

Abstract
We show that cytotoxic T lymphocytes (CTLs) infiltrating a kidney tumor recognize a peptide encoded by an alternative open reading frame (ORF) of the macrophage colony-stimulating factor (M-CSF) gene. Remarkably, this alternative ORF, which is translated in many tumors concurrently with the major ORF, is also translated in some tissues that do not produce M-CSF, such as liver and kidney. Such a dissociation of the translation of two overlapping ORFs from the same gene is unexpected. The antigenic peptide encoded by the alternative ORF is presented by human histocompatibility leukocyte antigen (HLA)-B*3501 and has a length of 14 residues. Peptide elution indicated that tumor cells naturally present this 14 mer, which is the longest peptide known to be recognized by CTLs. Binding studies of peptide analogues suggest that it binds by its two extremities and bulges out of the HLA groove to compensate for its length.
Keywords: renal cell carcinoma, HLA-B35, translation, peptide binding, natural peptide
Introduction
Antigens recognized by CD8-positive CTLs consist of peptides of 9 to 11 amino acids, which are presented by MHC class I molecules. The majority of those peptides originate from the degradation of intracellular proteins, which occurs in the cytosol and is exerted mainly by the proteasome (for a review, see reference 1). In recent years, an increasing number of peptides have been defined that do not simply result from the degradation of mature proteins, but rather result from nonclassical mechanisms acting at the level of transcription, splicing, translation, or processing 2. The biological significance of those processes had not been recognized before, presumably because they occur at a low level and produce limited amounts of materials that are difficult to detect. It is the exquisite sensitivity of CTLs that allowed the detection of the products of those processes and, by inference, the recognition of their actual significance. For the immunologist, the practical conclusion is that the complete spectrum of peptides presented by a given cell type cannot be deduced from the predicted sequence of the major products of the active genes.
Some of those nonclassical peptides result from posttranslational modifications, such as the conversion of an asparagine residue to an aspartic acid after N-linked glycosylation/deglycosylation reactions, or the oxidation of a cysteine residue through the formation of a disulfide bridge with a free cysteine 3 4 5. Other peptides are encoded by alternative transcripts starting on cryptic promoters located in intronic sequences 6 7. In one case, the alternative transcript corresponded to the antisense strand of the gene and therefore resulted from reverse strand transcription 7. Other peptides are encoded by retained introns resulting from incomplete splicing 8 9 10. Lastly, some peptides were found to result from the translation of alternative open reading frames (ORFs) 11 12 13 14. In those cases, the alternative ORF was translated concurrently with the major ORF present of the same mRNA, and the expression of both protein products appeared to parallel the expression of the corresponding mRNA.
Here we describe a new antigenic peptide, which is recognized by CTLs directed against human renal cell carcinomas (RCCs), and is encoded by an alternative ORF of the M-CSF gene. Remarkably, this alternative ORF is also translated in some tissues that do not express M-CSF at the protein level.
Materials and Methods
Cell Lines.
RCC line LB1047-RCC was derived from a metastatic lymph node lesion of patient LB1047 (HLA-A*0201, -A*2402.101, -B*3501, -B51, -Cw*0401, -Cw*1602). The lymphoblastoid cell line LB1047-EBV was established by standard techniques from the lymphocytes present in the culture supernatant of the same metastatic lymph node lesion. RCC line LE9211-RCC and EBV line LE9211-EBV have been described 15; RCC line HA7-RCC and lymphoblastoid line HA7-EBV were established in our laboratory from the primary tumor and peripheral blood lymphocytes of patient HA7 (provided by Drs. C.G. Stief [Hannover Medical School, Hannover, Germany] and J. Atzpodien [European Institute for Tumor Immunology and Prevention, Bonn, Germany); A110L is a lung carcinoma line, provided by Drs. M. Takenoyama and K. Yasumoto (University of occupational and Environmental Health, Kita Kyushu, Japan). Stable transfectant A110L-B35 was obtained by transfection of the HLA-B*3501 cDNA cloned into plasmid pEF-BOSpuro-PL3 16. Lines LB2046-PTEC and LB2043-PTEC were described previously 7. 293-EBNA cells were obtained from Invitrogen. CTL clones were derived from tumor-infiltrating lymphocytes (TILs) present in the primary culture of LB1047-RCC. The TILs were collected at day 3 after the initiation of tumor culture, expanded for 1 wk with IL-6 (103 U/ml) and IL-12 (10 ng/ml), and frozen at −80°C until establishment of the autologous tumor cell line. TILs were then used as effectors in an autologous mixed lymphocyte-tumor cell culture (MLTC) performed as described previously with minor modifications 17. In brief, 106 TIL-derived lymphocytes were mixed with 105 irradiated LB1047-RCC tumor cells and cultured in human serum with IL-6 (103 U/ml) and IL-12 (10 ng/ml). After 1 wk, 5 × 105 lymphocytes were stimulated again for 1 wk with irradiated tumor cells (105) with IL-7 (5 ng/ ml) and IL-2 (10 U/ml), and then 1 wk further with tumor cells and only IL-2 (25 U/ml). On day 21, lymphocytes were cloned by limiting dilution in the presence of tumor cells and allogeneic feeder cells in Iscove's medium supplemented with IL-2 (50 U/ml), and long-term culture of CTL clones was performed as described 17.
CTL Assays.
Chromium-release, TNF, and IFN-γ stimulation assays were performed as described previously 7. Where indicated, target cells were treated with 100 U/ml IFN-γ 24 h before the assay. For the peptide assay, labeled EBV-transformed B cells were incubated 30 min at 37°C with various concentrations of peptides diluted in X-VIVO 10 medium (Whittaker Bioproducts). Lines LB2046- and LB2043-PTEC were transiently transfected with the HLA-B*3501 cDNA using DMRIE-C (GIBCO BRL) before being used as stimulators for TNF assay. To test the fractions of peptides eluted from HLA class I molecules of LB1047-RCC, each fraction was dissolved in 200 μl X-VIVO 10 medium, and 50 μl were pulsed in duplicate onto A110L-B35 (10,000 cells) target cells for 30 min at 37°C. CTL 403A/9 was then added (5,000 cells) in a final volume of 110 μl of the same medium with 25 U/ml IL-2; IFN-γ was measured after 18 h.
Construction and Screening of the cDNA Library.
The cDNA library was constructed as described recently 16. In brief, mRNA was transcribed to cDNA with the Superscript Choice System (GIBCO BRL) using an oligo-(dT) primer containing a NotI site at its 5′ end. cDNA were then ligated to HindIII adaptors (Stratagene), phosphorylated, digested with NotI, size fractionated, and inserted into the HindIII and NotI sites of expression vector pCEP4 (Invitrogen). Recombinant plasmids were electroporated into Escherichia coli Top10F′ and selected with ampicillin (100 μg/ml). The library was divided into pools of about 100 cDNA clones. Plasmid DNA of each bacteria pool was extracted using the QIAprep 8 plasmid kit (QIAGEN). Pools of about 100 cDNAs were cotransfected into 293-EBNA cells as described 16. In brief, 5 × 104 293-EBNA cells were plated in flatbottom 96-well plates and transfected with 60 ng each of both the HLA-B*3501 and the HLA-Cw*0401 cDNAs cloned into plasmid pcDNA3 and with about 100 ng of plasmid DNA from each pool of the cDNA library of LB1047-RCC, using the Lipofectamin™ Reagent (GIBCO BRL). Transfected cells were tested in a CTL stimulation assay 24 h later.
Cloning of HLA cDNA from LB1047-RCC.
The HLA-B*3501 cDNA was cloned from the total RNA of line LB1047-RCC using standard reverse transcription (RT)-PCR with HLA class I–specific primers according to Ennis et al. 18 with Pfu DNA polymerase (Stratagene). The PCR products were digested thereafter with KpnI to eliminate HLA-A sequences, phosphorylated, and inserted into the EcoRV site of pcDNA3. A selected plasmid construct was sequenced and confirmed to contain the HLA-B*3501 cDNA. It was subcloned into plasmid pEF-BOSpuroPL3.
Construction of Minigenes.
Minigenes encoding alt.M-CSF were constructed by PCR using sense primer MPO29 (5′-actgggcggatcctgccctcccacgacatggct-3′; position 153–171 of M-CSF according to GenBank/EMBL/DDBJ accession no. NM-000757) and antisense primer MPO30 (5′-ACTGCCCGAATTCGTCACGAGGTCTCCATCTGA- CTG-3′; position 208–225 of M-CSF) or primer MPO39 (5′-ACTGCCCGAATTC TATCCTCGGTGATACTCCTG-3′; position 292–312 of M-CSF), containing a BamHI (MPO29) and an EcoRI (MPO30 and 39) site (underlined), respectively, and a stop codon (MPO39; bold). The PCR products were digested with BamHI and EcoRI, inserted into pcDNA3, and sequenced, to verify their identity.
DNA Sequence Analysis.
DNA sequencing reactions were performed with the ABI PRISM™ dRhodamine Terminator Cycle Sequencing Kit; the reaction products were separated on the ABI PRISM™ 310 Genetic Analyser (PerkinElmer).
M-CSF Detection.
M-CSF was quantified in the culture supernatant of 105 cells plated in 0.5 ml of medium in 48-well microtiter plates for 24 h, using a commercial ELISA kit (R&D Systems).
Immunohistochemistry.
Immunochemistry was carried out on sections of frozen or paraffin-embedded RCCs and normal organs. 7-μm frozen sections were fixed in formalin for 5 min at room temperature and washed in Tris-buffered saline (TBS), pH 7.4. For formalin-fixed paraffin-embedded tissues, 5-μm sections were deparaffinized in xylene, hydrated in a graded alcohol series, and washed in TBS, pH 7.4. Endogenous peroxidase activity was blocked by incubating all sections in Peroxidase Blocking Reagent (Dako) for 10 min at room temperature. Goat IgG (Sigma-Aldrich) diluted at 0.5 mg/ml in PBS/BSA 0.5% was used to block nonspecific Ig binding sites for 30 min. For alt.M-CSF detection, we used a polyclonal rabbit antiserum which was raised against the HPLC-purified synthetic alt.M-CSF peptide coupled to KLH (EUROGENTEC). The antiserum was purified by affinity chromatography on the synthetic alt.M-CSF peptide coupled to CNBr-activated sepharose®4B as described by the manufacturer (Amersham Pharmacia Biotech) and used at 2 μg/ml. As control we used 2 μg/ml rabbit IgG, provided by Dr. J.-P. Vaerman (Université Catholique de Louvain, Brussels, Belgium). Staining for M-CSF was done with 12.5 μg/ml of a mouse IgG2a mAb against human M-CSF (clone 26730.11; R&D Systems). As isotype control antibody we used the antimelanoma antibody KBA62 (Immunotech). Incubation with primary antibody and control antibody was done for 1 or 4 h for alt.M-CSF or M-CSF, respectively. Binding sites of primary antibodies were visualized using the mouse or rabbit EnVision™ + System, Peroxidase kit (Dako) according to the manufacturer's specifications. Enzymatic color development was performed using 3′3′9-aminoethyl carbazole (AEC) as substrate (Dako). Cells were then slightly counterstained with hematoxylin III according to Gill (Merck) and mounted with Faramount aqueous mounting medium (Dako). Slides were examined under a ZEISS Axioplan microscope equipped with a Hamamatsu C5810 color chilled 3 CCD camera, and images were captured by a Scion CG-7 frame grabber.
Extraction of HLA-associated Peptides and HPLC Fractionation.
Peptides were acid eluted from affinity-purified HLA class I molecules as described previously with modifications 19. In brief, 109 LB1047-RCC cells were lysed in buffer containing 20 mM Tris, pH 8.0, 150 mM NaCl, 1% NP-40, 1 mM PMSF, 5 μg/ml aprotinin, 10 μg/ml leupeptide, 10 μg/ml pepstatin A, 5 mM EDTA, and 0.04% sodium azide. After sample centrifugation at 100,000 g for 1 h, supernatant was filtered (0.22 μm) and precleared through a Sepharose column (Amersham Pharmacia Biotech). HLA molecules were then affinity-purified using mAb W6/32 coupled to CNBr-activated Sepharose. After elution of HLA class I molecules and associated peptides with 10% acetic acid, pH 2, dissociated peptides were separated from HLA molecules by centrifugation through Ultrafree-CL 5,000-kD filter (Millipore) at 3,500 g for 5 h. Peptide extracts were then fractionated by reverse-phase HPLC (rp-HPLC). Aliquots from 109 LB1047 cells were injected on a narrow bore 2 × 150 mm Deltapack C18 column (Waters) and eluted using a 60-min linear gradient of acetonitrile in water (0–60%) containing 0.1% trifluoroacetic acid at a flow rate of 200 μl/min. The column effluent was monitored with a UV detector at 210 nm. Fractions of 200 μl were collected, dried under vacuum, and stored at −20°C until use. Synthetic peptides were injected and eluted on the same C18 column under identical conditions.
Peptide Binding Assay.
Synthetic peptides were made by standard Fmoc chemistry using a peptide synthesizer Symphony/Multiplex™ (Protein Technologies Inc.); all peptides were >80% pure by analytical HPLC. Peptides were tested for binding to HLA-B*3501 in a competition assay as described recently 20. In brief, 721.221 cells transfected with HLA-B*3501 were stripped of HLA-bound peptides by mild acid treatment (pH 3.2), and incubated overnight at 4°C with a fluorescein (FL)-labeled reference peptide at a fixed concentration (250 nM), together with decreasing concentrations (50 to 0.09 μM) of competitor peptides. After washing, the ability of each peptide to compete the binding of FL-labeled reference peptide was assayed by measuring the amount of HLA-bound FL-labeled peptide with a FACScan™ (Becton Dickinson). The reference peptide was LPSC(FL)ADVEF, a Cys-derivative analogous to the tyrosinase antigenic peptide presented by HLA-B35, LPSSADVEF 21. The concentration of each peptide needed to inhibit 50% binding of the FL-labeled peptide (IC50) was calculated according to the respective exponential regression curve.
Results
CTL Clones Recognizing RCC Line LB1047-RCC.
By stimulating in vitro with autologous tumor cells the T lymphocytes infiltrating the kidney tumor of patient LB1047, we obtained a panel of CD8+ CTL clones that specifically lysed the autologous tumor cells, but not the autologous EBV-transformed B lymphocytes nor the NK-sensitive cells K562. Two allogeneic RCC lines expressing HLA-B35 were also recognized by CTL 403A/9, indicating that this CTL clone might recognize a shared antigen presented by HLA-B35 (Fig. 1).
Figure 1.

Lytic activity of CTL clone 403A/9. Targets were: autologous tumor cells LB1047-RCC, allogeneic HLA-B35–positive renal carcinoma cells HA7-RCC and LE9211-RCC, and the corresponding EBV-transformed B cell lines LB1047-EBV, HA7-EBV, and LE9211-EBV. NK target K562 was also used as control. All target cells were treated with 100 U/ml IFN-γ 24 h before the test. Chromium release was measured after 4 h.
Identification of the Antigen Recognized by CTL 403A/9.
To identify the antigen recognized by CTL 403A/9, we constructed a directional cDNA library with mRNA from tumor line LB1047-RCC as described recently 16. DNA from this library was transiently transfected into 293-EBNA cells together with DNA from the autologous HLA-B*3501 cDNA plasmid construct. We then screened the transfected cells for their ability to stimulate CTL 403A/9 by adding CTL to the microcultures and measuring the production of IFN-γ. We isolated a cDNA which was able to stimulate CTL 403A/9 when transfected with the HLA-B*3501 cDNA (Fig. 2).
Figure 2.

Isolation of a cDNA encoding the antigen recognized by CTL 403A/9. 293-EBNA cells were transfected transiently with cDNA 314 and with a plasmid construct encoding HLA-B*3501. CTL 403A/9 was added after 24 h and the amount of IFN-γ in the supernatant was measured 24 h later. LB1047-RCC cells were used as positive control.
This cDNA was 4 kb long, and its sequence corresponded to the full-length transcript of the gene encoding M-CSF, a cytokine also known as CSF-1, which promotes growth, differentiation, and activation of monocytes, macrophages, and osteoclasts 22 23. To identify the region of the M-CSF transcript that encodes the antigenic peptide, we transfected truncated variants of the cDNA, and found that fragments containing only the 5′-terminal 335 nucleotides were able to confer antigenic expression (data not shown). We tested a series of synthetic peptides derived from this region of the M-CSF protein sequence for recognition by CTL 403A/9, but they were all negative. We then analyzed the 5′ nucleotidic sequence in the two other reading phases and identified an alternative ORF encoding a putative polypeptide of 25 amino acids (alt.M-CSF; Fig. 3 A). To determine whether the antigenic peptide was derived from this polypeptide, we constructed minigenes encoding either the full length or the first 20 amino acids of alt.M-CSF, and transfected them with the HLA-B*3501 cDNA into 293 cells. We observed that CTL 403A/9 recognized cells transfected with both constructs, suggesting that the antigenic peptide derived from the first 20 amino acids of this alternative translation product of the M-CSF cDNA (Fig. 3 B).
Figure 3.



Identification of the peptide recognized by CTL 403A/9. (A) Partial sequence of the 5′ end of the M-CSF cDNA, showing the alternative ORF (boxed) coding for a 25-amino acid polypeptide (alt.M-CSF), and the initial part of the major ORF which encodes the leader sequence and the first residues of the mature M-CSF, which starts at position +1. The arrow indicates the boundary between exon 1 and 2 (reference 46). The antigenic peptide in the alt.M-CSF sequence is shown in bold. (B) CTL recognition of cells transfected with minigenes containing the alternative ORF encoding alt.M-CSF. 293-EBNA cells were transfected with plasmids containing HLA-B*3501 and minigenes encoding either the full-length alt.M-CSF product of 25 amino acids or a truncated product containing the first 20 amino acids. CTL 403A/9 was added after 24 h and production of IFN-γ was measured 24 h later. (C) Recognition of peptide LPAVVGLSPGEQEY by CTL 403A/9. Chromium-labeled LB1047-EBV B cells were incubated 30 min at 37°C with the indicated peptides at various concentrations. CTL 403A/9 was added at an effector/target ratio of 10, and chromium release was measured after 4 h.
We then tested several synthetic peptides encoded by the alt.M-CSF sequence, and found that peptide LPAVVGLSPGEQEY was able to sensitize autologous EBV-transformed B cells to lysis by CTL 403A/9 (Fig. 3 C). Although class I–binding peptides are usually only 8 to 11 amino acids long, this 14-amino acid peptide appeared to be optimal as truncation of either the NH2- or the COOH-terminal residue resulted in the loss of CTL recognition (Fig. 3 C).
Expression of alt.M-CSF.
M-CSF is primarily an inflammatory cytokine that is not produced in a constitutive manner but is induced in areas of inflammatory reactions. The only tissues that express M-CSF constitutively are the pregnant uterus, where M-CSF was found to act as a placental growth and differentiation factor 22 24, and the bone marrow, where low constitutive levels of M-CSF have been detected and appear to control the differentiation of the monocyte/macrophage lineage 25. Despite this restricted expression of M-CSF, its mRNA is present in many tissues, including normal liver, kidney, bladder, and breast (26 27 28; and data not shown). This suggests that M-CSF expression is regulated at a posttranscriptional level. This control appears to be disrupted in certain cancers, such as those of the urogenital tract, which frequently produce high amounts of M-CSF detectable either by immunohistochemistry on tissue sections or by ELISA on serum samples. These tumors include breast, ovarian, uterine, renal, bladder, and prostate carcinomas 22 29 30 31. In some cases, the tumor cells also express the M-CSF receptor, which is the product of protooncogene c-fms, thereby allowing an autocrine loop that is associated with a poor prognosis of the tumor disease 32.
A control mechanism also appears to regulate the translation of alt.M-CSF, the alternative ORF described here, as we observed that several cell lines expressed the M-CSF mRNA but were not recognized by our CTL (data not shown). This implies that the expression profile of this new antigen cannot be established by RT-PCR. Therefore, we raised a rabbit antiserum against a synthetic peptide of 25 amino acids corresponding to the entire alt.M-CSF. This polyclonal antiserum was purified by affinity chromatography, but it was found unable to detect the alt.M-CSF peptide on Western blots, presumably because of the small size of the peptide. However, the antiserum gave a nice specific signal by immunohistochemistry, and it was used to stain sections of different kidney tumors. We found that 6/10 RCCs translated alt.M-CSF, including the autologous tumor specimen LB1047-RCC (Fig. 4 B). To determine whether alt.M-CSF was also translated in normal kidney, we tested a series of sections derived from biopsies of normal kidney donors. We observed a strong staining in most cells of the proximal tubule epithelium (Fig. 4 E). This staining appeared specific, as it was abolished by blocking with an excess of alt.M-CSF peptide but not with an irrelevant peptide (data not shown). Interestingly, the tubular cells that were stained with the alt.M-CSF antiserum were not stained with an antibody directed against the M-CSF cytokine (Fig. 4 F). This suggests that the translation of the major ORF and that of the alternative ORF of M-CSF are regulated independently.
Figure 4.
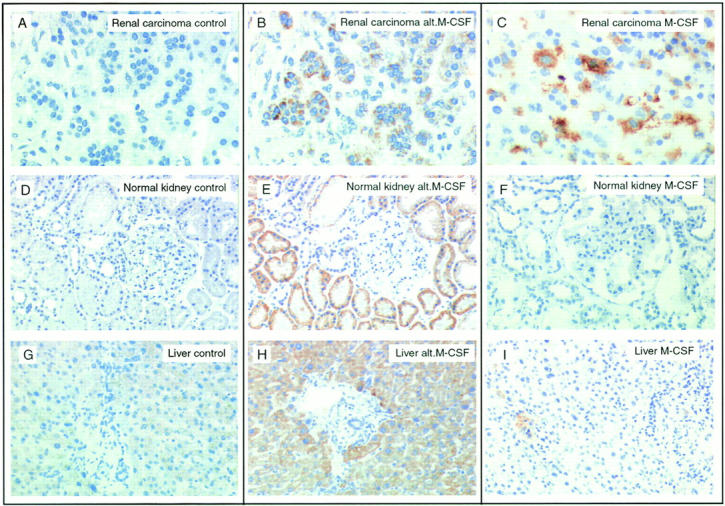
Expression of the alt.M-CSF product in tubular kidney cells and in hepatocytes. Paraffin sections from: the renal tumor of patient LB1047 (A and B); a normal kidney biopsy from a graft donor (D and E); or a normal liver (G and H) were stained with either a rabbit control IgG (A, D, and G) or a rabbit antiserum raised against the alt.M-CSF polypeptide (B, E, and H). Cryosections from: the renal tumor of patient EB56 (C), a normal kidney sample from patient LB2043 (F), or a normal liver (I) were stained with a monoclonal antibody against M-CSF. Original magnifications: ×200 (A–C) and ×100 (D–I).
To test directly the recognition of normal tubular kidney cells by our CTLs, we established short-term cultures from two different specimens of normal kidney cortex. These cultures, which were grown in the presence of epithelial growth factor, were mainly composed of proximal tubular cells, as determined by expression of aquaporin-1 7. Both lines were recognized by CTL403A/9 (Fig. 5). Although in vivo they do not produce the M-CSF cytokine (Fig. 4 F), the tubular cells grown in vitro secreted detectable amounts of M-CSF in their supernatant (Fig. 5). This is presumably related to the cellular activation that is inherent to in vitro cell culture in the presence of growth factors.
Figure 5.

Recognition of normal kidney cells by CTL 403A/9. Short-term cell lines LB2046-PTEC and LB2043-PTEC (10,000 cells/well), which derive from proximal tubular epithelial cells (PTEC), were incubated with CTL 403A/9 (10,000/well), and the production of TNF in the supernatant was measured after 24 h. In the absence of CTL, LB2043-PTEC, and LB2046-PTEC produced 2 and 3 pg/ml TNF, respectively. As indicated, both lines were transiently transfected with HLA-B*3501, to ensure expression of the presenting molecule. Line LB2046-PTEC, which was already recognized by CTL 403A/9 in the absence of transfection, was later confirmed to express HLA-B35 naturally. The amount of M-CSF produced by the same cell lines was meas-ured by ELISA on culture supernatants and is indicated.
Because of the constitutive expression of alt.M-CSF in normal renal tubular cells, we also investigated other normal tissues by immunohistochemistry. We observed a strong and homogenous staining of normal hepatocytes (Fig. 4 H). Normal thyroid, lung, stomach, colon, ovary, and breast were negative, but a few isolated cells of the duodenal submucosa were also stained (data not shown). Although they express alt.M-CSF, hepatocytes do not produce the M-CSF cytokine (Fig. 4 I). Again, these data indicate that the expression of the products of the two ORFs is differentially regulated, and that translation of alt.M-CSF can occur in the absence of translation of M-CSF.
Elution of the Natural Peptide Bound to HLA-B35.
Peptides presented by MHC class I molecules usually have a length between 8 and 11 amino acids. The optimal synthetic peptide recognized by our CTLs is 14 amino acids long. Because of this unusual length, we wanted to determine whether the natural ligand presented by HLA-B35 at the surface of the tumor cells was identical to the synthetic peptide of 14 amino acids. We therefore eluted the peptides bound to the HLA class I molecules purified from LB1047-RCC cells, and separated the peptides by hplc. Fractions were then pulsed onto HLA-B35–positive target cells, CTL 403A/9 was added, and IFN-γ production was measured to estimate CTL activation. As shown in Fig. 6 A, only fraction 28, which eluted from the HPLC column after 28 min, was able to stimulate the CTLs, indicating that it contained the epitope under its natural form. To determine whether the active material present in fraction 28 was identical to the synthetic 14-mer peptide, we ran the synthetic peptide on the same HPLC column under identical conditions. We observed that the retention time of the peptide, measured by UV absorption, was 28.5 min (Fig. 6 B). Furthermore, when the fractions obtained with the synthetic 14-mer peptide were pulsed onto target cells, CTL 403A/9 recognized fraction 28, and to a lower extent fraction 29, but no other fraction (Fig. 6 C). Synthetic peptides extended by one residue at the NH2 terminus or the COOH terminus were also run on the column and found by UV absorption to elute at 29.8 and 27.4 min, respectively. Peptides shorter than the 14-mer eluted before 26.5 min. These results demonstrate that the natural peptide is identical to the synthetic peptide, and has a length of 14 amino acids.
Figure 6.
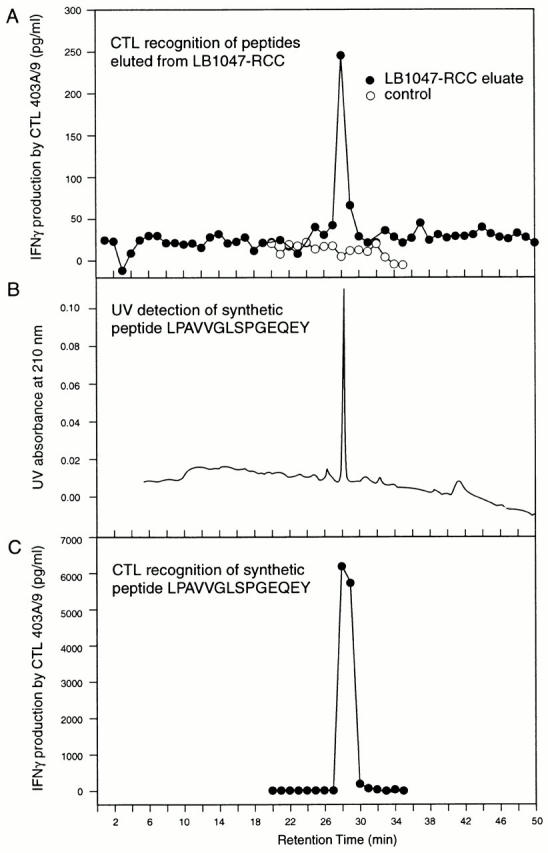
Elution of the natural peptide recognized by CTL 403A/9. (A) HPLC fractions of peptides eluted with acid from the HLA class I molecules of LB1047-RCC were tested for their ability to stimulate CTL 403A/9 after pulsing onto HLA-B35–positive target cells A110L-B35. The results of one representative experiment out of three are shown. (B) Synthetic peptide LPAVVGLSPGEQEY (150 pmol) was injected on the same HPLC column and its elution was monitored by UV absorption. (C) 1 pmol of the synthetic peptide was injected on the same column and the fractions were tested for CTL recognition as indicated in panel A.
Binding and Recognition of Peptide Analogues of LPAVVGLSPGEQEY.
The binding motif for HLA-B*3501 has a proline in position 2 and a tyrosine in position 9. The alt.M-CSF peptide has a proline in position 2 and a tyrosine at the COOH terminus. A priori, these two residues could therefore serve as anchor residues. However, because of its unusual length, the peptide needs to adopt a nonclassical conformation to fit into the groove of HLA-B35. For instance, it could either be anchored by its two extremities and bulge out of the groove in its middle part, or be anchored at one end and somewhere in the middle, with the other end overhanging the HLA cleft 33 34. To distinguish these possibilities, we tested a series of peptide analogues bearing different substitutions at various positions. The analogues were tested for their ability to bind to HLA-B35 in a competition assay with a fluorescent unrelated HLA-B35–binding peptide (Table ). Removal or alanine substitution of the COOH-terminal tyrosine completely abolished binding, indicating that the peptide is anchored by the COOH-terminal tyrosine. However, substitution of the proline in position 2 with either alanine, tryptophan, methionine, or glycine only marginally affected HLA-B35 binding, suggesting that this proline is not an anchor residue. In the binding motif for HLA-B35, the proline is located in position −7 with regard to the COOH-terminal tyrosine. At the corresponding position the alt.M-CSF peptide has a leucine. However, this leucine does not appear to play a role in binding, as its substitution with alanine did not affect binding. Another proline located in position −5 from the COOH terminus is apparently not involved either. Lastly, when the NH2-terminal leucine was changed into alanine, binding still occurred efficiently, whereas removal of this leucine completely abolished binding. Altogether, these results suggest that the peptide is bound to HLA-B35 via its two extremities: the side chain of the COOH-terminal tyrosine and the amino group of the NH2-terminal residue. We therefore speculate that the middle part of the peptide bulges out of the groove to compensate for the unusual length of the peptide. This would make the middle part of the peptide mostly accessible to the T cell receptor. In line with this prediction, when we tested the analogues for CTL recognition, we observed a complete loss of recognition of the analogues substituted at positions 7 or 9, although these peptides were very good binders (Table ). As amino acid changes in positions 1 and 2 also affected to some extent the strength of CTL recognition, we suggest that the core epitope interacting with the TCR is located at position 7–9, and the nature of residues 1 and 2 contributes to a proper orientation of the core epitope.
Table 1.
HLA Binding and CTL Recognition of Peptide Analogues
| Peptide sequence | Binding to HLA-B3501IC50 (μM) | Recognition by CTL 403A/9(pg/ml IFN-γ) |
|---|---|---|
| FPSDSWCYF | 4.1 | 0 |
| WTEAQRLDCWRGGQ | >50 | 0 |
| LPAVVGLSPGEQEY | 3.1 | 12,248 |
| LPAVVGLSPGEQE | >50 | 0 |
| LPAVVGLSPGEQEA | >50 | 0 |
| LAAVVGLSPGEQEY | 2.6 | 576 |
| LWAVVGLSPGEQEY | 4.7 | 2,461 |
| LMAVVGLSPGEQEY | 6.5 | 596 |
| LGAVVGLSPGEQEY | 17.4 | 21 |
| LPAVVGASPGEQEY | 1.3 | 0 |
| LPAVVGLSAGEQEY | 1.1 | 0 |
| LAAVVGLSAGEQEY | 1.7 | 0 |
| APAVVGLSPGEQEY | 2.8 | 674 |
| PAVVGLSPGEQEY | >50 | 0 |
Discussion
The antigenic peptide described here is encoded by an alternative ORF of the M-CSF gene. The other examples of peptides encoded by alternative ORF and recognized by antitumor CTLs were derived from the TRP1, NY-ESO1, and intestinal carboxyl esterase genes 11 12 13 14. In those cases, the translation of the alternative ORF appeared to parallel the translation of the major ORF, and there was a good correlation between the expression of the transcript and the translation of both ORFs 35. This is not the case here. The translation of the major and the alternative ORF of the M-CSF gene appears to be independently controlled. How this control mechanism works is unclear.
In the previous examples, where the translation of both ORFs was coordinated, the alternative ORF was also located within the major ORF, and it appeared likely that its translation resulted from a mechanism of leaky scanning, also named scanthrough 36 37. This mechanism, first described by Kozak et al., occurs when some ribosomes fail to initiate translation at the first ATG and continue to scan the mRNA 38 39. It is favored when the first ATG is not in an optimal context for efficient initiation. Such an optimal context, which consists of a purine in position −3 and/or a guanine in position +4, is absent from the ATG that opens the major ORF of M-CSF. However, translation of alt.M-CSF does not seem to result simply from leaky scanning, as it occurs in the absence of translation of the major ORF, at least in liver and kidney. A more complex mechanism appears to be involved. For instance, the translation of the major ORF, which is presumably cap dependent, could be inhibited by factors binding the 5′ untranslated region (5′UTR) of the mRNA. Such a mechanism has been proposed for TNF, whose translation is repressed in resting macrophages and induced under inflammatory conditions 40 41. In that scenario, the translation of alt.M-CSF should be cap independent and could result from mechanisms such as those involving internal ribosome entry sites (IRES), which would allow to overcome the blockade of cap-dependent translation. Alternatively, because alt.M-CSF is entirely encoded by exon 2, its translation in liver and kidney might still be cap dependent but occur on transcripts lacking the first exon and therefore lacking the putative translational repressor bound to the 5′UTR.
Contrary to many antigens recognized by CTLs on melanoma, all the shared antigens currently defined on kidney cancers are also expressed to some extent in normal tissues 42. The peptide described here is also not tumor specific, and it is therefore unclear whether it will be useful for immunotherapy of kidney cancer. The fact that CTLs against such antigens are present in tumor-infiltrating lymphocytes and were shown in some cases to be amplified in vivo suggests that they can take part in the antitumor response without obvious autoimmune toxicity 14. However, the study of animal models has indicated that effective tumor regressions mediated by such CTLs require the induction of very strong responses, which then can induce autoimmune damage 43.
A remarkable feature of the alt.M-CSF antigenic peptide is its exceptional length. As far as we know, it is the longest peptide known to be presented to CD8 T cells by MHC class I molecules. Given that its structure is closed at both ends, the groove of the class I molecules cannot accommodate such a long peptide in the extended conformation. The crystal structure of peptides of either 10 or 11 amino acids combined with class I molecules has indicated that such peptides can adopt two distinct conformations to fit in the class I groove. They can either bulge out of the cleft in their middle part or hang over the edge of the cleft at one extremity 33 34 44. Our results suggest that our 14-mer peptide adopts the first conformation, being anchored by its two extremities and protruding out of the groove in its middle part to compensate for its length. The fact that the TCR contact site appears to be located in the central part of the peptide is in line with this structure, which implies that the protruding part of the peptide is the most accessible for TCR recognition. The crystal structure of HLA-B*3501 associated with other peptides has been solved, and has led to the suggestion that B*3501 might not be able to accommodate peptides longer than 10 residues 45. This was based on the observation that the COOH terminus of peptides bound to B*3501 is placed closer to the A pocket than in other HLA class I alleles, so that a nonameric peptide already has a tendency to bulge out to fit in the groove. It was then speculated that longer peptides should protrude even more to fit, resulting in weaker binding interactions. Our results do not follow this prediction, as we observed for the 14-mer peptide a binding affinity similar to that of nonamer FPSDSWCYF, which is considered as a high-affinity binding peptide 20. Understanding the molecular basis for this high affinity will clearly require resolution of the crystal structure of HLA-B*3501 combined with the alt.M-CSF peptide to confirm the exact conformation of this 14-amino acids peptide in the B35 groove.
Acknowledgments
We thank D. Colau, D. Godelaine, L. Heidecker, L. Pilotte, M. Swinarska, and J. Van Snick for expert help at various steps of the project, and S. Depelchin for editorial assistance.
This work was supported by the Belgian Programme on Interuniversity Poles of Attraction initiated by the Belgian State, Prime Minister's Office, Science Policy Programing, and by grants from the Fonds J. Maisin (Belgium), the Fédération Belge contre le Cancer (Belgium), the FB Assurances, and VIVA (Belgium). M. Probst-Kepper was supported by a postdoctoral fellowship of the Deutsche Forschungsgemeinschaft (grant no. PR 554/1-1), and by the Schering Foundation, Germany.
Footnotes
Abbreviations used in this paper: FL, fluorescein; ORF, open reading frame; RCC, renal cell carcinoma; TIL, tumor-infiltrating lymphocyte.
B. Gaugler's present address is Laboratoire d'Immunologie des Tumeurs, Institut Paoli-Calmettes, Marseille 13009, France.
References
- Rock K.L., Goldberg A.L. Degradation of cell proteins and the generation of MHC class I-presented peptides. Annu. Rev. Immunol. 1999;17:739–779. doi: 10.1146/annurev.immunol.17.1.739. [DOI] [PubMed] [Google Scholar]
- Mayrand S.-M., Green W.R. Non-traditionally derived CTL epitopesexceptions that prove the rules? Immunol. Today. 1998;19:551–556. doi: 10.1016/s0167-5699(98)01342-5. [DOI] [PubMed] [Google Scholar]
- Skipper J.C.A., Kittlesen D.J., Hendrickson R.C., Deacon D.D., Harthun N.L., Wagner S.N., Hunt D.F., Engelhard V.H., Slingluff C.L.J. Shared epitopes for HLA-A3-restricted melanoma-reactive human CTL include a naturally processed epitope from Pmel-17/gp100. J. Immunol. 1996;157:5027–5033. [PubMed] [Google Scholar]
- Meadows L., Wang W., den Haan J.M., Blokland E., Reinhardus C., Drijfhout J.W., Shabanowitz J., Pierce R., Agulnik A.I., Bishop C.E. The HLA-A*0201-restricted H-Y antigen contains a posttranslationally modified cysteine that significantly affects T cell recognition. Immunity. 1997;6:273–281. doi: 10.1016/s1074-7613(00)80330-1. [DOI] [PubMed] [Google Scholar]
- Pierce R.A., Field E.D., den Haan J.M.M., Caldwell J.A., White F.M., Marto J.A., Wang W., Frost L.M., Blokland E., Reinhardus C. Cutting Edgethe HLA-A*0101-restricted HY minor histocompatibility antigen originates from DFFRY and contains a cysteinylated cysteine residue as identified by a novel mass spectrometric technique. J. Immunol. 1999;163:6360–6364. [PubMed] [Google Scholar]
- Guilloux Y., Lucas S., Brichard V.G., Van Pel A., Viret C., De Plaen E., Brasseur F., Lethé B., Jotereau F., Boon T. A peptide recognized by human cytolytic T lymphocytes on HLA-A2 melanomas is encoded by an intron sequence of the N-acetylglucosaminyltransferase V gene. J. Exp. Med. 1996;183:1173–1183. doi: 10.1084/jem.183.3.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Eynde B.J., Gaugler B., Probst-Kepper M., Michaux L., Devuyst O., Lorge F., Weynants P., Boon T. A new antigen recognized by cytolytic T lymphocytes on a human kidney results from reverse strand transcription. J. Exp. Med. 1999;190:1793–1799. doi: 10.1084/jem.190.12.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulie P.G., Lehmann F., Lethé B., Herman J., Lurquin C., Andrawiss M., Boon T. A mutated intron sequence codes for an antigenic peptide recognized by cytolytic T lymphocytes on a human melanoma. Proc. Natl. Acad. Sci. USA. 1995;92:7976–7980. doi: 10.1073/pnas.92.17.7976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins P.F., El-Gamil M., Li Y.F., Fitzgerald E.B., Kawakami Y., Rosenberg S.A. The intronic region of an incompletely spliced gp100 gene transcript encodes an epitope recognized by melanoma-reactive tumor-infiltrating lymphocytes. J. Immunol. 1997;159:303–308. [PubMed] [Google Scholar]
- Lupetti R., Pisarra P., Verrecchia A., Farina C., Nicolini G., Anichini A., Bordignon C., Sensi M., Parmiani G., Traversari C. Translation of a retained intron in tyrosinase-related protein (TRP)-2 mRNA generates a new cytotoxic T lymphocyte (CTL)-defined and shared human melanoma antigen not expressed in normal cells of the melanocytic lineage. J. Exp. Med. 1998;188:1005–1016. doi: 10.1084/jem.188.6.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.-F., Parkhurst M.R., Kawakami Y., Robbins P.F., Rosenberg S.A. Utilization of an alternative open reading frame of a normal gene in generating a novel human cancer antigen. J. Exp. Med. 1996;183:1131–1140. doi: 10.1084/jem.183.3.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.-F., Johnston S.L., Zeng G., Topalian S.L., Schwartzentruber D.J., Rosenberg S.A. A breast and melanoma-shared tumor antigenT cell responses to antigenic peptides translated from different open reading frames. J. Immunol. 1998;161:3596–3606. [PubMed] [Google Scholar]
- Aarnoudse C.A., van den Doel P.B., Heemskerk B., Schrier P.I. Interleukin-2-induced, melanoma-specific T cells recognize camel, an unexpected translation product of LAGE-1. Int. J. Cancer. 1999;82:442–448. doi: 10.1002/(sici)1097-0215(19990730)82:3<442::aid-ijc19>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Ronsin C., Chung-Scott V., Poullion I., Aknouche N., Gaudin C., Triebel F. A non-AUG-defined alternative open reading frame of the intestinal carboxyl esterase mRNA generates an epitope recognized by renal cell carcinoma-reactive tumor-infiltrating lymphocytes in situ. J. Immunol. 1999;163:483–490. [PubMed] [Google Scholar]
- Gaugler B., Brouwenstijn N., Vantomme V., Szikora J.-P., Van der Spek C.W., Patard J.-J., Boon T., Schrier P., Van den Eynde B.J. A new gene coding for an antigen recognized by autologous cytolytic T lymphocytes on a human renal carcinoma. Immunogenetics. 1996;44:323–330. doi: 10.1007/BF02602776. [DOI] [PubMed] [Google Scholar]
- Guéguen M., Patard J.-J., Gaugler B., Brasseur F., Renauld J.-C., Van Cangh P.J., Boon T., Van den Eynde B. An antigen recognized by autologous CTL on a human bladder carcinoma. J. Immunol. 1998;160:6188–6194. [PubMed] [Google Scholar]
- Hérin M., Lemoine C., Weynants P., Vessière F., Van Pel A., Knuth A., Devos R., Boon T. Production of stable cytolytic T-cell clones directed against autologous human melanoma. Int. J. Cancer. 1987;39:390–396. doi: 10.1002/ijc.2910390320. [DOI] [PubMed] [Google Scholar]
- Ennis P.D., Zemmour J., Salter R.D., Parham P. Rapid cloning of HLA-A,B cDNA by using the polymerase chain reactionfrequency and nature of errors produced in amplification. Proc. Natl. Acad. Sci. USA. 1990;87:2833–2837. doi: 10.1073/pnas.87.7.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt D.F., Henderson R.A., Shabanowitz J., Sakaguchi K., Michel H., Sevilir N., Cox A., Appela E., Engelhard V.H. Characterization of peptides bound to the class I MHC molecule HLA-A2.1 by mass spectrometry. Science. 1992;255:1261–1263. doi: 10.1126/science.1546328. [DOI] [PubMed] [Google Scholar]
- Mandruzzato S., Stroobant V., Demotte N., van der Bruggen P. A human CTL recognizes a caspase-8-derived peptide on autologous HLA-B*3503 molecules and two unrelated peptides on allogeneic HLA-B*3501 molecules. J. Immunol. 2000;164:4130–4134. doi: 10.4049/jimmunol.164.8.4130. [DOI] [PubMed] [Google Scholar]
- Morel S., Ooms A., Van Pel A., Wolfel T., Brichard V., van der Bruggen P., Van den Eynde B.J., Degiovanni G. A tyrosinase peptide presented by HLA-B35 is recognized on a human melanoma by autologous cytotoxic T lymphocytes. Int. J. Cancer. 1999;83:755–759. doi: 10.1002/(sici)1097-0215(19991210)83:6<755::aid-ijc10>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Roth P., Stanley E.R. The biology of CSF-1 and its receptor. Curr. Top. Microbiol. Immunol. 1992;181:141–167. doi: 10.1007/978-3-642-77377-8_5. [DOI] [PubMed] [Google Scholar]
- Fixe P., Praloran V. Macrophage colony-stimulating-factor (M-CSF or CSF-1) and its receptorstructure-function relationships. Eur. Cytokine Netw. 1997;8:125–136. [PubMed] [Google Scholar]
- Daiter E., Pampfer S., Yeung Y.G., Barad D., Stanley E.R., Pollard J.W. Expression of colony-stimulating factor-1 in the human uterus and placenta. J. Clin. Endocrinol. Metab. 1992;74:850–858. doi: 10.1210/jcem.74.4.1548350. [DOI] [PubMed] [Google Scholar]
- Besse A., Trimoreau F., Faucher J.L., Praloran V., Denizot Y. Prostaglandin E2 regulates macrophage colony stimulating factor secretion by human bone marrow stromal cells. Biochim. Biophys. Acta. 1999;1450:444–451. doi: 10.1016/s0167-4889(99)00048-8. [DOI] [PubMed] [Google Scholar]
- Ernst T.J., Ritchie A.R., Demetri G.D., Griffin J.D. Regulation of granulocyte- and monocyte-colony stimulating factor mRNA levels in human blood monocytes is mediated primarily at a post-transcriptional level. J. Biol. Chem. 1989;264:5700–5703. [PubMed] [Google Scholar]
- Roth P., Bartocci A., Stanley E.R. Lipopolysaccharide induces synthesis of mouse colony-stimulating factor-1 in vivo. J. Immunol. 1997;158:3874–3880. [PubMed] [Google Scholar]
- Ezure T., Ishiwata T., Asano G., Tanaka S., Yokomuro K. Production of macrophage colony-stimulating factor by murine liver in vivo. Cytokine. 1997;9:53–58. doi: 10.1006/cyto.1996.0135. [DOI] [PubMed] [Google Scholar]
- Scholl S.M., Lidereau R., de la Rochefordiere A., Le-Nir C.C., Mosseri V., Nogues C., Pouillart P., Stanley F.R. Circulating levels of the macrophage colony stimulating factor CSF-1 in primary and metastatic breast cancer patients. A pilot study. Breast Cancer Res. Treat. 1996;39:275–283. doi: 10.1007/BF01806155. [DOI] [PubMed] [Google Scholar]
- Chambers S.K., Kacinski B.M., Ivins C.M., Carcangiu M.L. Overexpression of epithelial macrophage colony-stimulating factor (CSF-1) and CSF-1 receptora poor prognostic factor in epithelial ovarian cancer, contrasted with a protective effect of stromal CSF-1. Clin. Cancer Res. 1997;3:999–1007. [PubMed] [Google Scholar]
- Steube K.G., Meyer C., Drexler H.G. Secretion of functional hematopoietic growth factors by human carcinoma cell lines. Int. J. Cancer. 1998;78:120–124. doi: 10.1002/(sici)1097-0215(19980925)78:1<120::aid-ijc19>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Kacinski B.M. CSF-1 and its receptor in ovarian, endometrial and breast cancer. Ann. Med. 1995;27:79–85. doi: 10.3109/07853899509031941. [DOI] [PubMed] [Google Scholar]
- Guo H.-C., Jardetzky T.S., Garrett T.P.J., Lane W.S., Strominger J.L., Wiley D.C. Different length peptides bind to HLA-Aw68 similarly at their ends but bulge out in the middle. Nature. 1992;360:364–366. doi: 10.1038/360364a0. [DOI] [PubMed] [Google Scholar]
- Collins E.J., Garboczi D.N., Wiley D.C. Three-dimensional structure of a peptide extending from one end of a class I MHC binding site. Nature. 1994;371:626–629. doi: 10.1038/371626a0. [DOI] [PubMed] [Google Scholar]
- Rimoldi D., Rubio-Godoy V., Dutoit V., Lienard D., Salvi S., Guillaume P., Speiser D., Stockert E., Spagnoli G., Servis C. Efficient simultaneous presentation of NY-ESO-1/LAGE-1 primary and nonprimary open reading frame-derived CTL epitopes in melanoma. J. Immunol. 2000;165:7253–7261. doi: 10.4049/jimmunol.165.12.7253. [DOI] [PubMed] [Google Scholar]
- Malarkannan S., Horng T., Shih P.P., Schwab S., Shastri N. Presentation of out-of-frame peptide/MHC class I complexes by a novel translation initiation mechanism. Immunity. 1999;10:181–690. doi: 10.1016/s1074-7613(00)80067-9. [DOI] [PubMed] [Google Scholar]
- Bullock T.N.J., Patterson A.E., Franlin L.L., Notidis E., Eisenlohr L.C. Initiation codon scanthrough versus termination codon readthrough demonstrates strong potential for major histocompatibility complex class I-restricted cryptic epitope expression. J. Exp. Med. 1997;186:1051–1058. doi: 10.1084/jem.186.7.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. The scanning model for translationan update. J. Biol. Cell. 1989;108:229–241. doi: 10.1083/jcb.108.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. Initiation of translation in prokaryotes and eukaryotes. Gene. 1999;234:187–208. doi: 10.1016/s0378-1119(99)00210-3. [DOI] [PubMed] [Google Scholar]
- Gueydan C., Droogmans L., Chalon P., Huez G., Caput D., Kruys V. Identification of TIAR as a protein binding to the translational regulatory AU-rich element of tumor necrosis factor α mRNA. J. Biol. Chem. 1999;274:2322–2326. doi: 10.1074/jbc.274.4.2322. [DOI] [PubMed] [Google Scholar]
- Piecyk M., Wax S., Beck A.R.P., Kedersha N., Gupta M., Maritim B., Chen S., Gueydan C., Kruys V., Streuli M., Anderson P. TIA-1 is a translational silencer that selectively regulates the expression of TNF-α. EMBO J. 2000;19:4154–4163. doi: 10.1093/emboj/19.15.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Eynde B., Probst-Kepper M. Antigens recognized by T lymphocytes on renal cell carcinoma Renal and Adrenal Tumours—Biology and Management OxfordUniversity Press, Oxford, UK. In press2001. [Google Scholar]
- Ludewig B., Ochsenbein A.F., Odermatt B., Paulin D., Hengartner H., Zinkernagel R.M. Immunotherapy with dendritic cells directed against tumor antigens shared with normal host cells results in severe autoimmune disease. J. Exp. Med. 2000;191:795–803. doi: 10.1084/jem.191.5.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speir J.A., Stevens J., Joly E., Butcher G.W., Wilson I.A. Two different, highly exposed, bulged structures for an unusually long peptide bound to rat MHC class I RT1-A. Immunity. 2001;14:81–92. doi: 10.1016/s1074-7613(01)00091-7. [DOI] [PubMed] [Google Scholar]
- Menssen R., Orth P., Ziegler A., Saenger W. Decamer-like conformation of a nona-peptide bound to HLA-B*3501 due to non-standard positioning of the C terminus. J. Mol. Biol. 1999;285:645–653. doi: 10.1006/jmbi.1998.2363. [DOI] [PubMed] [Google Scholar]
- Ladner M.B., Martin G.A., Noble J.A., Nikoloff D.M., Tal R., Kawasaki E.S., White T.J. Human CSF-1gene structure and alternative splicing of mRNA precursors. EMBO J. 1987;6:2693–2698. doi: 10.1002/j.1460-2075.1987.tb02561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]