

Abstract
The complement system enhances antibody responses to T-dependent antigens, but paradoxically, deficiencies in C1 and C4 are strongly linked to autoantibody production in humans. In mice, disruption of the C1qa gene also results in spontaneous autoimmunity. Moreover, deficiencies in C4 or complement receptors 1 and 2 (CR1/CR2) lead to reduced selection against autoreactive B cells and impaired humoral responses. These observations suggest that C1 and C4 act through CR1/CR2 to enhance humoral immunity and somehow suppress autoimmunity. Here we report high titers of spontaneous antinuclear antibody (ANA) in C4 −/ − mice. This systemic lupus erythematosus–like autoimmunity is highly penetrant; by 10 mo of age, all C4 − /− females and most males produced ANA. In contrast, titers and frequencies of ANA in Cr2 − /− mice, which are deficient in CR1 and CR2, never rose significantly above those in normal controls. Glomerular deposition of immune complexes (ICs), glomerulonephritis, and splenomegaly were observed in C4 − /− but not Cr2 − /− mice. C4 − /−, but not Cr2 − /−, mice accumulate activated T and B cells. Clearance of circulating ICs is impaired in preautoimmune C4 − /−, but not Cr2 − /−, mice. C4 deficiency causes spontaneous, lupus-like autoimmunity through a mechanism that is independent of CR1/CR2.
Keywords: complement, autoantibody, glomerulonephritis, splenomegaly, immune complex
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by generalized disturbances in T and B lymphocytes and inflammatory damage to many tissues 1 2. Activated, autoreactive B cells are present in patients with SLE and produce high titers of serum autoantibody to nuclear components. Serum antibody to double-stranded (ds)DNA is an important diagnostic marker for SLE. These antinuclear antibodies (ANAs) can directly attack tissues or form immune complexes (ICs) that elicit inflammation and damage tissues such as the kidney. Either individually or together, both processes can eventually cause organ failure 1 3 4.
The etiology and pathogenesis of SLE remain poorly understood. A variety of genetic factors have been linked to SLE 1 4 5, and among these, deficiencies in components of the classical pathway of complement carry perhaps the strongest association 1. SLE develops in most individuals with genetic deficiencies in C1 or C4 6 7 8 9. Although genetic deficiencies in complement are rare, acquired deficiencies in complement components are common 8 and characteristic of SLE flares in patients 10 11 12. These acquired deficiencies are presumed to result from the consumption of complement by ICs 10 11 12; their impact, if any, in SLE pathogenesis is unknown. Thus, in humans, components of the classical pathway for complement activation, especially C1 and C4, may suppress incipient autoimmunity.
Recently, a similar role for C1q in suppressing autoimmunity in mice was demonstrated by Botto et al. 13. C1qa − /− mice on a mixed B6/129 genetic background spontaneously produced high titers of ANA, whereas wild-type controls generated low levels of autoantibody; consequently, 25% of C1qa − /− mice also exhibited glomerulonephritis 13.
How C1q activity suppresses autoimmunity remains unknown. C1 is the first component in the classical pathway, and one subunit of C1q associates with two subunits of C1r and C1s to form the C1 macromolecular complex 14. C1q binds to the Fc portions of antibodies complexed with antigen. This binding induces enzymatic activity by C1r, leading to the sequential activation of C1s, C4, and C2 to form the C3 convertase 14 15. The split products of C4 and C3 can attach covalently to proteins 14 15, and several of these split products—C4b, C4d, iC3b, C3dg, and C3d—are ligands for the CR1 (CD35) and CR2 (CD21) complement receptors 16. Significantly, C4 often mediates the biological activities of C1. For example, phagocytosis and lysis of bacteria 17 are regulated by C1 activity in generating C4 fragments and the formation of the C3 convertase. This interdependence suggests that C1q might suppress autoimmunity through a mechanism also requiring C4, a notion supported by clinical evidence that deficiencies in either C1 or C4 are strongly linked to SLE 6 7 9 18.
How might C1 and C4 suppress the production of autoreactive antibody? Paradoxically, complement promotes specific immunity to T-dependent antigens 16; B lymphocytes express receptors for complement 19, and the temporary depletion of C3 reduces primary antibody responses 20. Also, impaired humoral immune responses are common in individuals with genetic deficiencies in some complement components 21. Recently, mice deficient in complement C1q, C4, and C3 were shown to have diminished antibody and germinal center (GC) responses 22 23. Diminished antibody and GC responses are also characteristic of Cr2 knockout mice that are deficient for CR1 and CR2 24 25 26. The observation that C4 − /− and Cr2 − /− mice generate identical patterns of humoral impairment suggests that C4 enhances B cell responses via CR1/CR2 16.
Three dominant models, not mutually exclusive, have been proposed to explain how C4 interacts with CR1/CR2 to promote humoral immunity. First, CR1/CR2 may focus complement-decorated ICs to follicular dendritic cells, promoting their support of GCs 27 28 29. Second, complement-decorated antigens may bridge the B cell antigen receptor (BCR) and CR2/CD19 coreceptor to enhance B cell responses 16 21 30. Third, complement-decorated antigens may aggregate CR2/CD19 complexes independently of the BCR and elicit CD19 signals that increase B cell responsiveness 31. Similar mechanisms may operate during the late stages of B cell development in bone marrow. Cr2 − /− and C4 − /− mice do not efficiently anergize B cells expressing autoreactive BCRs 32, and naive B cells from Cr2 − /− mice express patterns of VH gene segment usage different from Cr2 +/− controls 26. Similarly, lpr/lpr mice bred to be deficient in C4 or CR1/CR2 have accelerated autoimmune disease 32.
These models to explain complement's (C4) role in enhancing humoral immunity and suppressing autoantibody depend on CR1/CR2. This dependence on CR1/CR2 in systemic autoimmunity is consistent with the observation that leukocytes from SLE patients express lower amounts of CR1 and CR2 33. Similarly, CR1 and CR2 are progressively lost from the surfaces of B cells in MRL/lpr mice, even before the onset of autoimmune nephritis 34.
However, if complement's promotion of humoral immunity and suppression of SLE are mediated by CR1/CR2, the absence of clinical associations between genetic deficiencies of C3 and SLE 6 7 is perplexing, as C3 split products are principal ligands for CR1 and CR2 16 21. Indeed, whereas C3-deficient mice have poor primary antibody responses 16, they exhibit good selection against autoreactive B cells and no significant acceleration of lpr-induced autoimmunity 32. The weak association of C3 deficiency and autoimmunity does not preclude a role for CR1 and CR2 in self-tolerance, as C4 also generates ligands for CR1 and CR2 16. The absence of an association does, however, raise the possibility that C4 promotes autoantibody production independently of CR1/CR2 and by a mechanism distinct from complement's immunoenhancing activities.
In this study, we demonstrate that C4 − /− mice achieve high levels of spontaneous ANA, splenomegaly, and glomerulonephritis by 10 mo of age. Complete genetic penetrance of C4 deficiency occurs in female mice, but only two-thirds of age-matched males become autoimmune. In contrast, Cr2 − /− mice on the same genetic background did not produce significant levels of ANA nor exhibit kidney pathology. Thus, C4 deficiency elicits a lupus-like autoimmunity in mice by processes that do not depend on CR1 and CR2.
Materials and Methods
Mice.
Cr2 − /− 25 and C4 − /− 22 mice were originally established on the 129/Sv genetic background and subsequently bred onto a hybrid B6/129, homozygous Ighbbackground as described 26. In brief, Cr2 − /− and C4 − /− mice were crossed with C57BL/6 mice (The Jackson Laboratory), and F2 offspring were typed for Cr2 or C4 22 25 26 and Igh haplotypes 26. C57BL/6 and 129 mice share identical MHC haplotypes, the locus most prominently linked to autoimmunity. The two strains differ at Igh: B6 mice are Ighb and 129 mice are Igha. As heterogeneity at Igh could alter the potential B cell repertoires in these mice, we selected only cohorts of B6/129 mice and their littermates carrying mutant C4 or Cr2 alleles that were also homozygous for Ighb. We designate this background as B6/129.Ighb or wild type. More than 10 females and males in each cohort were bred to generate experimental and control animals. In some experiments, C57BL/6 mice were also used as normal controls. Mice with a mixed B6/129 genetic background have been used in studies on autoimmunity 13 35 36. In some studies, wild-type B6/129 mice exhibit slightly higher levels of background autoantibody than do B6 mice 13. In others, even old B6/129 mice do not have elevated levels of autoantibody 35 36. MRL-Faslpr mice were purchased from The Jackson Laboratory. All mice in this study were maintained under specific pathogen–free conditions at the Duke University Medical Center vivarium. Mice were bled at 2, 5–6, or 10 mo of age. All mice were killed at 10 mo of age.
Detection of ANA and Antibody Specific for Native DNA.
Slides containing HEp-2 cells and Crithidia luciliae were purchased from Sigma-Aldrich and Scimedx Corp., respectively. The presence of IgG ANA and IgG specific for native (n)DNA was determined by reactivity to HEp-2 37 or C. luciliae 38, respectively. Slides were rehydrated in PBS, pH 7.4, blocked with PBS containing 10% FCS and 0.1% Tween 20 (Sigma-Aldrich), and then washed with PBS containing 1% BSA and 0.1% Tween 20. Serum samples were diluted in this washing solution starting at 1:40 and 1:10 for ANA and anti-nDNA, respectively, and incubated with substrates for 1 h at room temperature. Bound serum IgG was revealed by FITC-conjugated goat anti–mouse IgG (Sigma-Aldrich). Slides were counterstained with Evans blue (Sigma-Aldrich), and examined blindly under a fluorescence microscope. All serum samples that were positive at the starting dilution were serially diluted (1:3 for ANA, 1:2 for nDNA) and titrated to endpoints.
ELISA for Anti-DNA Antibodies.
Double-stranded calf thymus DNA (dsDNA; Sigma-Aldrich) was purified by phenol–chloroform extraction and then treated with S1 nuclease (Life Technologies) as described 39 to remove single-stranded (ss)DNA contaminants. To prepare ssDNA, dsDNA was boiled in water for 10 min and diluted in ice cold 1× SSC buffer to 50 μg/ml. dsDNA was also diluted to 50 μg/ml in 1× SSC. Diluted ss- and dsDNA preparations were coated and plates blocked as described 40 41 42. Serum samples were diluted 1:100 and incubated on DNA-coated plates for 1 h at room temperature. Each ELISA plate included a standard of a serially diluted mAb, TG7-83 (IgG1/λ1; from T.F. Tedder, Duke University, Durham, NC) that avidly binds both ss- and dsDNA. After washing, bound IgG was revealed by horseradish peroxidase (HRP)-conjugated goat anti–mouse IgG (Sigma-Aldrich). HRP activity was determined and analyzed as described elsewhere 26. The TG7-83 standard bound immobilized ss- and dsDNA to an endpoint concentration of 25 ng/ml. Serum samples were considered positive if the OD at 1:100 dilution was greater than the TG7-83 endpoint OD on the same plate.
Histopathology of Kidney and Spleen.
Kidney and spleen sections were prepared as described 43. Immunohistochemistry for the detection of T and B cells and GCs in spleen sections has been described elsewhere 43. To identify IgG and C3 deposition on kidney sections, acetone-fixed sections were rehydrated in PBS for 20 min and blocked in PBS, pH 7.4, containing 10% normal goat serum (Life Technologies) and 0.1% Tween 20. The sections were then stained at room temperature for 1 h with FITC-conjugated goat anti–mouse IgG (Sigma-Aldrich) or goat anti–mouse C3 antibodies (ICN Biomedicals). After staining, slides were washed, counterstained with Evans blue, and examined by fluorescence microscopy. Some kidney sections were postfixed with 1% paraformaldehyde (Sigma-Aldrich), stained with hematoxylin and eosin (Sigma-Aldrich), and examined by microscopy. Glomerulonephritis was determined according to established criteria 44. Renal function was assessed by measurement of urea nitrogen in serum using a blood urea nitrogen (BUN) rate kit (Sigma-Aldrich).
Flow Cytometry.
Splenocyte suspensions were prepared and blocked for FcR-mediated binding 26. Cells were then stained with biotinylated antibodies, followed by staining with streptavidin and antibodies conjugated with fluorochromes. 7-aminoactinomycin D (7-AAD; Molecular Probes, Inc.) was used to identify dead cells. The following antibodies/conjugates were used: biotinylated monoclonal anti–Mac-1, -B220, -CD44, or –TCR-β (PharMingen); FITC-labeled monoclonal GL-7, anti–Gr-1, –TCR-β, or -B220 (PharMingen); PE-conjugated monoclonal anti-B220 or -Fas (PharMingen); and Red 613–labeled streptavidin (Life Technologies).
In Vivo Clearance of Circulating ICs.
The IgG2bλ1 mAb, P14.2.14 (from Dr. T. Imanishi-Kari, Tufts University, Boston, MA), which binds (4-hydroxy-3-nitrophenyl)acetyl (NP) with a K a ≈ 106 M−1, was used to make ICs. P14.2.14 was mixed with biotinylated NP16-BSA at a 3:1 molar ratio and incubated at 37°C for 1 h. After incubation, the reaction mixture was centrifuged at 12,500 g for 10 min; <4% of total protein precipitated. Soluble ICs were injected intravenously into mice. Each mouse received a preparation of ICs formed with 100 μg of antibodies and 13 μg of biotinylated NP16-BSA, arbitrarily designated as 100 relative units (RU). Mice were bled at different times after injection, and levels of IC in plasma were assessed by ELISA. In brief, streptavidin (Sigma-Aldrich) was coated on plates at 10 μg/ml in 0.1 M carbonate buffer, pH 8.8. Plates were then washed and blocked with PBS containing 0.1% Tween 20 and 1% BSA. Plasma samples were serially diluted, added to plates, and incubated for 30 min at room temperature. After washing, bound ICs were revealed with HRP-conjugated goat anti–mouse IgG2b or goat anti–mouse Igλ (Southern Biotechnology Associates, Inc.) at room temperature for 1 h. Plates were then washed, developed, and analyzed as above.
Results
C4−/−, not Cr2−/−, Mice Produce High Titers of Autoantibody.
Cohorts of C4 − /−, Cr2 − /−, and control mice were examined for IgG ANA. At 2 mo of age, IgG ANA was not detectable in C4 − /− mice (n = 12) or in age- and sex-matched wild-type controls (n = 8). By 5–6 mo of age, low titers of ANA were occasionally detected (≈20%) in B6/129.Ighband B6 mice (Fig. 1 A). However, more than half (9/17) of age-matched, female C4 − /− mice had developed titers of ANA that were comparable to ANA present in sera pooled from B6.MRL-Faslpr mice (Fig. 1 A). At 5–6 mo, some male C4 − /− mice exhibited higher levels of ANA than sex-matched controls, but the difference was not statistically significant (P > 0.05; χ2 test). Unlike the C4-deficient mice, 5–6-mo-old Cr2 − /− mice produced only occasional and low levels of serum ANA that could not be distinguished from that present in control groups (Fig. 1 A).
Figure 1.
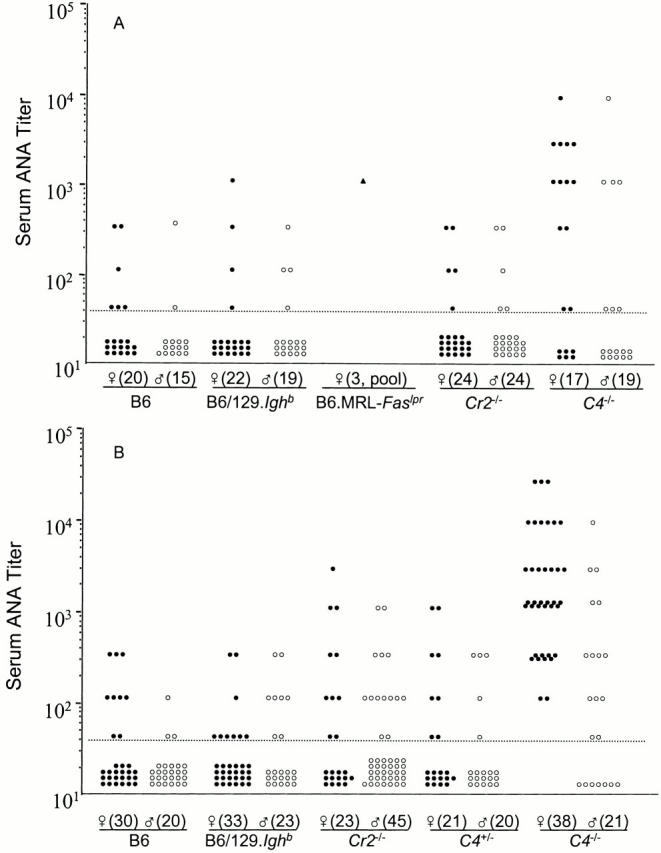
ANA titers in Cr2 − /−, C4 − /−, and control mice at 5–6 (A) or 10 mo (B) of age. Each point represents a serum sample from an individual mouse, except for B6.MRL-Faslpr, which is a serum pool from three mice. Numbers in parentheses following the symbols of sex are indicate numbers of mice. The broken line indicates the cut-off titer (1:40); placement of data points below this line does not indicate differences in titers.
By 10 mo of age, the incidence of ANA in both wild-type and Cr2 − /− mice increased by an insignificant amount (P > 0.05, χ2 test), and ANA titers remained equivalent in both groups as well (Fig. 1 B and Fig. 2). C4 +/− mice also exhibited background levels of ANA at 10 mo. In contrast, significant titers of ANA, some as high as 1:30,000, were present in all 10-mo-old, female C4 − /− mice (Fig. 1 B and Fig. 2). The majority (14/21) of 10-mo-old male C4 − /− mice were also positive for ANA, but ANA titers and the frequency of ANA+ males were significantly lower than those of female littermates (Fig. 1 B).
Figure 2.

Representative staining for IgG ANA and anti-nDNA serum antibody from B6.Ighb (left), Cr2 − /− (center), and C4 − /− (right) mice. HEp-2 cells were used as substrates for ANA (top panels, ×200 magnification), and C. luciliae were used to detect anti-nDNA antibody (bottom panels, ×1,000 magnification). Bound serum IgG was revealed by FITC-conjugated goat anti–mouse IgG (green). Cells were counterstained with Evans blue (red). Arrow indicates serum IgG binding to the kinetoplast of C. luciliae, where dsDNA is present in native form.
Antibody specific for DNA is a common component of ANA, and anti-DNA antibody is often used as a diagnostic marker for SLE 1. Therefore, we screened the sera of C4 − /−, Cr2 − /−, and control mice for the presence of antibody specific for ss- and dsDNA by ELISA 40 41 42 and for antibody specific for the native form of dsDNA, nDNA (Fig. 2 and Table and Table ) 38.
Table 1.
Spontaneous Autoantibody Production in 5–6-mo-old Mice
| B6 | B6/129.Igh b | Cr2 −/− | C4 −/− | |||||
|---|---|---|---|---|---|---|---|---|
| female | male | female | male | female | male | female | male | |
| n | 20 | 15 | 22 | 19 | 24 | 24 | 19 | 17 |
| ANA percent positive | 30 | 13 | 18 | 21 | 21 | 21 | 68 | 41 |
| Range (titer) | 0–360 | 0–360 | 0–1,080 | 0–360 | 0–360 | 0–360 | 0–9,720 | 0–9,720 |
| Median | 0 | 0 | 0 | 0 | 0 | 0 | 360 | 0 |
| χ2 (P) | ns | ns | – | – | ns | ns | <0.005 | ns |
| Anti-ssDNA percent positive | 0 | 0 | 18 | 5 | 46 | 13 | 74 | 41 |
| Range (μg/ml) | 0 | 0 | 0–50 | 0–5 | 0–14 | 0–4 | 0–342 | 0–48 |
| Median | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 0 |
| χ2 (P) | <0.05 | ns | – | – | <0.05 | ns | <0.0005 | <0.01 |
| Anti-dsDNA percent positive | 5 | 0 | 18 | 5 | 25 | 13 | 53 | 29 |
| Range (μg/ml) | 0–3 | 0 | 0–10 | 0–6 | 0–14 | 0–7 | 0–350 | 0–30 |
| Median | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 |
| χ2 (P) | ns | ns | – | – | ns | ns | <0.05 | ns |
| Anti-nDNA percent positive | 0 | 0 | 5 | 5 | 14 | 14 | 47 | 35 |
| Range (titer) | 0 | 0 | 0–20 | 0–10 | 0–20 | 0–20 | 0–80 | 0–40 |
| Median | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| χ2 (P) | ns | ns | – | – | ns | ns | <0.005 | <0.05 |
Table 2.
Spontaneous Autoantibody Production in 10-mo-old Mice
| B6 | B6/129.Igh b | Cr2 −/− | C4 +/− | C4 −/− | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| female | male | female | male | female | male | female | male | female | male | |
| n | 30 | 20 | 33 | 23 | 23 | 45 | 21 | 20 | 38 | 21 |
| ANA percent positive | 30 | 15 | 27 | 35 | 43 | 31 | 38 | 25 | 100 | 67 |
| Range (titer) | 0–360 | 0–120 | 0–360 | 0–360 | 0–1,080 | 0–360 | 0–1,080 | 0–360 | 120–29,160 | 0–9,720 |
| Median | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1,080 | 120 |
| χ2 (P) | ns | ns | – | – | ns | ns | ns | ns | <0.00001 | <0.05 |
| Anti-ssDNA percent positive | 13 | 0 | 18 | 13 | 22 | 9 | 10 | 0 | 84 | 29 |
| Range (μg/ml) | 0–11 | 0 | 0–150 | 0–32 | 0–38 | 0–10 | 0–4 | 0 | 0–286 | 0–357 |
| Median | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 13 | 0 |
| χ2 (P) | ns | ns | – | – | ns | ns | ns | ns | <0.00001 | ns |
| Anti-dsDNA percent positive | 7 | 0 | 15 | 4 | 26 | 7 | 14 | 5 | 76 | 14 |
| Range (μg/ml) | 0–12 | 0 | 0–150 | 0–30 | 0–55 | 0–12 | 0–5 | 0–3 | 0–318 | 0–260 |
| Median | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 0 |
| χ2 (P) | ns | ns | – | – | ns | ns | ns | ns | <0.00001 | ns |
| Anti-nDNA percent positive | 10 | 0 | 9 | 4 | 9 | 7 | 0 | 0 | 58 | 14 |
| Range (titer) | 0–20 | 0 | 0–20 | 0–10 | 0–20 | 0–20 | 0 | 0 | 0–160 | 0–80 |
| Median | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 10 | 0 |
| χ2 (P) | ns | ns | – | – | ns | ns | ns | ns | <0.00005 | ns |
In general, the frequencies of reactive sera and specific IgG titers were highest for ssDNA, followed by dsDNA and nDNA, respectively. At 5–6 mo of age, only 5% (1/20) of B6 mice expressed detectable serum titers of any form of anti-DNA antibody, a frequency too low to determine if females were more prone to autoantibody production than males (Table ). Levels of all three types of anti-DNA antibody were modestly higher in age-matched B6/129.Ighb mice than in B6 mice. Anti–ss- and dsDNA antibodies appear to be more common in female B6/129.Ighb mice (18%; 4/22) than in males (5%; 1/21) (Table ). At 5–6 mo, the frequency of female (46%; 11/24), but not male (13%; 3/24), Cr2 − /− mice with serum anti-ssDNA IgG was significantly higher than that of B6/129.Ighb controls. However, frequencies of Cr2 − /− mice with serum IgG reactive to ds- and nDNA did not differ from those in wild-type animals (Table ). In sharp contrast to the other cohorts of mice, both the frequencies and titers of anti-DNA IgG were generally elevated in female and male C4 − /− mice. Between 74% (14/19) and 47% (9/19) of 5–6-mo-old female C4 − /− mice developed significant levels of IgG antibody specific for ss-, ds-, and nDNA (Table ). At 5–6 mo of age, male C4 − /− mice also had increased levels of serum anti-ssDNA and -nDNA antibody, but the fraction of positive animals (41%; 7/17, and 35%; 6/17, respectively) and their antibody titers were below that of C4 − /− females (Table ).
With time, titers of serum autoantibody and the frequency of positive animals increased. These increases are not significant in 10-mo-old B6, B6/129.Ighb, and Cr2 − /− mice but are pronounced in female C4 − /− animals (Table ). Every (38/38) C4 − /− female mouse developed IgG ANA by 10 mo of age, and most also had significant titers of serum IgG that bound ssDNA (84%; 32/38), dsDNA (76%; 29/38), or nDNA (58%; 22/38). Autoantibody titers were also significantly higher in 10-mo-old C4 − /− females, with median concentrations of serum anti-DNA IgG in the range of ≈10 μg/ml (Table ). In contrast, 10-mo-old male C4 − /− mice exhibited only a modest increase in ANA frequencies and titers and had levels of anti-DNA antibodies that were not significantly different from controls (Table ).
Autoantibody production by female C4 − /− mice is not an artifact of the mixed genetic background present in these animals. Significant autoimmunity was not present in either female or male 10-mo-old C4 +/− controls (Table ). Thus, C4 deficiency alone is capable of inducing a potent autoimmune state in B6/129.Ighb mice. Realization of this autoimmunity is, however, regulated by sex-specific factors.
Although a significantly higher proportion of female Cr2 − /− mice exhibited anti-ssDNA IgG antibody at 5–6 mo (Table ), at 10 mo autoantibody levels in Cr2 − /− mice were not different from B6/129.Ighb controls (Table ). Thus, the absence of CR1 and CR2 is not sufficient to cause SLE-like autoimmunity in mice with a genetic background that is a mix of the B6 and 129 strains.
IC Deposition and Glomerulonephritis in C4−/− Mice.
Consistent with a pattern of SLE-like autoimmunity, 10-mo-old female C4 − /− mice manifest a striking glomerular pathology with a predominantly mesangial deposition of IgG and C3 and marked enlargement with hypercellularity (Fig. 3). This pattern of glomerulonephritis was detected in half (5/10) of female C4 − /− mice, but not in C4 − /− males (0/5). Despite the striking renal histopathology present in 10-mo-old female C4 − /− mice, we did not detect compromised kidney function. BUN levels in five randomly chosen 10-mo-old female C4 − /− and 129/B6.Ighb mice remained within normal ranges (22.5 ± 2.5 and 20.4 ± 1.6 mg/dl [mean ± SEM], respectively; P > 0.05, Student's t test). C4 − /− mice in this BUN test cohort were killed, and their kidneys were examined histologically; two exhibited the characteristic glomerular IgG and C3 deposition and mononuclear cell infiltration (Fig. 3). In contrast to C4 − /− mice, cohorts of 10 female Cr2 − /− and B6/129.Ighb animals exhibited normal glomerular structure. Significant deposits of IgG or C3 were not observed in the kidneys of wild-type controls and Cr2 − /− mice (Fig. 3).
Figure 3.

Glomerular IC deposition and glomerulonephritis in C4 − /− mice. Kidney sections from 10-mo-old, female B6/129.Ighb (left), Cr2 − /− (center), and C4 − /− (right) mice were stained with FITC-conjugated goat anti–mouse IgG (green, top panels, ×200 magnification) or goat anti–mouse C3 (green, center panels, ×200 magnification) and counterstained with Evans blue (red). Glomerular cellularity was examined in sections stained with hematoxylin and eosin (bottom panels, ×400 magnification). G, glomerulus; T, tubule.
Splenomegaly in C4-deficient Mice.
Like other strains of mice that develop spontaneous, systemic autoimmunity 45, female C4 − /− animals develop splenomegaly. The spleens of 2-mo-old C4 − /− mice (56 ± 2 mg, n = 12) are no larger than those of wild-type controls (62 ± 3 mg, n = 10). By 10 mo, average spleen weight in B6, B6/129.Ighb, and C4+/− mice uniformly increased by 40–50% to ∼90 mg (Fig. 4). The spleens of Cr2 − /− mice were comparably sized at 10 mo of age, with averages of 101 ± 6 and 108 ± 7 mg/spleen in males and females, respectively (Fig. 4). However, increases in 10-mo-old female C4 − /− mice were much larger, with an average spleen weight (368 ± 64 mg) about fourfold heavier than that of age-matched controls (90 ± 7 mg; P < 0.0005, Student's t test). More than two-thirds (18/26) of 10-mo-old C4 − /− female mice had spleen weights at least twice the wild-type average, and approximately one-third (8/26) had spleens greater than or equal to four times larger (Fig. 4). Splenomegaly was present in at least one 10-mo-old male C4 − /− mouse, but as a group, male C4−/− mice do not exhibit significant splenic enlargement in comparison to age-matched controls (P > 0.05, Student's t test). Indeed, at 10 mo, distributions of spleen weights in B6, B6/129.Ighb, C4 +/−, and Cr2 − /− mice were not significantly different (Fig. 4). Lymph nodes and Peyer's patches in C4 − /− mice appeared comparable in size to all control groups (data not shown).
Figure 4.

Mice deficient in C4 exhibit splenomegaly. Mice at 10 mo of age were killed and their spleens weighed. The broken line indicates 2 SD above the average weight of spleens in all B6, B6/129.Ighb, and C4 +/− mice. *P < 0.0005, Student's t test.
Analysis of Splenocyte Populations in C4−/− Mice.
To determine if splenomegaly in C4 − /− mice resulted from the expansion of splenocyte populations, we analyzed spleen cell compartments by flow cytometry. Spleens of 10-mo-old B6, B6/129.Ighb, and Cr2 − /− mice, strains that show no evidence of splenomegaly (Fig. 4), contain similar numbers of nucleated cells (average, 114 ± 14 × 106; range, 66–150 × 106). Numbers of nucleated splenocytes from five randomly chosen 10-mo-old female C4 − /− mice (178 ± 40 × 106 per spleen) were moderately (50–60%) increased over controls, even though spleen weights were increased an average of 300%. This observation suggests that vascular congestion plays a significant role in the splenomegaly of C4 − /− mice.
Spleens from C4 − /− and wild-type mice have comparable numbers of B220+ B cells (77 ± 23 × 106 versus 63 ± 8 × 106, respectively) and TCR-β+ T cells (33 ± 8 × 106 versus 30 ± 2 × 106) (Table ). However, macrophage (Mac-1+Gr-1−) and neutrophil populations (Mac-1+Gr-1+) 46 were, respectively, two- and fourfold larger in C4 − /− animals than in age-matched controls (Table ). This increase in inflammatory cells was not evident in Cr2 − /− mice.
Table 3.
Flow Cytometric Analysis of Splenocytes
| B6 | B6/129.Igh b | Cr2 −/− | C4 −/− | |||
|---|---|---|---|---|---|---|
| Age (mo) | 10 | 2 | 10 | 10 | 2 | 10 |
| Percent TCR-β1 | 23.33 ± 2.30 (ns) | 35.45 ± 0.45 | 26.53 ± 1.53 | 29.23 ± 2.01 (ns) | 37.65 ± 3.18 (ns) | 19.06 ± 1.82 (<0.01) |
| Percent B220+ | 55.03 ± 0.96 (ns) | 47.40 ± 2.83 | 55.15 ± 1.15 | 48.98 ± 4.00 | 46.05 ± 3.18 (ns) | 44.16 ± 6.50 (ns) |
| Percent MAC+Gr-1− | 6.85 ± 0.32 (ns) | 7.70 ± 0.84 | 6.09 ± 0.33 | 6.57 ± 0.58 (ns) | 7.07 ± 0.70 (ns) | 11.79 ± 1.12 (<0.005) |
| Percent MAC+Gr-1+ | 2.47 ± 0.23 (<0.05) | 1.86 ± 0.14 | 1.83 ± 0.21 | 2.05 ± 0.22 (ns) | 2.03 ± 0.41 (ns) | 6.57 ± 2.09 (<0.05) |
| Percent TCR-β1CD44high | 13.20 ± 0.87 (ns) | 5.88 ± 1.42 | 12.30 ± 0.24 | 9.65 ± 0.19 (<0.01) | 5.20 ± 1.46 (ns) | 16.58 ± 1.34 (<0.05) |
| Percent TCR-β1GL-7+ | 0.52 ± 0.13 (ns) | 0.26 ± 0.06 | 0.30 ± 0.01 | 0.24 ± 0.03 (ns) | 0.24 ± 0.10 (ns) | 1.56 ± 0.28 (<0.005) |
| Percent TCR-β1Fas+ | 2.04 ± 0.19 (ns) | 0.51 ± 0.09 | 1.62 ± 0.22 | 1.64 ± 0.19 (ns) | 0.56 ± 0.14 (ns) | 7.67 ± 0.93 (<0.0005) |
| Percent B220+CD44high | 1.59 ± 0.18 (ns) | 1.52 ± 0.23 | 1.33 ± 0.09 | 0.91 ± 0.09 (<0.01) | 0.96 ± 0.11 (ns) | 2.34 ± 0.41 (<0.05) |
| Percent B220+GL-7+ | 1.27 ± 0.34 (ns) | 0.45 ± 0.11 | 0.67 ± 0.07 | 0.46 ± 0.10 (ns) | 0.42 ± 0.10 (ns) | 2.63 ± 0.84 (<0.05) |
| Percent B220+Fas+ | 0.31 ± 0.05 (ns) | 0.12 ± 0.01 | 0.23 ± 0.03 | 0.26 ± 0.03 (ns) | 0.13 ± 0.02 (ns) | 0.84 ± 0.14 (<0.005) |
Percentage of TCR-β1, B220+, Mac-1+Gr-1+, or Mac-1+Gr-1− cells are enumerated on a gate of live (7-AAD−) cells. All of the other calculations are on a live lymphocyte gate. P values (Student's t test, compared to age-matched B6/129.Ighb mice) are shown in parentheses; ns, statistically not significant (P > 0.05) in comparison to age-matched B6/129.Ighb mice. Data represent the mean ± SEM from four to five mice at the age of 10 mo and two mice of 2 mo in each group, randomly chosen from two to three independent litters. All mice are female.
Whereas the numbers of splenic lymphocytes in C4 − /− mice do not differ from controls, T and B cell populations in 10-mo-old C4 − /− mice show evidence of cellular activation by increased expression of CD44, Fas, or GL-7 (Fig. 5 and Table ) 47. This activation is not constitutive in C4 − /− animals, as frequencies of activated lymphocytes in 2-mo-old C4 − /− mice are not significantly different from age-matched B6/129.Ighb controls (Table ). Notably, GL-7 is a marker of GC B cells 48, and we indeed detected spontaneous GC formation in the spleens of C4 − /− mice by immunohistochemistry (Chen, Z. and G. Kelsoe, unpublished data). This observation implies a role for the GC reaction in the production of autoantibody in these mice 49.
Figure 5.
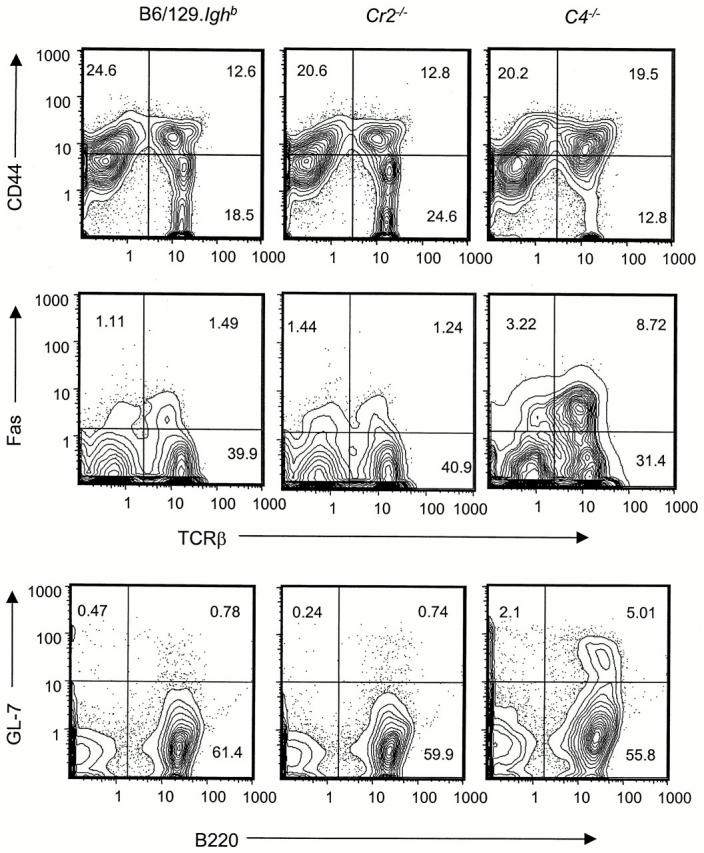
Increased frequencies of activated T and B cells in spleens of C4 − /− mice. Splenocytes from 10-mo-old female B6/129.Ighb (left), Cr2 − /− (center), and C4 − /− (right) mice were analyzed by flow cytometry. Profiles are generated from a gate for live lymphocytes and are representative of four to five mice in each group (see Table ).
Impaired Clearance of ICs in C4−/− Mice.
Complement is implicated in the clearance of ICs 8. Walport et al. 8 have proposed that autoimmunity in complement-deficient patients might result from failure to clear immunogenic ICs. Thus, we tested the role of C4 in the clearance of soluble ICs in vivo at an age (2 mo) when C4 − /− mice show no apparent signs of autoimmunity. Cohorts (n = 2–6) of 2-mo-old C4 − /−, C4 +/−, Cr2 − /−, and B6/129.Ighb mice were injected intravenously with 100 RU of soluble ICs. Levels of IC in plasma (∼30 RU/ml) were comparable in all groups at 2 min after injection (Fig. 6). From 2 to 10 min, IC concentrations in plasma decreased about threefold in C4 +/−, Cr2 − /−, and wild-type mice; plasma IC levels in C4 − /− mice, however, remained almost constant during this period. After 10 min, clearance rates were identical in all groups, including C4 − /−. The initial delay in IC clearance by C4 − /− mice resulted in a persistent, two- to threefold increase in circulating IC levels over that of Cr2 − /− mice and B6/129.Ighb controls. Higher levels of ICs in C4 − /− mice remained for as long as 70 min after IC administration (Fig. 6). Rapid clearance of ICs in Cr2 − /− mice suggests that CR1/CR2 plays a minor role in eliminating this type of IC from the blood circulation.
Figure 6.
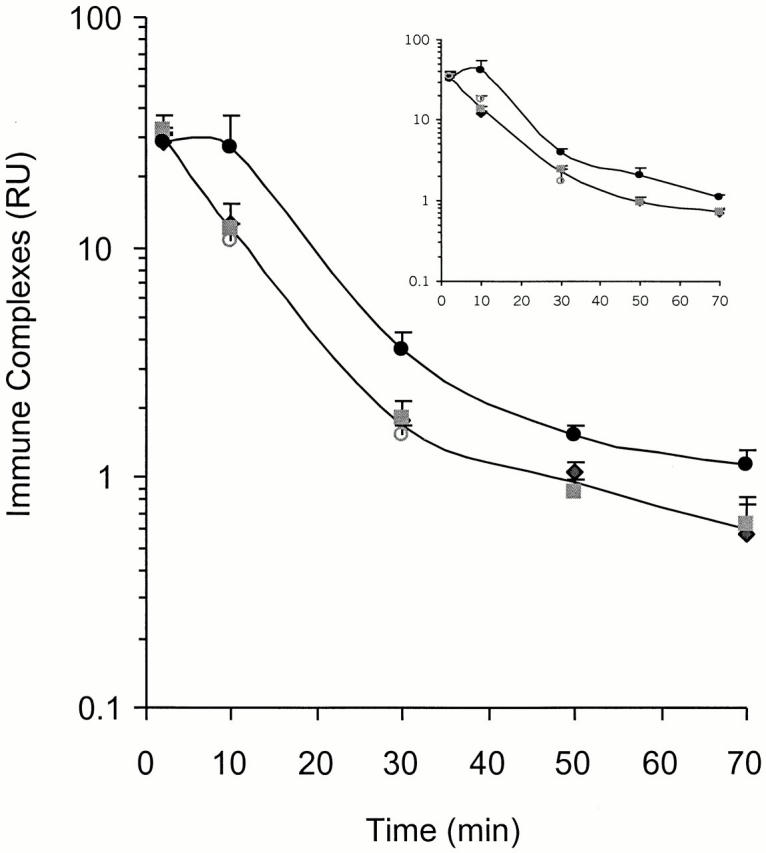
Impaired clearance of ICs in C4 − /− mice. Cohorts of B6/129.Ighb (♦), Cr2 − /− (▪), C4 +/− (○), and C4 − /− (•) mice were each injected with 100 RU of ICs through tail veins and bled from orbital sinus at 2, 10, 30, 50, or 70 min after IC injection. IC levels in plasma were assessed by ELISA. Data in the figure were determined with a goat anti–mouse IgG2b detector antibody; inset data were determined with goat anti–mouse Igλ. Upper curves represent the kinetics of IC clearance in C4 − /− mice, and lower curves are average clearance rates by B6/129.Ighb, C4 +/−, and Cr2 − /− mice; these values do not differ significantly from each other (P > 0.05; Student's t test). Individual clearance curves for B6/129.Ighb, C4 +/−, and Cr2 − /− mice are not shown, but IC RU (mean ± SEM) values for each group are presented; each point represents two to six mice. At the time points of 10, 30, 50, and 70 min, IC levels in C4 − /− animals differ significantly from B6.Ighb mice (P < 0.05, Student's t test).
Though unlikely, C4 binding to ICs could shield epitopes recognized by the goat anti–mouse IgG detector antibody and consequently reduce the apparent levels of plasma ICs in C4-sufficient mice (Fig. 6). Thus, we repeated the measurements of IC levels using a detector antibody specific for mouse λ L chain; IC clearance rates were virtually identical to those detected by anti-IgG (Fig. 6, inset). IC clearance was similarly impaired in both female and male C4 − /− mice (data not shown), implying that if high levels of ICs can break self-tolerance 8, gender-specific factors control the onset of pathological autoimmunity.
Discussion
How does the absence of complement components that enhance humoral immunity promote the production of autoantibody? The conundrum of complement deficiencies causing SLE is so paradoxical 50 that the role of complement per se has been questioned 51. It has been proposed that the genetic association of C4 and C2 with lupus only reflects the linkage of polymorphic MHC class I and II genes to the C4 and C2 genes 51. This proposal is not supported by the fact that systemic autoimmunity develops in the majority of patients with defective C1q, a locus not linked to the MHC 52. Furthermore, C1qa knockout mice with a hybrid B6/129 genetic background develop spontaneous autoimmunity, providing direct evidence that C1q deficiency results in the loss of self-tolerance 13. In this study, we demonstrate that C4 − /− mice, in contrast to control animals with the same genetic background, spontaneously produce ANA and DNA-specific autoantibody and exhibit histologic glomerulonephritis in an age- and sex-dependent manner. The H-2b haplotype and the genetic background of B6/129.Ighb stock are permissive for a humoral autoimmunity caused by C4 deficiency.
The autoantibody in C4-deficient mice is similar to that characteristic of SLE, and the genetic penetrance of autoimmunity in C4 − /− mice displays the strong bias for females present in SLE. Case reviews of patients with genetic deficiencies in C4 reveal that 92% (11/12) of C4-deficient females, in contrast to 44% (4/9) of males, were also diagnosed with SLE 6 7. These values are remarkably similar to the frequencies of 10-mo-old female and male C4 − /− mice with ANA (Table ). Factors that segregate with gender modify the suppressive effect of C4 on autoimmunity in both humans and mice (Fig. 1 and Fig. 4 and Table and Table ).
In the absence of significant autoimmunity in Cr2 − /− mice (Fig. 1 Fig. 2 Fig. 3 and Table and Table ), we conclude that C4 inhibits autoimmunity through a mechanism independent of CR1 and CR2. Cr2 − /− mice do not produce levels of serum ANA or anti-DNA antibody significantly above that of normal controls, they have histologically normal kidneys, and they exhibit no splenomegaly or evidence for generalized T and B cell activation (Fig. 1 Fig. 2 Fig. 3 Fig. 4 Fig. 5 and Table Table Table ). These observations are inconsistent with models of autoimmunity that rely on the ability of the CD19/CR2/CD81 coreceptor complex to regulate self-reactive B cells 16 32 50. In the absence of strong BCR signals, autoreactive B cells in the bone marrow of Cr2 − /− mice might not encounter sufficient antigen concentrations to affect negative selection by apoptotic deletion, receptor revision, or anergy 50. Indeed, the repertoire of peripheral B cells is altered even in Cr2 − /− mice with genetic backgrounds that do not promote autoimmunity 26. However, our experiments indicate that the presence of self-reactive B cells is not sufficient for the development of humoral autoimmunity.
What could account for the activation of self-reactive B cells in C4-deficient individuals? Deficiencies in C1q and C4 render mice less able to clear apoptotic cells/debris 13 53, and C1 was located on the surfaces of apoptotic cells 54. Botto et al. 13 have reported accumulations of TdT-mediated dUTP-biotin nick-end labeling (TUNEL)+ cells in histologically normal glomeruli in C1qa − /− mice. Phagocytosis of apoptotic cells by human macrophages is enhanced by complement-containing sera 55. Mevorach et al. 56 found that apoptotic materials are immunogenic and accelerate the production of autoantibody in mice not prone to autoimmunity. However, the significance of the immunogenicity of apoptotic debris is clouded by reports that phagocytosis of apoptotic cells by dendritic cells renders them tolerogenic 57 58. Nonetheless, apoptotic cellular debris, especially if associated with microbial products in the form of ICs 18, might activate autoreactive T and B lymphocytes and induce serum antibody specific for self-antigens. Indeed, nucleosome-specific, CD4+ T cells have been identified in lupus-prone mice. Moreover, immunization with nucleosomes enhanced autoantibody production 59.
Complement also acts in the clearance of ICs 18. Our work shows that the clearance of circulating ICs is delayed in C4 − /− mice even before the onset of detectable autoimmunity (Fig. 6). Although most ICs were eventually removed from circulation in the absence of C4 (Fig. 6), perhaps by Fc receptors 60 61, levels of ICs remained modestly elevated in C4 − /− mice for as long as 70 min. With time, this delay could cause significant accumulations of ICs.
Several plausible consequences of diminished IC clearance in C4 − /− mice come to mind. First, excessive IC deposition could cause inflammatory damage, especially in blood-filtering organs such as kidney and spleen (Fig. 3). Second, inflammatory damage might expose cryptic autoantigens and activate destructive T and B lymphocytes 8. Third, abundant ICs might enhance antigen uptake and activation by antigen-presenting cells, promoting lymphocyte activation, release of inflammatory cytokines, and the abrogation of anergy 8 18. Guinea pigs deficient in C4 exhibit delayed clearance of particulate ICs 62 and manifest signs of polyclonal B cell activation and a high incidence of IgM rheumatoid factors 63. The blood-filtering function of the spleen may localize IC-induced inflammation, explaining the absence of hypertrophy in lymph nodes and Peyer's patches. ICs and/or the inflammation they induce might also account for the activation of T and B cells in C4 − /− mice (Fig. 5 and Table ).
What mediates the role of C4 in immune clearance and autoimmunity? Only deficiencies in C1 6 7 9 13 18, C4 (6 7 9; Fig. 1 Fig. 2 Fig. 3, Table and Table ), and, to a lesser extent, C2 6 7, are strongly associated with spontaneous autoimmunity. In contrast, C3 deficiency does not significantly predispose to autoimmunity in either humans 6 7 or mice 32. Thus, do C1 and C4 inhibit autoimmunity via their tandem roles in the classical pathway of complement activation or do they act independently? C1 and C4 could act through a receptor(s) for C4 split products, with C1 catalyzing the formation of C4 ligands. Two complement receptors are known to bind C4 fragments, CR1 and CR2 16. Human CR1 participates in IC clearance; ICs are trapped by CR1 on erythrocytes and transported to the liver and spleen where they are phagocytosed by reticuloendothelial cells 18. C1q and C4b cooperatively increase immune adhesion mediated by human CR1 64. However, human CR1 contributes little to the phagocytosis of apoptotic cells by macrophages in vitro 55. In our study, mice deficient in CR1 and CR2 clear soluble ICs normally (Fig. 6). This observation carries the caveat that mice have two functional homologues of human CR1, murine CR1 and Crry 65. The crry product is proposed to function as a regulatory protein 65, but its homology to CR1 suggests that it might substitute for CR1 in Cr2 − /− mice.
If not CR1, CR2, or Crry, what other molecules could mediate the inhibition of autoimmunity by C4? C3-binding proteins distinct from CR1 and CR2 are present on mouse neutrophils and platelets. Although the identities and function(s) of these proteins are not characterized, they may mediate CR1/CR2-independent adherence of ICs. Whether they bind C4 fragments is unknown 66. Recently, CR3 and CR4, but not CR1, were shown to enhance phagocytosis of apoptotic cells 55. However, the primary ligand for CR3 and CR4 is iC3b 67. As C3 is not associated with autoimmunity 6 7 32, it is unlikely that CR3 and CR4 are. Alternatively, candidate C1q receptors 68, including the recently cloned C1qRP 69, might affect IC clearance in vivo and thereby inhibit autoimmunity. If this were the case, the association of C4 deficiency and autoimmunity could be explained by the ability of C4 (and perhaps C2) to anchor/stabilize C1 ligands on ICs and/or apoptotic cell debris and promote their adhesion 14 15 64. This hypothesis could explain the ordered relationship C1>C4>C2 of deficiencies in components of the classical pathway and risk for SLE 6 7 8 18. However, no direct evidence indicates whether these molecules act sequentially or independently to suppress humoral autoimmunity.
We propose a “multiple hit” hypothesis for the induction of humoral autoimmunity in C4 − /− mice. First, C4 deficiency may promote autoimmunity by impairing selection against autoreactive, immature/transitional B cells in the bone marrow; evidence suggests that this effect on the B cell repertoire is mediated by CR1/CR2 32. However, this altered B cell repertoire alone does not lead to serum autoantibody (Fig. 1 and Fig. 2 and Table and Table ), although it may accelerate incipient loss of self-tolerance 32. Thus, C4 also acts independently of CR1 and CR2 to promote systemic autoimmunity (Fig. 1 Fig. 2 Fig. 3 and Table and Table ). This effect becomes pronounced with aging and is moderated by sex-linked factors (Fig. 1 and Fig. 2 and Table and Table ). A plausible cause of autoimmunity in C4- and perhaps C1-deficient animals is impaired clearance of apoptotic cell debris 53 55 and ICs (Fig. 6; reference 8). Accumulation of these potential immunogens might break tolerance and activate humoral responses to self-components.
Acknowledgments
We are grateful to Dr. M. Carroll (Harvard University) for Cr2 − /− and C4 − /− mice and to Drs. T. Imanishi-Kari (Tufts University) and Dr. T.F. Tedder (Duke University) for hybridoma cell lines. We appreciate the technical assistance of Mss. P. Farless, M. Gendelman, and K. Young and editorial help from Ms. S.P. Mroz. We thank Drs. M. Frank and D. Pisetsky (Duke University) for their advice and critical reviews of this work.
This work was supported in part by U.S. Public Health Service grants AI-24335, AG-13789, and AG-10207.
Footnotes
Abbreviations used in this paper: ANAs, antinuclear antibodies; BCR, B cell antigen receptor; BUN, blood urea nitrogen; GC, germinal center; HRP, horseradish peroxidase; ICs, immune complexes; RU, relative unit; SLE, systemic lupus erythematosus.
References
- Pisetsky D.S. Systemic lupus erythematosus. Curr. Opin. Immunol. 1991;3:917–923. doi: 10.1016/s0952-7915(05)80014-7. [DOI] [PubMed] [Google Scholar]
- Kotzin B.L. Systemic lupus erythematosus. Cell. 1996;85:303–306. doi: 10.1016/s0092-8674(00)81108-3. [DOI] [PubMed] [Google Scholar]
- Craft J., Peng S., Fujii T., Okada M., Fatenejad S. Autoreactive T cells in murine lupusorigins and roles in autoantibody production. Immunol. Res. 1999;19:245–257. doi: 10.1007/BF02786492. [DOI] [PubMed] [Google Scholar]
- Wakeland E.K., Wandstrat A.E., Liu K., Morel L. Genetic dissection of systemic lupus erythematosus. Curr. Opin. Immunol. 1999;11:701–707. doi: 10.1016/s0952-7915(99)00039-4. [DOI] [PubMed] [Google Scholar]
- Vyse T.J., Kotzin B.L. Genetic susceptibility to systemic lupus erythematosus. Annu. Rev. Immunol. 1998;16:261–292. doi: 10.1146/annurev.immunol.16.1.261. [DOI] [PubMed] [Google Scholar]
- Figueroa J.E., Densen P. Infectious diseases associated with complement deficiencies. Clin. Microbiol. Rev. 1991;4:359–395. doi: 10.1128/cmr.4.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross S.C., Densen P. Complement deficiency states and infectionepidemiology, pathogenesis and consequences of neisserial and other infections in an immune deficiency. Medicine (Baltimore) 1984;63:243–273. [PubMed] [Google Scholar]
- Walport M.J., Davies K.A., Morley B.J., Botto M. Complement deficiency and autoimmunity. Ann. NY Acad. Sci. 1997;815:267–281. doi: 10.1111/j.1749-6632.1997.tb52069.x. [DOI] [PubMed] [Google Scholar]
- Hauptmann G., Tappeiner G., Schifferli J.A. Inherited deficiency of the fourth component of human complement. Immunodefic. Rev. 1988;1:3–22. [PubMed] [Google Scholar]
- De Bracco M.M., Manni J.A. Serum levels of C1q, C1r and C1s in normal and pathologic sera. Arthritis Rheum. 1974;17:121–128. doi: 10.1002/art.1780170204. [DOI] [PubMed] [Google Scholar]
- Sturfelt G., Sjoholm A.G., Svensson B. Complement components, C1 activation and disease activity in SLE. Int. Arch. Allergy Appl. Immunol. 1983;70:12–18. doi: 10.1159/000233266. [DOI] [PubMed] [Google Scholar]
- Swaak A.J., Aarden L.A., Statius van Eps L.W., Feltkamp T.E. Anti-dsDNA and complement profiles as prognostic guides in systemic lupus erythematosus. Arthritis Rheum. 1979;22:226–235. doi: 10.1002/art.1780220304. [DOI] [PubMed] [Google Scholar]
- Botto M., Dell'Agnola C., Bygrave A.E., Thompson E.M., Cook H.T., Petry F., Loos M., Pandolfi P.P., Walport M.J. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat. Genet. 1998;19:56–59. doi: 10.1038/ng0598-56. [DOI] [PubMed] [Google Scholar]
- Cooper N.R. The classical complement pathwayactivation and regulation of the first complement component. Adv. Immunol. 1985;37:151–216. doi: 10.1016/s0065-2776(08)60340-5. [DOI] [PubMed] [Google Scholar]
- Porter R.R. The complement components coded in the major histocompatibility complexes and their biological activities. Immunol. Rev. 1985;87:7–17. doi: 10.1111/j.1600-065x.1985.tb01142.x. [DOI] [PubMed] [Google Scholar]
- Carroll M.C. The role of complement and complement receptors in induction and regulation of immunity. Annu. Rev. Immunol. 1998;16:545–568. doi: 10.1146/annurev.immunol.16.1.545. [DOI] [PubMed] [Google Scholar]
- Tomlinson S. Complement defense mechanisms. Curr. Opin. Immunol. 1993;5:83–89. doi: 10.1016/0952-7915(93)90085-7. [DOI] [PubMed] [Google Scholar]
- Walport M.J., Davies K.A., Botto M. C1q and systemic lupus erythematosus. Immunobiology. 1998;199:265–285. doi: 10.1016/S0171-2985(98)80032-6. [DOI] [PubMed] [Google Scholar]
- Lay W.H., Nussenzweig V. Receptors for complement of leukocytes. J. Exp. Med. 1968;128:991–1009. doi: 10.1084/jem.128.5.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepys M.B. Role of complement in induction of the allergic response. Nat. New Biol. 1972;237:157–159. doi: 10.1038/newbio237157a0. [DOI] [PubMed] [Google Scholar]
- Fearon D.T., Carter R.H. The CD19/CR2/TAPA-1 complex of B lymphocyteslinking natural to acquired immunity. Annu. Rev. Immunol. 1995;13:127–149. doi: 10.1146/annurev.iy.13.040195.001015. [DOI] [PubMed] [Google Scholar]
- Fischer M.B., Ma M., Goerg S., Zhou X., Xia J., Finco O., Han S., Kelsoe G., Howard R.G., Rothstein T.L. Regulation of the B cell response to T-dependent antigens by classical pathway complement. J. Immunol. 1996;157:549–556. [PubMed] [Google Scholar]
- Cutler A.J., Botto M., van Essen D., Rivi R., Davies K.A., Gray D., Walport M.J. T cell–dependent immune response in C1q-deficient micedefective interferon gamma production by antigen-specific T cells. J. Exp. Med. 1998;187:1789–1797. doi: 10.1084/jem.187.11.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina H., Holers V.M., Li B., Fung Y., Mariathasan S., Goellner J., Strauss-Schoenberger J., Karr R.W., Chaplin D.D. Markedly impaired humoral immune response in mice deficient in complement receptors 1 and 2. Proc. Natl. Acad. Sci. USA. 1996;93:3357–3361. doi: 10.1073/pnas.93.8.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahearn J.M., Fischer M.B., Croix D., Goerg S., Ma M., Xia J., Zhou X., Howard R.G., Rothstein T.L., Carroll M.C. Disruption of the Cr2 locus results in a reduction in B-1a cells and in an impaired B cell response to T-dependent antigen. Immunity. 1996;4:251–262. doi: 10.1016/s1074-7613(00)80433-1. [DOI] [PubMed] [Google Scholar]
- Chen Z., Koralov S.B., Gendelman M., Carroll M.C., Kelsoe G. Humoral immune responses in Cr2−/− miceenhanced affinity maturation but impaired antibody persistence. J. Immunol. 2000;164:4522–4532. doi: 10.4049/jimmunol.164.9.4522. [DOI] [PubMed] [Google Scholar]
- Klaus G.G., Humphrey J.H., Kunkl A., Dongworth D.W. The follicular dendritic cellits role in antigen presentation in the generation of immunological memory. Immunol. Rev. 1980;53:3–28. doi: 10.1111/j.1600-065x.1980.tb01038.x. [DOI] [PubMed] [Google Scholar]
- Schriever F., Nadler L.M. The central role of follicular dendritic cells in lymphoid tissues. Adv. Immunol. 1992;51:243–284. doi: 10.1016/s0065-2776(08)60489-7. [DOI] [PubMed] [Google Scholar]
- Tew J.G., Wu J., Qin D., Helm S., Burton G.F., Szakal A.K. Follicular dendritic cells and presentation of antigen and costimulatory signals to B cells. Immunol. Rev. 1997;156:39–52. doi: 10.1111/j.1600-065x.1997.tb00957.x. [DOI] [PubMed] [Google Scholar]
- van Noesel C.J., Lankester A.C., van Lier R.A. Dual antigen recognition by B cells. Immunol. Today. 1993;14:8–11. doi: 10.1016/0167-5699(93)90316-d. [DOI] [PubMed] [Google Scholar]
- Tedder T.F., Inaoki M., Sato S. The CD19-CD21 complex regulates signal transduction thresholds governing humoral immunity and autoimmunity. Immunity. 1997;6:107–118. doi: 10.1016/s1074-7613(00)80418-5. [DOI] [PubMed] [Google Scholar]
- Prodeus A.P., Goerg S., Shen L.M., Pozdnyakova O.O., Chu L., Alicot E.M., Goodnow C.C., Carroll M.C. A critical role for complement in maintenance of self-tolerance. Immunity. 1998;9:721–731. doi: 10.1016/s1074-7613(00)80669-x. [DOI] [PubMed] [Google Scholar]
- Wilson J.G., Ratnoff W.D., Schur P.H., Fearon D.T. Decreased expression of the C3b/C4b receptor (CR1) and the C3d receptor (CR2) on B lymphocytes and of CR1 on neutrophils of patients with systemic lupus erythematosus. Arthritis Rheum. 1986;29:739–747. doi: 10.1002/art.1780290606. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Kozono Y., Waldschmidt T.J., Berthiaume D., Quigg R.J., Baron A., Holers V.M. Mouse complement receptors type 1 (CR1;CD35) and type 2 (CR2;CD21)expression on normal B cell subpopulations and decreased levels during the development of autoimmunity in MRL/lpr mice. J. Immunol. 1997;159:1557–1569. [PubMed] [Google Scholar]
- O'Keefe T.L., Williams G.T., Batista F.D., Neuberger M.S. Deficiency in CD22, a B cell–specific inhibitory receptor, is sufficient to predispose to development of high affinity autoantibodies. J. Exp. Med. 1999;189:1307–1313. doi: 10.1084/jem.189.8.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell D.A., Taylor P.R., Cook H.T., Moss J., Bygrave A.E., Walport M.J., Botto M. Cutting edgeC1q protects against the development of glomerulonephritis independently of C3 activation. J. Immunol. 1999;162:5676–5679. [PubMed] [Google Scholar]
- Rippey J.H., Carter S., Hood P., Carter J.B. Problems in ANA test interpretationa comparison of two substrates. Diagn. Immunol. 1985;3:43–46. [PubMed] [Google Scholar]
- Aarden L.A., de Groot E.R., Feltkamp T.E. Immunology of DNA. III. Crithidia luciliae, a simple substrate for the determination of anti-dsDNA with the immunofluorescence technique. Ann. NY Acad. Sci. 1975;254:505–515. doi: 10.1111/j.1749-6632.1975.tb29197.x. [DOI] [PubMed] [Google Scholar]
- Hande S., Notidis E., Manser T. Bcl-2 obstructs negative selection of autoreactive, hypermutated antibody V regions during memory B cell development. Immunity. 1998;8:189–198. doi: 10.1016/s1074-7613(00)80471-9. [DOI] [PubMed] [Google Scholar]
- Iliev A., Spatz L., Ray S., Diamond B. Lack of allelic exclusion permits autoreactive B cells to escape deletion. J. Immunol. 1994;153:3551–3556. [PubMed] [Google Scholar]
- Spatz L., Saenko V., Iliev A., Jones L., Geskin L., Diamond B. Light chain usage in anti-double-stranded DNA B cell subsetsrole in cell fate determination. J. Exp. Med. 1997;185:1317–1326. doi: 10.1084/jem.185.7.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putterman C., Diamond B. Immunization with a peptide surrogate for double-stranded DNA (dsDNA) induces autoantibody production and renal immunoglobulin deposition. J. Exp. Med. 1998;188:29–38. doi: 10.1084/jem.188.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob J., Kassir R., Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. I. The architecture and dynamics of responding cell populations. J. Exp. Med. 1991;173:1165–1175. doi: 10.1084/jem.173.5.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel L., Tian X.H., Croker B.P., Wakeland E.K. Epistatic modifiers of autoimmunity in a murine model of lupus nephritis. Immunity. 1999;11:131–139. doi: 10.1016/s1074-7613(00)80088-6. [DOI] [PubMed] [Google Scholar]
- Mohan C., Morel L., Yang P., Watanabe H., Croker B., Gilkeson G., Wakeland E.K. Genetic dissection of lupus pathogenesisa recipe for nephrophilic autoantibodies. J. Clin. Invest. 1999;103:1685–1695. doi: 10.1172/JCI5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagasse E., Weissman I.L. Flow cytometric identification of murine neutrophils and monocytes. J. Immunol. Methods. 1996;197:139–150. doi: 10.1016/0022-1759(96)00138-x. [DOI] [PubMed] [Google Scholar]
- Laszlo G., Hathcock K.S., Dickler H.B., Hodes R.J. Characterization of a novel cell-surface molecule expressed on subpopulations of activated T and B cells. J. Immunol. 1993;150:5252–5262. [PubMed] [Google Scholar]
- Han S., Zheng B., Schatz D.G., Spanopoulou E., Kelsoe G. Neoteny in lymphocytesRag1 and Rag2 expression in germinal center B cells. Science. 1996;274:2094–2097. doi: 10.1126/science.274.5295.2094. [DOI] [PubMed] [Google Scholar]
- Diamond B., Katz J.B., Paul E., Aranow C., Lustgarten D., Scharff M.D. The role of somatic mutation in the pathogenic anti-DNA response. Annu. Rev. Immunol. 1992;10:731–757. doi: 10.1146/annurev.iy.10.040192.003503. [DOI] [PubMed] [Google Scholar]
- Carroll M.C. The lupus paradox. Nat Genet. 1998;19:3–4. doi: 10.1038/ng0598-3. [DOI] [PubMed] [Google Scholar]
- Schur P.H. Inherited complement component abnormalities. Annu. Rev. Med. 1986;37:333–346. doi: 10.1146/annurev.me.37.020186.002001. [DOI] [PubMed] [Google Scholar]
- Campbell R.D., Carroll M.C., Porter R.R. The molecular genetics of components of complement. Adv. Immunol. 1986;38:203–244. doi: 10.1016/s0065-2776(08)60007-3. [DOI] [PubMed] [Google Scholar]
- Taylor P.R., Carugati A., Fadok V.A., Cook H.T., Andrews M., Carroll M.C., Savill J.S., Henson P.M., Botto M., Walport M.J. A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells In vivo. J. Exp. Med. 2000;192:359–366. doi: 10.1084/jem.192.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korb L.C., Ahearn J.M. C1q binds directly and specifically to surface blebs of apoptotic human keratinocytescomplement deficiency and systemic lupus erythematosus revisited. J. Immunol. 1997;158:4525–4528. [PubMed] [Google Scholar]
- Mevorach D., Mascarenhas J.O., Gershov D., Elkon K.B. Complement-dependent clearance of apoptotic cells by human macrophages. J. Exp. Med. 1998;188:2313–2320. doi: 10.1084/jem.188.12.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mevorach D., Zhou J.L., Song X., Elkon K.B. Systemic exposure to irradiated apoptotic cells induces autoantibody production. J. Exp. Med. 1998;188:387–392. doi: 10.1084/jem.188.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauter B., Albert M.L., Francisco L., Larsson M., Somersan S., Bhardwaj N. Consequences of cell deathexposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J. Exp. Med. 2000;191:423–434. doi: 10.1084/jem.191.3.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F.P., Platt N., Wykes M., Major J.R., Powell T.J., Jenkins C.D., MacPherson G.G. A discrete subpopulation of dendritic cells transports apoptotic intestinal epithelial cells to T cell areas of mesenteric lymph nodes. J. Exp. Med. 2000;191:435–444. doi: 10.1084/jem.191.3.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan C., Adams S., Stanik V., Datta S.K. Nucleosomea major immunogen for pathogenic autoantibody-inducing T cells of lupus. J. Exp. Med. 1993;177:1367–1381. doi: 10.1084/jem.177.5.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank, M.M., T.J. Lawley, M.I. Hamburger, and E.J. Brown. 1983. NIH Conference: immunoglobulin G Fc receptor-mediated clearance in autoimmune diseases. Ann. Intern. Med. 98:206–218. [DOI] [PubMed]
- Aderem A., Underhill D.M. Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- Ellman L., Green I., Judge F., Frank M.M. In vivo studies in C4-deficient guinea pigs. J. Exp. Med. 1971;134:162–175. doi: 10.1084/jem.134.1.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottger E.C., Hoffmann T., Hadding U., Bitter-Suermann D. Guinea pigs with inherited deficiencies of complement components C2 or C4 have characteristics of immune complex disease. J. Clin. Invest. 1986;78:689–695. doi: 10.1172/JCI112628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tas S.W., Klickstein L.B., Barbashov S.F., Nicholson-Weller A. C1q and C4b bind simultaneously to CR1 and additively support erythrocyte adhesion. J. Immunol. 1999;163:5056–5063. [PubMed] [Google Scholar]
- Molina H., Wong W., Kinoshita T., Brenner C., Foley S., Holers V.M. Distinct receptor and regulatory properties of recombinant mouse complement receptor 1 (CR1) and Crry, the two genetic homologues of human CR1. J. Exp. Med. 1992;175:121–129. doi: 10.1084/jem.175.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigg R.J., Alexander J.J., Lo C.F., Lim A., He C., Holers V.M. Characterization of C3-binding proteins on mouse neutrophils and platelets. J. Immunol. 1997;159:2438–2444. [PubMed] [Google Scholar]
- Brown E.J. Complement receptors and phagocytosis. Curr. Opin. Immunol. 1991;3:76–82. doi: 10.1016/0952-7915(91)90081-b. [DOI] [PubMed] [Google Scholar]
- Nicholson-Weller A., Klickstein L.B. C1q-binding proteins and C1q receptors. Curr. Opin. Immunol. 1999;11:42–46. doi: 10.1016/s0952-7915(99)80008-9. [DOI] [PubMed] [Google Scholar]
- Nepomuceno R.R., Henschen-Edman A.H., Burgess W.H., Tenner A.J. cDNA cloning and primary structure analysis of C1qR(P), the human C1q/MBL/SPA receptor that mediates enhanced phagocytosis in vitro. Immunity. 1997;6:119–129. doi: 10.1016/s1074-7613(00)80419-7. [DOI] [PubMed] [Google Scholar]