

Abstract
Minor histocompatibility antigens (mHAgs) present a significant impediment to organ and bone marrow transplantation between HLA-identical donor and recipient pairs. Here we report the identification of a new HLA-A*0201–restricted mHAg, HA-8. Designation of this mHAg as HA-8 is based on the nomenclature of Goulmy (Goulmy, E. 1996. Curr. Opin. Immunol. 8:75–81). This peptide, RTLDKVLEV, is derived from KIAA0020, a gene of unknown function located on chromosome 9. Polymorphic alleles of KIAA0020 encode the alternative sequences PTLDKVLEV and PTLDKVLEL. Genotypic analysis demonstrated that the HA-8–specific cytotoxic T lymphocyte (CTL) clone SKH-13 recognized only cells that expressed the allele encoding R at P1. However, when PTLDKVLEV was pulsed onto cells, or when a minigene encoding this sequence was used to artificially translocate this peptide into the endoplasmic reticulum, it was recognized by CTLs nearly as well as RTLDKVLEV. This indicates that the failure of CTLs to recognize cells expressing the PTLDKVLEV-encoding allele of KIAA0020 is due to a failure of this peptide to be appropriately proteolyzed or transported. Consistent with the latter possibility, PTLDKVLEV and its longer precursors were transported poorly compared with RTLDKVLEV by transporter associated with antigen processing (TAP). These studies identify a new human mHAg and provide the first evidence that minor histocompatibility differences can result from the altered processing of potential antigens rather than differences in interaction with the relevant major histocompatibility complex molecule or T cell receptor.
Keywords: minor histocompatibility antigens, antigen processing, graft versus host disease, transplantation, Fourier transform mass spectrometry
Introduction
Bone marrow transplantation (BMT) is a crucial therapy for hematological malignancies. Its therapeutic efficacy can be attributed in part to the beneficial graft versus leukemia effect in which residual host leukemic cells are eliminated by mature donor-derived T cells present in the bone marrow inoculum 1. Although mature donor-derived T cells facilitate graft acceptance, their reactivity against minor histocompatibility antigens (mHAgs) expressed by the recipient also leads to GVHD 2 3 4 5 6 7 8 9, and the administration of immunosuppressive drugs after allogeneic bone marrow transplants is essential to reduce morbidity and mortality 6 10 11.
mHAgs have been shown to be peptides derived from normal cellular proteins that are presented by MHC class I and II molecules 12 13. Over 50 different mHAg genetic loci have been defined among inbred strains of mice 14, whereas the number in humans is unknown. Due to the difficulty of identifying mHAg peptides, a full understanding of the basis for minor antigenic disparities has yet to be achieved. It is generally believed that identification of a more extensive array of mHAg peptides and their cognate genes will lead to improvements in pre-BMT donor and recipient typing 15 and, in the more distant future, immunomodulatory strategies to decrease GVHD incidence and severity as well as graft rejection.
The structure, genetics, and tissue distribution of several human mHAgs have been studied by using specific T cells isolated from allogeneic BMT patients 16 17 18 19 20 21. The use of such T cells, together with either direct peptide extraction and mass spectrometry 22 23 24 25 26 or cDNA cloning 21 27, has led to the successful identification of seven peptides corresponding to human mHAgs. In this report, we describe the identification of the amino acid and nucleotide sequences of an HLA-A*0201–restricted mHAg, termed HA-8, as well as its negative polymorphic counterparts. Our results provide the first evidence that the existence of mHAg can result from altered processing of immunologically similar peptides rather than differences in interaction with the relevant MHC molecule or T cell receptor.
Materials and Methods
Cell Culture.
The HA-8 mHAg-specific, HLA-A*0201–restricted CD8+ CTL clone (designated SKH-13 CTL) was derived from the PBLs of a male patient who received a BMT from his HLA-identical sister for chronic myelogenous leukemia. This CTL clone was used in cytotoxicity and epitope reconstitution assays either 11–15 d after stimulation as described previously 28, or 1–3 d after thawing a frozen aliquot. HLA-A*0201+ EBV–transformed B cell lines (BLCLs) include SDK (donor), SKH (recipient), JY, RSR, JDR, Ruppen, and C1R-A2. BLCLs from the Centre d'Étude du Polymorphisme Humain (CEPH) (Paris, France; reference family 1362) were a gift from Dr. S.M. Prescott and the Huntsman Cancer Institute, University of Utah (Salt Lake City, UT). The TxB cell hybrids T1 and T2, which are positive and negative for the transporter associated with antigen processing (TAP), respectively, were obtained from Dr. P. Cresswell (Yale University, New Haven, CT). BLCLs and hybrids were maintained in RPMI 1640 supplemented with 4 mM Hepes, 5% FCS, 0.125% SerXtend (Irvine Scientific), and 3 mM l-glutamine (BLCL medium). In some experiments, the CEPH BLCLs were infected with a recombinant vaccinia virus that encodes HLA-A*0201 (a gift of Dr. J. Yewdell, National Institute of Allergy and Infectious Diseases, Bethesda, MD) at a multiplicity of infection of 5:1, 18 h before use in cytotoxicity assays. High-level cell surface expression of HLA-A2 was confirmed with flow cytometry, and uninfected and wild-type vaccinia–infected cells were used as controls.
Extraction and HPLC Fractionation of Immunoaffinity-purified, HLA-A*0201–associated Peptides.
HLA-A*0201 molecules were immunoaffinity purified from JY cells with mAb BB7.2 29 and their associated peptides were extracted as described previously 25. The peptide extract was fractionated on a narrow bore HAISIL C18 column (2.1 × 40 mm, 5 μm particles, 300 Å pore size; Higgins) on 130A HPLC (Applied Biosystems). The elution gradient used was 0–10% solvent B in 10 min, 10–60% B in the next 55 min, and 60–100% B in the next 7 min, where solvent A was 0.1% TFA (HPLC grade; Applied Biosystems) in NANOpure water (Barnstead) and solvent B was 0.085% TFA in 60% acetonitrile (HPLC grade; Mallinckrodt) in NANOpure water. Fractions were collected every 40 s at a flow rate of 200 μl/min. Active fractions were pooled and rechromatographed with the identical column and gradient, but using 0.1% heptafluorobutyric acid (HFBA) as the ion-pairing agent. Half of the active second dimension material was used for a third dimension fractionation on a microcapillary 30 column (280 μm outer diameter [OD], 75 μm inner diameter [ID]) packed with 25 cm of 5 μm C18 beads (YMC). TFA was used as the ion-pairing agent in buffers A and B described above, and the column was eluted with a linear gradient of 0–100% B over 40 min at a flow rate of 300 nl/min.
Epitope Reconstitution Assays.
Aliquots of each HPLC fraction were incubated with 2,000 51Cr-labeled, TAP-deficient T2 target cells and 7.5 μg/ml human β2-microglubulin (Calbiochem) for 30 min at 37°C in 150 μl BLCL medium. CTLs were added in 100 μl BLCL medium at an E/T of 10:1 in a standard 51Cr-release assay 22. Synthetic peptides were assayed using the same protocol.
Peptide Analysis Using an Online Effluent Splitter and a Fourier Transform Mass Spectrometer.
Active third dimension HPLC fractions were analyzed by electrospray ionization (ESI) on a Fourier transform mass spectrometer (FTMS) equipped with nanoflow liquid chromatography and a modified online effluent splitter 31 32 33. Samples were fractionated using a microcapillary HPLC column (50 μm ID; reference 30) packed with 12 cm of 5 μm C18 beads (YMC) and a 45-min gradient of 0–86% solvent B (solvent A is 0.1 M acetic acid in water; solvent B is 0.1 M acetic acid in 70% acetonitrile) flowing at 825 nl/min. The online splitter directed 1/8 of the effluent to the FTMS for analysis and 7/8 into microtiter plate wells containing 25 μl NANOpure water, which was reserved for epitope reconstitution assays.
Sequence Analysis of Candidate Antigens.
Collision-activated dissociation (CAD) mass spectra were recorded on selected peptide candidates using a Finnigan LCQ ion trap MS equipped with sheathless nanoflow HPLC-ESI as described previously 32. Data were acquired by manually switching from MS-only mode to MS/MS mode after the chromatographic elution of a marker peptide. In MS/MS mode, the ion of interest was isolated using a 3.0 atomic mass unit isolation window and fragmented using 35% collision energy. CAD spectra were analyzed in a manual search of the GenBank/EMBL/DDBJ DNA and protein databases.
Synthetic Peptides.
Peptides were synthesized on an AMS 1400 multiple peptide synthesizer (Gilson Medical Electronics) using solid-phase FMOC chemistry and Wang resins. Peptides were HPLC purified to >98% on a C-8 column (Vydac). Purity and identity of all synthetic peptides were confirmed using ESI with an LCQ MS.
Class I Peptide-binding Affinity Assay.
Relative affinities of peptides for HLA-A*0201 molecules were measured as described 34. In brief, affinity-purified HLA-A*0201 molecules were incubated at room temperature with an 125I-labeled indicator peptide (FLPSDYFPSV) and graded doses of test peptides in PBS, pH 7.0, containing 0.05% NP-40, 1 μM human β2-microglubulin (Calbiochem), 1 mM PMSF, 1.3 mM 1,10-phenanthroline, 73 μM pepstatin A, 8 mM EDTA, and 200 μM Nα-p-tosyl-l-lysine chloromethyl ketone. After 48 h, class I peptide complexes were separated from free peptides by gel filtration, and the dose of individual test peptides that reduced the binding of indicator peptide by 50% (IC50) was calculated.
Reverse Transcription PCR Amplification and Sequencing of the KIAA0020 Region Encoding the HA-8 mHAg.
Poly(A)+ RNA was isolated with a QuickPrep Micro mRNA Purification kit (Amersham Pharmacia Biotech), and cDNA was synthesized using a First Strand cDNA synthesis kit (MBI Fermentas). Amplifications were performed with 0.5 μmol each of forward primer 5′-ATCAGAAGTTTTAAAGGCCAC-3′ and reverse primer 5′-GCTTCAATCATTTCTGATCTG-3′. Products were purified with the Wizard PCR Preps DNA purification system (Promega) and cloned using the AdvanTAge cloning kit (CLONTECH Laboratories, Inc.). At least five individual clones were sequenced bidirectionally for each cell line examined.
Genotyping of HA-8 mHAg Polymorphisms.
Genomic DNA was isolated from BLCLs with a Puregene kit (Gentra Systems). Adjacent intronic sequence required for genomic PCR analysis was obtained from a genomic DNA library constructed by TA cloning 35. Amplifications were performed with the allele-specific forward primers 5′-GTCAGCAGATCACCG-3′ (HA-8R) and 5′-GTCAGCAGATCACCC-3′ (HA-8P and HA-8PL), and a common reverse primer 5′-GGGCAACAGTTATGGA-3′. BLCLs were also genotyped using a modified PCR-RFLP technique 36. The forward primer 5′-ggatatacagcagagctttc-3′ was used with the antisense primer 5′- TCTAACACTTTGTCCCAGAATT-3′. The underlined A is normally G in the KIAA0020 sequence and was substituted such that an EcoRI site is created only when the primer anneals to the HA-8R allele. PCR products were digested with EcoRI and analyzed on a 2.5% agarose gel. HA-8P and HA-8PL polymorphisms produced a single 183-bp band, whereas the HA-8R allele produced 165- and 22-bp bands.
Expression Plasmids Encoding the HA-8 mHAg and Its Homologues.
Minigene expression constructs encoding RTLDKVLEV, PTLDKVLEV, or PTLDKVLEV, with or without the adenovirus E3-19K endoplasmic reticulum (ER) insertion sequence 37, were constructed using the pEAK10 vector (Edge Bio Systems). The constructs all encoded a Kozak sequence and initiator methionine (AGCTTCCACCATG) and a stop codon (TTA). All products were ligated into HindIII-NotI cut pEAK10 and verified by sequencing. Additional expression plasmids were constructed that encoded truncated proteins corresponding to the HA-8R, HA-8P, and HA-8PL alleles. These initiate from the native start codon, include the peptides corresponding to the HA-8 mHAg and its alleles at residues 149–157, and terminate at the predicted amino acid 223. Products containing these sequences were reverse transcription (RT)-PCR amplified using a 5′ primer containing a HindIII site, the native Kozak sequence and start codon, and a 3′ primer containing the KIAA0020 sequence up to nucleotide 1087, an in-frame stop codon, and a NotI site. All constructs were introduced into SDK BLCLs by electroporation, followed by incubation in BLCL medium for 48 h and selection with puromycin (0.7 μg/ml; Edge Bio Systems) for an additional 48 h before use in cytolytic assays.
Streptolysin O Peptide Transport Assay.
In vitro assays of TAP-mediated peptide transport were performed as described previously 38, with modifications. T1 cells (106/sample) were permeabilized on ice for 15 min with streptolysin O (15 U/ml; Murex Diagnostics) and incubated for 5 min at 37°C with 100 ng of the reporter peptide TVNKTERAY (reference 39; radiolabeled with Na125I using the chloramine T method; reference 40), 10 μl 100 mM ATP, and indicated dilutions of competitor peptides. The reporter peptide contains an N-linked glycosylation site (Asn-X-Thr/Ser) and will become glycosylated after translocation by TAP into the ER. Glycosylated reporter peptide was isolated using Con A–Sepharose (Amersham Pharmacia Biotech), eluted with 0.2 M methyl α-d-mannopyranoside (Sigma-Aldrich), and quantitated on a γ counter. Reporter peptide transport in TAP-negative T2 cells was assessed as a negative control. Samples were done in duplicate except for T2 negative control and T1 cells with no inhibitor, which were done in triplicate.
Results
Mass Spectrometric Identification of the HA-8 mHAg Epitope.
To identify the mHAg recognized by CTL clone SKH-13, HLA-A*0201–associated peptides were purified from the BLCL JY and fractionated by reverse phase (RP)-HPLC. Fractions were analyzed for their ability to reconstitute CTL recognition using TAP-deficient T2 cells as target cells and the SKH-13 CTLs as effector cells in a 51Cr-release assay. Active fractions were pooled and carried forward into another round of RP-HPLC under different conditions. Single peaks of reconstituting activity were observed through three rounds of fractionation (Fig. 1). Candidate masses for the HA-8 mHAg were identified by an online effluent splitter analysis of the third dimension active fractions using a combination of nanoflow liquid chromatography with ESI on an FTMS 26. By comparing the abundance of peptide ions in spectra from wells that showed epitope reconstituting activity with SKH-13 CTLs, five candidate peptides were identified (Fig. 1 D).
Figure 1.
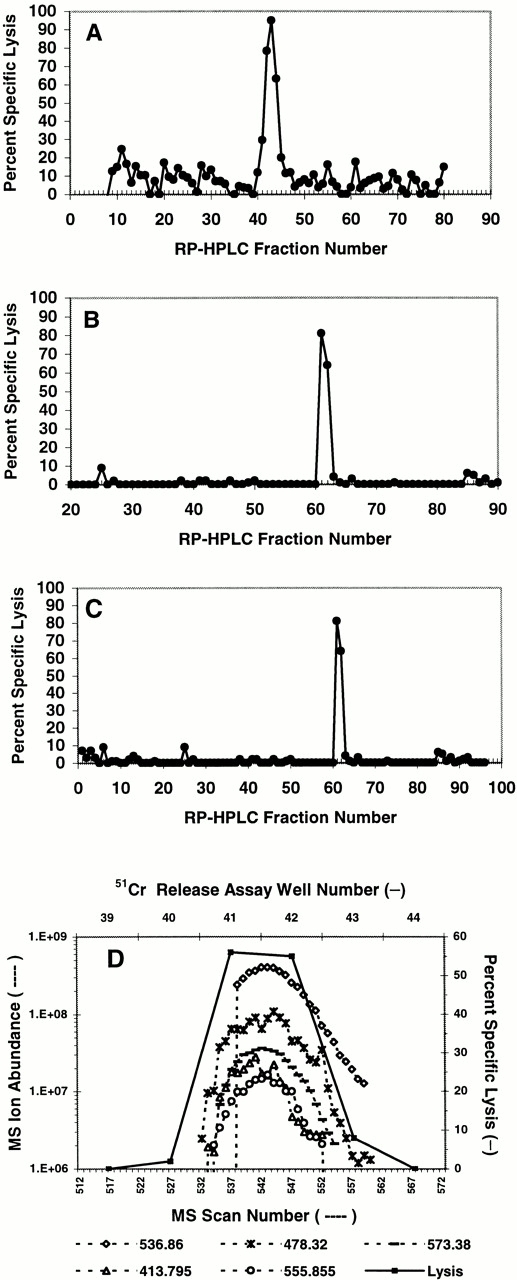
Reconstitution of the HA-8 mHAg epitope with HPLC-fractionated peptides extracted from HLA-A*0201 molecules. HLA-A*0201–associated peptides were purified from 5 × 1010 JY cells and fractionated by RP-HPLC as described in Materials and Methods. Aliquots of each fraction corresponding to 5 × 109 cell equivalents were preincubated with 51Cr-labeled T2 cells and tested for their ability to reconstitute epitope activity for the SKH-13 CTL clone. An E/T ratio of 10:1 was used. (A) First dimension separation of extracted peptides was achieved using TFA as the ion-pairing agent. (B) Fractions 42–44 from A were pooled and rechromatographed using HFBA as the ion-pairing agent. (C) Fractions 61 and 62 from B were pooled and rechromatographed on a microcapillary column using TFA as the ion-pairing agent. (D) Determination of candidate peptides via MS correlated with 51Cr-release assay. Aliquots of each splitter fraction corresponding to 1.5 × 109 cell equivalents were incubated with 51Cr-labeled T2 target cells and tested for their ability to reconstitute epitope activity as described in Materials and Methods. Ion abundances of candidate masses within the MS scan window 520–560 were plotted and correlated to the percent specific 51Cr release in that same region. Background lysis of T2 by the CTLs in the absence of any peptides was 7% in A, 2% in B, 1% in C, and 2% in D. Positive control lysis was 91% in A, 98% in B, 96% in C, and 98% in D.
The most abundant candidate ion (m/z 536.834+2) was analyzed by CAD on an LCQ MS, and its sequence was determined to be RTXDKVXEV (Fig. 2A and Fig. B). X represents either Leu or Ile, amino acids of identical mass that cannot be differentiated on the LCQ MS. To determine whether this sequence encoded the HA-8 mHAg, the peptide was synthesized with an equimolar mixture of Leu and Ile at each X position and tested for its ability to sensitize T2 target cells to lysis by SKH-13 CTLs. Target cells pulsed with RTXDKVXEV were lysed by SKH-13 CTLs, with half-maximal activity seen at a peptide concentration of ∼50 pM (Fig. 2 C). Of the four possible isomers containing either Leu or Ile in place of X in this sequence, only the form containing Leu at both P3 and P7 coeluted from a microcapillary HPLC column with the naturally processed peptide m/z 536.834+2 (data not shown). By comparing the signal intensity of the naturally occurring peptide with a known amount of synthetic RTLDKVLEV, we calculated that this mHAg is present at ∼10 copies per BLCL (data not shown). Target cells pulsed with the RTLDKVLEV peptide were lysed by SKH-13 CTLs, with half-maximal activity seen at a peptide concentration of 20 pM (Fig. 2 C). RTLDKVLEV additionally reconstitutes CTL lysis when incubated with SDK, a BLCL derived from the HLA-identical sibling donor (data not shown). Thus, RTLDKVLEV defines the HLA-A*0201–restricted HA-8 mHAg epitope.
Figure 2.
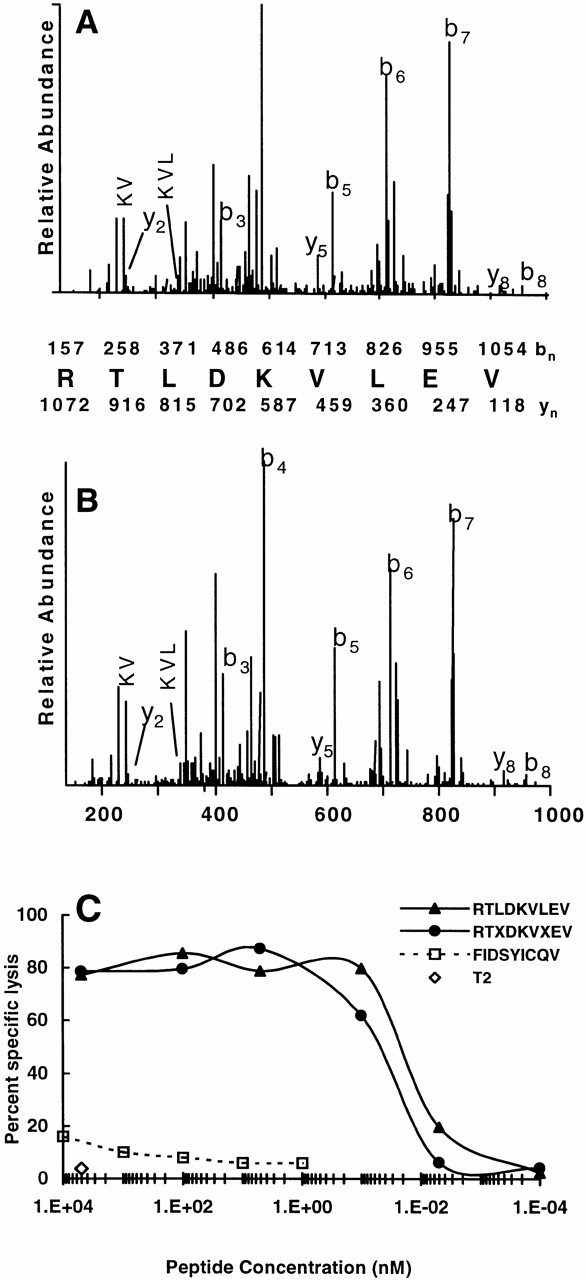
Identification of the HA-8 mHAg antigenic peptide. (A) CAD mass spectrum of candidate peptide (M+2H)2+ ion with monoisotopic m/z of 536.834+2 as eluted from JY BLCL. (B) CAD mass spectrum of synthetic peptide RTLDKVLEV. Mass spectra were recorded on a Finnigan LCQ ion trap MS operating with a 3.0 atomic mass unit isolation window and 35% collision energy. The b and y ions (reference 54) are labeled above and below the amino acid sequence, respectively. Ions observed in the spectrum are underlined. (C) HA-8 mHAg epitope reconstitution with synthetic peptides. A standard 51Cr-release assay was performed by incubating the indicated quantities of synthetic peptides with 51Cr-labeled T2 target cells and then adding the mHAg-specific CTL clone SKH-13. An E/T ratio of 10:1 was used. FIDSYICQV corresponds to the mHAg A2-HY (reference 25), and serves as a negative control. Background lysis of T2 by the CTLs in the absence of any peptides was 4%; positive control lysis was 87%.
The HA-8 mHAg Is Encoded by the Gene KIAA0020.
A search of the known protein and DNA sequence databases with the sequence RTLDKVLEV generated a perfect match with residues 149–157 of the predicted protein sequence of KIAA0020 (GenBank/EMBL/DDBJ accession no. D13645; Fig. 3) and also to a predicted sequence within a human expressed sequence tag (accession no. AA307160; data not shown). AA307160 is identical to KIAA0020 over 493 bp with the exception of four ambiguous nucleotides, and a single nucleotide insertion and deletion (data not shown). The latter appeared to be sequencing mistakes, since their effect was to encode multiple stop codons in the same reading frame as the epitope sequence upstream of the deletion and downstream of the insertion. Thus, we hypothesized that KIAA0020 represents the gene encoding the HA-8 mHAg. To definitively prove that the KIAA0020 gene encodes HA-8, a 669-bp region of KIAA0020 cDNA was cloned into a eukaryotic expression vector and transiently transfected into SDK BLCL as described in Materials and Methods. This transfectant was recognized by SKH-13 CTLs (Fig. 4), thus conclusively demonstrating that the HA-8 mHAg is encoded by KIAA0020.
Figure 3.

Alignment of the predicted protein sequence of the KIAA0020 cDNA with those of homologous sequences found in GenBank/EMBL/DDBJ. Predicted amino acid differences are indicated in bold type; bold X's represent nucleotide sequence ambiguity. Predicted epitope encoding and epitope analogous regions are indicated inside the rectangle. Black bars indicate the positions of RT-PCR primers used to amplify the 347-bp cDNA fragment described in the text and used subsequently for sequencing.
Figure 4.

The human HA-8 mHAg epitope is encoded by the KIAA0020 sequence represented in GenBank/EMBL/DDBJ under accession no. D13645. A eukaryotic expression plasmid containing a 669-bp region of the KIAA0020 cDNA encoding RTLDKVLEV was transiently transfected into SDK (mHAg− donor) BLCL as described in Materials and Methods and incubated with the SKH-13 CTL clone in a standard 51Cr-release assay. SKH is the BLCL of the mHAg+recipient.
Identification of KIAA0020-related Sequences That Encode Potential HA-8 mHAg Alleles.
To elucidate the basis for differential expression of HA-8 within the human population, we searched protein and DNA sequence databases with the entire KIAA0020 coding sequence, and identified three human homologues (Fig. 3). GenBank/EMBL/DDBJ accesssion no. AC012407 exhibits low homology to KIAA0020 in the epitope-flanking regions, and only 1/9 amino acids are identical in the epitope-analogous region. More importantly, it has been mapped to chromosome 18, whereas KIAA0020 maps to chromosome 9 41. Thus, AC012407 was excluded as a potential allele of KIAA0020, although it may be a member of a gene family that includes KIAA0020. In contrast, AA478933 and N90372 exhibited >97.6% identity over >84 amino acid residues and are predicted to encode RALDKVLEV and PTLDKVLEV, respectively, in the region homologous to the HA-8 mHAg epitope. Therefore, both AA478933 and N90372 were considered candidates for alleles of the KIAA0020 gene sequence.
RT-PCR primers (represented by black bars in Fig. 3) that amplified a 347-bp cDNA product surrounding the epitope were generated from sequences that were identical among KIAA0020, AA307160, and AA478933 but not conserved in the chromosome 18–encoded sequence. Consistent with the finding that the HA-8 mHAg sequence RTLDKVLEV is encoded by KIAA0020, sequences identical to KIAA0020 were amplified from all four BLCLs that were recognized by SKH-13 CTLs (Table ). The only RT-PCR products amplified from the three BLCLs not recognized by SKH-13 CTLs were identical to N90372 (with the exception of two ambiguous nucleotides in the N90372 sequence; Fig. 3). The absence of the KIAA0020 sequence, together with the presence of the N90372 sequence, is consistent with the hypothesis that these represent alleles of the same gene. The polymorphic nucleotide sequences corresponding to KIAA0020 and N90372 were designated HA-8R and HA-8P, respectively.
Table 1.
Correlation of KIAA0020 Sequence Polymorphisms with HA-8 Phenotype
| HLA-A2+ BLCLs | Polymorphism(s) encoded by sequenced DNA | Cytolysis by SKH-13 CTLs |
|---|---|---|
| JY | RTLDKVLEV, RTLDKVLEV | + |
| Ruppen | RTLDKVLEV, RTLDKVLEV | + |
| JDR | RTLDKVLEV, PTLDKVLEV | + |
| SKH (recipient) | RTLDKVLEV, PTLDKVLEL | + |
| SDK (donor) | PTLDKVLEV, PTLDKVLEL | − |
| RSR | PTLDKVLEV, PTLDKVLEV | − |
| C1R-A2 | PTLDKVLEV, PTLDKVLEV | − |
The only RT-PCR products amplified from the HA-8+ BLCL JY and Ruppen encoded HA-8R (Table ), whereas both HA-8R and HA-8P were amplified from the HA-8+ BLCL JDR. Interestingly, the amplification products of the HA-8− BLCL SDK included not only HA-8P, but also a second sequence with an additional nucleotide substitution. This sequence again resulted in the substitution of P for R at the P1 position in the epitope, whereas the additional change led to the substitution of L for V at P9 (Table ). This sequence was designated HA-8PL. HA-8PL has not been observed in any of the other HA-8− cells that we have genotyped. However, SDK is the sibling and bone marrow donor for SKH, and the products amplified from SKH BLCL included both HA-8R and HA-8PL. Collectively, these results are consistent with the hypothesis that HA-8R, HA-8P, and HA-8PL are alleles of a single genetic locus.
Further evidence for this hypothesis was obtained by screening all individuals in three consecutive generations of a large family for expression of the HA-8 mHAg, and correlating the results with their genotype at the KIAA0020 locus. BLCLs derived from each individual in CEPH reference family 1362 were infected with a recombinant vaccinia virus expressing the HLA-A*0201 gene and tested for HA-8 expression with SKH-13 CTLs (Fig. 5). The genotype of each individual at nucleotide 864 in the KIAA0020 cDNA sequence, which determines either an R or P in P1 of the epitope recognized by the SKH-13 CTL clone, was determined by testing genomic DNA derived from each BLCL with a PCR-RFLP assay that distinguishes between G and C at this position. All HA-8− members of the CEPH family 1362 exhibited the HA-8P sequence, whereas all HA-8+ members expressed both HA-8R and HA-8P (Fig. 5). These results confirm that the KIAA0020 gene has at least two alleles, of which only HA-8R leads to specific CTL recognition.
Figure 5.

HA-8 mHAg allelic segregation analysis. Segregation of the HA-8R and HA-8P alleles and correlation of the HA-8R allele with cytolysis by SKH-13 CTLs at an E/T ratio of 5:1 is shown using the BLCL of CEPH reference family 1362. Filled circles (females) or squares (males) indicate strong lysis by SKH-13 CTLs (phenotype positive); open symbols indicate minimal lysis (phenotype negative). Numbers below symbols were assigned to each family member by the University of Utah. R/P represents HA-8R/P heterozygous genotype; P/P represents HA-8P genotype (the HA-8P/liter polymorphism cannot be distinguished by these genotyping PCR primers). Genotyping of HA-8 mHAg alleles was determined by PCR-RFLP of genomic DNA as described in Materials and Methods.
Effect of KIAA0020 Polymorphisms on CTL Recognition and HLA-A*0201 Binding.
To gain further insight into the mechanisms governing the lack of CTL recognition of the cells of HA-8− individuals, we determined the relative binding affinities of the three observed polymorphic peptides for HLA-A*0201 (Fig. 6 A). RTLDKVLEV inhibited the binding of an iodinated indicator peptide by 50% (IC50) at a concentration of 40 nM, whereas PTLDKVLEV and PTLDKVLEL bound 50-fold and 250-fold less well, respectively. We next compared the ability of the three polymorphic peptides to reconstitute the epitope for the SKH-13 CTL clone when exogenously pulsed onto target cells (Fig. 6 B). SKH-13 CTL recognition of PTLDKVLEL required nearly 4,500-fold more peptide for half-maximal lysis (∼90 nM) than recognition of RTLDKVLEV (20 pM). This difference is larger than the difference in HLA-A*0201 binding affinity, suggesting that the substitutions at P1 and P9 have an additional detrimental effect on CTL recognition. However, recognition of PTLDKVLEV required only 15-fold more peptide (half-maximal lysis of 300 pM). Taking into account the differences in binding affinity, this suggests that SKH-13 CTL recognizes PTLDKVLEV as well as or better than RTLDKVLEV. This stands in contrast to the complete lack of recognition of donor-derived (self) SDK BLCL cells or cells that are homozygous for HA-8P (Table , Fig. 5, and data not shown). These results suggested that differences in MHC binding and CTL recognition did not account for the failure of SKH-13 CTLs to recognize cells that were homozygous for HA-8P and that differences in antigen processing might instead be responsible.
Figure 6.
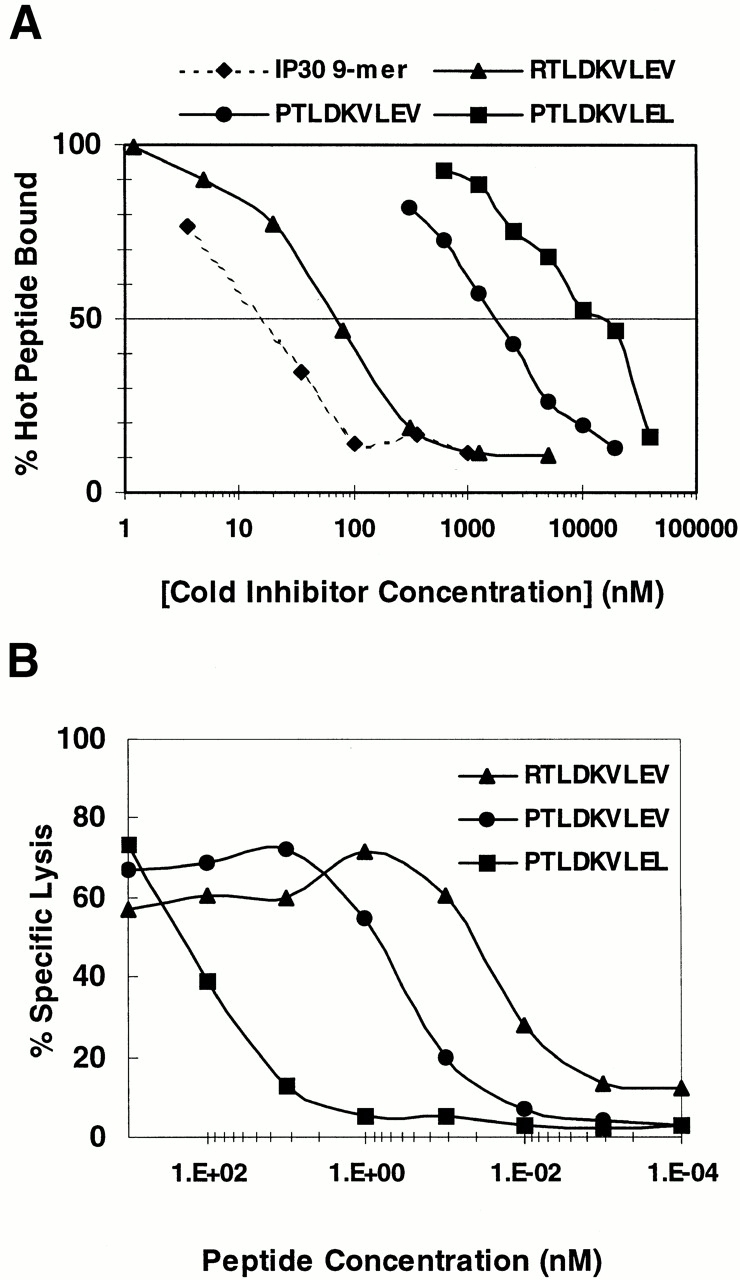
HLA-A*0201 binding affinity of HA-8 mHAg peptide variants and SKH-13 CTL recognition of exogenously pulsed peptide variants. (A) Binding of RTLDKVLEV, PTLDKVLEV, and PTLDKVLEL to HLA-A*0201. HPLC-purified synthetic peptides were assayed for their ability to inhibit the binding of the iodinated peptide FLPSDYFPSV (♦) to affinity-purified HLA-A*0201 molecules in a cell-free peptide binding assay (see Materials and Methods). (B) RTLDKVLEV, PTLDKVLEV, and PTLDKVLEL were tested for their ability to reconstitute the epitope for the SKH-13 CTL clone. Epitope reconstitution assay conditions are described in Materials and Methods. An E/T ratio of 10:1 was used. Background CTL lysis of T2 in the absence of any peptides was 4%; positive control lysis (SKH) was 80%.
Effect of KIAA0020 Polymorphisms on Antigen Processing.
To address whether antigen processing might influence recognition by SKH-13 CTLs, KIAA0020 minigenes encoding the HA-8R–, HA-8P–, and HA-8PL–derived peptides were expressed transiently in SDK BLCL cells. The minigene constructs used in these experiments resulted in the peptides being produced in the cytosol or cotranslationally translocated into the ER via the adenovirus E3-19K signal sequence. Of the cytosolically translated minigenes, only the one encoding the HA-8R peptide was recognized by SKH-13 CTLs (Fig. 7 A). A construct containing a 669-bp fragment that encoded the HA-8P or HA-8PL peptide in the context of a large cytosolically translated product (analogous to that shown in Fig. 4) demonstrated similar lack of recognition by SKH-13 CTLs (Fig. 7 B). These observations correlate well with the data observed in Table and Fig. 5, which demonstrate recognition of only those cells that express HA-8R. In contrast, SKH-13 CTLs recognized cells expressing both HA-8R and HA-8P peptides derived from minigenes that enabled direct translocation into the ER, although cells expressing HA-8PL in this format were still not recognized (Fig. 7 A). The lack of recognition of cells transfected with the E3-19K–directed HA-8PL construct likely reflects the lower HLA-A*0201 binding affinity of PTLDKVLEL relative to PTLDKVLEV. However, the recognition of PTLDKVLEV by SKH-13 CTLs only when it is translocated into the ER via the E3-19K signal sequence indicates that the lack of CTL recognition of cells expressing the HA-8P encoding sequence under normal circumstances is due to impaired TAP transport of this peptide or to differences in cytosolic proteolysis of its precursors.
Figure 7.
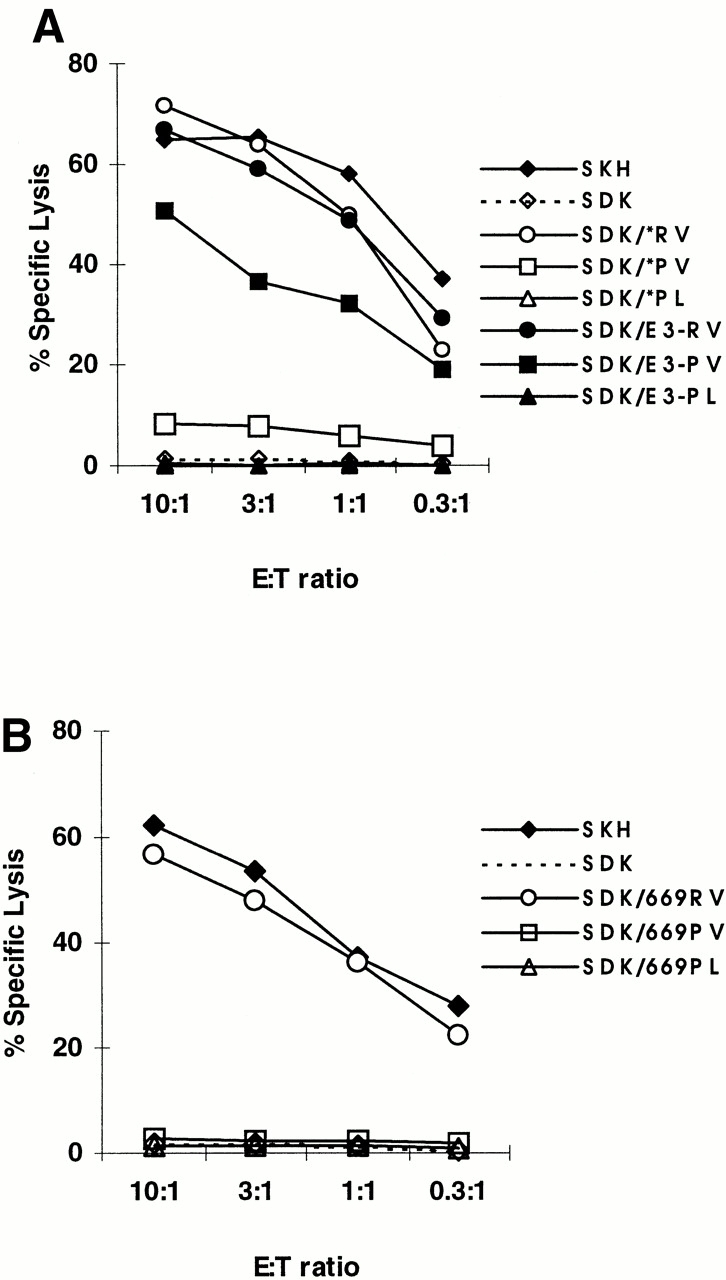
Comparative SKH-13 CTL recognition of transiently transfected KIAA0020 constructs. (A) CTL recognition of the donor-derived BLCL SDK is shown after transient transfection of experimental minigene constructs. Constructs tested contain a minigene cassette encoding the KIAA0020 polymorphic peptides either with (closed symbols) or without (open symbols) an E3-19K adenovirus leader peptide sequence, which directs translocation of the expressed peptide to the ER, bypassing normal TAP transport. CTL lysis of donor (SDK) and recipient (SKH) BLCL is also shown. (B) Eukaryotic expression plasmids expressing a truncated KIAA0020 protein initiating from the native start codon, encoding RTLDKVLEV, PTLDKVLEV, or PTLDKVLEL without an E3-19K adenovirus leader peptide sequence, were transiently transfected into SDK BLCL as described in Materials and Methods and incubated with the SKH-13 CTL clone in a standard epitope reconstitution assay. CTL lysis of recipient (SKH) and donor (SDK) BLCL cells is indicated.
To determine whether impaired TAP transport of PTLDKVLEV-containing peptides could account for their failure to be displayed by HLA-A*0201, we examined their ability to be transported by TAP in vitro. Because the exact peptides produced by cytosolic proteolysis are not known, RTLDKVLEV, PTLDKVLEV, and versions of each peptide containing one or five additional natural NH2-terminal flanking amino acids were tested as substrates for their ability to inhibit TAP-dependent transport of the radiolabeled reporter peptide TVNKTERAY in streptolysin O–permeabilized T1 cells (Fig. 8). RTLDKVLEV as well as the 10- and 14-mer peptides containing this sequence were all transported efficiently, exhibiting IC50 values in the range of 30–45 μM. In contrast, PTLDKVLEV, HPTLDKVLEV, and KSADHPTLDKVLEV were transported considerably less well, with IC50 values of 175, 355, and 102 μM, respectively. Taken together, these data are consistent with the hypothesis that impaired TAP translocation of HA-8P–derived peptides accounts for the failure of endogenously processed PTLDKVLEV to be recognized by HA-8 mHAg-specific CTLs.
Figure 8.

In vitro transport of HA-8 mHAg peptide variants. T1 cells were permeabilized with streptolysin O (15 U/ml) and incubated with radioiodinated reporter peptide TVNKTERAY plus the indicated dilution of test peptides. Reporter peptide transport in TAP-negative T2 cells was assessed as a negative control. Samples were done in duplicate except for T2 negative control and T1 cells with no inhibitor, which were done in triplicate.
Discussion
In this study, we identified the nonamer peptide RTLDKVLEV, represented within the KIAA0020 cDNA, as a new HLA-A*0201–restricted mHAg, HA-8. KIAA0020 is located on chromosome 9 and encodes a predicted protein of 508 amino acids. Sequencing of cDNA from the BLCLs of various HA-8+ and HA-8− individuals also identified KIAA0020-related polymorphic sequences HA-8P and HA-8PL, which encode the peptides PTLDKVLEV and PTLDKVLEL, respectively. Based on inheritance patterns, HA-8P is a bona fide allele of KIAA0020. We have yet to demonstrate definitively that HA-8PL is also an allele of KIAA0020, since this polymorphism was observed only in one sibling pair, but its expression in both siblings suggests that this is likely. In addition, database searches also identified an additional sequence with high homology to KIAA0020 that encoded the predicted peptide RALDKVLEV. Because expression of the gene encoding this sequence was not observed in any BLCL, it remains to be seen whether it also represents an allele of KIAA0020. Nonetheless, the KIAA0020 polymorphisms associated with HA-8+ and HA-8− phenotypic expression most likely represent at least a triallelic system. In contrast, only two alleles have been defined for the other known autosomal human mHAg HA-1 24, HB-1 27, and HA-2 (reference 22, and Pierce, R.A., and V.H. Engelhard, unpublished data).
In principle, any genetic polymorphism that qualitatively or quantitatively affects the display of self-peptides at the cell surface could give rise to an mHAg disparity in an MHC identical transplantation setting 13. These can be classified based on their impact on either T cell recognition or peptide presentation by MHC molecules. In the case of the human mHAgs B7-HY 23 and A1-HY 26, and mouse mHAg MTF 42, the mHAg− alleles encode polymorphic peptides that retain their ability to bind to the relevant MHC molecule. The existence of these mHAgs is thus dependent on the presence within any individual of TCR with an appropriate fine specificity to distinguish mHAg-expressing cells from their negative counterparts. In some cases, T cell discrimination may be augmented by the presence of posttranslational modifications on the mHAg+ allelic peptides 25 26. TCR discrimination between two presented peptides has been proposed to define the human HB-1 mHAg 27, the murine mHAgs H3a 43 and H13 44, and the rat MTF mHAg 45. However, endogenous presentation of the mHAg− peptide has not been formally demonstrated in any of these cases.
Two additional mechanisms in which the corresponding negative allelic peptides are not presented at the cell surface enable mHAgs to be distinguished by T cells. This type of differential display may enable a more robust T cell response, since it will not be limited to TCRs that can make fine distinctions among closely related peptides. In one case, mHAgs are distinguished due to polymorphisms that diminish or abolish the ability of the mHAg− peptide to bind to the relevant MHC molecule. This mechanism underlies the immunogenicity of the human mHAg HA-1 24 and the murine mHAg HY-Db 46. A second possibility is that the mHAg-positive and -negative peptides are antigenically similar, but are handled differently by the antigen processing machinery of the cell. Such a possibility has not been previously described for mHAgs.
A critical observation in the present study is that HLA-A*0201+ cells expressing HA-8P and HA-8PL genes or minigene fragments in the cytosol are not recognized by SKH-13 CTLs, yet PTLDKVLEV as an exogenously pulsed or ER translocated peptide is recognized nearly as well as the HA-8 mHAg peptide RTLDKVLEV. This observation could indicate that either PTLDKVLEV or its precursors are transported poorly by TAP, or that differences between the HA-8R and HA-8P protein products lead either to a failure to produce the PTLDKVLEV epitope or to its destruction by the proteasome or other cellular proteases. It is noteworthy that in comparing the cDNA sequences of a 347-bp region surrounding the epitope in the seven HA-8+ and HA-8− BLCLs listed in Table , the only polymorphisms were at the P1 and P9 positions within the epitope (data not shown). In addition, the ability of PTLDKVLEV to be recognized by the SKH-13 CTLs when targeted to the ER with an NH2-terminally fused E3-19K signal sequence argues against substantial degradation of this peptide by aminopeptidases in the ER 47 48. Differential proteolysis in the cytosol is one possible explanation for the failure of HA-8− cells to present PTLDKVLEV. Previous studies have demonstrated that subunit-independent proteasomal cleavage is augmented three residues to the COOH terminal side of a P 49 50. This could result in greater destruction of the PTLDKVLEV epitope. On the other hand, we observed that PTLDKVLEV, as well as PTLDKVLEV-containing peptides with additional natural NH2-terminal flanking amino acids, are translocated into the ER in vitro appreciably less well than RTLDKVLEV and its analogous NH2-terminally extended peptides. This is consistent with previous studies showing that peptides containing a P at P1 or P2 are generally transported poorly by human, mouse, and rat TAP 51 52 53, whereas positively charged residues, and particularly R at P1–P3, enhanced TAP interaction 52. It is interesting that even in comparing the 14-mer peptides KSADHPTLDKVLEV and KSADHRTLDKVLEV in this study, the presence of P at P6 also diminished peptide transport by TAP. Thus, while the exact epitope-containing substrates for TAP transport in vivo have not been identified, our data suggest that those derived from HA-8P and HA-8PL will be poorly transported by TAP. Taken together, our results suggest that the basis for lack of immunogenicity of endogenous PTLDKVLEV lies not in differential recognition of the peptide by the HA-8–specific TCR, but rather in its inability to be productively transported by TAP for presentation by HLA-A*0201 at the cell surface. Thus, HA-8R represents the first mHAg that has been shown to result from differential antigen processing and this may represent an important mechanism for the generation of mHAgs.
The elucidation of mHAg peptides and their cognate genes forms a critical foundation for future attempts to improve the outcome of MHC-identical, mHAg-mismatched BMT, or solid organ transplants. The HA-8 mHAg is present in the population at a phenotype frequency of ∼65%. Additionally, HA-8 is presented by the HLA-A*0201 restriction element, which has a high phenotype frequency in most populations (i.e., 49% in the Caucasian population). This suggests that BMT donor and recipient pairs will often be discordant for this mHAg. Although the significance of the HA-8 mHAg in GVHD or graft rejection remains to be determined, the genotyping reagents described in this study will allow prospective studies of association between HA-8 incompatibility and outcomes after transplantation. The prospect of using the DNA sequences encoding mHAgs in pre-BMT molecular typing to improve donor selection or as a prognostic indicator of GVHD/graft rejection risk becomes increasingly more attainable with the identification of each new mHAg peptide. In the more distant future, potential applications may include tolerance induction in solid organ and bone marrow transplant and the design of prophylaxis against GVHD and rejection.
Acknowledgments
This work was supported by the US Public Health Service grants AI20963 (to V.H. Engelhard), AI33993 (to D.F. Hunt), AI39501 (to L.C. Eisenlohr), CA76930 (to E.H. Warren), and CA18029 (to S.R. Riddell), a Clinical Investigator Award from the Cancer Research Fund of the Damon Runyon-Walter Winchell Foundation (to E.H. Warren), and support from the Leukemia and Lymphoma Society and American Cancer Society Grant RPG-98-036-01-CIM (to L.C. Eisenlohr). A.G. Brickner was supported by National Institutes of Health Immunology Training Grant AI07496, A.L. Zarling by Cancer Training Grant CA09109, and T.N. Golovina by the Cancer Research Institute/CIGNA Foundation Fellowship.
Footnotes
A. Brickner, E.H. Warren, J.A. Caldwell, and Y. Akatsuka contributed equally to this work.
Abbreviations used in this paper: BMT, bone marrow transplantation; CAD, collision-activated dissociation; CEPH, Centre d'Étude du Polymorphisme Humain; ER, endoplasmic reticulum; ESI, electrospray ionization; FTMS, Fourier transform MS; HFBA, heptafluorobutyric acid; ID, inner diameter; mHAg, minor histocompatibility antigen; MS, mass spectrometer; RP, reverse phase; RT, reverse transcription; TAP, transporter associated with antigen processing.
References
- Horowitz M.M., Gale R.P., Sondel P.M., Goldman J.M., Kersey J., Kolb H.J., Rimm A.A., Ringden O., Rozman C., Speck B. Graft-versus-leukemia reactions after bone marrow transplantation. Blood. 1990;75:555–562. [PubMed] [Google Scholar]
- Kernan N.A., Dupont B. Minor histocompatibility antigens and marrow transplantation. N. Engl. J. Med. 1996;334:323–324. doi: 10.1056/NEJM199602013340510. [DOI] [PubMed] [Google Scholar]
- Goulmy E., Termijtelen A., Bradley B.A., van Rood J.J. Alloimmunity to human H-Y. Lancet. 1976;2:1206. doi: 10.1016/s0140-6736(76)91727-x. [DOI] [PubMed] [Google Scholar]
- Goulmy E. Human minor histocompatibility antigens. Curr. Opin. Immunol. 1996;8:75–81. doi: 10.1016/s0952-7915(96)80108-7. [DOI] [PubMed] [Google Scholar]
- Goulmy E., Gratama J.W., Blokland E., Zwaan F.E., van Rood J.J. A minor transplantation antigen detected by MHC-restricted cytotoxic T lymphocytes during graft-versus-host disease. Nature. 1983;302:159–161. doi: 10.1038/302159a0. [DOI] [PubMed] [Google Scholar]
- Goulmy E. Minor histocompatibility antigens in man and their role in transplantation. In: Morris P.J., Tilney N.C., editors. Transplantation Reviews. Saunders; Philadelphia: 1988. pp. 29–44. [Google Scholar]
- de Bueger M., Goulmy E. Human minor histocompatibility antigens. Transpl. Immunol. 1993;1:28–38. doi: 10.1016/0966-3274(93)90056-e. [DOI] [PubMed] [Google Scholar]
- Pardoll D. Taming the sinister side of BMTDr. Jekyll and Mr. Hyde. Nat. Med. 1997;3:833–834. doi: 10.1038/nm0897-833. [DOI] [PubMed] [Google Scholar]
- Hakim F.T., Mackall C.L. The immune systemeffector and target of graft-versus-host disease. In: Ferrara J.L.M., Deeg H.J., Burakoff S.J., editors. Graft-vs.-Host Disease. Marcel Dekker, Inc.; New York: 1997. pp. 257–289. [Google Scholar]
- Beatty P.G., Herve P. Immunogenetic factors relevant to acute GVHD. In: Burakoff S.J., Deeg H.J., Ferrara S., Atkinson K., editors. Graft-versus-Host-DiseaseImmunology, Pathophysiology and Treatment. Marcel Dekker, Inc.; New York: 1989. pp. 415–423. [Google Scholar]
- Sullivan K.M. Graft-versus-host disease. In: Forman S.J., Blume K.G., Donnall T.E., editors. Bone Marrow Transplantation. Blackwell Scientific Publications; Boston: 1994. pp. 339–362. [Google Scholar]
- Wallny H.J., Rammensee H.G. Identification of classical minor histocompatibility antigen as cell-derived peptide. Nature. 1990;343:275–278. doi: 10.1038/343275a0. [DOI] [PubMed] [Google Scholar]
- Simpson E., Roopenian D.C. Minor histocompatibility antigens. Curr. Opin. Immunol. 1997;9:655–661. doi: 10.1016/s0952-7915(97)80045-3. [DOI] [PubMed] [Google Scholar]
- Doolittle D.P., Davisson M.T., Guidi J.N., Green M.C. Catalog of mutant genes and polymorphic loci. 3rd ed. In: Lyon M.F., Rastan S., Brown S.D.M., editors. Genetic Variants and Strains of the Laboratory Mouse. Oxford University Press; New York: 1996. pp. 17–854. [Google Scholar]
- Martin P.J. How much benefit can be expected from matching for minor antigens in allogeneic marrow transplantation? Bone Marrow Transplant. 1997;20:97–100. doi: 10.1038/sj.bmt.1700868. [DOI] [PubMed] [Google Scholar]
- Goulmy E. Human minor histocompatibility antigensnew concepts for marrow transplantation and adoptive immunotherapy. Immunol. Rev. 1997;157:125–140. doi: 10.1111/j.1600-065x.1997.tb00978.x. [DOI] [PubMed] [Google Scholar]
- de Bueger M., Rood J.J., Bakker A., van der Woude F., Goulmy E. Tissue distribution of human minor histocompatibility antigensubiquitous versus restricted tissue distribution indicates heterogeneity among human cytotoxic T lymphocyte defined non-MHC antigens. J. Immunol. 1992;149:1788–1794. [PubMed] [Google Scholar]
- Marijt W.A., Veenhof W.F., Goulmy E., Willemze R., van Rood J.J., Falkenburg J.H. Minor histocompatibility antigens HA-1-, -2-, and -4-, and HY-specific cytotoxic T-cell clones inhibit human hematopoietic progenitor cell growth by a mechanism that is dependent on direct cell-cell contact. Blood. 1993;82:3778–3785. [PubMed] [Google Scholar]
- de Bueger M., Bakker A., van Rood J.J., Goulmy E. Minor histocompatibility antigens, defined by graft-vs.-host disease-derived cytotoxic T lymphocytes, show variable expression on human skin cells. Eur. J. Immunol. 1991;21:2839–2844. doi: 10.1002/eji.1830211127. [DOI] [PubMed] [Google Scholar]
- Warren E.H., Greenberg P.D., Riddell S.R. Cytotoxic T-lymphocyte-defined human minor histocompatibility antigens with a restricted tissue distribution. Blood. 1998;91:2197–2207. [PubMed] [Google Scholar]
- Warren E.H., Gavin M., Simpson E., Chandler P., Page D.C., Disteche C., Stankey K.A., Greenberg P.D., Riddell S.R. The human HY gene encodes a novel HLA-B8-restricted H-Y antigen. J. Immunol. 2000;164:2807–2814. doi: 10.4049/jimmunol.164.5.2807. [DOI] [PubMed] [Google Scholar]
- den Haan J.M., Sherman N.E., Blokland E., Huczko E., Koning F., Drijfhout J.W., Skipper J.C., Shabanowitz J., Hunt D.F., Engelhard V.H. Identification of a graft versus host disease-associated human minor histocompatibility antigen. Science. 1995;268:1476–1480. doi: 10.1126/science.7539551. [DOI] [PubMed] [Google Scholar]
- Wang W., Meadows L.R., den Haan J.M., Sherman N.E., Chen Y., Blokland E., Shabanowitz J., Agulnik A., Hendrickson R.C., Bishop C.E. Human H-Ya male-specific histocompatibility antigen derived from the SMCY protein. Science. 1995;269:1588–1590. doi: 10.1126/science.7667640. [DOI] [PubMed] [Google Scholar]
- den Haan J.M., Meadows L., Wang W., Pool J., Blokland E., Bishop T.L., Reinhardus C., Shabanowitz J., Offringa R., Hunt D.F. The minor histocompatibility antigen HA-1a diallelic gene with a single amino acid polymorphism. Science. 1998;279:1054–1057. doi: 10.1126/science.279.5353.1054. [DOI] [PubMed] [Google Scholar]
- Meadows L.R., Wang W., den Haan J.M., Blokland E., Reinhardus C., Drijfhout J.W., Shabanowitz J., Pierce R., Agulnik A., Bishop C.E. The HLA-A*0201-restricted HY antigen contains a posttranslationally modified cysteine that significantly affects T cell recognition. Immunity. 1997;6:273–281. doi: 10.1016/s1074-7613(00)80330-1. [DOI] [PubMed] [Google Scholar]
- Pierce R.A., Field E.D., den Haan J.M., Caldwell J.A., White F.M., Marto J.A., Wang W., Frost L.M., Blokland E., Reinhardus C. The HLA-A*0101 restricted HY minor histocompatibility antigen originates from DFFRY and contains a cysteinylated cysteine residue as identified by a novel mass spectrometric technique. J. Immunol. 1999;163:6360–6364. [PubMed] [Google Scholar]
- Dolstra H., Fredrix H., Maas F., Coulie P.G., Brasseur F., Mensink E., Adema G.J., de Witte T.M., Figdor C.G., van de Wiel-van Kemenade E. A human minor histocompatibility antigen specific for B cell acute lymphoblastic leukemia. J. Exp. Med. 1999;189:301–308. doi: 10.1084/jem.189.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie S.J., Lewinsohn D.A., Patterson B.K., Jiyamapa D., Krieger J., Corey L., Greenberg P.D., Riddell S.R. In vivo migration and function of transferred HIV-1-specific cytotoxic T cells. Nat. Med. 1999;5:34–41. doi: 10.1038/4716. [DOI] [PubMed] [Google Scholar]
- Brodsky F.M., Parham P., Barnstable C.J., Crumpton M.J., Bodmer W.F. Monoclonal antibodies for analysis of the HLA system. Immunol. Rev. 1979;47:3–61. doi: 10.1111/j.1600-065x.1979.tb00288.x. [DOI] [PubMed] [Google Scholar]
- Kennedy R.T., Jorgenson J.W. Quantitative analysis of individual neurons by open tubular liquid chromatography with voltammetric detection. Anal. Chem. 1989;61:436–441. doi: 10.1021/ac00180a012. [DOI] [PubMed] [Google Scholar]
- Cox A.L., Skipper J.C., Chen Y., Henderson R.A., Darrow T.L., Shabanowitz J., Engelhard V.H., Hunt D.F., Slingluff C.L. Identification of a peptide recognized by five melanoma-specific human cytotoxic T cell lines. Science. 1994;264:716–719. doi: 10.1126/science.7513441. [DOI] [PubMed] [Google Scholar]
- Shabanowitz J., Settlage R.E., Marto J.A., Christian R.E., White F.M., Russo P.S., Martin S.E., Hunt D.F. Sequencing the primordial soup. In: Burlingame A.L., Carr S.A., Baldwin M.A., editors. Mass Spectrometry in Biology and Medicine. Humana Press; Towata, NJ: 1999. pp. 163–177. [Google Scholar]
- Martin S.E., Shabanowitz J., Hunt D.F., Marto J.A. Sub-femtomole MS and MS/MS peptide sequence analysis using nano-HPLC micro-ESI Fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem. 2000;72:4266–4274. doi: 10.1021/ac000497v. [DOI] [PubMed] [Google Scholar]
- Chen Y., Sidney J., Southwood S., Cox A.L., Sakaguchi K., Henderson R., Appella E., Hunt D.F., Sette A., Engelhard V.H. Naturally processed peptides longer than nine amino acid residues bind to the class I MHC molecule HLA-A2.1 with high affinity and in different conformations. J. Immunol. 1994;152:2874–2881. [PubMed] [Google Scholar]
- Akatsuka Y., Warren E.H., Brickner A.G., Engelhard V.H., Riddell S.R. Determination of intronic sequences adjacent to an exon using PCR and genomic library constructed by TA cloning. Anal. Biochem. 2001;In press doi: 10.1006/abio.2000.4897. [DOI] [PubMed] [Google Scholar]
- Mercier B., Ferec C., Dufosse F., Huart J.J. Improvement in HLA-DQB typing by PCR-RFLPintroduction of a constant restriction site in one of the primers for digestion control. Tissue Antigens. 1992;4:86–89. doi: 10.1111/j.1399-0039.1992.tb01964.x. [DOI] [PubMed] [Google Scholar]
- Bacik I., Cox J.H., Anderson R., Yewdell J.W., Bennink J.R. TAP (transporter associated with antigen processing)-independent presentation of endogenously synthesized peptides is enhanced by endoplasmic reticulum insertion sequences located at the amino- but not carboxyl-terminus of the peptide. J. Immunol. 1994;152:381–387. [PubMed] [Google Scholar]
- Yellen-Shaw A.J., Laughlin C.E., Metrione R.M., Eisenlohr L.C. Murine transporter associated with antigen presentation (TAP) preferences influence class I–restricted T cell responses. J. Exp. Med. 1997;186:1655–1662. doi: 10.1084/jem.186.10.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neisig A., Roelse J., Sijts A.J., Ossendorp F., Feltkamp M.C., Kast W.M., Melief C.J., Neefjes J.J. Major differences in transporter associated with antigen presentation (TAP)-dependent translocation of MHC class I-presentable peptides and the effect of flanking sequences. J. Immunol. 1995;154:1273–1279. [PubMed] [Google Scholar]
- Greenwood F.C., Hunter W.M., Glover J.S. The preparation of 131I labelled human growth hormone of high specific radioactivity. Biochem. J. 1963;89:114–123. doi: 10.1042/bj0890114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura N., Miyajima N., Sazuka T., Tanaka A., Kawarabayasi Y., Sato S., Nagase T., Seki N., Ishikawa K., Tabata S. Prediction of the coding sequences of unidentified human genes. I. The coding sequences of 40 new genes (KIAA0001-KIAA0040) deduced by analysis of randomly sampled cDNA clones from human immature myeloid cell line KG-1. DNA Res. 1994;1:27–35. doi: 10.1093/dnares/1.1.27. [DOI] [PubMed] [Google Scholar]
- Loveland B.E., Wang C.R., Yonekawa H., Hermel E., Lindahl K.F. Maternally transmitted histocompatibility antigen of micea hydrophobic peptide of a mitochondrially encoded protein. Cell. 1990;60:971–980. doi: 10.1016/0092-8674(90)90345-f. [DOI] [PubMed] [Google Scholar]
- Zuberi A.R., Christianson G.J., Mendoza L.M., Shastri N., Roopenian D. Positional cloning and molecular characterization of an immunodominant cytotoxic determinant of the mouse H3 minor histocompatibility complex. Immunity. 1998;9:687–698. doi: 10.1016/s1074-7613(00)80666-4. [DOI] [PubMed] [Google Scholar]
- Mendoza L., Paz P., Zuberi A.R., Christianson G., Roopenian D.C., Shastri N. Minors held by majors. The H13 minor histocompatibility locus defined as a peptide/MHC class I complex. Immunity. 1997;7:461–472. doi: 10.1016/s1074-7613(00)80368-4. [DOI] [PubMed] [Google Scholar]
- Bhuyan P.K., Young L.L., Lindahl K.F., Butcher G.W. Identification of the rat maternally transmitted minor histocompatibility antigen. J. Immunol. 1997;158:3753–3760. [PubMed] [Google Scholar]
- Greenfield A., Scott D., Pennisi D., Ehrmann I., Ellis P., Cooper L., Simpson E., Koopman P. An H-YDb epitope is encoded by a novel mouse Y chromosome gene. Nat. Genet. 1996;14:474–478. doi: 10.1038/ng1296-474. [DOI] [PubMed] [Google Scholar]
- Snyder H.L., Yewdell J.W., Bennink J.R. Trimming of antigenic peptides in an early secretory compartment. J. Exp. Med. 1994;180:2389–2394. doi: 10.1084/jem.180.6.2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes E.A., Ortmann B., Surman M., Cresswell P. The protease inhibitor, N-acetyl-l-leucyl-l-leucyl-leucyl-l-norleucinal, decreases the pool of major histocompatibility complex class I-binding peptides and inhibits peptide trimming in the endoplasmic reticulum. J. Exp. Med. 1996;183:1569–1578. doi: 10.1084/jem.183.4.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussbaum A.K., Dick T.P., Keilholz W., Schirle M., Stevanovic S., Dietz K., Heinemeyer W., Groll M., Wolf D.H., Huber R. Cleavage motifs of the yeast 20S proteasome β subunits deduced from digests of enolase 1. Proc. Natl. Acad. Sci. USA. 1998;95:12504–12509. doi: 10.1073/pnas.95.21.12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuttler C., Nussbaum A.K., Dick T.P., Rammensee H.G., Schild H., Hadeler K.P. An algorithm for the prediction of proteasomal cleavages. J. Mol. Biol. 2000;298:417–429. doi: 10.1006/jmbi.2000.3683. [DOI] [PubMed] [Google Scholar]
- Momburg F., Roelse J., Howard J.C., Butcher G.W., Hammerling G.J., Neefjes J.J. Selectivity of MHC-encoded peptide transporters from human, mouse and rat. Nature. 1994;367:648–651. doi: 10.1038/367648a0. [DOI] [PubMed] [Google Scholar]
- Uebel S., Kraas W., Kienle S., Wiesmuller K.-H., Jung G., Tampe R. Recognition principle of the TAP transporter disclosed by combinatorial peptide libraries. Proc. Natl. Acad. Sci. USA. 1997;94:8976–8981. doi: 10.1073/pnas.94.17.8976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Endert P.M., Riganelli D., Greco G., Fleischhauer K., Sidney J., Sette A., Bach J.-F. The peptide-binding motif for the human transporter associated with antigen processing. J. Exp. Med. 1995;182:1883–1895. doi: 10.1084/jem.182.6.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt D.F., Yates J.R., Shabanowitz J., Winston S., Hauer C.R. Protein sequencing by tandem mass spectrometry. Proc. Natl. Acad. Sci. USA. 1986;83:6233–6237. doi: 10.1073/pnas.83.17.6233. [DOI] [PMC free article] [PubMed] [Google Scholar]