

Abstract
The parasitic protozoan Trypanosoma cruzi employs multiple molecular strategies to invade a broad range of nonphagocytic cells. Here we demonstrate that the invasion of human primary umbilical vein endothelial cells (HUVECs) or Chinese hamster ovary (CHO) cells overexpressing the B2 type of bradykinin receptor (CHO-B2R) by tissue culture trypomastigotes is subtly modulated by the combined activities of kininogens, kininogenases, and kinin-degrading peptidases. The presence of captopril, an inhibitor of bradykinin degradation by kininase II, drastically potentiated parasitic invasion of HUVECs and CHO-B2R, but not of mock-transfected CHO cells, whereas the B2R antagonist HOE 140 or monoclonal antibody MBK3 to bradykinin blocked these effects. Invasion competence correlated with the parasites' ability to liberate the short-lived kinins from cell-bound kininogen and to elicit vigorous intracellular free calcium ([Ca2+]i) transients through B2R. Invasion was impaired by membrane-permeable cysteine proteinase inhibitors such as Z-(SBz)Cys-Phe-CHN2 but not by the hydrophilic inhibitor 1-trans-epoxysuccinyl-l-leucyl-amido-(4-guanidino) butane or cystatin C, suggesting that kinin release is confined to secluded spaces formed by juxtaposition of host cell and parasite plasma membranes. Analysis of trypomastigote transfectants expressing various cysteine proteinase isoforms showed that invasion competence is linked to the kinin releasing activity of cruzipain, herein proposed as a factor of virulence in Chagas' disease.
Keywords: Trypanosoma cruzi, bradykinin, cruzipain, cysteine proteinases, kinin receptors
Introduction
Chagas' disease, the chronic infection by the parasitic protozoan Trypanosoma cruzi, is a major cause of cardiomyopathy in rural Latin America. Transmitted by blood-sucking triatomine insects, the infective forms of T. cruzi (trypomastigotes) rapidly enter the bloodstream, from where they disseminate the infection to multiple tissues. After invading macrophages, muscle, and other nucleated cells, the trypomastigotes escape from endocytic vacuoles and migrate into the cytoplasm where they transform into round-shaped amastigotes, the replicating forms. Within 5–6 d, the host cells rupture, releasing large numbers of trypomastigotes and amastigotes into interstitial spaces. Acute pathology and parasite tissue load subside with the onset of immunity, but the pathogen is not eradicated. After years of asymptomatic infection, 10–24% of the patients develop a severe chronic cardiomyopathy characterized by myocarditis, fibrosis, microcirculatory lesions, cardiomegaly, and conduction system abnormalities 1 2 3.
At the cellular level, T. cruzi trypomastigotes invade nonphagocytic cells by a unique mechanism distinct from phagocytosis 4 5. Penetration by tissue culture trypomastigotes (TCTs) is preceded by energy-dependent adhesive interactions 6 involving the parasites' surface glycoproteins 7 8 and negatively charged host surface molecules 9. Depending on the host cell–parasite combination studied, invasion requires activation of the TGF-β signaling pathway 10 or stimulation of host cell receptors coupled to heterotrimeric G proteins 11 12. Efforts to characterize the hitherto unknown Ca2+-signaling agonist pointed to a crucial role of a cytosolic parasitic serine protease of 80 kD, oligopeptidase B 13. Although null mutants generated by targeted deletion of the oligopeptidase B gene were poorly infective 14, purified or recombinant oligopeptidase B alone failed to induce intracellular free calcium ([Ca2+]i) transients in the mammalian cells 13. Because addition of recombinant oligopeptidase B to null parasite extracts reconstituted [Ca2+]i signaling, it was suggested that the agonistic activity was generated by oligopeptidase B–mediated processing of a cytoplasmic T. cruzi precursor molecule 14.
Other clues to understand the role of T. cruzi proteases in host cell invasion emerged from in vitro assays performed with synthetic inhibitors of cruzipain 15, the parasite's major cysteine proteinase 16 17 18. Encoded by multiple polymorphic genes 19 20, this cathepsin L–like proteinase is the most extensively characterized isoform expressed by replicating forms of the parasite 16 17 18 21. Given the broad pH range of the activity profile and the high stability of cruzipain 17, the finding of antigen deposits of this molecule in foci of myocardial inflammation 22 suggested that this proteinase may contribute to pathology. Our findings that the substrate specificity of cruzipain resembles that of tissue kallikrein and that cruzipain releases the bradykinin (BK)-like vasoactive peptide lysyl-bradykinin (“kallidin”) from its large precursor forms, high (H-) and low (L-) molecular weight kininogens 23, suggested that T. cruzi may directly trigger the kinin system through the activity of this cysteine proteinase.
Here we demonstrate that the short-lived kinin peptides and their cognate G protein–coupled cellular receptors 24 are engaged in the signaling mechanisms leading to T. cruzi invasion. We also show that invasion of cells that overexpress the constitutive B2 subtype of BK receptor is critically modulated by the kinin-degrading activity of host kininase II, also known as the angiotensin I–converting enzyme (ACE). The finding that activation of the proinflammatory kinin cascade by trypomastigotes potentiates invasion may shed light on the molecular basis of Chagas' disease pathophysiology.
MATERIALs and METHODS
Cells and Parasites.
Chinese hamster ovary (CHO) cells transfected with the cDNA encoding the rat B2 type of BK receptor (B2R; CHO-B2R) or mock-transfected CHO cells (CHO-mock) were used 25. Subclone rB2CHO12/4 showed a maximum 3H-BK binding activity of 1.3 pmol/mg of protein at passage 2. CHO cells were cultured in HAM's F12, each supplemented with 10% (vol/vol) of FCS at 37°C in a humidified atmosphere containing 5% CO2. Vero cells were cultivated in DMEM with 10% FCS. Human primary umbilical vein endothelial cells (HUVECs) were obtained by treatment of umbilical veins with a 0.1% (wt/vol) collagenase IV solution (Sigma-Aldrich). Primary HUVECs were seeded in 25-cm2 flasks (Corning) coated with 2% porcine skin gelatin, and grown in M199 medium supplemented with 2 mM glutamine, 2.5 μg/ml amphotericin B, 100 μg/ml penicillin, 100 μg/ml gentamycin, 0.13% sodium bicarbonate, and 20% FCS. Cells were maintained at 37°C in a humidified 5% CO2 atmosphere until they reached confluency. After treatment with 0.02% trypsin/0.02% EDTA, HUVECs were seeded into 24-well plates with gelatin-coated glass coverslips and cultivated at 37°C for several days before being used in invasion assays.
T. cruzi epimastigotes (Dm28c clone) were cultivated at 28°C in LIT medium containing 10% FCS. TCTs were harvested from the supernatants of infected Vero cultures maintained in DMEM supplemented with 2% FCS (TCT-FCS). TCT transfectants overexpressing Dm28c genes encoding the major cruzipain 18 isoform (for simplicity, hereafter designated cruzipain-1) or cruzipain-2 20 23 were obtained by cloning full-length copies of each of these into the SmaI-HindIII sites of pTEX plasmid 26. Log phase Dm28c epimastigotes were transfected by electroporation with a single pulse of 450 kV, 500 μF in an electroporator (Bio-Rad Laboratories). The parasites were selected for growth in LIT medium containing 10% FCS and 200 μg/ml of geneticin (Sigma-Aldrich) for six consecutive weeks and reselected at 800 μg/ml of geneticin for four additional weeks. Metacyclogenesis was done by incubating stationary phase–transfected epimastigotes in Grace's medium, pH 5.5, including 800 μg/ml of geneticin for 7 d at 27°C. TCT transfectants were collected from Vero cell supernatants 3–4 d after infection with the metacyclics. Plasmid contents were stable for at least 7 wk of culture in the absence of the selecting drug; TCT transfectants were tested in invasion assays after a 3-wk passage. The cysteine proteinase activity contained in cell lysates from transfected or wild-type parasites was measured as the rate of hydrolysis of ε-l-NH2-Cap-l-(SBz)C-MCA (20 μM) in Na2HPO4, 50 mM Na2HPO4, 200 mM NaCl, and 5 mM EDTA, pH 7.0, supplemented with 2.5 mM dithiothreitol (DTT), at 37°C. To prepare the parasite cell lysates, freshly released TCTs were washed twice in HBSS and resuspended in 300 μl of PBS, pH 7.2, containing 2 mM EDTA. Then, parasites were subjected to freeze and thaw cycles (two times), followed by the addition of Triton X-100 to 1%. Samples were kept on ice for 10 min and soluble material was recovered by centrifugation at 13,000 g. Protein concentration was determined by the Dc-protein kit (Bio-Rad Laboratories). Peptidase activity was measured in lysates normalized to 2 μg/ml protein (final concentration). Enzyme stability tests were performed by mixing 2 μl of lysates (1 mg/ml) to 100 μl of 0.1 M glycine, pH 12, for 5 s. Assay buffer was added to 1 ml and the peptidase activity was measured as described above.
Cell Invasion Assays.
CHO-B2R, CHO-mock, or native HUVECs were plated on 13-mm round coverslips at a density of 2.5 × 104 cells/cm2 in appropriate medium supplemented with 10% FCS and cultivated in 24-well plates for 48 h at 37°C in a 5% CO2 atmosphere. Before addition of TCTs, coverslips with attached cells were washed three times with HBSS and kept in serum-free medium containing 1 mg/ml BSA. The parasites added to the wells were freshly released from cultures of infected Vero cells cultivated in DMEM-FCS (2%). After removing cellular debris by low speed centrifugation (169 g), the parasite suspension was diluted three times in HBSS, spun down at 2,000 g, and gently resuspended in DMEM-BSA or M199-BSA (1 mg/ml each). Invasion assays with CHO cells or HUVECs were done in a total volume of 0.6 ml of DMEM-BSA or M199-BSA, respectively, at a parasite/host cell ratio of 2:1 for 3 h at 37°C, unless otherwise specified. When required, the medium was supplemented with 25 μM of captopril (CAP medium). The effects of receptor antagonists or antibodies were assayed by adding to CAP medium 0.1 μM HOE 140 (Aventis), 5–100 nM BK (Calbiochem), or 200 nM of mAbs. In some experiments, CAP medium was supplemented with purified H-kininogen (9 nM) 5 min before adding TCTs to HUVECs. Likewise, the effects of protease inhibitors were tested by adding 10 or 75 μM 1-trans-epoxysuccinyl-l-leucylamido-(4-guanidino) butane (E-64), 10 μM leupeptin (Sigma-Aldrich), 10 μM Z-(SBz)Cys-Phe-CHN2 15, or 1 μM recombinant human cystatin C (from Dr. Magnus Abrahamson, Lund University, Lund, Sweden) to the CAP medium. The effect of cruzipain-1 on invasion was evaluated by adding 10-fold dilutions of the activated protease to wells (0–10 nM final concentration) immediately after the addition of the parasites. The interaction was stopped by removing TCTs and washing cells three times with HBSS. Monolayers were fixed with Bouin and stained with Giemsa. Invasion was quantified by counting the number of intracellular parasites in a total of 100 cells per coverslip. Values represent means ± SD of at least three independent experiments, each done in triplicate under “blinded” conditions. Statistical analysis was done by one-way analysis of variance at a P = 0.05 significance level.
Digital Imaging Fluorescence Microscopy.
The changes in [Ca2+]i were determined using the fluorescent dye Fura 2-AM (Molecular Probes). Customized chambers used in these experiments were designed as follows: 30-mm plastic Petri dishes were drilled leaving 1-cm diameter holes in the center covered at the bottom by thin glass coverslips (0.7 mm) mounted with silicone glue. The plates were extensively washed and sterilized under a UV lamp for 20 min before use; coverslips for HUVECs contained 2% porcine gelatin. CHO cells and HUVECs were plated at 2 × 105 cells per dish in the appropriate medium supplemented with 10 and 20% of FCS, respectively, and cultivated for 24 h at 37°C in 5% CO2. The monolayers were washed three times with HBSS and incubated for 20 min at 37°C in a 5% CO2 atmosphere using serum-free HAM's F12 medium, pH 7.4, supplemented with 12.5 mM Hepes, 1 mg/ml BSA, 2.5 mM probenecid (Sigma-Aldrich), 25 μM captopril, and 6 μM Fura 2-AM. After rinsing three times to remove extracellular dye, the cells were maintained at 37°C for 15 min in the same medium without Fura 2-AM. Fura-loaded cells were analyzed in an Axiovert 100 microscope under an oil immersion 40× objective (ZEISS). Fluorescence images were collected by a digital CCD camera using a 510-nm filter. [Ca2+]i was monitored at 36–37°C by alternating the excitation wavelengths between 334 and 380 nm using the Attofluor Ratio System (ZEISS). Raw fluorescence images were digitalized to a pixel assay, point density readings were taken for each image, and a visual display of the 340/380-nm ratio was produced. Before adding parasites or purified proteases, the variation of [Ca2+]i of all cells in the field was monitored for 2 min; 20–30 cells which did not present spontaneous [Ca2+]i transients were chosen as regions of interest. After initial monitoring, the cellular responsiveness to kinin receptor agonists was assessed by adding BK (5–50 nM) to CHO-B2R or HUVECs preloaded with Fura 2-AM, using CAP medium. Vigorous [Ca2+]i transients were observed for nearly 100% of the monitored cells. Specificity of BK-induced transients was checked by adding 100 nM of HOE 140 to the monolayers before 50-nM agonist exposure. CHO-mock failed to induce significant [Ca2+]i elevations up to 50 nM BK. When indicated, 100 nM of HOE 140 was applied to the cells before the application of enzyme or parasites. Assays with TCTs were carried out at a parasite/host cell ratio of 10:1. Responding cells were observed under light phase to confirm interaction with the parasites. The Attograph software was used to generate tracings representing [Ca2+]i transients of individual cell responses, as well as average responses of 20–30 cells (n = 30). Purified cruzipain-1 was tested at 5 nM, after a 15-min activation of a stock solution (10-fold) with 2.5 mM DTT in PBS, pH 7.2. Controls included the addition of DTT-containing buffer alone or activated cruzipain-1 pretreated with 75 μM E-64 for 30 min. The specificity of HOE 140 was tested by pretreating HUVECs with 100 nM of the B2R antagonist before stimulating the cells with 14 nM α-thrombin (Dr. Russolina Zingali, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil) or with 50 nM BK. In the last series of experiments, the [Ca2+]i concentrations were determined by the two-point calibration in vivo method 27 after sequentially adding 20 μM ionomycin (Sigma-Aldrich) and 10 mM EGTA to the cultures.
Purified Proteins and Abs.
Cruzipain (GP57/51) was isolated from crude aqueous extracts of Dm28c epimastigotes as described 28. Recombinant cruzain (cruzipain-1) lacking the COOH-terminal extension 18 was expressed in Escherichia coli (Dr. J.H. McKerrow, University of California at San Francisco, San Francisco, CA). Recombinant cruzipain-2 devoid of the COOH terminus 20 was expressed in Saccharomyces cerevisiae 23 and partially purified by affinity chromatography on thiolpropyl–Sepharose 6B (Amersham Pharmacia Biotech); SDS-PAGE revealed a major band of 29 kD. Purified H-kininogen was from Calbiochem. mAbs MBK3 (IgG1) recognizing the BK epitope in domain D4 of human and bovine H-/L-kininogens, HKL16 directed to domain D6H of human H-kininogens 29, mAb212 directed to cruzipain-1 17, and IgG1 myeloma protein (MOPC 31c; Sigma-Aldrich) were used. Antiserum to cruzipain-2 (rab 222) was raised in rabbit chimeric protein, where a fragment (Tyr 147 to Trp 187) from the central domain of cruzipain-2 20 had been fused to glutathione transferase.
Cruzipain Isoforms Released by Trypomastigotes.
TCTs were harvested from supernatants of infected Vero cells, washed with HBSS, and resuspended at 2 × 107 cells/ml in DMEM without FCS. After incubation at 37°C for 2 h, the cell suspension was centrifuged at 3,000 g, and the supernatant was filtered through 0.2-μm Millipore filters. 200 μl of the supernatant was tested for proteolytic activity (see above). For the control, 30 μM E-64 (final concentration) was added to the supernatants. Immunoabsorption of cruzipain isozymes was carried out by treating 600 μl of the TCT supernatant for 2 h at room temperature with 10% of protein A–agarose beads (Sigma-Aldrich) previously coated with 10 μl of mAb212 ascites or with 50 μl of rab 222 antiserum. For controls, irrelevant mAb or preimmune serum was used. After washing with PBS, the hydrolytic activity associated to the resin was measured with 50 μM fluorogenic substrate in the presence or absence of E-64.
Kinin Release Assays.
The kinin concentration in reaction mixtures of human H-kininogen and cruzipain was measured by competitive ELISA (Markit-M; Dainippon). After activating stock solutions of Dm28c cruzipain-1 (50 nM) with 2.5 mM DTT, dilutions of the protease were mixed (2.5–10 nM) with H-kininogen (10 nM) in 200 μl of a reaction buffer containing 50 mM Na2HPO4, pH 6.5, 200 mM NaCl, 5 mM EDTA, and 0.25 mM DTT. After incubation for 30 min at 37°C, the reaction was stopped by adding 75 μM E-64, 1 mg/ml BSA, and 25 μM captopril; samples were deproteinized with ice-cold TCA. After diluting the soluble fractions with the supplier's buffer, the samples were applied to the ELISA plate. A standard curve was constructed with synthetic BK. Assays with recombinant cruzipain isozymes (20 nM) were performed as described above. Controls performed with enzyme buffer alone did not induce kinin release. The kinin-releasing activity of trypomastigotes was measured after incubating 10 nM of H-kininogen with TCTs (3 × 106 cells) in 250 μl of Ham's F12, 12.5 mM Hepes, 25 μM captopril, and 1 mg/ml BSA, pH 6.5, for 30 min at 37°C. For the control, TCTs were preincubated with 75 μM E-64 for 30 min at 37°C before the addition of H-kininogen. The reaction was stopped by adding E-64 (final concentration of 10 μM) to the cell suspension. Parasites were centrifuged at 3,000 rpm for 10 min, the supernatants were filtered through 0.2-μm Millipore filters, and kinin release was quantified as above. Experiments were done in triplicates.
Results
Trypomastigotes Induce Ca2+ Transients in Mammalian Cells Overexpressing the B2R.
Given the evidence that the major cysteine proteinase from T. cruzi, cruzipain, acts as a kininogenase 23, we set out to investigate if short-lived kinins were also generated when trypomastigotes interact with mammalian cells in vitro. As the B2Rs are preferentially coupled to G proteins of the Gq subtype, we reasoned that target cells that express elevated levels of this receptor should swiftly respond to kinins released upon trypomastigote contact. We addressed this question by first examining if the TCT could trigger [Ca2+]i transients in Fura-2–loaded CHO cells that were transfected with CHO-B2R. These assays were run in CAP medium to prevent rapid degradation of the short-lived kinins. Asynchronous and repetitive [Ca2+]i transients were promptly induced ∼50 s after TCTs had been added to CHO-B2R (Fig. 1A, Fig. C, and Fig. E). Average responses (Fig. 1 A) involved 67% of the CHO-B2R exposed to the parasites. The data registered with two single CHO-B2R cells (Fig. 1C and Fig. E) show that TCT induces multiple peaks of [Ca2+]i, although the intensity and frequency of the peaks varied from one cell to another, presumably due to the varying number of productive contacts established by the parasites. By contrast, [Ca2+]i transients evolved much slower in CHO-mock and were of low frequency and intensity (Fig. 1G and Fig. H), involving only a small fraction (7%) of the cell population (Fig. 1 H, average responses). The overall response of CHO-B2R was markedly reduced when the B2R-selective antagonist HOE 140 was added to the CAP medium before the addition of TCTs (Fig. 1, individual cell responses in D and F, average responses in B), indicating that the B2R may play an important role for the TCT-induced [Ca2+]i transients. We next evaluated if this signaling pathway was also engaged when the parasites interact with nontransfected host cells expressing high levels of endogenous B2R, such as primary HUVECs. The addition of TCTs to CAP medium likewise induced [Ca2+]i transients in HUVECs (Fig. 2, single cell tracing in A and average cell responses in D). The response, which involved 29% of the host cells, was markedly reduced by HOE 140 (Fig. 2B and Fig. E). Control experiments showed that HOE 140 did not reduce the [Ca2+]i transients induced by α-thrombin (Fig. 2G and Fig. H). Estimates of the cytosolic [Ca2+]i by in vivo calibration methods 28 indicated that the responses induced by α-thrombin and BK (in both cases involving nearly 100% of the HUVECs) peaked at ∼500 and 770 nM, respectively, starting from a base level of ∼70 nM. By contrast, HOE 140 attenuated the vigorous [Ca2+]i transients which BK, applied at 250 nM, elicited in HUVECs (Fig. 2I and Fig. K). These results confirmed that HOE 140 acts as a specific antagonist of B2R in the HUVEC system. The addition of epimastigotes failed to elicit significant transients in these cells (Fig. 2C and Fig. F). Collectively, these data suggest that Dm28c TCT signals in HUVECs through the constitutively expressed B2R.
Figure 1.
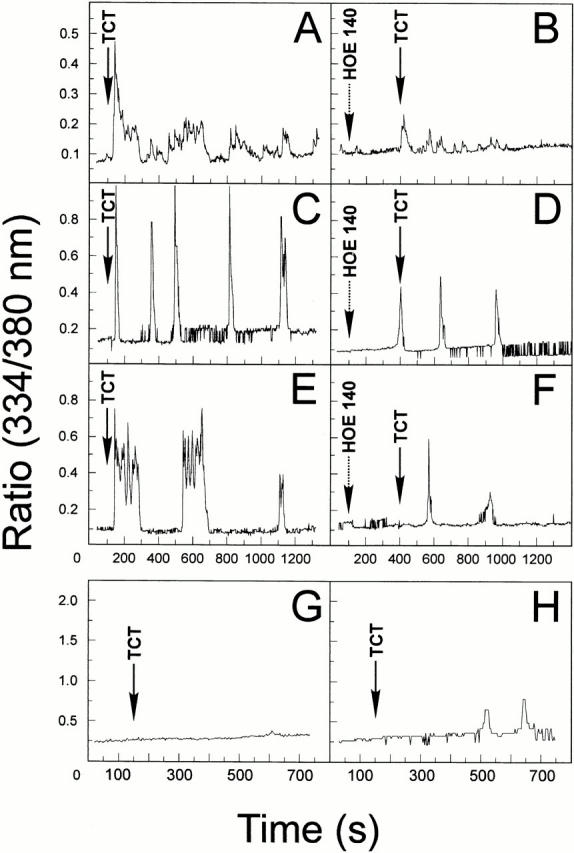
Trypomastigotes induce [Ca2+]i transients in CHO cells through the B2R. The changes of [Ca2+]i were determined on CHO-mock or CHO-B2R preloaded with Fura 2-AM in Ham's F12 medium supplemented with 1 mg/ml of BSA, 12.5 mM Hepes, and 25 μM captopril. The tracings represent cytosolic [Ca2+]i transients that addition of TCTs (parasite/host cell ratio of 10:1) induces in either a single cell or average responses of 20–30 cells. (A) TCTs (arrow) added to CHO-B2R, average responses. (B) TCTs added to CHO-B2R that were pretreated for 200 s with 100 nM HOE 140 (left arrow), average responses. (C and E) TCTs added to CHO-B2R, individual cell response. (D and F) TCTs added to CHO-B2R pretreated with HOE 140, individual cell responses. (G) TCTs added to CHO-mock, individual cell responses. (H) TCTs added to CHO-mock, average responses. CHO-mock failed to induce significant [Ca2+]i elevations upon BK application (up to 50 nM).
Figure 2.

TCTs induce cytosolic [Ca2+]i transients in HUVECs through the B2R. The changes in [Ca2+]i were determined by adding TCTs (parasite/host cell ratio of 10:1) to monolayers of HUVECs preloaded with Fura 2-AM, loaded, plated on gelatin-coated glass coverslips, maintained in M199 medium with 12.5 mM Hepes, 1 mg/ml BSA, and 25 μM captopril. (A) TCTs added to HUVECs, average responses; (D) individual cell response; (B) TCTs added to HUVECs that were previously treated with 100 nM HOE 140, average cell responses; (E) individual cell response; (C) epimastigotes (EPI) added to HUVECs, average responses; (F) epimastigotes added to HUVECs, individual cell responses. (G–I) Internal controls showing the sensitivity of HUVECs to BK and selectivity of the B2R antagonist HOE 140. Tracings represent average cell responses. (G) Cytosolic [Ca2+]i changes induced by 14 nM α-thrombin; (H) effect of 100 nM HOE 140 on thrombin-induced responses; (I) responses induced by 5 nM BK; (K) effect of 100 nM HOE 140 on responses induced by BK.
Signaling through B2R Increases Host Cell Susceptibility to T. cruzi Invasion.
As it is well established that the induction of [Ca2+]i transients is crucial for invasion by T. cruzi of many different cell types, we reasoned that the [Ca2+]i responses which TCTs stimulated in CHO-B2R cells may have increased their susceptibility to infection. Consistent with this prediction, the invasion index for CHO-B2R in the presence of CAP medium was nearly 4.5-fold higher than that for CHO-mock (Fig. 3 A), whereas captopril had no effect on the invasion of CHO-mock (not shown). Similarly to the inhibitory effects of HOE 140 on TCT-induced [Ca2+]i transients, addition of the B2R antagonist decreased the invasion of CHO-B2R in CAP medium, whereas it had no effect on the invasion of CHO-mock (Fig. 3 A). Similar effects were noted when we attempted to downregulate B2R by applying high concentrations of the agonist, i.e., 0.1 μM BK, to these cultures. Using primary cultures of HUVECs, we noted that parasite invasion was enhanced threefold in the presence of 25 μM captopril, whereas absence of captopril or inclusion of HOE 140 in CAP medium significantly attenuated invasion (Fig. 3 B). Although HUVECs were less sensitive to invasion than CHO-B2R, TCT invasion of HUVECs in the presence of CAP medium was likewise reduced by the addition of high concentrations (0.1 μM) BK (Fig. 3 B). In conclusion, TCT invasion of target cells that overexpress B2R is markedly enhanced in the presence of captopril, suggesting that the short-lived kinin agonist is protected from degradation by the kininase II inhibitor.
Figure 3.

T. cruzi invasion is enhanced in mammalian cells overexpressing B2R. (A) Invasion assays were performed with CHO-B2R or CHO-mock in HAM's F12 medium containing 1 mg/ml of BSA in the presence or absence of 25 μM captopril. Dm28c TCTs were added at a ratio of 2:1 (parasite vs. CHO) and the interaction proceeded for 3 h at 37°C in the presence or absence of 100 nM HOE 140 or BK, as indicated. (B) TCTs were added to HUVECs (ratio of 2:1) in M199 medium containing 1 mg/ml BSA. The interaction proceeded for 3 h in the presence or absence of 25 μM captopril. HOE 140 or BK, 100 nM each, was added to the CAP medium immediately before addition of the parasites. Values represent the number of intracellular parasites per 100 cells as means ± SD of three independent experiments. Significant differences (P < 0.05) between paired bars are marked by asterisks (*/*; **/**).
Processing of Cell-bound Kininogens Is Required for TCT Invasion.
The fact that TCT invasion is potentiated by kininase II inhibitors and reduced in cultures exposed to specific B2R antagonist or to high levels of exogenous BK pointed to the possibility that a low amount of plasma kininogen either remains associated with the cell surfaces of target cells and/or is displayed by the parasites, in either way serving as a source for precursors of kinin-signaling peptides. Consistent with this concept, the addition of an exogenous supply of H-kininogen (9 nM) to the serum-free CAP medium resulted in a greater than threefold increase in parasite invasion (Fig. 4 A). To test the hypothesis that HUVEC susceptibility to TCT invasion is influenced by the levels of kinin precursors and to further substantiate our notion that kinins are indeed involved in the signaling mechanism, we tested the effects of an mAB (MBK3) to BK that cross-reacts with the kinin segment of human and bovine kininogens 27. Our data show that preincubation of HUVECs with 300 nM MBK3 in CAP medium significantly reduced the potentiating effects of captopril, whereas the addition of unrelated Ab (IgG1) failed to interfere with TCT invasion, and HKL16, an mAb directed to an epitope of kininogen domain D6H, had only marginal inhibitory activity (Fig. 4 B). The results suggest that MBK3 compromises parasite invasion of HUVECs, either by blocking the processing of cell-bound kininogens and/or by “neutralizing” the liberated effector kinin peptide.
Figure 4.

H-kininogen potentiates parasite invasion of HUVECs. (A) The effects of addition of H-kininogen on parasite invasion. Interaction was carried out in CAP medium at a parasite/host cell ratio of 2:1 for 3 h in the presence or absence of 9 nM H-kininogen. Data are expressed as means ± SD of three independent experiments. The statistical significance between pairs of data is indicated by asterisks (P < 0.05). (B) The effects of mAbs directed against different kininogen epitopes. CAP medium was supplemented with 200 nM MBK3, IgG1 myeloma protein (MOPC 31c), or HKL6, and the interaction was carried out at a ratio of 5:1 (parasites/host cells) for 3 h. Results are expressed as percentage of inhibition of invasion, in relation to values obtained in cultures containing CAP medium alone.
E-64–sensitive Proteases Mediate Kinin Release by Living Trypomastigotes.
Given the indications that the major cysteine proteinase isoform, cruzipain-1, releases kinins from H-kininogen 23, and that an accessible BK epitope is required for parasite invasion (Fig. 4 B), we sought to examine if living trypomastigotes are able to release kinins from purified H-kininogen. To this end, washed TCTs were resuspended in CAP medium and 10 nM of purified H-kininogen was added to the suspension. ELISA measurements of the kinin concentrations in culture supernatants revealed the presence of this peptide within 30 min (Fig. 5 A). The processing reaction, which evolved in the absence of reducing agents, converted a small fraction (∼10%) of available H-kininogens into immunoreactive kinins over a limited time period. Significantly, the kinin-releasing activity was completely abolished by pretreating the parasites with an irreversible inhibitor of cysteine proteinases, E-64. These data suggest that TCTs engage cysteine proteinases in releasing kinins from soluble H-kininogens. As trypomastigotes are poorly endocytic 30, the membrane-impermeable E-64 inhibitor either binds to secreted cysteine proteases or acts on activated enzymes as they reach the parasites' flagellar pocket 17.
Figure 5.

Kinin-releasing activity of TCTs and recombinant cruzipain isoforms. (A) Freshly released parasites, harvested from supernatants of Vero cultures maintained in DMEM-FCS, were washed and resuspended in Ham's F12 medium containing 12.5 mM Hepes, 1 mg/ml of BSA, and 25 μM captopril at pH 6.5. Purified H-kininogen (10 nM, final concentration) was added to 250 μl of the parasites (3 × 106 cells), and the suspension was incubated at 37°C for 30 min. Involvement of cysteine proteases was examined by preincubating the TCTs in medium supplemented with E-64 (75 μM). After removing the parasites by centrifugation (3,000 rpm for 15 min at 4°C), the supernatants were filtered, deproteinized with ice-cold TCA, and the kinin concentrations were determined by a competitive ELISA. (B) Kinin-releasing activity of recombinant cruzipain-1 or cruzipain-2. The reaction was carried out by incubating each of the recombinant proteases (20 nM) with H-kininogen (10 nM) in 200 μl of 50 mM Na2HPO4, pH 6.5, 200 mM NaCl, 5 mM EDTA, and 0.25 mM DTT for 2 h at 37°C. Controls were carried out by preinactivating the proteinases with E-64. Values represent the mean ± SD of three independent assays.
Trypomastigotes Release Different Cruzipain Isoforms.
As pointed out in a previous study 23, the Met-Lys and Arg-Ser flanking site bonds of BK are cleaved at different efficiencies by two distinct recombinant cruzipain isoforms, namely cruzain (cruzipain-1) and cruzipain-2. Assays performed with each of these genetically engineered proteinases (both truncated on the COOH-terminal extension) confirmed that they are able to release immunoreactive kinins from purified H-kininogen, albeit with different efficiencies (Fig. 5 B). Consistent with the demonstration of kinin-releasing activity by living trypomastigotes, analysis of the supernatants from parasite suspensions revealed the presence of peptidases that were activated by DTT and inhibited by E-64 (Fig. 6 A). Immunoprecipitation using isoform-specific Abs mAb212 (anti–cruzipain-1) and rab 222 (anti–cruzipain-2) followed by fluorogenic substrate assay suggested that both isoforms were present in the supernatants (Fig. 6 B). Hence, TCTs released cysteine protease isoforms that may be involved in parasite-induced kinin release.
Figure 6.
Differential infectivity of TCTs overexpressing cruzipain isoforms. (A) Analysis of the cysteine protease activity present in supernatants from wild-type Dm28c TCTs. Parasite suspensions (2 × 107 cells/ml) were incubated at 37°C for 2 h in DMEM without FCS. The filtered supernatants were tested for peptidase activity at 37°C using 20 μM ε-NH2-Cap-Leu-(SBz)Cys-MCA in 50 mM Na2HPO4, pH 6.5, 200 mM NaCl, 5 mM EDTA, and 5 mM DTT. The graph depicts enzyme activity found in trypomastigote supernatants (Supnt) activated with DTT (▪) or pretreated with 30 μM E-64 (•). (B) Protein A–agarose beads were loaded with anti–cruzipain-1 or anti–cruzipain-2 and the immunobeads were added to supernatants obtained from wild-type TCT suspensions. After 2 h of incubation at room temperature, the immune complexes associated with the beads were washed with PBS and the initial rates of hydrolysis by enzymes associated with the solid phase were monitored in the presence or absence of 30 μM E-64, as specified above. Results (initial rates) are represented by the mean of three independent experiments. (C) The alkaline pH stability of the cysteine proteases from wild-type (WT) Dm28c TCTs or TCT-cz2 was assessed by mixing 2 μl of the Triton X-100 lysates (1 mg/ml of protein) with 100 μl of 0.1 M glycine buffer, pH 12. After 5 s, the lysates were diluted in the reaction buffer and the residual E-64–sensitive peptidase activity was measured for TCT-cz2 transfectants or wild-type parasites with ε-l-NH2-Cap-l-(SBz)C-MCA. Gray bars depict rates obtained after alkaline treatment of lysates. The values for initial rates of hydrolysis are expressed as means ± SD of three independent experiments. (D) Invasion assays performed by adding the TCT-cz1 or TCT-cz2 transfectants to monolayers of CHO-B2R (black bars) or CHO-mock (gray bars) in Ham's F12 medium containing 1 mg/ml of BSA in the presence of 25 μM captopril for 3 h at a parasite/host cell ratio of 2:1. Assays were carried out in the presence or absence of 100 nM HOE 140 as indicated, and the number of intracellular parasites per 100 cells was calculated. Results are given as means ± SD of three independent experiments. Statistical significance (P < 0.05) is indicated by asterisks.


The Kinin Pathway of Parasite Invasion Is Linked to the Expression of Cruzipain-1.
As both recombinant cruzipain-1 and cruzipain-2 act as kininogenases in vitro, we investigated if the engagement of B2R by the TCT may be linked to differential expression of these isoforms. To this end, we used the episomal pTEX plasmid to generate TCT transfectants containing full-length copies of the gene encoding for the major cruzipain-1 isoform (TCT-cz1) or for the cruzipain-2 gene (TCT-cz2). Consistent with the higher stability of recombinant cruzipain-2 at alkaline pH (Lima, A.P.C.A., unpublished observations), we found that a significant proportion of the overall cysteine proteinase activity present in TCT-cz2 lysates (30%) was attributed to enzymes that resist inactivation at pH 12 (Fig. 6 C), whereas only a minor fraction (8%) of alkaline-resistant cysteine peptidases were present in TCT-cz1 (not shown) or wild-type cells (Fig. 6 C). Hence, the profile of isoforms expressed by TCT-cz2, although still dominated by the alkaline-sensitive major cruzipain-1 isoform, is skewed towards increased production of the cruzipain-2–like isoforms. Remarkably, invasion assays performed with target cells that do not overexpress B2R, i.e., CHO-mock or Vero cells, revealed that TCT-cz1 was consistently less infective (exemplified for CHO-mock; Fig. 6 D, gray bar at left) compared with TCT-cz2 (Fig. 6 D, gray bar at right) or wild-type parasites (Fig. 3 A). TCT-cz1 efficiently invaded CHO-B2R cells, and this effect was reduced almost to the reference level of CHO-mock by the addition of HOE 140 (Fig. 6 D, black bars at right), indicating that the B2R pathway is used by the cz1 transfectants. By contrast, overexpression of cruzipain-2 did not significantly enhance invasion of CHO-B2R cells compared with CHO-mock (Fig. 6 D, right). Furthermore, HOE 140 did not reduce TCT-cz2 infectivity for CHO-B2R (Fig. 6 D, right), suggesting that these parasites signal through kinin-independent pathways. Collectively, these data suggest that T. cruzi's ability to engage the kinin signaling pathway of invasion is preferentially linked to a dominant expression of the major cysteine proteinase isoform (cruzipain-1).
Purified Cruzipain Triggers [Ca2+]i Transients in Cells Expressing B2R.
Given the indications that cruzipain-1 is found in the supernatants from wild-type TCTs and that living trypomastigotes release kinins through E-64–sensitive protease(s), we tested the ability of purified cruzipain-1 to induce [Ca2+]i transients. Indeed, we found that the parasite proteinase triggered robust [Ca2+]i transients in CHO-B2R loaded with Fura-2 (Fig. 7 B). This response was almost nullified by adding HOE 140 at 50 nM (Fig. 7 D) to the CAP medium, whereas [des Arg9-Leu8-BK], an antagonist of the B1R subtype, failed to interfere (data not shown). Importantly, the catalytic activity of cruzipain-1 was essentially required for the B2R stimulation because pretreatment with E-64 abrogated the [Ca2+]i response (Fig. 7 C). These data suggest that cruzipain-1 generates a kinin-like signaling factor by processing kininogen displayed by CHO cells. Unlike the strong [Ca2+]i transients in CHO-B2R, cruzipain-1 induced only a minor [Ca2+]i response in CHO-mock (Fig. 7 A). This low response was not inhibited by HOE 140 or by [des Arg9-Leu8-BK] (data not shown), suggesting that the minor [Ca2+]i transients induced in CHO-mock resulted from a kinin-independent signaling pathway.
Figure 7.

Cruzipain induces [Ca2+]i transients in CHO cells through the B2R. Activated cruzipain-1 or E-64–treated cruzipain-1 were added to monolayers of CHO cells in Ham's F12 medium supplemented with 1 mg/ml of BSA, 25 μM captopril. The protease was diluted 10-fold to 5 nM. (A) CHO-mock; (B) CHO-B2R exposed to cruzipain; (C) CHO-B2R exposed to E-64–treated cruzipain; (D) CHO-B2R pretreated with 100 nM HOE 140 for 120 s and then exposed to cruzipain-1 (indicated by arrows). Addition of the proteinase buffer alone did not elicit any [Ca2+]i transients in CHO cells. The y-axis represents the ratio of fluorescent absorbances at 334 and 380 nm. The tracings represent average cell responses.
Parasite Invasion of HUVECs Is Enhanced by BK or by Cruzipain.
To further investigate the interrelation between cruzipain activity and the parasite's ability to invade target cells by the kinin pathway, dose–response curves were separately established for BK (up to 100 nM) or for purified cruzipain. Parasite invasion peaked at 1 nM BK, whereas higher concentrations significantly reduced invasion to levels even below baseline, most likely due to receptor desensitization (Fig. 8 A). Thus, stimulation of B2R by kinin concentrations in the low nanomolar range significantly increases HUVECs' susceptibility to invasion by T. cruzi, whereas higher concentrations of the same peptide protected the host cells. Given that cruzipain-1 is able to release kinins from soluble H-kininogen (Fig. 8 B) and that it induces potent [Ca2+]i transients through B2R (Fig. 7), we checked if variations in the levels of activated protease could likewise influence the effectiveness of the invasion process. HUVEC invasion was modulated upon addition of increasing concentrations of cruzipain-1 to CAP medium, peaking at 5 nM activated protease (Fig. 8 C). The similar concentration range observed for peak activities of exogenous BK and purified cruzipain-1 suggests that the parasite competence to generate the kinin agonist may be influenced by quantitative and/or qualitative differences of cysteine protease expressed by the parasite. As a further attempt to characterize the involvement of the kinin-releasing cysteine proteases in the invasion process, the effects of synthetic or natural cysteine protease inhibitors were investigated. Unexpectedly, addition of E-64, an irreversible inhibitor of papain-like enzymes, failed to protect HUVECs from infection even when tested at 75 μM (<1% inhibition; Fig. 8 D). Likewise, invasion was not impaired in cultures either supplemented with leupeptin (10 μM) or by cystatin C (1 μM), a tight binding protein inhibitor present in human plasma. Considering the facts that (a) E-64 prevents kinin release from H-kininogen by TCTs (Fig. 5 A), and that (b) trypomastigotes are poorly endocytic in cell suspensions 30, we reasoned that the failure of membrane-impermeable cruzipain inhibitors to impair invasion could be an indication that kinin liberation occurs within the secluded sites formed by the juxtaposition of host and parasite plasma membranes. To address this possibility, we tested the effects of a membrane-permeable, irreversible inhibitor of cruzipain, Z-(SBz)Cys-Phe-CHN2 15 and found that it effectively inhibited (85%) HUVEC invasion, the extent of protection conferred by this compound being higher than that observed with HOE 140 (Fig. 8 D).
Figure 8.
The involvement of cruzipain in parasite invasion. (A) TCTs were added to HUVECs (parasite/host cell ratio of 5:1) in M199 CAP medium containing BK (0, 10, 50, and 100 nM) and incubated at 37°C for 3 h. (B) The graph illustrates the dose dependence of kinin-releasing activity of cruzipain-1. Enzyme stocks (50 nM) preactivated with DTT were subsequently diluted to the appropriate concentration in 50 mM Na2HPO4, pH 6.5, 5 mM EDTA, and 0.25 mM DTT supplemented with 10 nM H-kininogen. After a 30-min incubation at 37°C, the reaction was stopped by the addition of 75 μM E-64 and 25 μM captopril. Kinin release by activated cruzipain-1 (•) or E-64–treated cruzipain-1 (♦) was determined by competitive ELISA. Controls carried out with buffer in the absence of cruzipain did not lead to kinin release. (C) Invasion assays were carried out by adding aliquots of preactivated cruzipain-1 (50 nM stocks) to CAP medium before addition of TCTs to HUVECs (parasite/host cell ratio of 5:1, 1 h). (D) Invasion assays carried out at a parasite/host cell ratio of 5:1 for 3 h in CAP medium supplemented with Z-(SBz)Cys-Phe-CHN2 (10 μM), E-64 (75 μM), leupeptin (10 μM), cystatin C (1 μM), and HOE 140 (100 nM). The addition of DMSO vehicle (1%) did not interfere with invasion. The activity of each compound was expressed as the percentage of inhibition of parasite invasion with reference to assays performed in CAP medium alone. Data are expressed as means ± SD of at least three independent experiments. The statistical analysis was done by one-way analysis of variance; significance was considered at *P < 0.05.


Discussion
In this study, we demonstrate that trypomastigote invasion of host cells expressing BK B2Rs is drastically increased due to signaling by kinin peptides. The presence of captopril in the culture medium was a crucial prerequisite for the increased invasiveness of target cells expressing B2R, most likely because BK was protected from rapid degradation by ACE/kininase II 31. The effects induced by captopril suggest that T. cruzi tissue tropism may be influenced by variable levels and activities of kinin-degrading peptidases expressed in different host tissues. Indeed, highly vascularized tissues such as kidney parenchyma and lungs abundantly expressing ACE 32 are virtually spared from T. cruzi parasitism, whereas other tissues undergo massive infection and destruction during acute infection 33. Thus, variable expression levels of ACE may modulate the severity of lesions and fibrosis induced by this pathogen in the cardiovascular system of chagasic patients.
Because the presence of captopril or HOE 140 did not interfere with parasite invasion of CHO-mock (this study), Vero cells (not shown), or L6E9 myoblasts (Docampo, R., personal communication), parasites likely invade these cell lines by alternative mechanisms such as the TGF-β–dependent transducing pathway 7 or oligopeptidase B–mediated production of [Ca2+]i signaling agonists 13 14. Whereas the latter pathway is thought to involve proteolytic processing of a cytoplasmic precursor protein of the parasite 14, the kinin-mediated signal transduction route described herein likely depends on the processing of a host-derived precursor, namely kininogen(s). Preliminary analysis of a limited panel of well-established T. cruzi strains and clones indicates that invasion through the kinin-transducing pathway is not ubiquitous (Scharfstein, J., unpublished observations), indicating that clones from this highly diverse parasite species 34 35 36 37 may vary with respect to the ability to release proinflammatory kinins.
One of the conundrums of this study is the source of kininogens involved in the invasion process. Kininogens are secretory proteins displaying high affinity binding sites for heparan sulfate proteoglycans exposed on most cell surfaces 38, and thus we speculate that serum-derived kininogens attach to HUVECs or CHO cells during cultivation in vitro. At first sight, the finding that purified cruzipain is able to release kinins from cell-bound kininogens seems paradoxical because papain-like enzymes are rather sensitive to inhibition by the cystatin-like domains of kininogens 39. However, it is well known that the cystatin inhibitory sites in domain D3 of kininogen's heavy chain overlaps with the cell binding site that docks it to endothelial cells 40, and therefore may not be available for cruzipain inhibition. Our finding that FITC-labeled cruzipain failed to bind to HUVECs (data not shown) is consistent with this interpretation. Further, our demonstration that activated cruzipain stimulates vigorous Ca2+ transients in CHO-B2R, and that HOE 140 and E-64 block this effect, points to proteolytic release of kinin agonists, either directly by secreted forms of the proteinase or indirectly by the activation of a kallikrein-like zymogen 23. We have previously shown that cruzipain-2 efficiently cleaves the NH2-terminal flanking site of BK in kininogen domain D4, whereas the major isoform, cruzain, herein referred as cruzipain-1, preferentially cleaves the COOH-terminal flanking site 23. In this study, we show the presence of both isozymes in TCT supernatants; however, invasion assays performed with TCT transfectants showed that overexpression of the major isoform cruzipain-1, but not of cruzipain-2, favors the engagement of the kinin-mediated invasion pathway. Hence, our findings point to a causal link between parasitic protease cruzipain-1 and host kinin signaling pathway.
Previous studies have revealed that [Ca2+]i transients induced by G protein–coupled receptor agonists stimulate the recruitment and fusion of lysosomes to the plasma membrane at sites where trypomastigotes are attached to host cells 4 5. Considering that adhesive interactions between TCTs and host cells favor their reciprocal activation 41, it is conceivable that kinin signaling is most effective when the processing reaction is restricted to sites formed by juxtaposition of host and parasite cell membranes. Hence, signals emanating from host contacts with a few trypomastigotes, e.g., in CHO-mock or in HOE 140–treated CHO-B2R, may promote the delivery of host lysosomal cargos to secluded intercellular spaces, and this incipient activation process may generate a reduced environment in these segregated sites thereby sustaining activation of cruzipain-type kininogenases. The finding that membrane-permeable cysteine proteinase inhibitor Z-(SBz)Cys-Phe-CHN2, but not hydrophilic inhibitor E-64, conveyed partial albeit significant protection of HUVECs, is consistent with such a “dual signaling” model.
As B2R is constitutively expressed by endothelial cells, smooth muscle, fibroblasts, epithelial cells, and neuronal cells 24, and the B1 subtype of kinin receptors is upregulated during inflammation 42, increased cellular invasion may be just one example of a physiopathological response that kinins and their metabolites may induce in tissues exposed to T. cruzi. For example, triggering of kinin receptors from vascular endothelial cells may facilitate the transmigration of the trypomastigotes across capillaries, modulate the expression of vascular adhesion molecules, and/or promote plasma leakage into interstitial spaces, and thus contribute to the microvascular changes observed in patients with chronic cardiomyopathy 43. Unraveling the delicate interplay between the parasite cysteine proteinases and the multiple components of the kinin cascade system may provide fresh insights into the molecular pathogenesis of Chagas' disease.
Acknowledgments
We thank Leila F.C. Duarte and Alda M. Alves for technical assistance, and Dr. Anibal Gil Lopes for free access to digital imaging fluorescence microscopy.
This work was supported by grants from Pronex II, Conselho Nacional de Pesquisas, Fundacão de Amparo à Pesquisa do Rio de Janeiro, Fundacão Universitária José Bonif ácio, and the Deutsche Forschungsgemeinschaft. The authors dedicate this work to the memory of the late Prof. Carlos Chagas Filho.
Footnotes
Abbreviations used in this paper: ACE, angiotensin I–converting enzyme; BK, bradykinin; [CA2+]i, intracellular free calcium; CHO, Chinese hamster ovary; DTT, dithiothreitol; H-kininogen, high molecular weight kininogen; HUVEC, human primary umbilical vein endothelial cell; TCT, tissue culture trypomastigote.
References
- Andrade Z.A. Immunopathology of Chagas' disease Mem. Inst. Oswaldo Cruz. 94Suppl. I1999. 71 80 [DOI] [PubMed] [Google Scholar]
- Rossi M. Microvascular changes as a cause of chronic cardiomyopathy in Chagas' disease. Am. Heart J. 1990;120:233–236. doi: 10.1016/0002-8703(90)90191-y. [DOI] [PubMed] [Google Scholar]
- Morris S.A., Tanowitz H., Wittner M., Bilezikjan J.P. Pathophysiological insights into the cardiomyopathy of Chagas' disease. Circulation. 1990;82:1900–1909. doi: 10.1161/01.cir.82.6.1900. [DOI] [PubMed] [Google Scholar]
- Tardieux I., Webster P., Ravesloot J., Boron W., Lunn J.A., Andrews N.W. Lysosome recruitment and fusion are early events required for Trypanosoma invasion of mammalian cells. Cell. 1992;71:1117–1130. doi: 10.1016/s0092-8674(05)80061-3. [DOI] [PubMed] [Google Scholar]
- Burleigh B.A, Andrews N.W. The mechanisms of Trypanosoma cruzi invasion of mammalian cells. Annu. Rev. Microbiol. 1995;49:175–200. doi: 10.1146/annurev.mi.49.100195.001135. [DOI] [PubMed] [Google Scholar]
- Schenckman S., Robbins E.S., Nussenzweig V. Attachment of Trypanosoma cruzi to mammalian cells requires parasite energy and invasion can be independent of the target cell cytoskeleton. Infect. Immun. 1991;59:645–654. doi: 10.1128/iai.59.2.645-654.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenckman S., Jiang M.S., Hart G.W., Nussenzweig V. A novel cell surface trans-sialidase of Trypanosoma cruzi generates a stage-specific epitope required for invasion of mammalian cells. Cell. 1991;65:1117–1125. doi: 10.1016/0092-8674(91)90008-m. [DOI] [PubMed] [Google Scholar]
- Giordano R., Chammas R., Veiga S.S., Colli W., Alves M.J. An acidic component of the heterogeneous Tc-85 protein family from the surface of Trypanosoma cruzi is a laminin binding glycoprotein. Mol. Biochem. Parasitol. 1994;65:85–94. doi: 10.1016/0166-6851(94)90117-1. [DOI] [PubMed] [Google Scholar]
- Herrera E.M., Ming M., Ortega-Barria E., Pereira M.E. Mediation of Trypanosoma cruzi invasion by heparan sulfate receptors on host cells and penetrin counter-receptors on the trypanossomes. Mol. Biochem. Parasitol. 1994;65:73–83. doi: 10.1016/0166-6851(94)90116-3. [DOI] [PubMed] [Google Scholar]
- Ming M., Ewen M.E., Pereira M.E. Trypanosoma invasion of mammalian cells requires activation of the TGF-β signaling pathway. Cell. 1995;82:287–296. doi: 10.1016/0092-8674(95)90316-x. [DOI] [PubMed] [Google Scholar]
- Tardieux I., Nathanson M.H., Andrews N.W. Role in host cell invasion of Trypanosoma cruzi–induced cytosolic free Ca2+ transients. J. Exp. Med. 1994;179:1017–1022. doi: 10.1084/jem.179.3.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docampo R., Moreno S.N.J. The role of Ca2+ in the process of cell invasion by intracellular parasites. Parasitol. Today. 1996;12:61–65. doi: 10.1016/0169-4758(96)80656-9. [DOI] [PubMed] [Google Scholar]
- Burleigh B.A., Caler E.V., Webster P., Andrews N.W. A cytosolic serine endopeptidase from Trypanosoma cruzi is required for the generation of Ca2+ signaling in mammalian cells. J. Cell Biol. 1997;136:609–620. doi: 10.1083/jcb.136.3.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caler E.V., de Avalos S.V., Haynes P.A, Andrews N.W., Burleigh B.A. Oligopeptidase B-dependent signaling mediates host cell invasion by Trypanosoma cruzi . EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:4975–4986. doi: 10.1093/emboj/17.17.4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meirelles M.N., Juliano L., Carmona E., Silva S.G., Costa E.M., Murta A.C., Scharfstein J. Inhibitors of the major cysteinyl proteinase (GP57/51) impair host cell invasion and arrest the intracellular development of Trypanosoma cruzi in vitro. Mol. Biochem. Parasitol. 1992;52:175–184. doi: 10.1016/0166-6851(92)90050-t. [DOI] [PubMed] [Google Scholar]
- Cazzulo J.J, Cousi R., Raimondi A., Wernstedt C., Hellman U. Further characterization and partial amino acid sequence of a cysteine protease (cruzipain) from Trypanosoma cruzi . Mol. Biochem. Parasitol. 1989;33:33–42. doi: 10.1016/0166-6851(89)90039-x. [DOI] [PubMed] [Google Scholar]
- Murta A.C., Persechini P.M., Souto-Padron T., de Souza W., Guimarães J.A., Scharfstein J. Structural and functional identification of GP57/51 antigen of Trypanosoma cruzi as a cysteine proteinase. Mol. Biochem. Parasitol. 1990;43:27–38. doi: 10.1016/0166-6851(90)90127-8. [DOI] [PubMed] [Google Scholar]
- Eakin A.E., Mills A.A, Harth G., McKerrow J.H., Craik C.S. The sequence, organization and expression of the major cysteine protease (cruzain) from Trypanosoma cruzi . J. Biol. Chem. 1992;267:7411–7420. [PubMed] [Google Scholar]
- Campetella O., Henrickson J., Aslund L., Frasch A.C.C., Petterson U., Cazzulo J.J. The major cysteine protease (cruzipain) of Trypanosoma cruzi is encoded by polymorphic tandemly repeated organized genes located in different chromosomes. Mol. Biochem. Parasitol. 1992;50:225–234. doi: 10.1016/0166-6851(92)90219-a. [DOI] [PubMed] [Google Scholar]
- Lima A.P.C.A., Tessier D.C., Thomas D.Y., Scharfstein J., Storer A.C., Vernet T. Identification of new cysteine protease gene isoforms in Trypanosoma cruzi . Mol. Biochem. Parasitol. 1994;6:333–338. doi: 10.1016/0166-6851(94)00144-8. [DOI] [PubMed] [Google Scholar]
- McGrath M.E., Eakin A.E., Engel J.C., McKerrow J.H., Craik C.S., Fletterick R.J. The crystal structure of cruzaina therapeutic target for Chagas' disease. J. Mol. Biol. 1995;247:251–259. doi: 10.1006/jmbi.1994.0137. [DOI] [PubMed] [Google Scholar]
- Morrot A., Strickland D.S., Higuchi M.L., Reis M., Pedrosa R., Scharfstein J. Human T cell response against the major cysteine proteinase (cruzipain) of Trypanosoma cruzirole of the multifunctional α2-macroglobulin receptor in antigen presentation by monocytes. Int. Immunol. 1997;9:825–834. doi: 10.1093/intimm/9.6.825. [DOI] [PubMed] [Google Scholar]
- Del Nery E., Juliano M.A., Lima A.P.C.A., Scharfstein J., Juliano L. Kininogenase activity by major cysteinyl proteinase (cruzipain) from Trypanosoma cruzi . J. Biol. Chem. 1997;272:25713–25718. doi: 10.1074/jbc.272.41.25713. [DOI] [PubMed] [Google Scholar]
- Bhoola K.D, Figueroa C.D., Worthy K. Bioregulation of kininskallikreins, kininogens, and kininases. Pharmacol. Rev. 1992;44:1–80. [PubMed] [Google Scholar]
- Quitterer U., Schroeder C., Müller-Esterl W., Rehm H. Effects of bradykinin and endothelin-1 on the calcium homeostasis of mammalian cells. J. Biol. Chem. 1995;270:1992–1999. doi: 10.1074/jbc.270.5.1992. [DOI] [PubMed] [Google Scholar]
- Kelly J.M., Das P., Thomas A.M. An approach to functional complementation by introduction of large fragments into Trypanosoma cruzi and Leishmania donovani using a cosmid shuttle vector. Mol. Biochem. Parasitol. 1994;65:51–62. doi: 10.1016/0166-6851(94)90114-7. [DOI] [PubMed] [Google Scholar]
- Kaufmann J., Haasemann M., Modrow S., Müller-Esterl W. Structural dissection of the multi-domain kininogens. Fine mapping of the target epitopes of antibodies interfering with their functional properties. J. Biol. Chem. 1993;268:9079–9091. [PubMed] [Google Scholar]
- Grynkiewicz G., Poenie M., Tsien R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Lima A.P.C.A., Scharfstein J., Storer A.C., Menard R. Temperature-dependent substrate inhibition of the cysteine proteinase (GP57/51) from Trypanosoma cruzi . Mol. Biochem. Parasitol. 1992;56:335–338. doi: 10.1016/0166-6851(92)90183-k. [DOI] [PubMed] [Google Scholar]
- de Souza W. Structural organization of the cell surface of pathogenic protozoa. Micron. 1995;26:405–430. doi: 10.1016/0968-4328(95)00010-0. [DOI] [PubMed] [Google Scholar]
- Ferreira S.H., Greene L.J., Alabaster V.A., Bakhle Y.S., Vane J.R. Activity of various fractions of bradykinin potentiating factor against angiotensin I converting enzyme. Nature. 1970;225:379–380. doi: 10.1038/225379a0. [DOI] [PubMed] [Google Scholar]
- Danilov S.M., Faerman A.I., Yu Printseva O., Martynov A.V, Yu Sakharov I., Trakht I.N. Immunohistochemical study of angiotensin-converting enzyme in human tissues using monoclonal antibodies. Histochemistry. 1987;87:487–490. doi: 10.1007/BF00496822. [DOI] [PubMed] [Google Scholar]
- Lenzi H.L., Oliveira D.N., Lima M.T., Gattass C.R. Trypanosoma cruzipaninfectivity of CL strain during murine acute infection. Exp. Parasitol. 1996;84:16–27. doi: 10.1006/expr.1996.0086. [DOI] [PubMed] [Google Scholar]
- Miles M.A., Souza A., Povoa M., Shaw J.J., Lainson R., Toye P.J. Isozymic heterogeneity of Trypanosoma cruzi in the first autochtonous patients with Chagas' disease in Amazonia Brazil. Nature. 1978;272:819–821. doi: 10.1038/272819a0. [DOI] [PubMed] [Google Scholar]
- Tibayrenc M., Ward P., Moya A., Ayala F.J. Natural populations of Trypanosoma cruzi, the agent of Chagas' disease, have a complex multiclonal structure. Proc. Natl. Acad. Sci. USA. 1986;83:115–119. doi: 10.1073/pnas.83.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otavio-Souto R.P., Fernandes O., Macedo A.M., Campbell D.A., Zingales B. DNA markers define two major phylogenetic lineages of Trypanosoma cruzi . Mol. Biochem. Parasitol. 1996;83:141–152. doi: 10.1016/s0166-6851(96)02755-7. [DOI] [PubMed] [Google Scholar]
- de Diego J.A., Palau M.T., Gamallo C., Penin P. Relationship between histopathological findings and phylogenetic divergence in Trypanosoma cruzi . Trop. Med. Int. Health. 1998;3:222–233. doi: 10.1111/j.1365-3156.1998.tb00275.x. [DOI] [PubMed] [Google Scholar]
- Renné T., Dedio J., David G., Müller-Esterl W. H-kininogen utilizes heparan sulfate proteoglycans for accumulation on endothelial cells. J. Biol. Chem. 2000;275:33688–33696. doi: 10.1074/jbc.M000313200. [DOI] [PubMed] [Google Scholar]
- Stoka V., Nycander M., Lenarcic B., Labriola C., Cazzulo J.J., Björk I., Turk V. Inhibition of cruzipain, the major cysteine proteinase of the protozoan parasite, Trypanosoma cruzi, by proteinase inhibitors of the cystatin superfamily. FEBS Lett. 1995;370:101–104. doi: 10.1016/0014-5793(95)00798-e. [DOI] [PubMed] [Google Scholar]
- Herwald H., Hasan A.A.K., Godovac-Zimmermann J., Schmaier A.H., Müller-Esterl W. Identification of an endothelial cell binding site on kininogens' domain D3. J. Biol. Chem. 1995;270:14634–14642. [PubMed] [Google Scholar]
- Moreno S.N.J., Silva J., Vercesi A.E., Docampo R. Cytosolic-free calcium elevation in Trypanosoma cruzi . J. Exp. Med. 1994;180:1535–1540. doi: 10.1084/jem.180.4.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marceau F. Kinin B1 receptorsa review. Immunopharmacology. 1995;30:1–26. doi: 10.1016/0162-3109(95)00011-h. [DOI] [PubMed] [Google Scholar]
- Higuchi M.L., Fukasawa S., de Brito T., Parzianello L.C., Belloti G., Ramires J.A.F. Different microcirculatory and interstitial matrix patterns in idiopathic dilated cardiomyopathy and Chagas' diseasea three dimensional confocal microscopy study. Heart. 1999;8:1–6. doi: 10.1136/hrt.82.3.279. [DOI] [PMC free article] [PubMed] [Google Scholar]