We therefore tested each of these molecules in our in vivo system of TNF-α–induced LC departure. As shown in Fig. 7 A, intradermal administration of increasing amounts of PGD2 significantly inhibits LC migration in a dose-dependent manner. In contrast, PGF2α, PGE2, 5-HETE, and 15-HETE did not prevent the mobility of LCs after TNF-α treatment, although at 100 nM, 5- and 15-HETE partially increased the LC density compared with animals that received DMSO alone. Similarly, LTB4 and LTC4 had no effect (not shown). Although we do not exclude the possibility that other lipophilic compounds may also be involved, this strongly suggests that schistosomula may exert its inhibitory effect on LC mobility through the production of PGD2. To confirm this, we used a synthetic analogue of PGD2 that is highly specific for the PGD2 receptor (DP receptor; reference 37). As seen in Fig. 7 B, compared with DMSO-treated animals, BW245C (10 nM) also abrogates the migration of LCs induced by TNF-α. As PGD2, and particularly BW245C, are known to increase the level of intracellular cAMP via its specific binding to the adenylate cyclase (AC)-coupled DP receptor 37, we hypothesized that cAMP may be the major signaling pathway involved in LC blockade. To this end, we tested the effect of the AC activator forskolin. As represented in Fig. 7 C, we found that forskolin (10 μM) abrogates the TNF-α–induced emigration of LCs. Taken together, our data suggest that the retention of LCs in the epidermis is likely mediated by a cAMP-dependent mechanism specifically triggered by PGD2.
Figure 7.

Effect of (A) the major schistosomula ES eicosanoids and (B) of the PGD2 analogue BW245C and the cAMP-elevating agent forskolin on the TNF-α–induced LC migration in vivo. (B) Mice received increasing concentrations of PGF2α, PGE2, PGD2, 15-HETE, 5-HETE, or vehicle alone (DMSO), or (B) they received BW245C (10 nM), forskolin (10 μM), or DMSO. After 20 min, mice were treated with 50 ng of TNF-α and epidermal sheets were prepared 1 h later. The number of LC/mm2 was determined by immunohistochemistry. This data is representative of three experiments (n = 4). Significant differences are designated by * (P < 0.001).
The DP Receptor Mediates the Inhibition of LC Migration Induced by Schistosoma.
We next verified by RT-PCR that the DP receptor is expressed on murine epidermal cells. For this purpose, as purification of freshly isolated LCs and KCs from mouse epidermis is extremely difficult to realize, we used the LC (XS52) and KC (Pam212) lines. As depicted in Fig. 8 A, we detected mRNA for the DP receptor in total epidermal cells, in the LC and, to a lesser extent, in the KC line. To demonstrate that the parasite-induced inhibitory effect on LC migration is due to the specific binding of PGD2 to the DP receptor, we treated mice with the highly specific DP receptor antagonist BW A868C 15 min before infection. 6 h later, the LC frequency was established in DMSO- and in BW A686C-treated animals. As shown in Fig. 8 B, BW A868C dose dependently restores the ability of LCs to leave the epidermis in infected mice. Altogether, these results show that targeting of PGD2 to the KC- and/or to the LC-expressed DP receptor is responsible for the blockade of LC emigration from the epidermis during infection by Schistosoma.
Figure 8.

(A) Expression of mRNA for the DP receptor in total epidermal cells and in the LC (XS52) and KC (Pam212) lines as assessed by RT-PCR. (B) Effect of the DP receptor antagonist BW A868C on LC emigration after infection with S. mansoni. 15 min before the infection, mice were injected intradermally with increasing amounts of BW A868C. 6 h later, epidermis were stained using anti-MHC class II Abs. This data is representative of three experiments (n = 4). Significant differences are designated by * (P < 0.001).
BW245C Inhibits CHS Responses Elicited by FITC.
To confirm our finding, we tested the effect of the DP receptor agonist BW245C in a model of contact sensitization induced by the hapten FITC 35. Compared with unsensitized mice, the number of LCs was reduced in the epidermis of sensitized mice 18 h after FITC painting (Fig. 9 A). In contrast, we found that LC migration is significantly impaired in BW245C-treated mice compared with sensitized control mice (DMSO/FITC). As assessed by flow cytometry, this defect in LC departure was associated with a dramatic reduction of the number of CD11c, FITC double-positive cells in the SLNs 24 h after sensitization (Fig. 9 B). Finally, to investigate whether the activation of the DP receptor during the sensitization phase results in an altered development of LC-dependent immune response, we measured the CHS response 5 d after FITC challenge. As expressed as ear swelling, BW245C-treated mice developed a profoundly reduced CHS responses (80% inhibition) compared with controls.
Figure 9.
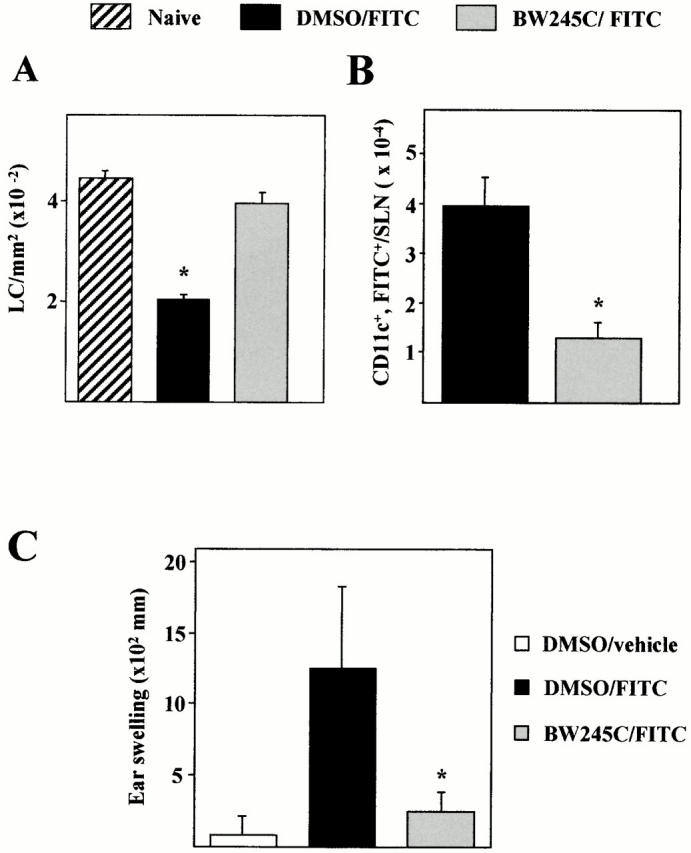
Effect of the PGD2 analogue BW245C on the FITC-induced migration of LCs and on CHS responses. Mice were injected intradermally into ear pinnae with BW245C (100 nM) 15 min before and 5 h after FITC topical application. (A) Epidermal LC density was analyzed 18 h after FITC painting and (B) the number of CD11c+FITC+ cells present in the SLNs determined 24 h after FITC application. (C) 5 d after sensitization, mice were challenged and 24 h later, ear thickness was measured. Results are expressed as means ± SD and are representative of three independent experiments (n = 7). Significant differences are designated by * (P < 0.001) for A and C and (P < 0.05) for B.
Discussion
Migration of LCs from the epidermis to the SLNs is a tightly regulated multistep process requiring inflammatory cytokines, chemokines, and adhesion molecules. However, very few studies have been devoted to investigate the molecular mechanisms which negatively regulate LC departure from the skin, especially during infections. In this report, we show for the first time that PGD2 directly inhibits the migration of LCs from the epidermis. This finding suggests a novel and unexpected function for this PG member in the control of LC homeostasis in the skin.
During cutaneous infections, skin-penetrating pathogens (in)directly activate LCs to migrate to the SLNs and may eventually use them to invade the hosts 21 38. During schistosomiasis, little is known about the immunological consequences of the interactions between schistosomes and cutaneous cells. Sato et al. suggested that resident LCs may participate in the initiation of the primary immune response in S. mansoni–infected guinea pigs, but the major APCs involved are rather newly recruited blood-born skin LCs/DCs 39 40. In another study, Riengrojpitak et al. hypothesized that infiltrating dermal APCs are important in the T cell priming in the SLNs 41. In this report, we investigated the possibility that S. mansoni may affect LC functions in vivo during murine infection. After checking that LCs did not undergo apoptosis after parasite penetration (data not shown), we showed that LCs exhibited evident signs of activation characterized by modifications of both LC morphology and phenotype, particularly for cells in the vicinity of the parasites or of their “ghosts”. Furthermore, we assessed the frequency of LCs in epidermal sheets from freshly isolated or from explanted infected skins. In both cases, we found that LCs are retained in the epidermis at all time points after infection (except at 120 h for the explants). Moreover, immunohistochemical analysis revealed no detectable DC accumulation in the SLNs from infected mice (1 to 10 d after infection, not shown). These results do not support those of Sato et al. who reported a significant LC depletion 12 h after infection in the guinea pig model 39. This may be attributed to differences in the animal models, in the schistosome species, or in the protocol of infection used. Interestingly enough, our observation was confirmed in a model of LC migration provoked by TNF-α. Indeed, we show that parasites transiently inhibit the TNF-α–induced release of LCs from the epidermis and the subsequent accumulation of DCs in the SLNs. Although not yet understood, this phenomenon has recently been described as a possible mechanism that could prevent and/or control the activation of immune responses against skin tumors 42 or retroviruses 43. Consequently, therapeutics able to antagonize the inhibitory effects which block LC migration (caused by abnormal cells or by pathogens) would be of great value. Whether or not Schistosoma used this stratagem to control and/or to orientate the cutaneous immune response is an open question which deserves further investigation. Indeed, despite the herein described inhibitory effect, we cannot exclude the possibility that some antigen-bearing LCs may migrate to the SLNs to initiate the response. Similarly, other cutaneous APCs (dermal DCs; reference 39) or SLN resident APCs 41 may also be important in initiating the immune response during schistosomiasis. Moreover, by transiently affecting the migration of LCs, Schistosoma may not only delay the induction of the immune response but also, by “exhausting” LCs, favor the priming of type 2 and nonpolarized T cells, as recently suggested by Langenkamp et al. 44.
We next explored the mechanisms that lead to the retention of LCs in the epidermis and attempted to identify the responsible factor(s). We first assessed by RT-PCR the expression of cytokines known to be involved in LC migration. We hypothesized that the antiinflammatory cytokine IL-10 may be implicated. Indeed, and in accordance with a recent report 45, we showed that IL-10–specific mRNA is strongly increased in S. mansoni–infected skin. Despite this, using IL-10 KO mice or WT mice treated with neutralizing anti–IL-10 Abs, we still observed the inhibition of LC migration in infected epidermis. Similarly, RT-PCR analysis suggest that the chemokine receptors CCR-1 and CCR-7 do not appear to be implicated in the herein described inhibitory effects (not shown). We then proceeded to the hypothesis that factors released by parasites while penetrating the skin may inhibit the TNF-α–induced signals involved in LC migration. We found that injection of lipophilic factors from the SESP mimicked the inhibitory effects observed during infection. We then tested the effects of the major cyclooxygenase and lipoxygenase products found in the SESP on the TNF-α–triggered migration of LC. Among them, we found that PGD2 specifically induces the retention of LCs in the skin after TNF-α treatment. We propose that PGD2 activates LCs by interacting with the AC-coupled DP receptor and that the resulting signaling pathway interferes with the TNF-α–induced signals implicated in LC departure. This later assumption is important as PGD2, as well as its metabolite 15d-PGJ2, can also activate the peroxisome proliferative-activated receptors, a family of nuclear receptors recently shown to inhibit the chemoattractant-induced migration of various cells 46 47. In the same manner, a novel receptor for PGD2 has recently been described 48. Interestingly, this seven transmembrane G protein–coupled receptor (termed CRTH2) is expressed on human T helper type 2 lymphocytes, eosinophils, and basophils and is involved in their recruitment to allergic inflammatory sites. We eliminate the possibility that CRTH2 may be involved in the inhibition of LC migration as (a) we did not find mRNA CRTH2 expression in the LC line XS52 (not shown), (b) BW245C is a poor agonist for CRTH2 48, and (c) CRTH2 is not coupled to an AC system but, on the contrary, induces Ca2 + mobilization in activated cells. Therefore, the dual action of PGD2 in either favoring or inhibiting cell migration is probably due to a selective expression of CRTH2 or DP receptor on target cells.
To our knowledge, PGD2 is the first molecule described to impair LC migration in vivo by directly affecting LC motility. Indeed, although other mechanisms may also take place 18 19, compounds known to block LC migration act rather by diminishing the synthesis or the release of inflammatory cytokines 49 50 51 or by interfering with their activities 52. For instance, in UVB- or enterotoxin-treated mice, agents that block protein kinase C or G protein–associated kinases inhibit LC departure in part by preventing the release of TNF-α or IL-1α in the epidermis 50 53 54. In this report, we show that, in a TNF-α–induced model of LC depletion, activation of the cAMP-mediated pathway inhibits LC departure from the skin. In this model, the exact mechanisms by which cAMP inhibits the TNF-α–induced migratory abilities of LCs are not elucidated but probably involve remodelling of the actin network and the reinforcement of contact between LCs and KCs. In these processes, different protein targets for cAMP may be involved including the small GTP-binding proteins rho, tyrosine kinases, or adhesion molecules 55. For instance, the sustained E-cadherin expression on LCs in S. mansoni–infected skins (even after TNF-α treatment; not shown), may be one of these.
In addition to its role in the development and/or the modulation of acute and chronic inflammation 56 57 58, PGD2 has multiple effects on the immune system. It enhances the release of mediators by eosinophils and mast cells, reduces the production of superoxide in neutrophils, and suppresses T cell mitogenesis 59 60. Here, we describe a novel function for PGD2 in that, during immune/inflammatory reactions, it may also control the migration of APCs from the site of antigen capture to the LNs. In the skin, PGD2 is among the major arachidonic acid metabolites produced (together with PGE2 and HETEs), particularly in the epidermis. Besides its role in KC proliferation and differentiation and in inflammatory responses 61, we propose that PGD2 may also act as an upstream component in a cascade of events that regulate the emigration of LCs from the skin, by a feedback mechanism. This hypothesis is supported by data showing that increased production of PGD2 is observed in the skin after UVB irradiation or antigen challenge 62. During infections, host- as well as pathogen-derived PGD2 synthase (the enzyme which transforms PGH2 to PGD2) may therefore play a key role in the maintenance of LC homeostasis in the skin. In our model, we have recently identified the parasite enzyme responsible for PGD2 synthesis in schistosomes. This PGD2 synthase is massively excreted by parasites while penetrating through the skin (unpublished data). Consequently, in addition to the endogenously produced PGD2, it is likely that Schistosoma may exploit the lipid metabolism of the host to convert fatty acid precursors into PGD2.
Our findings may have important consequences in the improvement of therapeutic treatments which aim to control skin diseases. Indeed, using a CHS model system, we have confirmed the potent ability of DP receptor agonists to inhibit LC migration out of the skin and to impair DC accumulation in the LNs. Furthermore, this defect in LC migration after hapten sensitization was associated with defective CHS responses after challenge. At present, we are testing the efficiency of PGD2 analogues (agonists and/or antagonists of the DP receptor) as well as modulators of the PGD2 synthase activity in diseases where reduction of immune cutaneous response is sought, such as eczematous and atopic dermatitis or, conversely, in diseases where stimulation of LC migration would be beneficial, such as in certain skin cancers (carcinomas) and infectious pathologies.
Acknowledgments
The authors thank S. Lister (Glaxo Wellcome, UK) for the donation of BW A868C as well as A. Takashima (University of Texas, Dallas, TX) and S. Yuspa (National Cancer Institute, National Institutes of Health, Bethesda, MD) for providing the XS52 and Pam212 cell lines, respectively. Drs. D. Sacks, J. Belkaid (National Institutes of Health), D. Staumont, E. Delaporte (CHRU-Lille), and A. Charbonnier (INSERM U416) are acknowledged for stimulating discussions and Drs. J. Khalife and D. Dombrowicz (INSERM U547) for correcting this manuscript.
V. Angeli and C. Faveeuw are respectively supported by a Ministere de l'Education Nationale, de la Recherche et de la Technologie (MENRT) and a Fondation pour la Recherche Medicale fellowship. F. Trottein is a member of the Centre National de la Recherche Scientifique.
Footnotes
Abbreviations used in this paper: AC, adenylate cyclase; CHS, contact hypersensitivity; CCR, CC chemokine receptor; DC, dendritic cell; DP receptor, PGD2 receptor; EIA, enzyme immunoassay; ES, excreted/secreted; HETE, 5-hydroxyeicosatetranoic acid; KC, keratinocyte; KO, knockout; LC, Langerhans cell; LT, leukotriene; RT, reverse transcription; SESP, schistosomula ES products; SLN, skin-draining LN; WT, wild-type.
References
- Banchereau J., Steinman R.M. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Tang A., Amagai M., Granger L.G., Stanley J.R., Udey M.C. Adhesion of epidermal Langerhans cells to keratinocytes is mediated by E-cadherin. Nature. 1993;361:82–85. doi: 10.1038/361082a0. [DOI] [PubMed] [Google Scholar]
- Kimber I., Dearman R.J., Cumberbatch M., Huby R.J. Langerhans cells and chemical allergy. Curr. Opin. Immunol. 1998;10:614–619. doi: 10.1016/s0952-7915(98)80078-2. [DOI] [PubMed] [Google Scholar]
- Moodycliffe A.M., Kimber I., Norval M. Role of TNFα in ultraviolet B light-induced dendritic cell migration and suppression of contact hypersensitivity. Immunology. 1992;81:79–84. [PMC free article] [PubMed] [Google Scholar]
- Ban E., Dupre I., Hermann E., Rohn W., Vendeville C., Quatannens B., Ricciardi-Castagnoli P., Capron A., Riveau G. CpG motifs induce Langerhans cell migration in vivo . Int. Immunol. 2000;12:737–744. doi: 10.1093/intimm/12.6.737. [DOI] [PubMed] [Google Scholar]
- Weinlich G., Heine M., Stossel H., Zanella M., Stoitzner P., Ortner U., Smolle J., Koch F., Sepp N.T., Schuler G., Romani N. Entry into afferent lymphatics and maturation in situ of migrating murine cutaneous dendritic cells. J. Invest. Dermatol. 1998;110:441–448. doi: 10.1046/j.1523-1747.1998.00161.x. [DOI] [PubMed] [Google Scholar]
- Kimber I., Cumberbatch M. Stimulation of Langerhans cell migration by TNFα. J. Invest. Dermatol. 1992;99:48–50. doi: 10.1111/1523-1747.ep12668986. [DOI] [PubMed] [Google Scholar]
- Enk A.H., Angeloni V.L., Udey M.C., Katz S.I. An essential role for Langerhans cell-derived IL-1β in the initiation of primary immune responses in skin. J. Immunol. 1993;150:3698–3704. [PubMed] [Google Scholar]
- Schwarzenberger K., Udey M.C. Contact allergens and epidermal proinflammatory cytokines modulate Langerhans cell E-cadherin expression in situ . J. Invest. Dermatol. 1996;106:553–558. doi: 10.1111/1523-1747.ep12344019. [DOI] [PubMed] [Google Scholar]
- Winzler C., Rovere P., Rescigno M., Granucci F., Penna G., Adorini L., Zimmermann V.S., Davoust J., Ricciardi-Castagnoli P. Maturation stages of mouse dendritic cells in growth factor–dependent long-term culture. J. Exp. Med. 1997;185:317–328. doi: 10.1084/jem.185.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeki H., Moore A.M., Brown M.J., Hwang S.T. Secondary lymphoid-tissue chemokine (SLC) and CC chemokine receptor 7 (CCR7) participate in the emigration pathway of mature dendritic cells from the skin to regional lymph nodes. J. Immunol. 1999;162:2472–2475. [PubMed] [Google Scholar]
- Gunn M.D., Kyuwa S., Tam C., Kakiuchi T., Matsuzawa A., Williams L.T., Nakano H. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J. Exp. Med. 1999;189:451–460. doi: 10.1084/jem.189.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F., Schaerli P., Loetscher P., Schaniel C., Lenig D., Mackay C.R., Qin S., Lanzavecchia A. Rapid and coordinated switch in chemokine receptor expression during dendritic cell maturation. Eur. J. Immunol. 1998;28:2760–2769. doi: 10.1002/(SICI)1521-4141(199809)28:09<2760::AID-IMMU2760>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Maestroni G.J. Dendritic cell migration controlled by α1b-adrenergic receptors. J. Immunol. 2000;165:6743–6747. doi: 10.4049/jimmunol.165.12.6743. [DOI] [PubMed] [Google Scholar]
- Kondo S., Pastore S., Fujisawa H., Shivji G.N., Mc- Kenzie R.C., Dinarello C.A., Sauder D.N. Interleukin-1 receptor antagonist suppresses contact hypersensitivity. J. Invest. Dermatol. 1995;105:334–338. doi: 10.1111/1523-1747.ep12320329. [DOI] [PubMed] [Google Scholar]
- Takayama K., Yokozeki H., Ghoreishi M., Satoh T., Katayama I., Umeda T., Nishioka K. IL-4 inhibits the migration of human Langerhans cells through the downregulation of TNF receptor II expression. J. Invest. Dermatol. 1999;113:541–546. doi: 10.1046/j.1523-1747.1999.00629.x. [DOI] [PubMed] [Google Scholar]
- Wang B., Zhuang L., Fujisawa H., Shinder G.A., Feliciani C., Shivji G.M., Suzuki H., Amerio P., Toto P., Saunder D.N. Enhanced epidermal Langerhans cell migration in IL-10 knockout mice. J. Immunol. 1999;162:277–283. [PubMed] [Google Scholar]
- Kobayashi Y., Matsumoto M., Kotani M., Makino T., Kobayashi Y., Matsumoto M., Kotani M., Makino T. Possible involvement of matrix metalloproteinase-9 in Langerhans cell migration and maturation. J. Immunol. 1999;163:5989–5993. [PubMed] [Google Scholar]
- Randolph G.J., Beaulieu S., Pope M., Sugawara I., Hoffman L., Steinman R., Muller W.A. A physiologic function for p-glycoprotein (MDR-1) during the migration of dendritic cells from skin via afferent lymphatic vessels. Proc. Natl. Acad. Sci. USA. 1998;95:6924–6929. doi: 10.1073/pnas.95.12.6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B., Aùmerio O., Sauder D. Role of cytokines in epidermal Langerhans cell migration. J. Leukoc. Biol. 1999;66:33–39. doi: 10.1002/jlb.66.1.33. [DOI] [PubMed] [Google Scholar]
- Arnoldi J., Moll H. Langerhans cell migration in murine cutaneous leishmaniasisregulation by TNFα, IL-1β, and MIPα. Dev. Immunol. 1998;6:3–11. doi: 10.1155/1998/21095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy K., Salafsky B., Potluri S., He Y.X., Li J.W., Shibuya T. Secretion of an anti-inflammatory, immunomodulatory factor by schistosomulae of Schistosoma mansoni . J. Inflamm. 1995;46:13–22. [PubMed] [Google Scholar]
- Trottein F., Descamps L., Nutten S., Dehouck M.P., Angeli V., Capron A., Cecchelli R., Capron M. Schistosoma mansoni activates host microvascular endothelial cells to acquire an anti-inflammatory phenotype. Infect. Immun. 1999;67:3403–3409. doi: 10.1128/iai.67.7.3403-3409.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trottein F., Nutten S., Angeli V., Delerive P., Teissier E., Capron A., Staels B., Capron M. Schistosoma mansoni schistosomula reduce E-selectin and VCAM-1 expression in TNFα-stimulated lung microvascular endothelial cells by interfering with the NF-κB pathway. Eur. J. Immunol. 1999;29:3691–3701. doi: 10.1002/(SICI)1521-4141(199911)29:11<3691::AID-IMMU3691>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Macatonia S.E., Patterson S., Knight S.C. Suppression of immune responses by dendritic cells infected with HIV. Immunology. 1989;67:285–289. [PMC free article] [PubMed] [Google Scholar]
- Van Overtvelt L., Vanderheyde N., Verhasselt V., Ismaili J., De Vos L., Goldman M., Willems F., Vray B. Trypanosoma cruzi infects human dendritic cells and prevents their maturationinhibition of cytokines, HLA-DR, and co-stimulatory molecules. Infect. Immun. 1999;67:4033–4040. doi: 10.1128/iai.67.8.4033-4040.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salio M., Cella M., Suter M., Lanzavecchia A. Inhibition of dendritic cell maturation by herpes simplex virus. Eur. J. Immunol. 1999;29:3245–3253. doi: 10.1002/(SICI)1521-4141(199910)29:10<3245::AID-IMMU3245>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Reis e Sousa C., Yap G., Schulz O., Rogers N., Schito M., Aliberti J., Hieny S., Sher A. Paralysis of dendritic cell IL-12 production by microbial products prevents infection-induced immunopathology. Immunity. 1999;11:637–647. doi: 10.1016/s1074-7613(00)80138-7. [DOI] [PubMed] [Google Scholar]
- Urban B.C., Ferguson D.J., Pain A., Willcox N., Plebanski M., Austyn J.M., Roberts D.J. Plasmodium falciparum-infected erythrocytes modulate the maturation of dendritic cells. Nature. 1999;400:73–77. doi: 10.1038/21900. [DOI] [PubMed] [Google Scholar]
- Fusco A.C., Salafsky B., Kevin M.B. Schistosoma mansonieicosanoid production by cercariae. Exp. Parasitol. 1985;59:44–50. doi: 10.1016/0014-4894(85)90055-4. [DOI] [PubMed] [Google Scholar]
- Xu S., Bergstresser P.R., Takashima A. Phenotypic and functional heterogeneity among murine epidermal-derived dendritic cell clones. J. Invest. Dermatol. 1995;105:831–836. doi: 10.1111/1523-1747.ep12326625. [DOI] [PubMed] [Google Scholar]
- Roop D.R., Hawley-Nelson P., Cheng C., Huspa S.H. Expression of keratin genes in mouse epidermis and normal and malignantly transformed epidermal cells in culture. J. Invest. Dermatol. 1983;81:144–149. doi: 10.1111/1523-1747.ep12540939. [DOI] [PubMed] [Google Scholar]
- Shelley W.B., Juhlin L. Selective uptake of contact allergens by the Langerhans cell. Arch. Dermatol. 1977;113:187–192. [PubMed] [Google Scholar]
- Larsen C.P., Steinman R.M., Witmer-Pack M., Hankins D.F., Morris P.J., Austyn J.M. Migration and maturation of Langerhans cells in skin transplants and explants. J. Exp. Med. 1990;172:1483–1493. doi: 10.1084/jem.172.5.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kripke M.L., Munn C.G., Jeevan A., Tang J.M., Bucana C. Evidence that cutaneous antigen-presenting cells migrate to regional lymph nodes during contact sensitization. J. Immunol. 1990;145:2833–2838. [PubMed] [Google Scholar]
- Salafsky B., Fusco A.C. Schistosoma mansonia comparison of secreted vs. non-secreted eicosanoids in developing schistosomulae and adults. Exp. Parasitol. 1987;64:361–367. doi: 10.1016/0014-4894(87)90048-8. [DOI] [PubMed] [Google Scholar]
- Narumiya S., Sugimoto Y., Ushikubi F. Prostanoid receptorsstructures, properties, and functions. Physiol. Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- Wu S.J., Grouard-Vogel G., Sun W., Mascola J.R., Brachtel E., Putvatana R., Louder M.K., Filgueira L., Marovich M.A., Wong H.K. Human skin Langerhans cells are targets of dengue virus infection. Nat. Med. 2000;6:816–820. doi: 10.1038/77553. [DOI] [PubMed] [Google Scholar]
- Sato H., Kamiya H. Role of epidermal Langerhans cells in the induction of protective immunity to Schistosoma mansoni in guinea-pigs. Immunology. 1995;84:233–240. [PMC free article] [PubMed] [Google Scholar]
- Sato H., Kamiya H. Accelerated influx of dendritic cells into the lymph nodes draining skin sites exposed to attenuated cercariae of Schistosoma mansoni in guinea-pigs. Parasite Immunol. 1998;20:337–343. doi: 10.1111/j.1365-3024.1998.158.x. [DOI] [PubMed] [Google Scholar]
- Riengrojpitak S., Anderson S., Wilson R.A. Induction of immunity to Schistosoma mansoniinteraction of schistosomula with accessory leucocytes in murine skin and draining lymph nodes. Parasitology. 1998;117:301–309. doi: 10.1017/s0031182098003187. [DOI] [PubMed] [Google Scholar]
- Lucas A.D., Halliday G.M. Progressor but not regressor skin tumors inhibit Langerhans cell migration from epidermis to local lymph nodes. Immunology. 1999;97:130–137. doi: 10.1046/j.1365-2567.1999.00751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich D.I., Woods G.M., Patterson S., Harvey J.J., Knight S.C. Retrovirus-induced immunosuppression via blocking of dendritic cell migration and down-regulation of adhesion molecules. Immunology. 1994;82:82–87. [PMC free article] [PubMed] [Google Scholar]
- Langenkamp A., Messi M., Lanzavecchia A., Sallusto F. Kinetics of dendritic cell activationimpact on priming of Th1, Th2 and nonpolarized T cells. Nat. Immunol. 2000;1:311–316. doi: 10.1038/79758. [DOI] [PubMed] [Google Scholar]
- Ramaswamy K., Kumar P., He Y.X. A role for parasite-induced PGE2 in IL-10-mediated host immunoregulation by skin stage schistosomula of Schistosoma mansoni . J. Immunol. 2000;165:4567–4574. doi: 10.4049/jimmunol.165.8.4567. [DOI] [PubMed] [Google Scholar]
- Goetze S., Xi X.P., Kawano H., Gotlibowski T., Fleck E., Hsueh W.A., Law R.E. PPARγ-ligands inhibit migration mediated by multiple chemoattractants in vascular smooth muscle cells. J. Cardiovasc. Pharmacol. 1999;33:798–806. doi: 10.1097/00005344-199905000-00018. [DOI] [PubMed] [Google Scholar]
- Kintscher U., Goetze S., Wakino S., Kim S., Nagpal S., Chandraratna R.A., Graf K., Fleck E., Hsueh W.A., Law R.E. Peroxisome proliferator-activated receptor and retinoid X receptor ligands inhibit monocyte chemotactic protein-1-directed migration of monocytes. Eur. J. Pharmacol. 2000;401:259–270. doi: 10.1016/s0014-2999(00)00461-1. [DOI] [PubMed] [Google Scholar]
- Hirai H., Tanaka K., Yoshie O., Ogawa K., Kenmotsu K., Takamori Y., Ichimasa M., Sugamura K., Nakamura M., Takano S., Nagata K. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J. Exp. Med. 2001;193:255–261. doi: 10.1084/jem.193.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumberbatch M., Dearman R.J., Kimber I. Inhibition by dexamethasone of Langerhans cell migrationinfluence of epidermal cytokine signals. Immunopharmacology. 1999;41:235–243. doi: 10.1016/s0162-3109(99)00037-5. [DOI] [PubMed] [Google Scholar]
- Shankar G., Johnson J., Kuschel L., Richins M., Burnham K. Protein-kinase-specific inhibitors block Langerhans cell migration by inhibiting IL-1α release. Immunology. 1999;96:230–235. doi: 10.1046/j.1365-2567.1999.00680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumberbatch M., Dearman R.J., Uribe-Luna S., Headon D.R., Ward P.P., Conneely O.M., Kimber I. Regulation of epidermal Langerhans cell migration by lactoferrin. Immunology. 2000;100:21–28. doi: 10.1046/j.1365-2567.2000.00014.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday G.M., Lucas A.D. Protein kinase C transduces the signal for Langerhans cell migration from the epidermis. Immunology. 1993;79:621–626. [PMC free article] [PubMed] [Google Scholar]
- Burnham K., Pickard S., Hudson J., Voss T. Requirements for Langerhans cell depletion following in vitro exposure of murine skin to ultraviolet-B. Immunology. 1993;79:627–632. [PMC free article] [PubMed] [Google Scholar]
- Pickard S., Shankar G., Burnham K. Langerhans cell depletion by staphylococcal superantigens. Immunology. 1994;83:568–572. [PMC free article] [PubMed] [Google Scholar]
- Ydrenius L., Majeed M., Rasmusson B.J., Stendahl O., Sarndahl E. Activation of cAMP-dependent protein kinase is necessary for actin rearrangements in human neutrophils during phagocytosis. J. Leukoc. Biol. 2000;67:520–528. doi: 10.1002/jlb.67.4.520. [DOI] [PubMed] [Google Scholar]
- Gilroy D.W., Colville-Nash P.R., Willis D., Chivers J., Paul-Clark M.J., Willoughby D.A. Inducible cyclooxygenase may have anti-inflammatory properties. Nat. Med. 1999;5:698–701. doi: 10.1038/9550. [DOI] [PubMed] [Google Scholar]
- Ajuebor M.N., Singh A., Wallace J.L. Cyclooxygenase-2-derived prostaglandin D2 is an early anti-inflammatory signal in experimental colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2000;279:238–244. doi: 10.1152/ajpgi.2000.279.1.G238. [DOI] [PubMed] [Google Scholar]
- Matsuoka T., Hirata M., Tanaka H., Takahashi Y., Murata T., Kabashima K., Sugimoto Y., Kobayashi T., Ushikubi F., Aze Y. Prostaglandin D2 as a mediator of allergic asthma. Science. 2000;287:2013–2017. doi: 10.1126/science.287.5460.2013. [DOI] [PubMed] [Google Scholar]
- Raible D.G., Schulman E.S., DiMuzio J., Cardillo R., Post T.J. Mast cell mediators prostaglandin D2 and histamine activate human eosinophils. J. Immunol. 1992;148:3536–3542. [PubMed] [Google Scholar]
- Kanamori Y., Niwa M., Kohno K., Al-Essa L.Y., Matsuno H., Kozawa O., Uematsu T. Migration of neutrophils from blood to tissuealteration of modulatory effects of prostanoid on superoxide generation in rabbits and humans. Life Sci. 1997;60:1407–1417. doi: 10.1016/s0024-3205(97)00086-6. [DOI] [PubMed] [Google Scholar]
- Ikai K., Imamura S. Prostaglandin D2 in the skin. Int. J. Dermatol. 1988;27:141–149. doi: 10.1111/j.1365-4362.1988.tb04917.x. [DOI] [PubMed] [Google Scholar]
- Massey W.A., Hubbard W.C., Liu M.C., Kagey-Sobotka A., Cooper P., Lichtenstein L.M. Profile of prostanoid release following antigen challenge in vivo in the skin of man. Br. J. Dermatol. 1991;125:529–534. doi: 10.1111/j.1365-2133.1991.tb14789.x. [DOI] [PubMed] [Google Scholar]