

Abstract
The effect of infection history is ignored in most animal models of infectious disease. The attachment protein of respiratory syncytial virus (RSV) induces T helper cell type 2–driven pulmonary eosinophilia in mice similar to that seen in the failed infant vaccinations in the 1960s. We show that previous influenza virus infection of mice: (a) protects against weight loss, illness, and lung eosinophilia; (b) attenuates recruitment of inflammatory cells; and (c) reduces cytokine secretion caused by RSV attachment protein without affecting RSV clearance. This protective effect can be transferred via influenza-immune splenocytes to naive mice and is long lived. Previous immunity to lung infection clearly plays an important and underestimated role in subsequent vaccination and infection. The data have important implications for the timing of vaccinations in certain patient groups, and may contribute to variability in disease susceptibility observed in humans.
Keywords: viral immunology, murine model, eosinophils, major histocompatibility complex tetramers, mucosal immunology
Introduction
The maturation of the immune system may partly be determined by microbial exposure early in life 1 2. The effect of early childhood infections on subsequent development of asthma and atopy has been widely discussed 3 4. Early exposure to mycobacteria 5, measles 6, and hepatitis A 7 may decrease the incidence of Th2-driven diseases, and the absence of gut flora leads to life-long inhibition of Th1 responses 8. Infants generally produce lower levels of IFN-γ, but this gradually increases with age 9. Collectively, these findings have led to the formulation of the “hygiene theory,” which suggests that reduced exposure to microbes deprives us of essential immune-educational input 10. Research into the effect of infection on immune responses to other antigens has mainly utilized agents such as ovalbumin (for an example, see reference 11). However, the effect of immunity to previous acute, successfully cleared pathogens on other infectious diseases has received little attention. We now use the murine model of respiratory syncytial virus (RSV) infection to address the impact of sequential infections on immunopathology.
In the 1960s, vaccination of infants with formalin-inactivated RSV resulted in a catastrophic enhancement of illness during subsequent natural encounter with this virus, leading to eosinophilic lung infiltrates and excess mortality 12. RSV is the most common cause of viral bronchiolitis in infants and young children in the Western world 13, and may also lead to asthma and atopy later in life 14. In BALB/c mice, a primary infection induces mild illness and a Th1 profile of immunity with resolution of infection in 7–10 d. Sensitization to the RSV attachment protein (G) before RSV infection leads to enhanced illness, mimicking that seen in the vaccine trials. Unlike a primary infection, the G protein induces pulmonary eosinophilia, which depends on CD4+ T cells producing type 2 cytokines (IL-4 and IL-5 [15, 16]) but is inhibited by overexuberant CD8+ T cells and IFN-γ 17. G-primed mice also display enhanced weight loss and illness (cachexia, ruffled fur, and reduced mobility) after RSV challenge.
The G-primed RSV infection model utilizes young inbred mice housed under strict pathogen-free conditions. However, these conditions may not be applicable to real life situations, as hosts are indeed exposed to numerous pathogens that may influence the response to subsequent infections. In the following study, we assess the influence of previous exposure to influenza virus (which induces strong IFN-γ responses from CD4+ and CD8+ T cells) on the Th2-driven eosinophilia and illness seen in the G-primed RSV mouse model. We demonstrate that such infection history benefits the host and has profound, long-lasting influences on subsequent responses to an additional mucosal infection. These findings have important implications for the timing and nature of vaccinations.
Materials and Methods
Mice and Virus Stocks.
10–12-wk-old female BALB/c mice (Harlan Olac Ltd.) were kept in pathogen-free conditions, according to institutional and Home Office guidelines. RSV (A2 strain) and recombinant vaccinia virus (rVV) expressing the attachment protein of RSV (rVV-G) or control β-galactosidase (rVV–β-gal) were grown in HEp-2 cells and assayed for infectivity 18. Influenza A X31 (hemagglutinin [HA] titer 1024) virus was supplied by Prof. Alan Douglas (National Institute for Medical Research, London, UK). All stocks were free of mycoplasma infection (determined by DNA hybridization; Gen-Probe Inc.).
Mouse Infection.
On day 0, mice were intranasally infected with 3 × 106 PFU of human RSV or 50 HA units of influenza (in 50 μl) and left to recover for 3–5 wk. Mice were then scarified on the rump with 3 × 106 PFU rVV-G or rVV–β-gal (in 10 μl). 14 d after vaccinia infection, mice were challenged intranasally with 3 × 106 PFU human RSV (in 50 μl). For brevity, the sequence of viral infections has been simplified in the remainder of the text. For example, intranasal RSV infection followed by rVV-G scarification and then finally RSV infection is represented by RSV–G-RSV (see Table ). The appearance and weight of mice were monitored daily. Mice were killed 7 d after the final viral infection by the injection of 3 mg pentobarbitone and exsanguinated via the femoral vessels.
Table 1.
Schematic Diagram of the Infection Sequence in BALB/c Mice
| Referencegroup | Main experimental groups | Control groups | ||||
|---|---|---|---|---|---|---|
| Designation | HEp2–G-RSV | RSV–G-RSV | Flu–G-RSV | HEp2–β-RSV | RSV–β-RSV | Flu–β-RSV |
| First infection: day 0 | Control (HEp-2) | RSV | Influenza | Control (HEp-2) | RSV | Influenza |
| Priming protein: day 21 | G protein | G protein | G protein | Control (β-gal) | Control (β-gal) | Control (β-gal) |
| Final infection: day 35 | RSV | RSV | RSV | RSV | RSV | RSV |
Mice were intranasally infected with RSV, influenza, or HEp-2 lysate on day 0, and after 21 d scarified with rVV-G or rVV–β-gal. On day 35, mice were infected intranasally with whole RSV and sacrificed 7 d later.
Illness Severity Score.
Mice were scored by an independent observer using a standard grading system as follows: 0, healthy; 1, barely ruffled fur; 2, ruffled fur but active; 3, ruffled fur and inactive; 4, ruffled fur, inactive and hunched; and 5, dead.
Cell Recovery.
Bronchoalveolar lavage (BAL), lung tissue, and serum were harvested using methods described previously 19. In brief, the lungs of each mouse were inflated six times with 1 ml of 12 mM lignocaine in Eagle's minimum essential medium and kept on ice. 100 μl of BAL fluid was cytocentrifuged onto glass slides and stained with hematoxylin and eosin. The remainder of BAL fluid was centrifuged, the supernatant was retained (stored at −80°C), and the pellet was resuspended at 106 cells/ml.
Production of MHC Class I Tetramer.
The gene encoding the Kd allele was cloned from mouse splenocytes using PCR and cloned into a T7 expression vector containing the biotin-[acetyl-ConA-carboxylase]synthetase (BirA) substrate peptide 20. Tagged Kd protein was purified using prokaryotic expression and refolded with human β2-microglobulin and peptide using standard methods 21. The peptides used were the immunodominant peptide TYQRTRALV from influenza nucleoprotein (NP) or SYIGINNI from the second matrix protein (M2) of RSV. The protein concentrate was biotinylated with BirA, and refolded monomers were purified using gel filtration and ion exchange before addition of streptavidin linked to PE. Tetramers were tested for their ability to stain appropriate antigen-specific T cell clones (data not shown) before use in experimental assays.
Flow Cytometric Analysis of Intracellular and Cell Surface Antigens.
Cells were stained with Quantum red (QR)-conjugated anti-CD8 or anti-CD4, FITC-conjugated CD45RB (both from BD PharMingen), and PE-conjugated MHC H-2Kd tetramers for 30 min on ice. Samples were then fixed for 20 min at room temperature with 2% formaldehyde. To detect intracellular cytokines, 106 cells/ml were incubated with 50 ng/ml PMA (Sigma-Aldrich), 500 ng/ml ionomycin (Calbiochem), and 10 mg/ml brefeldin A (Sigma-Aldrich) for 4 h at 37°C. Cells were then stained for CD8-QR and/or PE-conjugated MHC tetramers as described above and fixed. After permeabilizing with 0.5% saponin in PBS containing 1% BSA and 0.1% azide for 10 min, FITC-conjugated anti–IFN-γ (XMG1.2; BD PharMingen) or FITC-conjugated anti–IL-10 (JES 2A5; BD PharMingen) diluted 1:40 in saponin buffer was added. After 30 min, all samples were washed with PBS containing 1% BSA and 0.1% sodium azide and analyzed on a Coulter EPICS Elite flow cytometer collecting data on at least 40,000 lymphocytes. Directly conjugated isotype-matched control antibodies were used to set limits of background fluorescence.
Enumeration of Eosinophils.
Eosinophils were enumerated as granulocytes by flow cytometry, using their distinctive forward and side scatter properties. Identification was confirmed by counting eosinophils in hematoxylin and eosin–stained cytocentrifuge preparations.
Histology.
Mice were injected with 3 mg pentobarbitone and exsanguinated. The trachea was exposed and 1 ml of 2% formalin was instilled into the lungs. The inflated lungs were removed and placed in tubes containing 5 ml of 2% formal saline. The next day, samples were embedded in paraffin and sections were stained with hematoxylin and eosin for histological analysis.
Lung Virus Titer.
Clearance of RSV was assessed in lung homogenates from four mice per group at days 2, 4, and 7 after virus challenge. After centrifugation at 4,000 rpm for 4 min, supernatant was titrated in doubling dilutions on HEp-2 cell monolayers. 24 h later, monolayers were washed and incubated with peroxidase-conjugated goat anti-RSV antibody (Biogenesis). Infected cells were detected using 3-amino-9-ethylcarbazole (AEC), with infectious units being enumerated by light microscopy.
Assessment of the Effect of Previous Infection on Vaccinia Virus Replication.
Mice were infected intranasally with influenza, RSV, or HEp-2 antigen. After 3 wk, mice were then scarified on the rump with rVV-G. On days 2, 4, and 7 after scarification, the inguinal lymph nodes (draining the site of scarification) and a 1-cm square of scarified skin were removed. The inguinal lymph nodes were homogenized through a 100-μm nylon cell strainer (Falcon; Becton Dickinson), and the cells were counted and then surface stained for CD4-PE, CD8-QR, and CD45RB-FITC for 30 min on ice. After fixation in formaldehyde, expression of these markers was assessed by flow cytometry.
Both the external and internal surfaces of the scarified skin were scraped with a scalpel blade into serum-free medium, the suspension was centrifuged at 500 rpm to remove large debris, and 200 μl was applied to a HEp-2 monolayer in 2-ml tissue culture wells in log10 dilutions from neat to 10−5. After 2 h at 37°C, an additional 1.8 ml of R10F was added and the cultures were incubated for 24 h. The monolayers were then carefully washed and two drops of Gentian Violet was added for 20 min. After washing and drying, the number of virus plaques per well was counted by light microscopy. This number was multiplied by the dilution factor and then by five to adjust to plaques per milliliter (as only 200 μl of virus was added initially). The average number of plaques per milliliter for each dilution was then determined.
ELISA for RSV-specific Antibody.
ELISA coating antigen was prepared by infecting HEp-2 cells with RSV strain A2 at 1 PFU/cell. When significant sloughing of the monolayer was observed, the infected cells were harvested, centrifuged, resuspended in 3 ml distilled water, and then subjected to 2 min of sonication (Ultrawave Ltd.). 50-μl aliquots were stored at −20°C until required. Control antigen consisted of identically treated, uninfected HEP-2 cells. Microtiter plates were coated overnight at 37°C with 100 μl of a 1:100 dilution of either sonicated RSV or HEp-2 cells. After blocking with 2% normal rabbit serum for 2 h, dilutions of test samples (diluted in PBS containing 1% HEp-2 lysate) were added for an additional 1 h at room temperature. Bound antibody was detected using peroxidase-conjugated rabbit anti–mouse Ig and O-phenylenediamine (Sigma-Aldrich). Reactions were stopped with 50 μl of 2.5 M sulfuric acid. Optical densities were read at 490 nm (MR 5000; Dynatech Medical Products). The optical densities from RSV-coated wells were subtracted from those in HEp-2–coated wells for each sample.
Cytokine ELISA.
IL-4, IL-5, IFN-γ, and TNF were assessed in lung lavage supernatants by ELISA according to the manufacturers' instructions (BD PharMingen/Becton Dickinson). In brief, Immunosorb ELISA plates (Nunc) were coated with capture antibody and left overnight at 4°C. Wells were then washed five times with PBS/0.05% Tween 20 and blocked with PBS/10% fetal bovine serum for 1 h at room temperature. 100 μl of sample (neat or diluted 1:2) or standard was added to blocked wells for 2 h at room temperature. Bound cytokine was detected using biotinylated anticytokine antibody, avidin horseradish peroxidase, and tetramethylbenzidine. Color development was blocked with 2 N H2SO4, and optical densities were read at 450 nm. The concentration of cytokine in each sample was determined from the standard curve.
Purification of Influenza NP Tetramer–positive Cells.
10 mice were infected intranasally with influenza and scarified with rVV-G 3 wk later. After an additional 5 wk, mice were infected intranasally with RSV and 6 d later, lung and mediastinal lymph nodes were removed, disrupted into a single cell suspension, and counted. Individual nodes and lung were then incubated with biotinylated influenza NP-containing MHC tetramers for 30 min on ice. After washing, samples were incubated with avidin-coated MACS microbeads (20 μl/107 cells) and applied to a cooled Minimax column (Miltenyi Biotec). Samples of flow-through and column-bound cells were stained with QR-conjugated avidin (Sigma-Aldrich) and assessed for tetramer-positive cells by flow cytometry. Less than 1% of flow-through, but >90% of column-bound cells stained with the influenza tetramer.
CTL Assay.
Mice were infected with RSV or influenza, and after 14 d, spleens were removed and disrupted through a nylon mesh. Red blood cells were removed with ammonium chloride and 2 × 107 cells were added to a 25-cm3 flask in R10F containing 1% nonessential amino acids and β2-mercaptoethanol. 5 × 106 of the remaining splenocytes were incubated with 1 PFU/ml RSV or 1,000 HA influenza/107 cells. After 2 h, these were added to individual flasks containing splenocytes from either RSV- or influenza-primed animals. After 5 d at 37°C, the cells were counted and added as effectors in a chromium-release assay containing 104 RSV- or influenza-infected P815 cells (targets). In one experiment, purified MHC tetramer–binding cells were used directly ex vivo as effectors. Maximal and minimal lysis was determined by the addition of 2% Triton X-100 to targets or by incubating targets without effectors, respectively. Plates were then centrifuged at 800 rpm for 5 min and incubated at 37°C. After 4 h, plates were recentrifuged and 50 μl of supernatant was analyzed in a microplate scintillation counter (Top count; Packard).
Statistics.
Statistics were performed using a two-tailed analysis of variance assuming unequal variance.
Results
Homologous and Heterologous Lung Infection Abrogates Enhanced Illness in G-primed, RSV-challenged Mice.
In a standard experiment, mice are sensitized to the attachment G protein of RSV by dermal scarification with a vaccinia virus construct. 2–3 wk later, these mice are intranasally challenged with whole RSV (this infection regime is denoted by G-RSV). Mice treated in this way rapidly lose weight, become cachexic, and develop lung eosinophilia. In the following experiments, we determined what happens to the illness and eosinophilia caused by G-RSV if those mice had experienced a previous lung viral infection. The various groups of mice and sequences of infection are summarized in Table . As RSV is grown and titered in HEp-2 cells, a lysate of these cells is used as a control for the first infection. Mice infected with HEp-2 material alone consistently show no weight loss or lung inflammation (data not shown). Vaccinia virus–expressing β-gal controls for the vaccinia construct expressing the G protein. Therefore, mice sequentially infected with HEp-2–β-gal–RSV effectively experience a primary RSV infection and show mild weight loss (typically 5–10%). Sequential infection of mice with HEp-2–G-RSV represents a secondary infection to the G protein leading to 15–20% weight loss (Fig. 1 A), ruffled fur, and cachexia, similar to previous studies. These two sequences of infection represent those to which others are compared. Mice infected with RSV–β-gal–RSV (secondary infection to the whole virus) or with Flu–β-gal–RSV display the same weight loss as a primary RSV infection. Prior infection with RSV (RSV–G-RSV) and, remarkably, with influenza virus (Flu–G-RSV), significantly reduces G-induced weight loss (Fig. 1 A).
Figure 1.

Previous infection prevents illness during secondary responses to the attachment protein of RSV. (A) Weight loss was monitored on the day of, and during, the final RSV infection. The mean and standard deviation from five to six mice per group are shown. (B) Illness was monitored by a blinded observer using a standard grading system based on the degree of cachexia, ruffled fur, and mobility. The results from five to six mice per group per day were added (cumulative disease severity score). Mice experiencing secondary responses to the G protein are represented by the black bars; those preinfected with RSV and influenza are represented by the gray and white bars, respectively. (C) Eosinophils were enumerated in hematoxylin and eosin–stained cytocentrifuge preparations of cells recovered by BAL. The results represent individual mice from two independent experiments. The groups are designated as follows: A, HEp-2–G-RSV; B, HEp-2–β-gal–RSV; C, RSV–β-gal–RSV; D, Flu–β-gal–RSV; E, RSV–G-RSV; and F, Flu–G-RSV.
The beneficial effect of previous infection is also apparent when illness severity is scored and by measuring lung eosinophilia, which closely mirrors weight loss profile. All mice are healthy on the day of the final infection (illness score of 0). A primary RSV infection induces minimal illness (illness score of <1; data not shown) and no eosinophilia. Mice sensitized with rVV-G followed by RSV (HEp-2–G-RSV) experience immediate illness at day 1 (which is prolonged for 7 d after the final infection; Fig. 1 B) and extensive pulmonary eosinophilia (Fig. 1 C). Eosinophilia is not due to nonviral antigens (e.g., HEp-2 cells), as it is not induced in the RSV–β-gal–RSV or the HEp-2–β-gal–RSV group. Previous exposure to RSV or influenza (RSV–G-RSV or Flu–G-RSV) significantly reduces the illness duration and severity caused by the G protein. Both influenza- and RSV-preprimed animals appeared completely healthy by day 5 after the final RSV infection (Fig. 1 B). Previous infection with RSV or influenza also abolishes G-induced eosinophilia (Fig. 1 C). This dramatic effect of prior infection can be seen in hematoxylin and eosin–stained sections of lung. Mice sequentially infected with HEp-2–G-RSV have extensive lymphocytic and eosinophilic infiltrate surrounding both blood vessels and airways (Fig. 2, right). However, similar mice previously infected with RSV (Fig. 2, middle) or influenza (Fig. 2, left) have a different pattern of illness where infiltrates are reduced, restricted to the blood vessels, and contain no eosinophils.
Figure 2.

Previous lung infection changes the nature of the illness induced in rVV-G–primed, RSV-challenged mice. Lungs were inflated with 2% formalin in buffered saline, removed, and embedded in paraffin. Representative sections from HEp-2–G-RSV– (right), RSV–G-RSV– (middle), and influenza–G-RSV– (left) infected animals were stained with hematoxylin and eosin. The top panels show the extent of inflammatory infiltrate in representative sections of lung (original magnification: ×100); the bottom panels show the presence of eosinophils (indicated by arrows; original magnification: ×1,000, oil immersion).
Previous studies have shown that prior infection may alter the replication of vaccinia virus 22, which may explain the reduced eosinophilia and illness seen in our study. To test this, mice were primed with HEp-2 antigen, RSV, or influenza. After 3 wk, all mice were scarified with rVV-G and at different time points the skin and draining inguinal lymph nodes were removed. The titers of vaccinia recovered from the skin lesions were identical at all time points regardless of infection history (Fig. 3 A). There was also no difference in the expansion (Fig. 3 B) and cellular composition (CD4 or CD8; data not shown) or activation (CD45RB; Fig. 3 C) of cells in the draining inguinal lymph nodes. In addition, administration of influenza virus after the vaccinia virus has been cleared reduced eosinophilia, weight loss, and illness, similar to mice infected with influenza before rVV-G (data not shown). The results are also not explained by residual inflammatory activity to, or immune suppression by, the first infection. Mice had recovered their original weight and displayed histology and BAL cellular content similar to uninfected littermates. Proliferation assays to polyclonal stimulants or RSV showed no evidence of immunosuppression regardless of infection history (data not shown).
Figure 3.
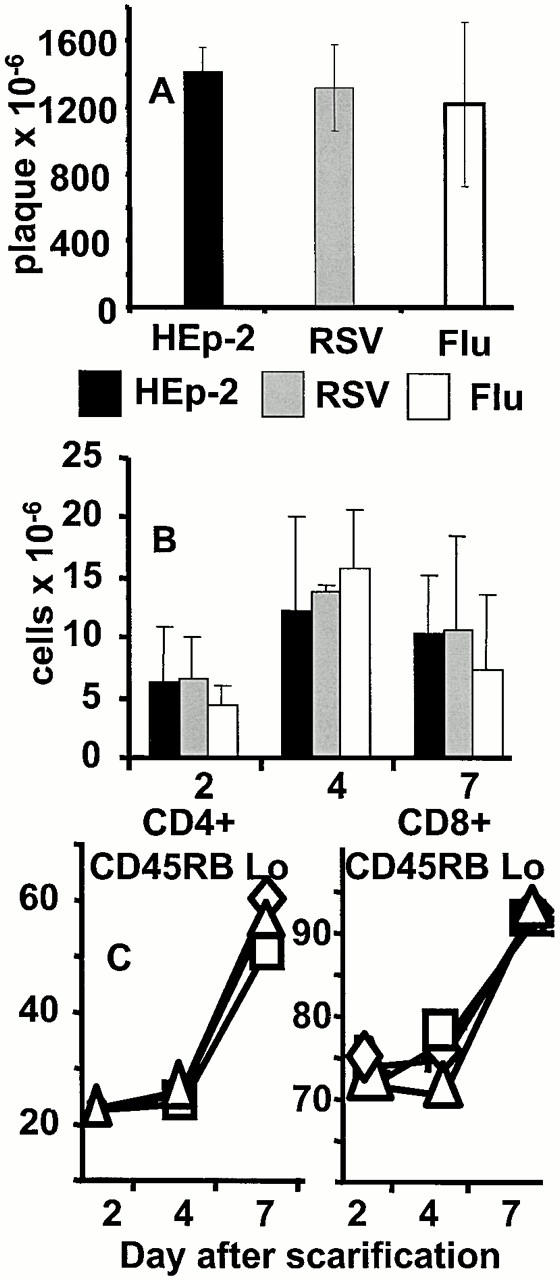
Previous lung infection does not alter the replication of rVV-G or the recruitment of cells to the inguinal lymph node. (A) Mice were intranasally infected with HEp-2, RSV, or influenza as shown and scarified with rVV-G after 3 wk. The number of plaques in the scarified area of skin was assessed after 24 h in HEp-2 monolayers. (B) Inguinal lymph nodes were also recovered and mechanically disrupted. Total viable cell recovery on days 2, 4, and 7 is shown. (C) The proportion of CD45RBlow CD4 (left), and CD8 (right) T cells was determined by flow cytometry from HEp-2- (□), RSV- (▵), and influenza- (⋄) preprimed mice. The results represent the mean and standard deviation of three to four mice per group.
Previous Influenza Virus Infection Reduces Antibody to the Attachment Protein (G) but Does Not Effect RSV Clearance.
RSV-specific antibody in serum was assessed by ELISA. Mice undergoing a secondary infection to either whole RSV (RSV–β-gal–RSV) or the G protein (HEp-2–G-RSV) displayed enhanced antibody (endpoint titer of 1/1,024) compared with mice undergoing a primary infection to RSV (HEp-2–β-gal–RSV and Flu–β-gal–RSV; endpoint of 1/32). Prior infection of G-primed, RSV-challenged mice with RSV (RSV–G-RSV) enhanced antibody titers further (endpoint of 1/4,096). However, previous experience of influenza virus (Flu–G-RSV) lowered antibody titers compared with G-primed, RSV-exposed mice (endpoint of 1/128; data not shown).
G protein priming enhances the elimination of a subsequent RSV infection. Previous infection of these mice with RSV or influenza did not alter RSV clearance as assessed by plaque assay on lung homogenates. Despite lower antibody titers in the Flu–G-RSV group, clearance of virus from the lungs was unaffected. The recoverable RSV titer on day 2 after the final infection was 4 ± 1.3 × 105, 4.8 ± 1.1 × 105, and 3.9 ± 1.9 × 105 in HEp-2–G-RSV, Flu–G-RSV, and RSV–G-RSV groups, respectively. No plaques were recovered on day 4 after infection compared with 12.2 ± 1.6 × 105 PFU/lung in mice undergoing a primary RSV infection. All mice undergoing a secondary infection to the G protein cleared virus by day 4 after the final intranasal infection regardless of infection history. As observed previously, all mice undergoing a primary RSV infection showed a slower clearance of virus from the lungs (by day 7 after final infection).
Mechanisms for Reduction in Illness and Pathology.
The G protein does not induce CD8+ T cells in BALB/c mice. We 17 and others 23 have shown that CD8+ T cells secreting IFN-γ induced by other RSV proteins inhibit G-primed lung eosinophilia. Therefore, we examined the phenotype of CD8+ T cells recruited to the lung by staining with H-2Kd tetramers containing the immunodominant epitopes from NP and RSV M2. Less than 0.5% influenza tetramer–positive cells were present in the lung before RSV challenge and <1% of these expressed IFN-γ. CD8+ T cells from mice sequentially infected with RSV–G-RSV do not bind the influenza NP MHC tetramer (Fig. 4 A) or an RSV M2–specific cell line (data not shown). However, the M2-specific cell line does bind the MHC class II M2–specific tetramer (Fig. 4 B). Sequential infection with Flu–G-RSV resulted in the recruitment of 16.9 ± 2.7% of CD8+ T cells that bound the influenza tetramer (Fig. 4 C). 39.4 ± 3.8% of these expressed IFN-γ (Fig. 4 E) whereas, in a separate experiment, 49.6 ± 6.4% were CD45RBlow (data not shown). The observation that intracellular IFN-γ increases dramatically from 1% previously to 39.4% after the final RSV challenge indicates activation of influenza-specific CD8+ T cells. In addition, influenza tetramer–positive cells in the spleen and lung of Flu-G–primed mice before RSV challenge were mostly CD45RBhigh (data not shown). In Flu–G-RSV mice, 6.8 ± 2.1% of CD8+ T cells bound the RSV tetramer, which is similar to mice undergoing a primary RSV infection (HEp-2–β-gal–RSV; data not shown). In mice sequentially infected with RSV–G-RSV, 13.8 ± 1.7% of CD8+ T cells bound the RSV tetramer (Fig. 4 D). Gating on these cells, we observed that 41.1 ± 6.2% of RSV-specific CD8+ T cells expressed intracellular IFN-γ (Fig. 4 F). Less than 0.5% of CD8+ T cells in these mice bound the influenza tetramer. It is interesting to note that the intensity of expression of tetramer varies between experiments. As T cell receptor levels are sensitive to activation, we believe this represents natural downregulation. The intensity of IFN-γ is also stronger in the RSV–G-RSV group (Fig. 4 F), which probably reflects activation by homologous rather than heterologous antigen. Intracellular IL-10 was also observed in MHC tetramer–stained cells (∼10%), the majority of which is present in cells that also produce IFN-γ (data not shown).
Figure 4.

RSV- and influenza-specific CD8+ T cells are recruited back to the lung during the final RSV infection. (A) BAL cells from mice sequentially primed with RSV–G-RSV do not bind the influenza NP–specific MHC tetramer. This sample was used to set the position of the quadrant. (B) M2-specific cell lines bind the RSV M2–specific MHC tetramer. The quadrant position was determined by staining the same sample with the influenza NP–specific tetramer. This cell line was generated from the spleen of rVV-M2–primed and RSV-challenged mice that had been grown in vitro for 3 wk. (C and D) The presence of NP- and M2-specific CD8+ T cells in mice sequentially infected with Flu–G-RSV and RSV–G-RSV, respectively. BAL was collected 7 d after the final RSV infection and samples were stained with MHC tetramers and antibodies to CD8. 40,000 lymphocytes were analyzed by flow cytometry. The quadrant positions were determined from staining cells in the RSV–G-RSV group with the NP-specific tetramer. (E and F) Anti-CD8– and tetramer-stained cells were then permeabilized with saponin and stained with FITC-conjugated antibody to IFN-γ. The proportion of CD8+, NP (E), and M2 (F) tetramer–stained cells expressing IFN-γ is shown. These results are representative of four to five mice per group.
TNF and IL-4 were significantly reduced in the BAL fluid (measured by ELISA) in Flu–G-RSV–infected mice. RSV–G-RSV–infected mice also displayed reduced IL-4 and IL-5 in lavage supernatants, whereas TNF and IFN-γ remained unchanged compared with the HEp-2–G-RSV group. The reduction in IL-4 and IL-5 may account for the reduced eosinophil recruitment by previous infections (Fig. 5).
Figure 5.
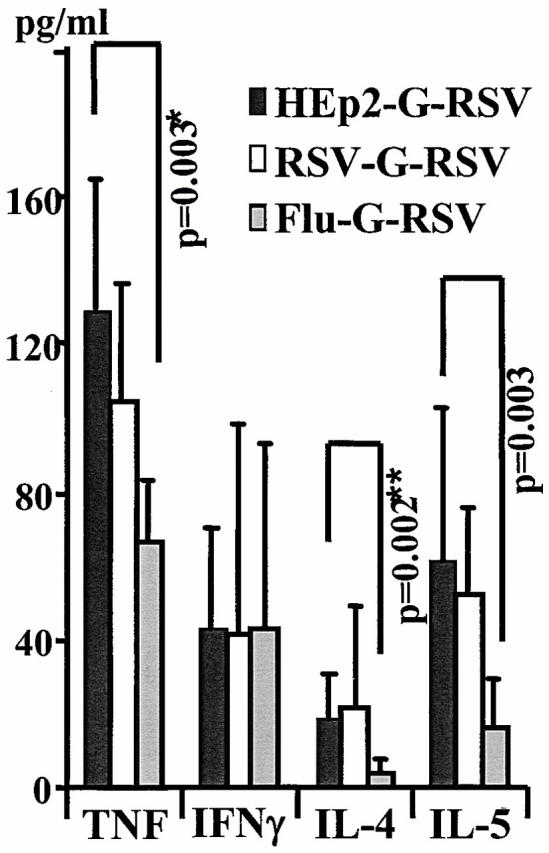
Previous infection reduces Th2 cytokines during secondary responses to the G protein. Mice were infected with HEp-2, RSV, or influenza, scarified with rVV-G 21 d later, and finally infected with RSV after 14 d. 7 d later, cytokine levels in lavage fluid were examined by ELISA and concentrations were calculated using a standard curve and linear regression analysis. The mean and standard deviation from individual mice in two independent experiments are shown.
Transfer of RSV- and Influenza-immune Splenocytes Partially Protects Mice from G-induced Eosinophilia and Is Long Lived.
To determine whether protection from illness can be transferred, mice were primed with RSV or influenza and splenocytes were recovered after 21 and 35 d. The cells were then adoptively transferred into naive recipients, which were then sensitized with rVV-G and challenged intranasally with RSV 2 wk later. Weight loss, illness severity score (data not shown), and lung eosinophilia (Fig. 6) were improved in mice given either RSV- or influenza-immune compared with naive splenocytes.
Figure 6.

Transfer of splenocytes from RSV- and influenza-immune mice partially protects G-primed, RSV-challenged mice from illness. Mice were intranasally infected with RSV and influenza and left for 21–35 d. 3 × 107 spleen cells were then transferred intravenously into naive animals. 1 d later, these mice were scarified with rVV-G and intranasally infected with RSV after an additional 14 d. Eosinophils were assessed in lung lavage samples in Giemsa-stained cytocentrifuge preparations. The shaded part represents naive mice (▪) or those infected with influenza (□), and left for 149 d before G priming and RSV infection.
The protective effect of previous infection on G-induced eosinophilia remains when the time between the first and second infection is increased from 21 to 149 d. Eosinophilia is significantly reduced in G-primed, RSV-challenged mice previously infected with influenza (Fig. 6, shaded area). Remarkably, protection from weight loss and illness severity was not maintained (data not shown). The observed increase in CD8+CD45RBlow cells in the BAL of influenza-primed mice may account for the reduction in eosinophilia (35.2 ± 5.4% in HEp-2–G-RSV mice compared with 50.2 ± 9.7 in Flu–G-RSV mice, P < 0.01; data not shown). Also, total intracellular IFN-γ was raised by previous influenza infection of rVV-G–primed RSV-challenged mice (20.66 ± 2 compared with 12.48 ± 4.73, P < 0.01). The mechanisms associated with eosinophilia and illness therefore disassociate as the time between sequential infections increases.
The effect of influenza infection on subsequent immunity and pathology to RSV does not appear to be due to cross-reactive CD8 T cell epitopes. Splenocytes from influenza- or RSV-infected mice were cultured with autologous APCs infected with RSV or influenza. After 5 d, cells were used in a cytotoxicity assay with RSV- or influenza-infected P815 cells. We were unable to test cytotoxic activity of influenza-immune splenocytes cultured with RSV, as the majority of the cells died during the 5-d incubation period, and those that were left did not stain with the influenza-specific MHC tetramer. Similar cell death occurred when RSV-immune splenocytes were cultured with influenza virus. However, influenza-immune splenocytes cultured with APCs infected with homologous virus expanded rapidly. These cells only lysed targets infected with influenza and not RSV (Fig. 7 A). To determine whether cross-reactivity was occurring in vivo, influenza tetramer–positive cells were purified using avidin-coated magnetic beads from the lung and mediastinal lymph nodes of influenza and rVV-G–infected mice 6 d after intranasal RSV challenge. These cells were added directly to a cytotoxicity assay containing RSV- and influenza-infected P815 cells (Fig. 7 B). Although cells purified from 10 mice were pooled, the maximum E/T ratios tested were 25:1. Remarkably, at this E/T ratio significant lysis of influenza- but not RSV-infected P815 cells was observed. Purified influenza tetramer–positive cells were also assessed in triplicate for proliferation to homologous or heterologous antigen. Proliferative responses to RSV were not significantly above the medium control (1,200 cpm with RSV compared with 1,384 cpm in medium alone, P > 0.09). This was not due to a defect in the sorted cells, as extensive incorporation of tritiated thymidine was observed in the presence of influenza-infected APCs (16,098 ± 1,034 cpm).
Figure 7.
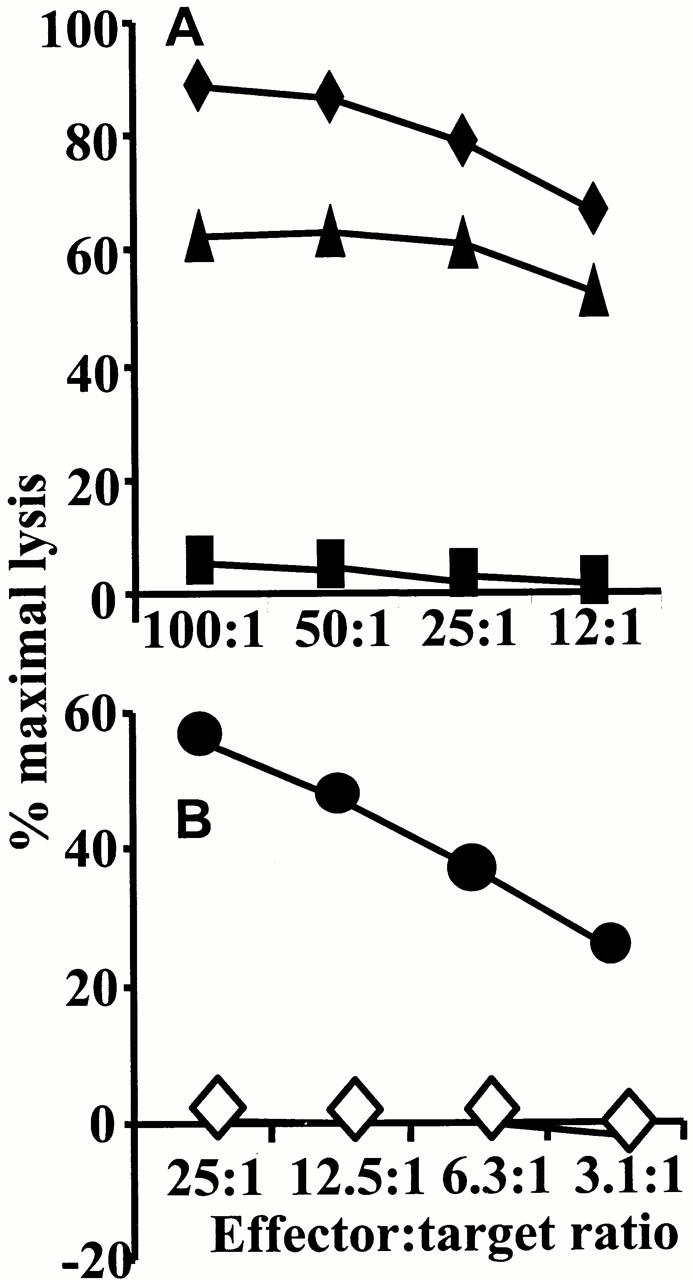
Splenocytes from influenza-primed mice do not kill RSV-infected targets. Spleens from RSV- or influenza-infected mice were removed after 14 d and disrupted to a single cell suspension. After 5 d in the presence of 2 PFU/cell of RSV or 1 HA unit of influenza virus, remaining cells were assessed for cytotoxic activity. RSV-immune mice effectively killed RSV-infected targets (♦). Influenza-immune splenocytes similarly killed influenza (▴) but not RSV-infected (▪) targets. (B) Influenza tetramer–positive cells were purified from the lung and mediastinal lymph nodes of 10 influenza-infected and RSV-challenged mice using avidin-coated magnetic beads and MACS. A cytotoxicity assay with influenza- (•) or RSV- (⋄) infected P815 cells was then performed. Spontaneous release has been subtracted from all results and never exceeded 10% of maximal chromium release.
Discussion
The effect of exposure to one organism on the pathogenesis of subsequent unrelated infections may have major biological consequence. In this study, we demonstrate that previous infection with influenza A virus dramatically protects mice from eosinophilia, weight loss, and illness caused by the G protein of RSV. Although the reduction in eosinophilia by influenza virus was variable, it was nonetheless statistically significant in all experiments and may reflect differences in the extent of the influenza-specific immune response induced in individual animals. An important observation is that previous influenza virus infection reduces G-induced eosinophilia for extended periods of time, whereas the influence on weight loss and illness wanes. This would indicate that the mechanisms for eosinophilia and illness are distinct. The possible mechanisms for alleviation of eosinophilia and illness include: (a) specific cross-reactivity in RSV T and B cell epitopes by influenza-primed cells; (b) bystander activation of influenza-specific cells; (c) “immunological imprinting” (skewing towards a Th1 phenotype); and (d) remodeling of the lung by the first infection.
Cross-reactive Immunity.
The beneficial effect of prior RSV exposure (RSV–G-RSV) is mediated primarily by specific recognition of epitopes within the homologous challenge virus. Cross-reactive memory cytotoxic T cell recognition of heterologous viruses has been described in some studies 24 25. Although individual CD4+ and CD8+ T cells may react with up to one million peptides 26, we have not observed cross-reactive immunity in proliferation or cytotoxicity assays. Incubating influenza-immune splenocytes with RSV in vitro for 5 d did not expand a population of cells capable of proliferating to, or lysing RSV-infected targets. Similarly, influenza tetramer–positive cells did not lyse RSV-infected targets. However, we cannot totally exclude cross-reactivity in vivo, as the lung microenvironment and the virus-infected APCs may support cross-reactivity. Prior RSV infection protects from eosinophilia induced by formalin-inactivated RSV vaccination 27. We show that this protection can be transferred and confers long-lasting strong protection. This may explain the absence of enhanced disease in older vaccinees during the early human vaccine trials, as older children would presumably have been exposed to protective natural lung infections before vaccination.
The effects described in this study are not explained by increased RSV or vaccinia virus clearance. Selin et al. show that prior immunity to lymphocyte choriomeningitis virus (LCMV), Pichinde virus, and murine cytomegalovirus is protective against a subsequent vaccinia virus infection 22. We have not detected differences in vaccinia virus replication in mice previously exposed to influenza or RSV in our study, which may reflect the acute mild nature of the infection and that replication is restricted to the lung. In addition, in preliminary experiments, the variability in the reduction of eosinophilia is identical when influenza virus is administered after the vaccinia virus.
G priming induces strong immunity with RSV clearance 4 d after infection. Although enhanced RSV-specific antibody occurred in all groups undergoing a secondary infection to either whole RSV or the G protein, viral clearance still took 4 d. Influenza actually reduced RSV-specific antibody without changing viral clearance. It is possible that recruited influenza-immune cells contribute to an environment in the lung that assists RSV clearance. CD8+ T cells can inhibit antibody production via production of IFN-γ 17 28. The production of IFN-γ by influenza-specific CD8+ T cells may therefore inhibit cytokines promoting B cells, a hypothesis supported by the cytokine ELISA data.
Bystander Activation.
The biological significance of bystander activation is hard to establish and should be treated with caution. Cycling, cytolytically active CD8+ T cells remain long after resolution of LCMV infection 29, providing a means for rapid elimination of subsequent infection with homologous virus. This may also be true for activated CD8+ T cells specific for heterologous viruses. Using MHC class I tetramers, we show that influenza NP–specific CD8+ T cells return to the lung and express intracellular IFN-γ and decreased CD45RB during RSV challenge. It is interesting to note that the intensity of IFN-γ expression is lower in influenza-specific compared with RSV-specific CD8+ T cells responding to homologous antigen, which may reflect differences in activation signals.
G protein–induced eosinophilia is sensitive to IFN-γ 17, and the return of influenza-specific CD8+ T cells to the lung may therefore tip the balance away from an environment conducive to eosinophilia. As memory cells are less dependent on costimulation compared with naive T cells 30, express higher levels of adhesion molecules, and are activated by IL-2, TNF, and IL-6 31 32, influenza-specific T cells may experience bystander activation. Bystander protection against heterologous virus has previously been demonstrated but the biological consequence was thought to be irrelevant 32 33 34. One problem with the bystander theory is that if influenza-specific CD8+ T cells are undergoing bystander production of IFN-γ, why do memory T cells resident in non–influenza-primed mice also not prevent eosinophilia? Memory responses to commensal organisms and environmental antigens do not appear to prevent G-induced eosinophilia. Influenza virus is particularly powerful at expanding T cell memory responses with Th1 cytokine profiles. Influenza-specific memory cells may therefore prevent eosinophilia due to their recent activation in the lung, their much increased frequency, and their production of IFN-γ but not Th2 cytokines.
Unlike the positive influence shown in the current study, bystander activation has been linked to immunopathology. Mice previously infected with LCMV show enhanced acute fatty necrosis after subsequent vaccinia virus infection that may be mediated by excess IFN-γ production 22 and are more sensitive to bacterial LPS 35. Coinfection of mice with Fasciola hepatica exacerbates disease to Bordetella pertussis 36, and Schistosoma mansoni egg–induced granulomas cause vaccinia virus persistence 37. Both viral and bacterial infections can interfere with peripheral tolerance 38, and bystander activation may induce and maintain autoimmunity 39 40 41.
Immunological Imprinting and Lung Remodelling.
Immune responses in children are skewed to a Th2 phenotype at birth and become more Th1-biased as they experience subsequent infections 1. In our study, the IFN-γ observed by intracellular staining and the reduction in IL-4 protein in lavage fluid indicate a shift towards a Th1 phenotype. It should be noted that although intracellular IFN-γ was observed in influenza-specific CD8+ T cells, IFN-γ protein in BAL supernatants was not significantly different between groups. The effect on eosinophilia may therefore represent a change in the ratio between Th1 and Th2 cytokines, as IL-4 and IL-5 were decreased in influenza-immune mice. IL-10 plays a major regulatory role in the course of inflammatory responses by downregulating cell recruitment and cytokine synthesis. The role of CD4+ T cells producing IL-10 and/or TGF-β has been extensively examined in the gut (for example, see references 42 43 44). Regulatory T cells producing IL-10 and/or TGF-β even suppress immune responses to other antigens in the same environment 45. TNF, on the other hand, has been linked to illness and weight loss during infection with several viruses 46. When released in large quantities, it enters the bloodstream and is associated with weight loss and wasting 47 48. The reduction of TNF and the increase in IL-10 in MHC tetramer–positive cells by previous infection may explain why illness is less severe than in naive G protein–primed, RSV-challenged mice.
Decreased TNF-mediated lysis of antigen-activated or virally infected cells 49 50 may also explain the results of this study. Such cell death in the large inflammatory infiltrate, or the infected respiratory epithelium, may result in the release of proinflammatory or damaging chemicals and lead to the illness observed during RSV infection. Alternatively, RSV-induced illness is thought to be a consequence of the immune response rather than uncontrolled viral replication. A reduction in inflammatory infiltrate may explain the reduced illness severity.
When influenza virus infection was given 149 d before G protein priming, eosinophilia but not illness and weight loss were abrogated. This divergence in different parameters of illness is easily explained by the effect on cytokines described above. The impact of previous influenza infection on TNF levels may subside with time. On the other hand, memory cells may still be recruited to the lung and influence eosinophilia via the production of IFN-γ. We did observe enhanced recruitment of activated CD8+ T cells to the lung in influenza-primed mice. Finally, the effect of previous infection on lung remodelling warrants further investigation. However, we did not observe an increase in vasculature or other architectural changes in lung tissue sections.
In summary, our results imply that infection history can have a significant positive biological effect on the immunopathology of subsequent unrelated infection. Clearly, vaccination against infectious diseases is a priority. However, animals naive to previous infections may not be the correct “role model” for infection-experienced humans. Furthermore, the composition of vaccines may need to be tailored to each age group. Infants will have encountered fewer infectious agents than adults, immunocompromised patients may lose the “memory” of infection, and the elderly experience immune senescence. Interactions between unrelated microorganisms have so far mostly been overlooked, but they clearly need further research, both in laboratory-based animal and in human studies.
Acknowledgments
The authors wish to thank Prof. Brigitte Askonas and Dr. Andrew Godkin for invaluable discussion, Prof. Alan Douglas for providing influenza virus X31, and Dr. Sharon Kendall for help and development of the photographs.
This work was supported by an Allan and Hanburys/South African Pulmonology Society Research Fellowship, Medical Research Council (South Africa) Overseas Scholarship (G. Walzl), a Wellcome Trust program grant (054797/Z/98/Z; P.J.M. Openshaw), and a Medical Research Council Career Establishment Grant (T. Hussell).
Footnotes
Abbreviations used in this paper: BAL, bronchoalveolar lavage; β-gal, β-galactosidase; HA, hemagglutinin; LCMV, lymphocyte choriomeningitis virus; M2, second matrix protein; NP, nucleoprotein; QR, Quantum red; RSV, respiratory syncytial virus.
References
- Prescott S.L., Macaubas C., Smallacombe T., Holt B.J., Sly P.D., Holt P.G. Development of allergen-specific T-cell memory in atopic and normal children. Lancet. 1999;353:196–200. doi: 10.1016/S0140-6736(98)05104-6. [DOI] [PubMed] [Google Scholar]
- Romagnani S. Induction of TH1 and TH2 responsesa key role for the ‘natural’ immune response? Immunol. Today. 1992;13:379–381. doi: 10.1016/0167-5699(92)90083-J. [DOI] [PubMed] [Google Scholar]
- Martinez F.D., Wright A.L., Taussig L.M., Holberg C.J., Halonen M., Morgan W.J. Asthma and wheezing in the first six years of life. N. Engl. J. Med. 1995;332:133–138. doi: 10.1056/NEJM199501193320301. [DOI] [PubMed] [Google Scholar]
- Holt P.G., Macaubas C. Development of long-term tolerance versus sensitization to environmental allergens during the perinatal period. Curr. Opin. Immunol. 1997;9:782–787. doi: 10.1016/s0952-7915(97)80178-1. [DOI] [PubMed] [Google Scholar]
- Shirakawa T., Enomoto T., Shimazu S., Hopkin J.M. The inverse association between tuberculin responses and atopic disorder. Science. 1997;275:77–79. doi: 10.1126/science.275.5296.77. [DOI] [PubMed] [Google Scholar]
- Shaheen S.O., Aaby P., Hall A.J., Barker D.J., Heyes C.B., Shiell A.W., Goudiaby A. Measles and atopy in Guinea-Bissau. Lancet. 1996;347:1792–1796. doi: 10.1016/s0140-6736(96)91617-7. [DOI] [PubMed] [Google Scholar]
- Matricardi P.M., Rosmini F., Ferrigno L., Nisini R., Rapicetta M., Chionne P., Stroffolini T., Pasquini P., D'Amelio R. Cross sectional retrospective study of prevalence of atopy among Italian military students with antibodies against hepatitis A virus. Br. Med. J. 1997;314:999–1003. doi: 10.1136/bmj.314.7086.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudo N., Sawamura S., Tanaka K., Aiba Y., Kubo C., Koga Y. The requirement of intestinal bacterial flora for the development of an IgE production system fully susceptible to oral tolerance induction. J. Immunol. 1997;159:1739–1745. [PubMed] [Google Scholar]
- Prescott S.L., Macaubas C., Holt B.J., Smallacombe T.B., Loh R., Sly P.D., Holt P.G. Transplacental priming of the human immune system to environmental allergensuniversal skewing of initial T cell responses toward the Th2 cytokine profile. J. Immunol. 1998;160:4730–4737. [PubMed] [Google Scholar]
- Rook G.A., Stanford J.L. Give us this day our daily germs. Immunol. Today. 1998;19:113–116. doi: 10.1016/s0167-5699(97)01204-8. [DOI] [PubMed] [Google Scholar]
- Erb K.J., Holloway J.W., Sobeck A., Moll H., Le G.G. Infection of mice with Mycobacterium bovis-Bacillus Calmette-Guerin (BCG) suppresses allergen-induced airway eosinophilia. J. Exp. Med. 1998;187:561–569. doi: 10.1084/jem.187.4.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh K., Fishaut J.M. Immunopathologic mechanisms in lower respiratory tract disease of infants due to respiratory syncytial virus. Prog. Med. Virol. 1980;26:94–118. [PubMed] [Google Scholar]
- Heilman C.A. Respiratory syncytial and parainfluenza viruses. J. Infect. Dis. 1990;161:402–406. doi: 10.1093/infdis/161.3.402. [DOI] [PubMed] [Google Scholar]
- Sigurs N., Bjarnason R., Sigurbergsson F., Kjellman B., Bjorksten B. Asthma and immunoglobulin E antibodies after respiratory syncytial virus bronchiolitisa prospective cohort study with matched controls. Pediatrics. 1995;95:500–505. [PubMed] [Google Scholar]
- Alwan W.H., Kozlowska W.J., Openshaw P.J.M. Distinct types of lung disease caused by functional subsets of antiviral T cells. J. Exp. Med. 1994;179:81–89. doi: 10.1084/jem.179.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alwan W.H., Record F.M., Openshaw P.J.M. Phenotypic and functional characterization of T cell lines specific for individual respiratory syncytial virus proteins. J. Immunol. 1993;150:5211–5218. [PubMed] [Google Scholar]
- Hussell T., Baldwin C.J., O'Garra A., Openshaw P.J.M. CD8+ T-cells control Th2-driven pathology during pulmonary respiratory syncytial virus infection. Eur. J. Immunol. 1997;27:3341–3349. doi: 10.1002/eji.1830271233. [DOI] [PubMed] [Google Scholar]
- Bangham C.R.M., Cannon M.J., Karzon D.T., Askonas B.A. Cytotoxic T-cell response to respiratory syncytial virus in mice. J. Virol. 1985;56:55–59. doi: 10.1128/jvi.56.1.55-59.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussell T., Spender L.C., Georgiou A., O'Garra A., Openshaw P.J.M. Th1 and Th2 cytokine induction in pulmonary T-cells during infection with respiratory syncytial virus. J. Gen. Virol. 1996;77:2447–2455. doi: 10.1099/0022-1317-77-10-2447. [DOI] [PubMed] [Google Scholar]
- Schatz P.J. Use of peptide libraries to map the substrate specificity of a peptide-modifying enzymea 13 residue consensus peptide specifies biotinylation in Escherichia coli . Biotechnology (NY). 1993;11:1138–1143. doi: 10.1038/nbt1093-1138. [DOI] [PubMed] [Google Scholar]
- Altman J.D., Moss P.H., Goulder P.R., Barouch D.H., McHeyzer W.M., Bell J.I., McMichael A.J., Davis M.M. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996;274:94–96. doi: 10.1126/science.274.5284.94. [DOI] [PubMed] [Google Scholar]
- Selin L.K., Varga S.M., Wong I.C., Welsh R.M. Protective heterologous antiviral immunity and enhanced immunopathogenesis mediated by memory T cell populations. J. Exp. Med. 1998;188:1705–1715. doi: 10.1084/jem.188.9.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikiatkhachorn A., Braciale T.J. Virus specific CD8+ T lymphocytes downregulate T helper cell type 2 cytokine secretion and pulmonary eosinophilia during experimental murine respiratory syncytial virus infection. J. Exp. Med. 1997;186:421–432. doi: 10.1084/jem.186.3.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selin L.K., Nahill S.R., Welsh R.M. Cross-reactivities in memory cytotoxic T lymphocyte recognition of heterologous viruses. J. Exp. Med. 1994;179:1933–1943. doi: 10.1084/jem.179.6.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selin L.K., Vergilis K., Welsh R.M., Nahill S.R. Reduction of otherwise remarkably stable virus-specific cytotoxic T lymphocyte memory by heterologous viral infections. J. Exp. Med. 1996;183:2489–2499. doi: 10.1084/jem.183.6.2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason D. A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol. Today. 1998;19:395–404. doi: 10.1016/s0167-5699(98)01299-7. [DOI] [PubMed] [Google Scholar]
- Waris M.E., Tsou C., Erdman D.D., Zaki S.R., Anderson L.J. Respiratory syncytial virus infection in BALB/c mice previously immunized with formalin-inactivated virus induces enhanced pulmonary inflammatory response with a predominant Th2-like cytokine pattern. J. Virol. 1996;70:2852–2860. doi: 10.1128/jvi.70.5.2852-2860.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemeny D.M., Noble A., Holmes B.J., Diaz-Sanchez D. Immune regulationa new role for the CD8+ T cell. Immunol. Today. 1994;15:107–110. doi: 10.1016/0167-5699(94)90152-X. [DOI] [PubMed] [Google Scholar]
- Selin L.K., Welsh R.M. Cytolytically active memory CTL present in lymphocytic choriomeningitis virus-immune mice after clearance of virus infection. J. Immunol. 1997;158:5366–5373. [PubMed] [Google Scholar]
- Croft M., Bradley L.M., Swain S.L. Naive versus memory CD4 T cell response to antigen. Memory cells are less dependent on accessory cell costimulation and can respond to many antigen-presenting cell types including resting B cells. J. Immunol. 1994;152:2675–2685. [PubMed] [Google Scholar]
- Unutmaz D., Pileri P., Abrignani S. Antigen-independent activation of naive and memory resting T cells by a cytokine combination. J. Exp. Med. 1994;180:1159–1164. doi: 10.1084/jem.180.3.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehl S., Hombach J., Aichele P., Hengartner H., Zinkernagel R.M. Bystander activation of cytotoxic T cellsstudies on the mechanism and evaluation of in vivo significance in a transgenic mouse model. J. Exp. Med. 1997;185:1241–1251. doi: 10.1084/jem.185.7.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarozinski C.C., Welsh R.M. Minimal bystander activation of CD8 T cells during the virus-induced polyclonal T cell response. J. Exp. Med. 1997;185:1629–1639. doi: 10.1084/jem.185.9.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murali K.K., Altman J.D., Suresh M., Sourdive D., Zajac A., Ahmed R. In vivo dynamics of anti-viral CD8 T cell responses to different epitopes. An evaluation of bystander activation in primary and secondary responses to viral infection. Adv. Exp. Med. Biol. 1998;452:123–142. doi: 10.1007/978-1-4615-5355-7_14. [DOI] [PubMed] [Google Scholar]
- Nguyen K.B., Biron C.A. Synergism for cytokine-mediated disease during concurrent endotoxin and viral challengesroles for NK and T cell IFN-gamma production. J. Immunol. 1999;162:5238–5246. [PubMed] [Google Scholar]
- Brady M.T., O'Neill S.M., Dalton J.P., Mills K.H. Fasciola hepatica suppresses a protective Th1 response against Bordetella pertussis . Infect. Immun. 1999;67:5372–5378. doi: 10.1128/iai.67.10.5372-5378.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Actor J.K., Marshall M.A., Eltoum I.A., Buller R.M., Berzofsky J.A., Sher A. Increased susceptibility of mice infected with Schistosoma mansoni to recombinant vaccinia virusassociation of viral persistence with egg granuloma formation. Eur. J. Immunol. 1994;24:3050–3056. doi: 10.1002/eji.1830241220. [DOI] [PubMed] [Google Scholar]
- Ehl S., Hombach J., Aichele P., Rulicke T., Odermatt B., Hengartner H., Zinkernagel R., Pircher H. Viral and bacterial infections interfere with peripheral tolerance induction and activate CD8+ T cells to cause immunopathology. J. Exp. Med. 1998;187:763–774. doi: 10.1084/jem.187.5.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans C.F., Horwitz M.S., Hobbs M.V., Oldstone M.B. Viral infection of transgenic mice expressing a viral protein in oligodendrocytes leads to chronic central nervous system autoimmune disease. J. Exp. Med. 1996;184:2371–2384. doi: 10.1084/jem.184.6.2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S.D., Vanderlugt C.L., Begolka W.S., Pao W., Yauch R.L., Neville K.L., Katz L.Y., Carrizosa A., Kim B.S. Persistent infection with Theiler's virus leads to CNS autoimmunity via epitope spreading. Nat. Med. 1997;3:1133–1136. doi: 10.1038/nm1097-1133. [DOI] [PubMed] [Google Scholar]
- Horwitz M.S., Bradley L.M., Harbertson J., Krahl T., Lee J., Sarvetnick N. Diabetes induced by Coxsackie virusinitiation by bystander damage and not molecular mimicry. Nat. Med. 1998;4:781–785. doi: 10.1038/nm0798-781. [DOI] [PubMed] [Google Scholar]
- Morrissey P.J., Charrier K., Braddy S., Liggitt D., Watson J.D. CD4+ T cells that express high levels of CD45RB induce wasting disease when transferred into congenic severe combined immunodeficient mice. Disease development is prevented by cotransfer of purified CD4+ T cells. J. Exp. Med. 1993;178:237–244. doi: 10.1084/jem.178.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powrie F., Carlino J., Leach M.W., Mauze S., Coffman R.L. A critical role for transforming growth factor-beta but not interleukin 4 in the suppression of T helper type 1–mediated colitis by CD45RBlow CD4+ T cells. J. Exp. Med. 1996;183:2669–2674. doi: 10.1084/jem.183.6.2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn R., Lohler J., Rennick D., Rajewsky K., Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- Groux H., O'Garra A., Bigler M., Rouleau M., Antonenko S., de Vries J.E., Roncarolo M.G. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997;389:737–742. doi: 10.1038/39614. [DOI] [PubMed] [Google Scholar]
- Orange J.S., Wolf S.F., Biron C.A. Effects of IL-12 on the response and susceptibility to experimental viral infections. J. Immunol. 1994;152:1253–1264. [PubMed] [Google Scholar]
- Fong Y., Tracey K.J., Moldawer L.L., Hesse D.G., Manogue K.B., Kenney J.S., Lee A.T., Kuo G.C., Allison A.C., Lowry S.F. Antibodies to cachectin/tumor necrosis factor reduce interleukin 1 beta and interleukin 6 appearance during lethal bacteremia. J. Exp. Med. 1989;170:1627–1633. doi: 10.1084/jem.170.5.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber J., Sgonc R., Hu Y.H., Beug H., Wick G. Thymocyte apoptosis induced by elevated endogenous corticosterone levels. Eur. J. Immunol. 1994;24:1115–1121. doi: 10.1002/eji.1830240516. [DOI] [PubMed] [Google Scholar]
- Wallach D., Varfolomeev E.E., Malinin N.L., Goltsev Y.V., Kovalenko A.V., Boldin M.P. Tumor necrosis factor receptor and Fas signaling mechanisms. Annu. Rev. Immunol. 1999;17:331–367. doi: 10.1146/annurev.immunol.17.1.331. [DOI] [PubMed] [Google Scholar]
- Lenardo M., Chan K.M., Hornung F., McFarland H., Siegel R., Wang J., Zheng L. Mature T lymphocyte apoptosis—immune regulation in a dynamic and unpredictable antigenic environment. Annu. Rev. Immunol. 1999;17:221–253. doi: 10.1146/annurev.immunol.17.1.221. [DOI] [PubMed] [Google Scholar]