

Abstract
Signal transducer and activator of transcription (STAT)-induced STAT inhibitor 1 (SSI-1) is known to function as a negative feedback regulator of cytokine signaling, but it is unclear whether it is involved in other biological events. Here, we show that SSI-1 participates and plays an important role in the insulin signal transduction pathway. SSI-1–deficient mice showed a significantly low level of blood sugar. While the forced expression of SSI-1 reduced the phosphorylation level of insulin receptor substrate 1 (IRS-1), SSI-1 deficiency resulted in sustained phosphorylation of IRS-1 in response to insulin. Furthermore, SSI-1 achieves this inhibition both by binding directly to IRS-1 and by suppressing Janus kinases. These findings suggest that SSI-1 acts as a negative feedback factor also in the insulin signal transduction pathway through the suppression of IRS-1 phosphorylation.
Keywords: insulin, IRS-1, SSI-1/SOCS1 signaling, Janus kinase, diabetes
Introduction
Insulin is a central regulator of various metabolic processes including cell growth, the uptake and metabolism of glucose, the synthesis and storage of neutral fat, and protein synthesis. Insulin mediates its biological effects through its specific receptor, the insulin receptor (IR). Like many growth factor receptors, IR has an intrinsic tyrosine kinase activity and activation of this activity takes place upon insulin binding 1 2. Insulin receptor substrate (IRS) proteins are substrate for IR and phosphorylated at multiple sites by treatment not only with insulin but also with insulin-like growth factor (IGF)-1 or IL–4 1 3. Phosphorylated IRS functions as a docking protein for the Grb2 or p85 regulatory subunit of phosphatidylinositol 3-kinase (PI3K) and endows the soluble factors, including insulin, with various biological activities 1 2. Both IRS-1– and IRS-2–deficient mice showed insulin resistance 4 5 6, indicating that IRS proteins are key molecules in insulin signaling. Interestingly, it has been suggested that Janus kinases (JAKs) are activated by insulin or IGF-1 treatment 7 8.
Proteins of the signal transducer and activator of transcription (STAT)-induced STAT inhibitor (SSI), also known as suppressor of cytokine signaling (SOCS) or cytokine-inducible src homology (SH)2 domain–containing protein (CIS) family, have been identified as negative feedback regulators of the JAK-STAT signal transduction pathway 9 10 11. So far, eight members of the SSI family gene have been identified 12 13 14. All member proteins of this family contain an SH2 domain in their central portion and conserved SC motif, also known as a SOCS box, in their COOH terminus 12 13 14. SSI-1 binds to the phosphotyrosine residue of JAK through its SH2 domain, and inhibits subsequent STAT activation by suppressing JAK activity 12. Previously, SSI-1–deficient mice have been generated and analyzed 15. SSI-1–deficient mice were normal at birth, but showed severe growth retardation with aging. They also showed atrophy in the thymus, a reduction in the number of lymphocytes, and infiltration of mononuclear cells in several organs, and died within 3 wk after birth 15.
In this report, we present evidence that SSI-1 is involved in the insulin signal transduction pathway. Lower blood sugar level was observed in SSI-1–deficient mice, indicating that deficiency of SSI-1 results in hypersensitivity to insulin action. In addition, forced expression of SSI-1 repressed the tyrosine phosphorylation of IRS-1, whereas lack of SSI-1 prolonged IRS-1 phosphorylation after insulin treatment. Furthermore, enhanced differentiation into adipocytes was demonstrated in embryonic fibroblasts of SSI-1–deficient mice in response to insulin. These results suggest that SSI-1 plays an important role in the negative regulation of the insulin signal transduction pathway through suppression of IRS-1 phosphorylation. This SSI-1 is involved in a signaling pathway other than cytokine signaling in vivo.
Materials and Methods
Blood Sugar, Serum Insulin, and Urine C-Peptide Level of SSI-1−/− Mice.
All observations were performed on 7–10-d-old SSI-1+/+ and SSI-1−/− mice. Blood was drawn from hearts, and blood sugar level was determined. Serum insulin level was also measured by RIA. To measure the C peptide level, urine was collected for 24 h, and the C peptide level was determined by ELISA.
Glucose Uptake.
105 cells were seeded on 6-well plates and stimulated with insulin at the indicated concentration for 60 min. 2-deoxy-d-[1-3H] glucose (2DOG) and 0.1 mM unlabeled 2DOG were added, and incubation was continued for a further 20 min. After washing with ice-cold PBS twice, cells were solubilized with 0.5 N NaOH and 0.1% SDS. Aliquots were assessed for protein content and radioactivity.
Cell Culture.
L929 stably expressed SSI-1, SSI-3, or SOCS5, and 293T cells were cultured in DMEM supplemented with 10% FCS and 500 μg/ml of G418 (Nacalai Tesque). SSI-1–expressing 3T3-L1 cells were also maintained in G418 containing DMEM. Murine embryonic fibroblast (MEF) cells were grown in DMEM containing 10% FCS, nonessential amino acids (GIBCO BRL), penicillin G, and streptomycin.
Insulin Stimulation In Vivo.
4-wk-old mice were fasted for 16 h, and then 5 U of human insulin were injected intraperitoneally. At 10 min after injection, adipose tissue was dissected immediately, lysed, and immunoprecipitatin was performed as described below.
Induction of Adipocyte Differentiation.
Induction of adipocyte differentiation was performed as described elsewhere 16. In brief, MEFs collected from 13.5 days post coitum (dpc) embryos were seeded onto 48-well plates, and propagated to confluence. After 48 h, the culture medium was replaced by the induction medium (DMEM containing 10% FCS, 0.5 mM 3-isobutyl-1-methyl xanthine, 0.5 mM dexamethasone, and 5 μg/ml of insulin). 2 d later, the induction medium was replaced with a maintenance medium (DMEM supplemented with 10% FCS and 5 μg/ml of insulin), which was renewed every other day. At day 6 of differentiation, adipose drops in the cells were stained with Oil Red-O and observed under a microscope. We used three independent pairs of SSI-1−/− and control mice from the same littermate and obtained almost identical results.
Reverse Transcription PCR.
L929 cells were stimulated with insulin and total RNA was isolated with the Total RNA Isolation System (Promega). Reverse transcription (RT) reaction was performed by using oligo dT primer and Moloney murine leukemia virus reverse transcriptase (Stratagene). The PCR reaction consisted of the following steps: 95°C for 1 min, 65°C for 1 min, 72°C for 1 min, 37 cycles for SSI-1; 95°C for 1 min, 62°C for 1 min, 72°C for 1 min, 38 cycles for SSI-3 and 32 cycles for SOCS5; and 95°C for 1 min, 60°C for 1 min, 72°C for 1 min, and 21 cycles for glyceraldehyde 3-phosphate dehydrogenase (G3PDH). The primer pairs were: SSI-1, 5′-CACCTTCTTGGTGCGCGACA-3′ and 5′-GCAGCTCGAAAAGGCAGTCG-3′; SSI-3, 5′-CCATGGTCACCCACAGCAAG-3′ and 5′-GTAAGCCCTCAAGGACCTAG-3′; SOCS5, 5′-CAGACAGGGAGCTTGGAAAG-3′ and 5′-GTTGAAGTCGAAGCTACGGG-3′; G3PDH, 5′-ACCACAGTCCATGCCATGCCATCAC-3′ and 5′-TCCACCACCCTGTTGCTGTA-3′.
Antibodies.
Anti–SSI-1 monoclonal antibody, 1262B 17, anti–SSI-3, and anti-SOCS5 polyclonal antibodies (Santa Cruz Biotechnology, Inc.) were used. Anti-JAK1 and anti-Tyk2 monoclonal antibodies were purchased from Transduction Laboratory, anti-IR, anti-JAK2, and anti–IRS-1 polyclonal antibodies from Santa Cruz Biotechnology, Inc., and antiphosphotyrosine monoclonal antibody, 4G10, from Upstate Biotechnology. Horseradish peroxidase (HRP)-conjugated anti–rabbit and anti–mouse IgG (Amersham Pharmacia Biotech) and HRP-conjugated anti–goat IgG (Southern Biotechnology Associates, Inc.) were used as second antibodies for Western blot analysis.
Immunoprecipitation and Western Blot Analysis.
Serum-starved L929 cells were treated with 10 nM insulin or 10 nM IGF-1 for the times indicated. The cells were lysed with an NP-40 lysis buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% NP-40, 10 mM Na2VO4, 0.5 mM dithiothreitol, 1 μg/ml pepstatin A, 1 μg/ml leupeptin, and 1 mM PMSF) and clarified by centrifugation. The supernatants were collected and incubated with the antibodies indicated for 16 h at 4°C. After incubation with protein A–sepharose 4B (Amersham Pharmacia Biotech) for 1 h, the beads were washed with the NP-40 lysis buffer four times, and then immunecomplex was resolved by 8% SDS-PAGE. The proteins were transferred onto a nitrocellulose filter (Schleicher & Schuell). After blocking with 5% skim milk, the filter was incubated with one of the first antibodies, and then with the appropriate second antibody. After washing, the signals were visualized by the ECL system (Amersham Pharmacia Biotech).
Transient Transfection.
293T cells were transfected with 2 μg of pEF-BOS-SSI-1, 2 μg of pEF-BOS-SSI-3, 2 μg of pEF-BOS-SOCS5, 5 μg of pEF-BOS-JAKs, 5 μg of pGEM3 SV HIR-IR, 3 μg of pCIBSD-IRS-1, or a combination of these expression plasmids with the standard calcium coprecipitation method. After 36 h, cells were harvested and subjected to Western blot analysis as described above with the indicated antibodies.
Results
Deficiency of SSI-1 Resulted in Hypoglycemia.
To characterize the physiological role of SSI-1 in vivo, we have previously generated mice which lack the SSI-1 gene (SSI-1−/− mice; reference 15). SSI-1−/− mice showed marked apoptosis in their lymphocytes and died within 3 wk after birth. Through the biochemical analyses of SSI-1−/− mice, we noticed that the blood sugar level of SSI-1 −/− mice was significantly lower than that of SSI-1+/+ mice (Fig. 1 A; +/+: 136.2 ± 26.5 mg/dl, n = 7; −/−: 84.9 ± 13.3 mg/dl, n = 7). However, the urine C peptide level of SSI-1−/− mice was no higher than that of SSI-1+/+ mice (Fig. 1 B; 396 ± 99.57 vs. 401.2 ± 97.19 ng/day, n = 6). Consistent with this, the serum insulin level of SSI-1−/− mice was also no higher than that of SSI-1+/+ mice (Fig. 1 C; +/+: 0.71 ± 0.40 ng/ml, n = 5; −/−: 0.56 ± 0.38 ng/ml, n = 5). These results indicated that the reduction in blood sugar level of SSI-1−/− mice was not due to the insulin level itself but to a change in sensitivity to insulin action. We speculated that SSI-1 also might act as a negative regulator of insulin signal transduction as well as of cytokine signaling and that SSI-1−/− mice might become hypersensitive to insulin action because of the lack of a suppression mechanism.
Figure 1.
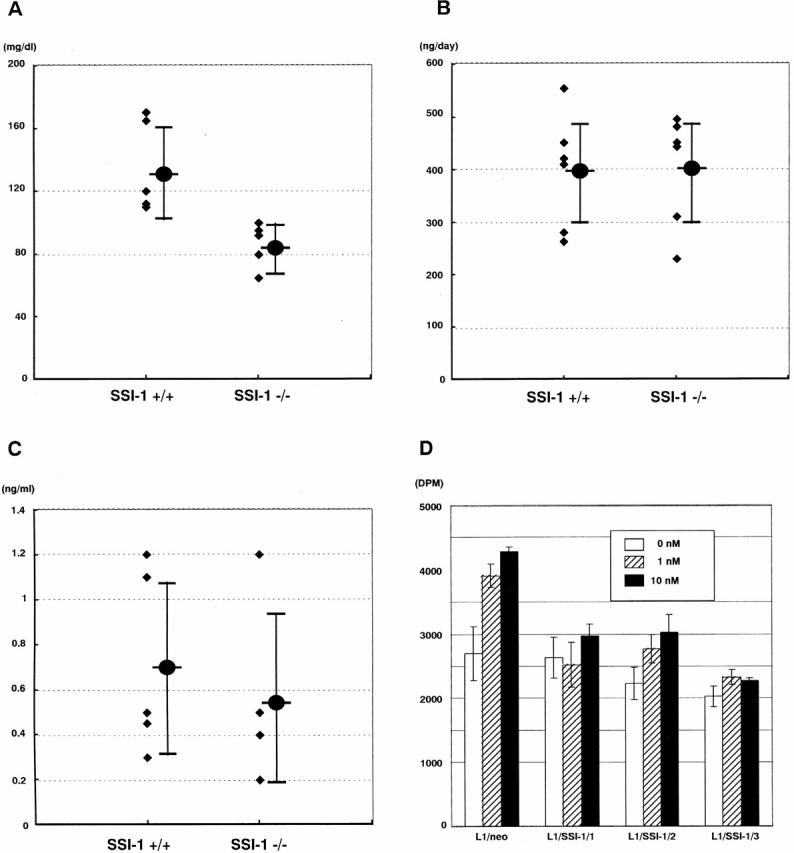
SSI-1−/− mice show low blood sugar level. (A) Blood sugar level, (B) urine c-peptide level, and (C) serum insulin level were measured in 7–10-d-old mice. ♦, raw data. Mean values ± SE are indicated as filled circles and vertical bars. (D) 3T3-L1/neo and three independent clones of 3T3-L1/SSI-1 cells were stimulated with insulin at 0 (white bar), 1 (hatched bar), and 10 nM (black bar) for 60 min, and incubated with 2DOG for a further 20 min. Each value is the mean ± SE of triplicate determinations.
To confirm this idea, we established SSI-1–expressing 3T3 L1 cells (L1/SSI-1) and performed a 2DOG uptake experiment (Fig. 1 D). L1/neo cells were facilitated on uptaking 2DOG in response to insulin, but in three independent clonal cell lines, L1/SSI-1/1, L1/SSI-1/2, and L1/SSI-1/3, 2DOG uptake was decreased compared with the parental cell line. It is noteworthy that the basal level of 2DOG uptake was also decreased in L1/SSI-1 cells, maybe due to the unresponsiveness to serum containing insulin in L1/SSI-1 cells. These results suggest that the expression level of SSI-1 affects the insulin action.
SSI-1 Inhibits the Phosphorylation of IRS-1 in Response to Insulin.
To elucidate how SSI-1 suppresses the insulin signal transduction, we first examined the effect of the SSI-1 protein on insulin signaling. SSI-1 is thought to bind the phosphotyrosine residue and block the phosphorylation cascade. Therefore, we expected that the forced expression of SSI-1 would alter the protein phosphorylation pattern after insulin treatment. We established the cell line L929/SSI-1 which stably expressed SSI-1 in L929 mouse fibroblast cells 20. Examination of the tyrosine phosphorylation pattern of total cellular proteins after insulin stimulation showed that phosphorylation of an ∼180-kD protein was significantly reduced in the L929/SSI-1 cells compared with L929/neo which was transfected with an empty vector (Fig. 2 A, indicated by arrow). Insulin stimulation induces the tyrosine phosphorylation of IRS-1 with a molecular mass of ∼180 kD 1 2. Therefore, we examined whether the reduced phosphorylation protein in L929/SSI-1 cells was the same as IRS-1. We also included SSI-3 and SOCS5 in this experiment because it has been reported that SSI-3 is induced by leptin or prolactin treatment and suggested that SSI-3 might be involved in metabolic regulation 18 19; Emanuelli et al. 21 showed that SSI-3 was induced by insulin, bound to IR, and inhibited STAT5 activation, and SOCS5 is induced after insulin stimulation as described below. To do this, we also established the cell lines L929/SSI-3 and L929/SOCS5, which expressed SSI-3 and SOCS5, respectively. Insulin treatment induced strong phosphorylation of IRS-1 in L929/neo cells (Fig. 2 B, top, lanes 1–4), whereas it was significantly reduced in L929/SSI-1 cells (Fig. 2. B, top, lanes 5–8). L929/SSI-3 cells also showed suppression of IRS-1 phosphorylation, but their inhibitory effect was rather weak compared with L929/SSI-1 cells (Fig. 2. B, top, lanes 9–12). In contrast to L929/SSI-1 and L929/SSI-3 cells, strong phosphorylation of IRS-1, almost the same as seen in L929/neo cells, was observed in L929/SOCS5 cells (Fig. 2 B, top, lanes 13–16). IRS-1 is also phosphorylated by treatment with IGF-1 2. Therefore, we analyzed the effect of SSI family proteins on IGF-1–stimulated IRS-1 phosphorylation and obtained almost the same result as with insulin (Fig. 2 B, bottom). Then, we analyzed whether SSI-1 deficiency led to augmentation of IRS-1 phosphorylation as a result of insulin treatment. Strong induction of IRS-1 phosphorylation was detected after 10 min of insulin stimulation, and it gradually declined at 60 and 180 min after stimulation in SSI-1+/+ MEFs (Fig. 2 C, top, lanes 1–4). In contrast, intense phosphorylation of IRS-1 in SSI-1−/− MEFs lasted, at least, up to 180 min (Fig. 2 C, top, lanes 5–8). These results suggest that SSI-1 and SSI-3 are capable of suppressing insulin signaling at the level of IRS-1 phosphorylation.
Figure 2.

SSI family proteins modulate the phosphorylation state of IRS-1. (A) L929/neo (lanes 1–4) and L929/SSI-1 (lanes 5–8) cells were stimulated with insulin for 0 (lanes 1 and 5), 10 (lanes 2 and 6), 30 (lanes 3 and 7), and 60 min (lanes 4 and 8). Total cell lysates were subjected to Western blot analysis with antiphosphotyrosine antibody. Arrow indicates reduced phosphorylation band in L929/SSI-1 cells. (B) L929/neo (lanes 1–4), L929/SSI-1 (lanes 5–8), L929/SSI-3 (lanes 9–12), or L929/SOCS5 (lanes 13–16) cells were treated with insulin (top and second panels) or IGF-1 (third and bottom panels) for the indicated times. Tyrosine phosphorylation of IRS-1 was analyzed (top and third panels) by immunoprecipitation followed by Western blot analysis. The second and bottom panels show that equal amounts of IRS-1 were immunoprecipitated. (C) MEFs derived from SSI-1+/+ (lanes 1–4) and SSI-1−/− (lanes 5–8) were treated with insulin and the phosphorylation state of IRS-1 was detected as in B. IP, immunoprecipitation; WB, Western blot analysis; pY, phosphotyrosine.
SSI-1 Suppresses the Phosphorylation of IRS-1 by Directly Binding.
We next studied the precise molecular mechanism by which SSI-1 and SSI-3 inhibit IRS-1 phosphorylation induced by insulin. Since SSI-1 is known to bind and inhibit JAKs 11 17, we considered that SSI-1 might act on IR in the same way. To this end, IR, SSI-1, or SSI-3 was transiently expressed in 293T cells, and the association of IR with SSI-1 or SSI-3 and the kinase activity of IR after insulin stimulation were analyzed. We found that neither SSI-1 nor SSI-3 either bound to IR or inhibited IR kinase activity (Fig. 3 A), which was consistent with our previously published data showing that SSI-1 could not suppress IR kinase activity in SSI-1–expressing M1 cells 9. Subsequently, we examined whether SSI-1/3 inhibited the phosphorylation of IRS-1 by binding to IRS-1 directly. 293T cells were transfected with IRS-1, SSI-1, or SSI-3 expression vector and cell extracts were prepared. After immunoprecipitation with anti–IRS-1 antibody, association of SSI-1/3 with IRS-1 was detected by Western blot analysis with anti–SSI-1/3 antibody. Binding of SSI-1 to IRS-1 was barely observed before insulin stimulation (Fig. 3 B, left, lane 3). However, when cells were stimulated with insulin, a substantial level of SSI-1 was coprecipitated with IRS-1 (Fig. 3 B, left, lane 4). This result indicated that SSI-1 preferentially bound to phosphorylated IRS-1, which suppressed further phosphorylation. On the other hand, we could not detect the association between SSI-3 and IRS-1 even after insulin stimulation in 293T cells (Fig. 3 B, right).
Figure 3.

Inhibition mechanism of IRS-1 phosphorylation by SSI-1 and SSI-3. (A) 293T cells were transfected with the expression vector carrying IR (lane 1), SSI-1 or SSI-3 (lane 2), or a combination of IR and SSI-1 or SSI-3 (lane 3), treated with insulin for 10 min before harvesting (lane 4), and then immunoprecipitated with an anti-IR antibody. Association of SSI-1 and SSI-3 with IR (top panel) and the kinase activity of IR (second panel) were analyzed by Western blot analysis using anti-SSI antibody and antiphosphotyrosine antibody, respectively. (B) 293T cells were transfected with the expression vector as indicated. After 36 h, cells were lysed and immunoprecipitated with an anti–IRS-1 antibody. Then Western blot analysis was performed with anti-SSI antibody (top). (C) IR-transfected 293T cells (left) were untreated (lane 1) or treated with insulin for 10 min (lane 2) and immunoprecipitated with anti–IRS-1 antibody. The immunecomplex was subjected to Western blot analysis with anti-JAK1 (top), anti-JAK2 (second panel), and anti–IRS-1 antibody (bottom). 4-wk-old mice were also stimulated with 5 U of insulin for 10 min. Fat cells were removed and processed as above (right). (D) 293T cells were transfected with IRS-1 (lanes 1, 6, and 11) or cotransfected IRS-1 with JAKs (JAK1, lanes 1–5; JAK2, lane 6–10; Tyk2, lanes 11–15), with JAKs and SSI-1 (lanes 3, 8, and 13), with JAKs and SSI-3 (lanes 4, 9, and 14), or with JAKs and SOCS5 (lanes 5, 10, and 15) expression vectors. Cells were lysed, immunoprecipitated with anti–IRS-1 antibody, and then subjected to Western blot analysis phosphotyrosine antibody to examine the phosphorylation state of IRS-1 (top panel). *IgG light chain.
SSI-1 Inhibits IRS-1 Phosphorylation Also by Inhibiting JAKs.
In spite of the lack of association of SSI-3 with IRS-1, SSI-3 still inhibits the IRS-1 phosphorylation (Fig. 2 B). Therefore, we further studied the inhibition mechanism of IRS-1 phosphorylation. Based on the fact that IL-4, IL-9, IL-13, and oncostatin M, whose receptors do not contain an intrinsic kinase activity, evoke the phosphorylation of IRS-1, and on the recent reports which demonstrated that JAKs are phosphorylated by insulin treatment 7 8, we examined the involvement of JAKs in insulin signaling. First, we demonstrated the association of JAKs with IRS-1. The interaction between IRS-1 and JAK1 or JAK2 (Fig. 3 C, lanes 1 and 2) but not Tyk2 (data not shown) was detected only after insulin treatment. Furthermore, we also found that JAK1 bound to IRS-1 in the adipose tissue in vivo after insulin stimulation (Fig. 3 C, lanes 3 and 4). Unfortunately, we could not detect the association of JAK2 and IRS-1 in the adipose tissue, maybe because of the low expression level of JAK2 in adipose tissue. These results indicated that JAKs are capable of binding to IRS-1 with insulin stimulation–dependent manner. Moreover, JAK1 and JAK2, but not Tyk2, bound to IR in 293T cells (data not shown).
We next investigated whether IRS-1–bound JAKs could phosphorylate IRS-1 and whether SSI family proteins could inhibit this process. It is known that the forced expression of JAKs results in their autophosphorylation and activation. Therefore, IRS-1 and JAKs together with SSI-1, SSI-3, or SOCS5, were coexpressed in 293T cells. Coexpression with JAK1 resulted in a significant level of IRS-1 phosphorylation. However, the degree of IRS-1 phosphorylation was greatly reduced by the coexpression of SSI-1 or SSI-3 with JAK1. Coexpression of SOCS5 with JAK1, on the other hand, did not affect JAK1 activity on IRS-1 phosphorylation (Fig. 3 D, top, lanes 1–5), which is consistent with the inhibition specificity of IRS-1 phosphorylation in L929 cell lines (Fig. 2 B). Furthermore, we obtained almost the same results for JAK2 as for JAK1 (Fig. 3 D, top, lanes 6–10). In contrast, the effect of Tyk2 on IRS1 phosphorylation was very weak compared with JAK1 and JAK2 (Fig. 3 D, top, lanes 11–15), which is parallel to the binding specificity of IRS-1 to JAKs and suggested that JAK1 and JAK2 might be involved in insulin signaling.
SSI-1 −/− MEFs Show Enhanced Differentiation into Adipocytes by Insulin.
For further confirmation of involvement of SSI-1 in insulin signaling, we used an established adipocyte differentiation system in which MEFs convert into mature adipocyte in the presence of adipogenic hormones such as insulin 16. We expected that if SSI-1 acts as a negative regulator in the insulin signaling, SSI-1 deficiency should lead to the enhancement of differentiation. As expected, MEFs derived from SSI-1−/− mice showed burst induction of differentiation into adipocytes compared with SSI-1+/+ MEFs after 6 d of insulin treatment (Fig. 4A and Fig. B). An additional 6 d of induction with insulin resulted in almost the same proportion of MEFs derived from SSI-1+/+ mice as those derived from SSI-1−/− mice differentiating into adipocytes (Fig. 4C and Fig. D). These results clearly indicate that SSI-1 can function as a negative regulator of insulin signaling.
Figure 4.
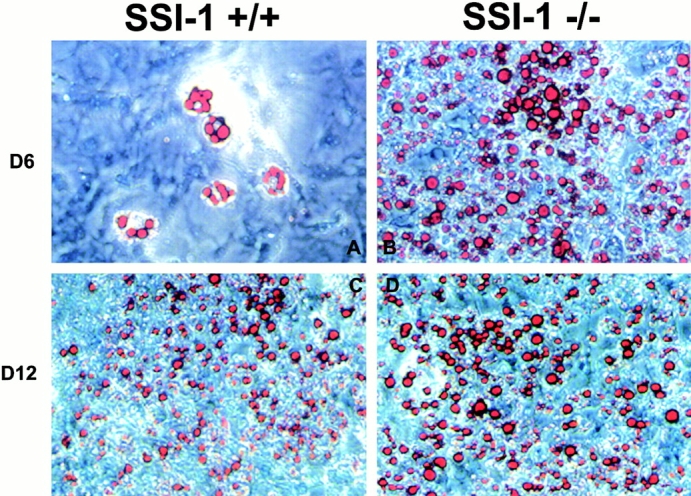

SSI-1−/− MEFs show enhanced induction of differentiation into adipocytes by insulin. (A and C) MEFs derived from SSI-1+/+ mice or (B and D) SSI-1−/− mice were induced to differentiate into adipocytes as described in Materials and Methods. Three independent littermate pairs were used for experiments, and representative results are shown. The lipid drops in the cells were stained red by Oil Red-O. Original magnification: ×20. (E) L929 cells were treated with insulin and total RNA was isolated. RT-PCR analysis was then performed as described in Materials and Methods.
Induction of SSI Family mRNAs in Response to Insulin.
Finally, we examined the induction of the SSI family mRNAs after insulin stimulation to determine whether SSI families function as a feedback regulator for insulin. The result indicated that SSI-1, SSI-3, and SOCS-5 mRNAs were all induced after insulin stimulation (Fig. 4 E).
Discussion
SSI-1 has been thought to function as an inhibitor of the cytokine signal transduction pathway. This report demonstrates that SSI-1 is also involved in the insulin signal transduction pathway in vivo. SSI-1 suppresses the insulin signal transduction pathway at the level of the phosphorylation of IRS-1 by interaction with IRS-1 and inhibiting JAKs activated by insulin and does not affect IR kinase activity. On the other hand, SSI-3 suppresses IRS-1 phosphorylation only by inhibiting JAKs. These differential inhibition mechanisms of IRS-1 phosphorylation between SSI-1 and SSI-3 are consistent with Fig. 2 B, demonstrating that the inhibitory effect of SSI-1 was stronger than that of SSI-3. Although it is shown that SSI-3 directly bound to IR and inhibited STAT5 activation in response to insulin 20, it is evident that SSI-1 may play an important role in vivo because SSI-1−/− mice show low blood sugar level even in the presence of SSI-3. In addition, we could not observe direct binding of SSI-1 and SSI-3 to IR in 293T cells but demonstrated that they suppressed IRS-1 phosphorylation.
Our study showed that SSI-1 might be capable of inhibiting all effector molecules activated by JAKs. Since the identification of SSI-1, it has been thought to act as a negative regulator specific for STAT activation by inhibiting the activity of JAKs in vivo. However, our results suggest that SSI-1 may have the potential to suppress effector molecules other than STATs. For example, IL-4 stimulation induces not only STAT6 activation but also phosphorylation of IRS proteins. Therefore, SSI-1 might inhibit both of these transduction pathways in IL-4 signaling. In addition, we demonstrated that SSI-1 associated with IRS-1 and suppressed further phosphorylation of IRS-1. Although it has been shown that SSI-1 could interact with various signaling molecules including c-kit, vav, and Grb-2 22, the biological significance of this interaction is not yet clear. Therefore, our findings established a novel concept of SSI-1 function.
In recent years, several reports suggested that TNF-α or INF-γ antagonizes insulin action and generates an insulin-resistant state 23 24. These phenomena raise a clinical point that patients with cytokine treatment frequently suffer from diabetes. Our present study suggests a possibility that SSI-1 might be involved in cytokine-induced diabetes because both TNF-α and INF-γ induce SSI-1 21. Furthermore, our unpublished data indicate that SSI-1 is indispensable to the cross-talk inhibition between several cytokines, for example, INF-γ stimulation does not antagonize IL-4 signaling in SSI-1–deficient T cells (Naka, T., H. Tsutsui, Y. Kawazoe, H. Kohzaki, Y. Morita, R. Nakagawa, M. Narazaki, K. Adachi, T. Yoshimoto, K. Nakanishi, and T. Kishimoto, unpublished observation). Therefore, our study suggests that SSI-1 might regulate the cross-talk not only of cytokine signaling but also between cytokine and endocrine hormones, and SSI-1 may be applicable to insulin-resistant diabetes.
Acknowledgments
We thank Drs. S. Nagata (Osaka University), R. Fukunaga (Osaka University), J. Krolewski (Columbia University, New York, NY), T. Asano (University of Tokyo, Tokyo, Japan), and T. Kobayashi (Toyama Medical University, Toyama, Japan) for providing pEF-BOS vector, JAK1 cDNA, Tyk2 cDNA, IRS-1 expression vector, and IR expression vector, respectively. We also thank Ms. A. Saitho and R. Harada for their secretarial assistance.
This work was supported by a Grant-in-Aid from the Ministry of Education, Science, and Culture, at Japan.
References
- White M.F. The insulin signalling system and the IRS proteins Diabetologia. 40Suppl.1997. S2 S17 [DOI] [PubMed] [Google Scholar]
- Myers M.G., Jr., Sun X.J., White M.F. The IRS-1 signaling system. Trends Biochem. Sci. 1994;7:289–293. doi: 10.1016/0968-0004(94)90007-8. [DOI] [PubMed] [Google Scholar]
- Keegan A.D., Nelms K., White M., Wang L.M., Pierce J.H., Paul W.E. An IL-4 receptor region containing an insulin receptor motif is important for IL-4-mediated IRS-1 phosphorylation and cell growth. Cell. 1994;5:811–820. doi: 10.1016/0092-8674(94)90356-5. [DOI] [PubMed] [Google Scholar]
- Araki E., Lipes M.A., Patti M.E., Bruning J.C., Haag B., III, Johnson R.S., Kahn C.R. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature. 1994;6502:186–190. doi: 10.1038/372186a0. [DOI] [PubMed] [Google Scholar]
- Tamemoto H., Kadowaki T., Tobe K., Yagi T., Sakura H., Hayakawa T., Terauchi Y., Ueki K., Kaburagi Y., Satoh S. Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature. 1994;6502:182–186. doi: 10.1038/372182a0. [DOI] [PubMed] [Google Scholar]
- Withers D.J., Gutierrez J.S., Towery H., Burks D.J., Ren J.M., Previs S., Zhang Y., Bernal D., Pons S., Shulman G.I. Disruption of IRS-2 causes type 2 diabetes in mice. Nature. 1998;6670:900–904. doi: 10.1038/36116. [DOI] [PubMed] [Google Scholar]
- Giorgetti-Peraldi S., Peyrade F., Baron V., Van Obberghen E. Involvement of Janus kinases in the insulin signaling pathway. Eur. J. Biochem. 1995;2:656–660. doi: 10.1111/j.1432-1033.1995.656_b.x. [DOI] [PubMed] [Google Scholar]
- Gual P., Baron V., Lequoy V., Van Obberghen E. Interaction of Janus kinases JAK-1 and JAK-2 with the insulin receptor and the insulin-like growth factor-1 receptor. Endocrinology. 1998;3:884–893. doi: 10.1210/endo.139.3.5829. [DOI] [PubMed] [Google Scholar]
- Naka T., Narazaki M., Hirata M., Matsumoto T., Minamoto S., Aono A., Nishimoto N., Kajita T., Taga T., Yoshizaki K. Structure and function of a new STAT-induced STAT inhibitor. Nature. 1997;6636:924–929. doi: 10.1038/43219. [DOI] [PubMed] [Google Scholar]
- Starr R., Willson T.A., Viney E.M., Murray L.J., Rayner J.R., Jenkins B.J., Gonda T.J., Alexander W.S., Metcalf D., Nicola N.A. A family of cytokine-inducible inhibitors of signalling. Nature. 1997;6636:917–921. doi: 10.1038/43206. [DOI] [PubMed] [Google Scholar]
- Endo T.A., Masuhara M., Yokouchi M., Suzuki R., Sakamoto H., Mitsui K., Matsumoto A., Tanimura S., Ohtsubo M., Misawa H. A new protein containing an SH2 domain that inhibits JAK kinases. Nature. 1997;6636:921–924. doi: 10.1038/43213. [DOI] [PubMed] [Google Scholar]
- Naka T., Fujimoto M., Kishimoto T. Negative regulation of cytokine signalingSTAT-induced STAT inhibitor. Trends Biochem. Sci. 1999;10:394–398. doi: 10.1016/s0968-0004(99)01454-1. [DOI] [PubMed] [Google Scholar]
- Hilton D.J. Negative regulators of cytokine signal transduction. Cell. Mol. Life Sci. 1999;12:1568–1577. doi: 10.1007/s000180050396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura A. The CIS/JAB familynovel negative regulators of JAK signaling pathways. Leukemia. 1998;12:1851–1857. doi: 10.1038/sj.leu.2401238. [DOI] [PubMed] [Google Scholar]
- Naka T., Matsumoto T., Narazaki M., Fujimoto M., Morita Y., Ohsawa Y., Saito H., Nagasawa T., Uchiyama Y., Kishimoto T. Accelerated apoptosis of lymphocytes by augmented induction of Bax in SSI-1 (STAT-induced STAT inhibitor-1) deficient mice. Proc. Natl. Acad. Sci. USA. 1998;26:15577–15582. doi: 10.1073/pnas.95.26.15577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T., Yoshida N., Kishimoto T., Akira S. Defective adipocyte differentiation in mice lacking the C/EBPβ and/or C/EBPδ gene. EMBO (Eur. Mol. Biol. Organ.) J. 1997;24:7432–7443. doi: 10.1093/emboj/16.24.7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narazaki M., Fujimoto M., Matsumoto T., Morita Y., Saito H., Kajita T., Yoshizaki K., Naka T., Kishimoto T. Three distinct domains of SSI-1/SOCS-1/JAB protein are required for its suppression of interleukin 6 signaling. Proc. Natl. Acad. Sci. USA. 1998;22:13130–13134. doi: 10.1073/pnas.95.22.13130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorbaek C., El-Haschimi K., Frantz J.D., Flier J.S. The role of SOCS-3 in leptin signaling and leptin resistance. J. Biol. Chem. 1999;42:30059–30065. doi: 10.1074/jbc.274.42.30059. [DOI] [PubMed] [Google Scholar]
- Pezet A., Favre H., Kelly P.A., Edery M. Inhibition and restoration of prolactin signal transduction by suppressors of cytokine signaling. J. Biol. Chem. 1999;35:24497–24502. doi: 10.1074/jbc.274.35.24497. [DOI] [PubMed] [Google Scholar]
- Morita Y., Naka T., Kawazoe Y., Fujimoto M., Narazaki M., Nakagawa R., Fukuyama H., Nagata S., Kishimoto T. Signals transducers and activators of transcription (STAT)-induced STAT inhibitor-1 (SSI-1)/suppressor of cytokine signaling-1 (SOCS-1) suppresses tumor necrosis factor α induced cell death in fibroblasts. Proc. Natl. Acad. Sci. USA. 2000;10:5405–5410. doi: 10.1073/pnas.090084797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuelli B., Peraldi P., Filloux C., Sawka-Verhelle D., Hilton D., Van Obberghen E. SOCS-3 is an insulin-induced negative regulator of insulin signaling. J. Biol. Chem. 2000;21:15985–15991. doi: 10.1074/jbc.275.21.15985. [DOI] [PubMed] [Google Scholar]
- De Sepulveda P., Okkenhaug K., Rose J.L., Hawley R.G., Dubreuil P., Rottapel R. Socs1 binds to multiple signalling proteins and suppresses steel factor-dependent proliferation. EMBO (Eur. Mol. Biol. Organ.) J. 1999;4:904–915. doi: 10.1093/emboj/18.4.904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil G.S. Mechanisms of TNF-α induced insulin resistance. Exp. Clin. Endocrinol. Diabetes. 1999;2:119–125. doi: 10.1055/s-0029-1212086. [DOI] [PubMed] [Google Scholar]
- Paz K., Hemi R., LeRoith D., Karasik A., Elhanany E., Kanety H., Zick Y. A molecular basis for insulin resistance. Elevated serine/threonine phosphorylation of IRS-1 and IRS-2 inhibits their binding to the juxtamembrane region of the insulin receptor and impairs their ability to undergo insulin-induced tyrosine phosphorylation. J. Biol. Chem. 1997;47:29911–29918. doi: 10.1074/jbc.272.47.29911. [DOI] [PubMed] [Google Scholar]