

Abstract
Matrix metalloproteinase (MMP)9/gelatinase B is increased in various nephropathies. To investigate its role, we used a genetic approach. Adult MMP9-deficient (MMP9−/−) mice showed normal renal histology and function at 3 mo. We investigated the susceptibility of 3-mo-old mice to the accelerated model of anti-glomerular basement membrane nephritis, in which fibrin is an important mediator of glomerular injury and renal impairment. Unexpectedly, nephritis was more severe in MMP9−/− than in control mice, as attested by levels of serum creatinine and albuminuria, and the extent of crescents and fibrin deposits. Circulating or deposited immunoglobulin G, interleukin (IL)-1β, or IL-10 were the same in MMP9−/− and MMP9+/+ mice. However, we found that fibrin is a critical substrate for MMP9, and in its absence fibrin accumulated in the glomeruli. These data indicate that MMP9 is required for a novel protective effect on the development of fibrin-induced glomerular lesions.
Keywords: matrix metalloproteinase, kidney, fibrin, crescent, proteinuria
Introduction
Matrix metalloproteinase (MMP) 9/gelatinase B has a restricted pattern of expression in developmental and adult tissues (for reviews, see references 1 and 2). It is expressed at the first step of renal development, in the mesenchyme of 11-d embryonic kidneys 3. This enzyme is required for kidney maturation in vitro since blocking MMP9 activity with specific antibodies or its natural inhibitor, the tissue inhibitor of metalloproteinase (TIMP)1, inhibits kidney branching morphogenesis and growth 3. In the adult kidney, MMP9 is mainly expressed in glomerular visceral epithelial cells and in collecting duct cells 4 5, and its expression is increased in a variety of human renal diseases and in animal models of nephropathies such as membranous nephropathy, polycystic kidney disease, and renal fibrosis (reference 6; for a review, see reference 7).
We took advantage of the availability of MMP9-deficient (MMP9−/−) mice 8 to investigate the renal phenotype of these mice and their susceptibility to anti-glomerular basement membrane (GBM) nephritis, a murine model of crescentic proliferative glomerulonephritis. We compared the accelerated model of anti-GBM nephritis in MMP9−/− mice and MMP9+/+ control mice bred on the same genetic background. This model is characterized by a rapid development of severe crescentic proliferative glomerulonephritis in which fibrin is an important mediator of glomerular injury and renal impairment 9. We expected that MMP9 deficiency would have a protective effect since MMP9 can degrade almost all GBM components (for a review, see reference 1), thus favoring the issue of fibrin in the urinary space of the glomerulus. On the contrary, renal disease was more severe in MMP9−/− mice than in their control mates. In particular, the extent of crescent formation and fibrin deposition was greater, which led us to show a novel effect of MMP9 on fibrin degradation.
Materials and Methods
Mice.
MMP9−/− 129SV mice were generated as described previously 8 and were used after eight backcrosses to C57BL/6 background. Control MMP9+/+ mice were also obtained by backcrossing +/− 129SV mice with wild-type C57BL/6 mice. Mouse studies followed the Institutional Animal Care and Use Guidelines. Renal phenotype was compared at 1, 3, 6, and 12 mo. 15 MMP9−/− and 15 MMP9+/+ female mice were used at each time point.
Antibodies and Reagents.
Sheep preimmune serum and sheep anti-rat GBM serum were supplied by B. Fouqueray (Hôpital Tenon) in our laboratory 10. Mac-1 and F4/80 hybridomas were provided by R. Monteiro (Institut National de la Santé et de la Recherche Médicale U25, Paris, France). Rabbit polyclonal anti–IL-1β was obtained from Endogen. Biotinylated mouse anti–rat antibodies were purchased from The Jackson ImmunoResearch Laboratories, affinity purified rabbit anti–mouse IgG conjugated to FITC or peroxidase, rabbit anti–sheep IgG conjugated to FITC, and goat anti–rat fibrin IgG conjugated to FITC were obtained from Biosys. Goat anti–rabbit IgGs conjugated to alkaline phosphatase were purchased from Amersham Pharmacia Biotech. Recombinant mouse IL-1β was obtained from Sigma-Aldrich. Recombinant mouse MMP9 and human recombinant TIMP1 were a gift from G. Murphy, University of East Anglia, Norwich, UK 11 and P. Zaoui, Centre Hospitalies Universitaire, Grenoble, France 3, respectively. Fibrin was prepared from human plasma provided by E. Verdy (Hôpital Tenon). Human cryodessicated fibrinogen was purchased from Clottagen and mouse IL-10 ELISA kit from Biosource International.
Induction of Glomerulonephritis.
For induction of glomerulonephritis, three sets of 10 MMP9−/− and 10 MMP9+/+ 3-mo-old female mice were sensitized by intraperitoneal injection of 200 μg of sheep preimmune serum in the same volume of Freund's Complete Adjuvant (Sigma-Aldrich), followed by intravenous injection of 200 μl of sheep anti-GBM serum 7 d later. To avoid anaphylactic reaction, mice received an intraperitoneal injection of 100 μl of anti-GBM serum 1 h before the intravenous injection. They were studied 24 h and 15 d after anti-GBM injection.
Antibody Response.
Circulating mouse IgGs directed against injected sheep anti-GBM antibody were measured by ELISA as described previously 12 in the sera of 11 MMP9−/− and 11 MMP9 +/+ female mice collected 24 h and 15 d after anti-GBM injection. In brief, Costar 96-well plates were coated overnight with 1 μg/well of sheep IgG, washed, and blocked with 2% BSA. Then, wells were incubated for 3 h with dilutions of mouse serum followed by species-specific rabbit anti–mouse IgG conjugated to peroxidase for 2 h at a dilution of 1:2,000. Peroxidase was revealed by O-phenylenediamine for 1 h, and absorbance was read at 450 nm on a Titertek Multiskan MCC/340 (Labsystem Group). Serum from each mouse was tested in serial dilutions from 1:25 to 1:10,000, with serum from five nonimmunized mice used as negative controls.
GBM deposits of injected sheep anti-GBM antibody or of mouse IgGs directed against injected sheep antibody were assessed by immunofluorescence on 3-μm kidney cryosections fixed in acetone for 4 min at 4°C. Binding was detected after a 30-min incubation with species-specific FITC-conjugated anti–sheep or anti–mouse IgG diluted 1:400, respectively.
Assessment of Renal Function and Proteinuria.
Mice were placed for 48 h in metabolic cages and urine was collected for the last 24 h. During this time, they were supplied with food and tap water ad libitum. Serum and urine creatinine (alkaline picric acid method) and urine total proteins (Biuret method) were measured on an autoanalyzer (Instrumentation Laboratory). Urine albumin levels were determined after electrophoresis of total proteins on Paragon gels (Beckman Coulter) followed by scanning on a Multiscan RC (Labsystem Group).
Assessment of Glomerular Injury.
Renal tissue was fixed in Dubosq-Brazil's fixative, embedded in paraffin, cut into 3-μm sections, and stained with periodic acid-Schiff (PAS) stain and Masson's Trichrome for histological analysis. Crescent formation (defined as the presence of three or more layers of cells in Bowman's space) and fibrin deposition were determined on 100 glomeruli/kidney. 11 MMP9−/− and 11 MMP9+/+ female mice killed 15 d after anti-GBM injection were used for these studies.
Fibrin deposition was also detected by immunofluorescence on cryosections fixed with acetone and incubated for 30 min with FITC-conjugated goat anti–rat fibrin IgG (1 μg/ml).
For detection of macrophages, 3-μm acetone-fixed kidney cryosections were stained using a three-layer immunoperoxidase technique. Number of macrophages was determined on 30 glomeruli/kidney. Kidney sections were obtained from five MMP9−/2 and five MMP9+/+ mice. They were washed in PBS, and endogenous avidin and biotin were saturated using an avidin-biotin blocking kit (Vector Laboratories). Sections were incubated for 30 min with the rat monoclonal anti–mouse macrophage Mac-1 (CD11b) or F4/80 followed for 30 min by a biotinylated mouse anti–rat IgG (4 μg/ml in PBS supplemented with 2% normal mouse serum). Avidin-biotin horseradish peroxidase complex (ABC reagent; Vector Laboratories) was then added for 30 min and peroxidase was revealed by 3-amino-9-ethyl carbazole for 15 min. Sections were counterstained with hematoxylin for 1 min.
Measurement of IL-10 Production.
Glomeruli were isolated from six MMP9+/+ and six MMP9−/− female mice (killed 48 h, 7 d, and 15 d after anti-GBM injection) and from the same number of noninjected (+/+) and (−/−) mice. Isolation was performed by graded sieving through no. 150, 106, and 75 meshes as described previously 13. 104/ml glomeruli were maintained in RPMI 1640 supplemented with 1% FCS and 1% penicillin/streptomycin for 24 h (37°C, 5% CO2). Supernatants were collected and IL-10 was measured by ELISA using a polyclonal antibody derived against the entire IL-10 protein (Biosource International). The detection limit of the assay was 13 pg/ml. Results were normalized to total protein in glomerular lysate measured by protein assay (Bio-Rad Laboratories) or to 1,000 glomeruli.
Analysis of IL-1β Degradation.
Isolated glomeruli from 11 MMP9+/+ and 11 MMP9−/− female mice killed 15 d after anti-GBM injection were directly lysed in SDS-PAGE sample buffer (50 mM Tris, 1% SDS, 5% glycerol, 0.002% bromophenol blue). 100 ng recombinant mouse IL-1β incubated or not with 150 ng activated recombinant mouse MMP9 for 1 h at 37°C, and 100 μg of glomerular lysate total protein were submitted to SDS-PAGE under reducing conditions in a 15% polyacrylamide gel and electro transferred to nitrocellulose (Schleicher & Schuell) for 2 h at a constant current of 190 mA. Afterwards, nitrocellulose sheet was saturated with 5% dry milk in 0.1% PBS-Tween for 1 h at 37°C, extensively washed in 0.1% PBS-Tween, and incubated overnight at 4°C with rabbit anti–mouse IL-1β IgG (4 μg/ml) or rabbit anti–mouse IL-1β IgG preincubated with a 50× excess of recombinant mouse IL-1β as control. Nitrocellulose was then incubated for 2 h at room temperature with goat anti–rabbit IgG conjugated to alkaline phosphatase (0.02 μg/ml; Amersham Pharmacia Biotech). Alkaline phosphatase activity was revealed by adding the NBT/BCI substrate (nitroblue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate complex in 100 mM Tris-HCl, 100 mM NaCl, and 5 mM MgCl2, pH 9.5). The reaction was stopped in 20 mM Tris-HCl, 5 mM EDTA, pH 8.0. Blots were analyzed with NIH Image 1.61 and converted to a graphical format.
Characterization of Fibrinolytic Effect of MMP9.
The fibrinolytic activity of MMP9 was demonstrated by fibrin zymography, by SDS-PAGE analysis of fibrinogen, and fibrin digestion products in the presence of MMP9, and by degradation of fibrin deposits in situ.
For zymographic analysis, recombinant mouse MMP9 11 was preactivated with 1 mM 4-aminophenylmercuric acetate (Sigma-Aldrich) at 37°C for 3 h and different quantities (300, 150, and 50 ng) were loaded onto 8% SDS polyacrylamide gels (Merck). Electrophoresis was performed under nonreducing conditions at 20 mA. Gels were washed twice for 30 min in 2.5% Triton X-100 to remove SDS and applied to a fibrin gel (50 mg of human fibrinogen, 4 U of thrombin, 10 μl of 10% Triton X-100 per gel) performed in gelatinase substrate buffer (50 mM Tris-HCl, 5 mM CaCl2, 1 μM ZnCl2, 0.01% NaN3, pH 7.5) for 48 h at 37°C. The MMP9 fibrinolytic activity was detected by a proteolytic band in the fibrin gel. In some experiments, metalloproteinase inhibitors (10 μg/ml of recombinant human TIMP1) were supplied in the fibrin gel.
For fibrin and fibrinogen degradation studies, 10 μg of human fibrinogen was dissolved in 20 μl gelatinase substrate buffer, then incubated for 24 h at 37°C with 300 ng activated recombinant mouse MMP9. Fibrinogen degradation by plasmin was assessed by incubating 10 μg of human fibrinogen for 24 h at 37°C with 10 mU of plasmin. Control experiments included omission of activated recombinant mouse MMP9 or plasmin and addition of a metalloproteinase inhibitor (1 mM 1,10-phenanthroline) or of a plasmin inhibitor (8 U aprotinin). Fibrinogen degradation was then estimated after electrophoresis on 8% SDS polyacrylamide gels under reducing conditions. Gels were stained with 0.5% Coomassie blue. Fibrin was prepared from human plasma according to the technique of Schwartz et al. 14. In brief, human plasma was incubated with thrombin (vol/vol; 1,000 U/ml thrombin) for 2 h at room temperature, then rinsed in PBS for 48 h at 4°C. The 3-μg fibrin pellet was incubated with activated recombinant mouse MMP9 (300 ng diluted in 5 μl gelatinase substrate buffer) for 72 h at 37°C. Control experiments included omission of activated recombinant mouse MMP9, addition of a metalloproteinase inhibitor (1 mM 1,10-phenanthroline) or of a plasmin inhibitor (8 U aprotinin) to the gelatinase buffer. The fibrin pellet was then solubilized in 25 μl urea buffer (9 M urea, 3% SDS, 3% mercaptoethanol in 0.04 M sodium phosphate buffer, pH 7.1) for 12 h at 37°C, and loaded on 6% SDS polyacrylamide gels under reducing conditions. Gels were stained with 0.5% Coomassie blue.
For in situ fibrin degradation studies, serial 3-μm kidney sections cut from frozen tissue obtained from three MMP9−/− mice (killed 15 d after anti-GBM injection) were incubated with activated recombinant mouse MMP9 (300, 150, and 50 ng) for 24 h at 37°C. In a second set of experiments designed to demonstrate MMP9 fibrinolytic activity in vivo, serial sections of kidney were incubated for 72 h at 37°C with glomerular extracts obtained from MMP9+/+ mice killed 15 d after anti-GBM injection and treated with a plasmin inhibitor (1,000 U/ml aprotinin). Control experiments included omission of glomerular extracts, addition of a metalloprotease inhibitor (1 mM 1,10 phenanthroline) and a plasmin inhibitor to glomerular extracts obtained from MMP9+/+ mice, and use of glomerular extracts obtained from MMP9−/− mice killed 15 d after anti-GBM injection and treated with a plasmin inhibitor. Negative immunofluorescent control was obtained by incubating the section with untreated glomerular extracts obtained from MMP9+/+ mice. Fibrin deposits on glomerular sections were detected by immunofluorescence as described above.
Statistics.
Results are expressed as mean ± SEM. Significances of differences between MMP9−/− and MMP9+/+ mice were determined by analysis of variance (ANOVA).
Results
MMP9 Deficiency Decreases Renal Function and Increases Proteinuria in Mice with Anti-GBM Nephritis.
We used 2–3-mo-old MMP9−/− mice and their age-matched MMP9+/+ mates to induce the accelerated model of anti-GBM glomerulonephritis. At 3 mo of age, the function and histology of the MMP9−/− kidneys were not different from MMP9+/+ kidneys (data not shown), as described previously 15.
We observed no significant difference in renal function and histological features between MMP9−/− and MMP9+/+ mice at the heterologous phase that occurs 24 h after the injection of sheep anti-GBM antibody (data not shown). At the autologous phase, 15 d after the injection of the anti-GBM antibody, both control and MMP9−/− mice developed a crescentic proliferative glomerulonephritis. Surprisingly, the renal failure was more severe in MMP9−/− mice (Fig. 1 A). These mice also showed a markedly increased urinary protein loss, expressed as albumin over creatinine ratio or daily proteinuria, compared with MMP9+/+ mates (Fig. 1 B). However, serum total proteins and serum albumin did not differ between the two groups.
Figure 1.
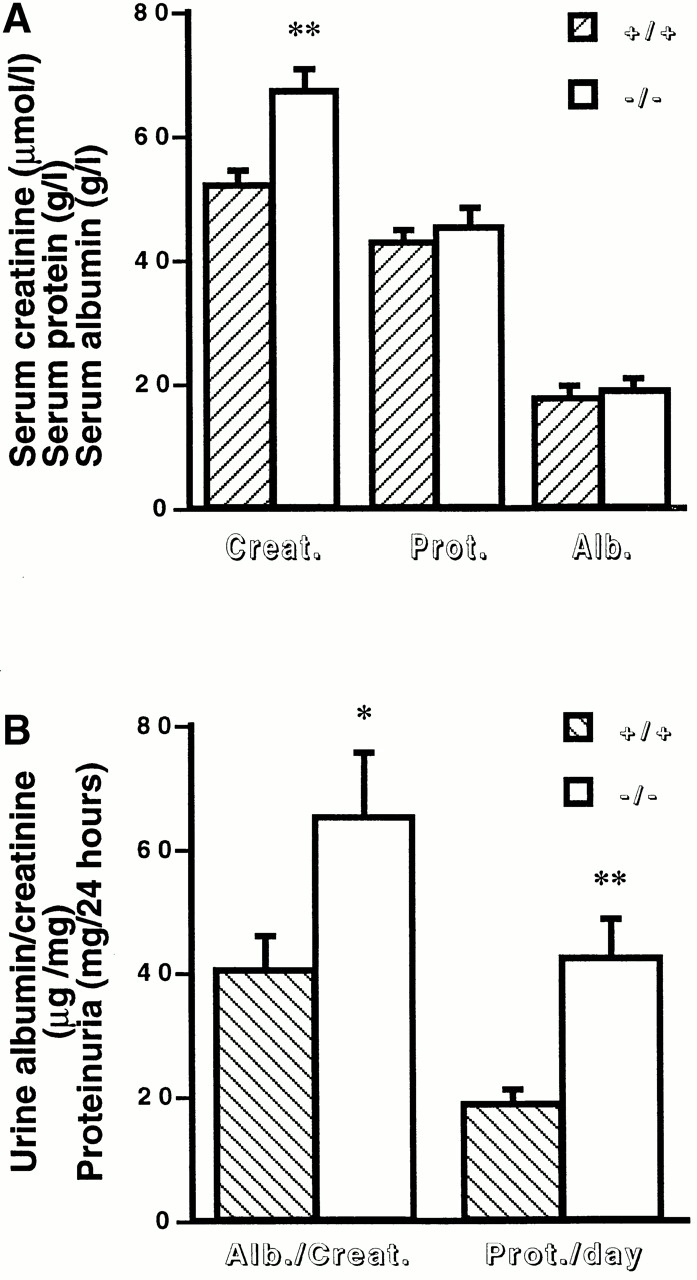
Effect of MMP9 deficiency on renal function in anti-GBM glomerulonephritis. (A) Serum creatinine (Creat.), protein (Prot.), and albumin (Alb.) levels in MMP9+/+ (hatched bars) and MMP9−/− (white bars) mice. n = 25. (B) Urinary parameters. Urine was collected for 24 h from MMP9+/+ (hatched bars) and MMP9−/− (white bars) mice housed in metabolic cages, and results were expressed as the ratio of albumin to creatinine (Alb./Creat.) or as daily proteinuria (Prot./day). Sera and urine were collected 15 d after injection of anti-GBM antibody. Values in noninjected MMP9+/+ mice were: albumin/creatinine = 10.23 ± 0.97; proteinuria/day = 4.01 ± 2.16. Values in noninjected MMP9−/− mice were almost identical to control MMP9+/+ mice: albumin/creatinine = 9.76 ± 1.02; proteinuria/day = 4.21 ± 1.36. Values are mean ± SEM; *P < 0.02, **P < 0.01 versus MMP9+/+ control mice. n = 15.
MMP9 Deficiency Aggravates Histological Features of Anti-GBM Nephritis.
Decreased renal function in MMP9−/− versus control mice was attributable to increased severity of lesions. Both MMP9−/− and MMP9+/+ mice developed proliferative glomerulonephritis but glomerular lesions were more severe in MMP9−/− mice as shown by the greater extent of fibrin deposits by immunofluorescent staining (compare Fig. 2 B with A and Fig. 3 A). Fibrin could also be observed in paraffin-embedded sections stained with PAS (Fig. 2 E). Crescent formation was moderate in MMP9+/+ kidneys and involved only 6% of glomeruli (Fig. 3 A). Crescents were larger in MMP9−/− kidneys and were observed in ∼50% of glomeruli (Fig. 3 A). However, the number of intraglomerular, periglomerular, and total macrophages was not significantly increased in MMP9−/− compared with MMP9+/+ kidneys (Fig. 3 B). Kidneys from MMP9−/− mice also showed numerous dilated tubules filled with casts which were not observed in control MMP9+/+ mice (compare Fig. 2 D with C). These casts apparently contained proteins or red blood cells attesting to the severity of glomerular disease.
Figure 2.
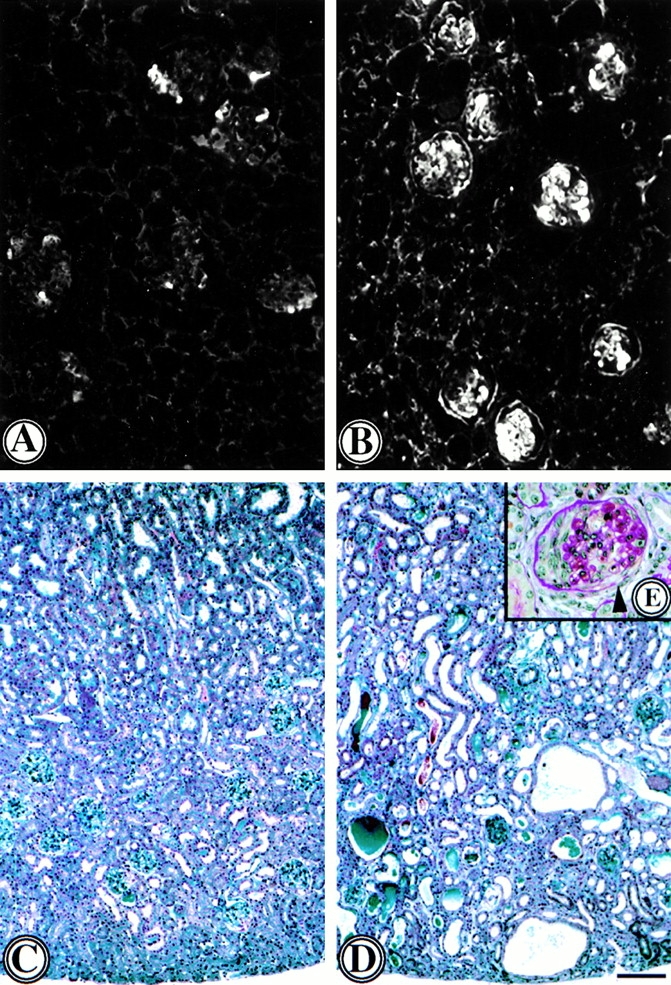
Photomicrographs of representative kidney sections from mice with anti-GBM nephritis 15 d after anti-GBM injection. (A and B) Fibrin deposition in glomeruli of (A) MMP9+/+ and (B) MMP9−/− mice assessed by immunofluorescence of cryostat frozen sections with antifibrin antibody. Intense glomerular fibrin deposits were observed in MMP9−/− kidney sections where they affected most of the glomeruli. Fibrin deposits were weaker and segmental in most glomeruli from MMP9+/+ mice. Bar, 70 μm. (C–E) Paraffin kidney sections from (C) MMP9+/+ and (D and E) MMP9−/− mice stained with (C and D) Masson's Trichrome or (E) PAS. Note numerous dilated tubules filled with proteinaceous and red blood cells containing casts in MMP9−/− kidneys, contrasting with the lack of tubule lesions in control MMP9+/+ kidneys. A representative glomerulus from an MMP9−/− kidney is shown in E. Note an extensive crescent in the left half of the glomerulus and massive fibrin deposition mainly in the right half, with rupture of the Bowman's capsule (arrow). Such severe glomerular lesions were unusually observed in MMP9+/+ control kidneys. Bar: (C and D) 170 μm; (E) 40 μm.
Figure 3.
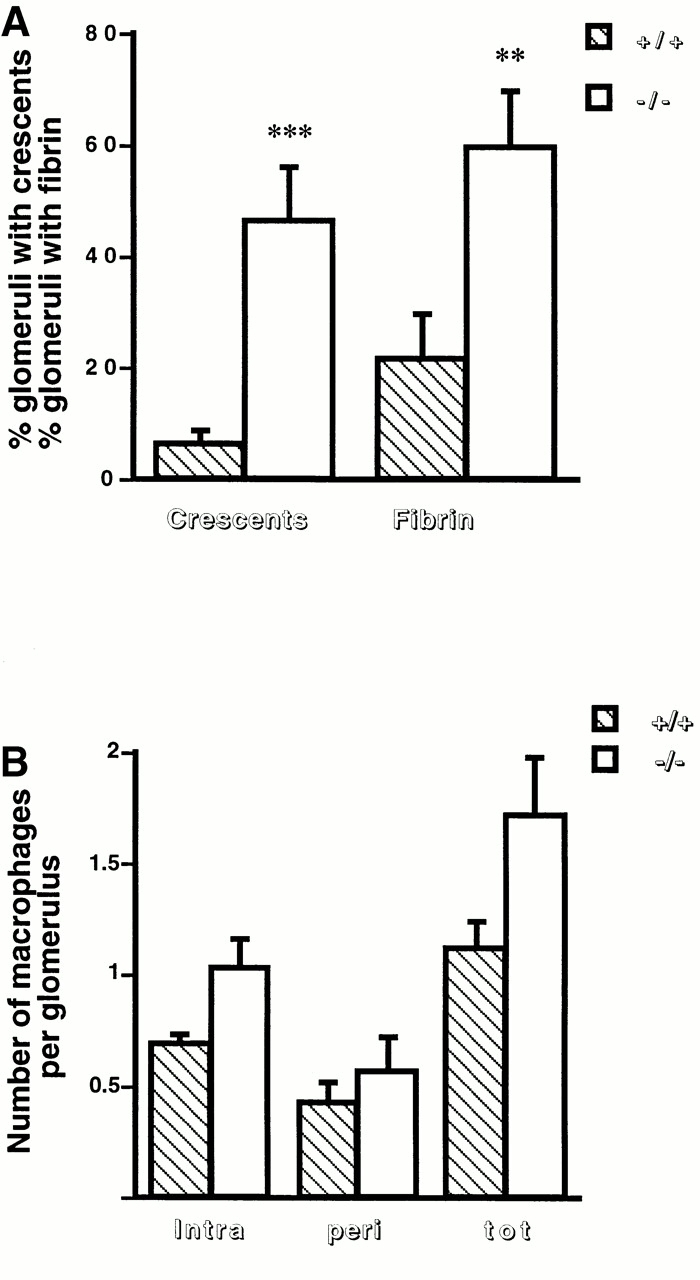
Glomerular fibrin deposition, crescent formation, and glomerular macrophages 15 d after anti-GBM injection. (A) The percentages of glomeruli with crescent or fibrin deposition in MMP9+/+ (hatched bars) and MMP9−/− (white bars) kidneys were evaluated on Masson's Trichrome (fibrin) and PAS (crescent) stains of paraffin sections. They were determined on 100 glomeruli per kidney; 11 kidneys were examined. Values are mean ± SEM; **P < 0.01, ***P < 0.001 versus MMP9+/+ control mice. (B) Numbers of intraglomerular (intra), periglomerular (peri), and total (tot) macrophages per glomerulus in MMP9+/+ (hatched bars) and MMP9−/− (white bars) mice were counted after staining the macrophages with F4/80 antibody as described in Materials and Methods. They were determined on 30 glomeruli per kidney; five kidneys were examined. Values are mean ± SEM. No statistical difference was observed.
MMP9 Deficiency Does Not Alter Immune Parameters, IL-1β, or IL-10.
Since the extent of immune response to sheep anti-GBM antibody is important in determining the severity of injury in this model of glomerulonephritis, we measured circulating levels of mouse anti–sheep IgG antibodies. Antibody titers, measured by ELISA at dilution of sera from 1:25 to 1:10,000, were not significantly different for MMP9−/− mice compared with their (+/+) control group. Results obtained at a serum dilution of 1:200 are shown in Fig. 4 A. We then used immunofluorescence to evaluate whether the greater severity of the nephritis in MMP9−/− mice could result from larger amounts of immune deposits. We observed no difference in GBM deposition of injected sheep anti-GBM antibody (Fig. 4B and Fig. C), or mouse anti–sheep IgG (Fig. 4D and Fig. E) in MMP9+/+ (Fig. 4B and Fig. D) compared with MMP9−/− (Fig. 4C and Fig. E) mice.
Figure 4.
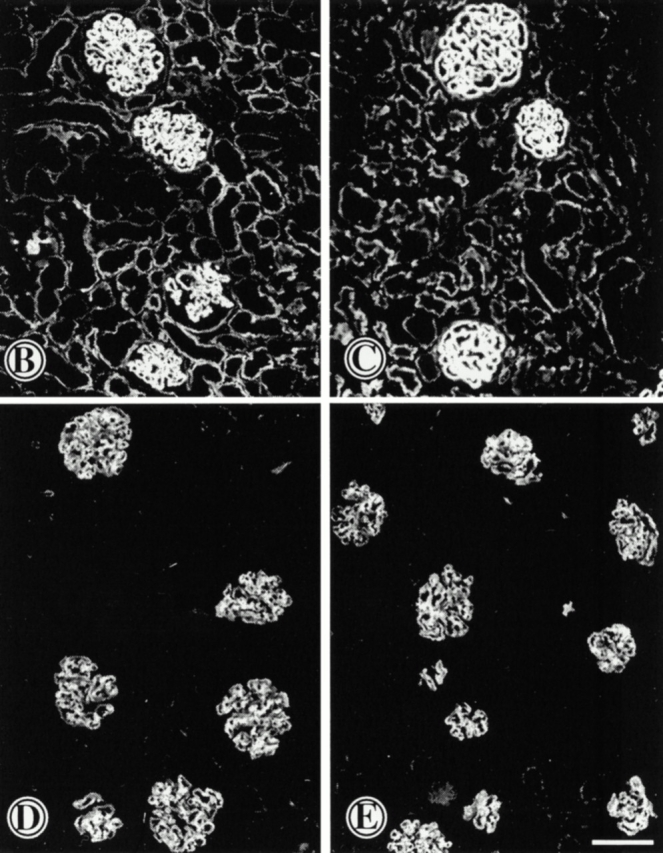

Assessment of immune response. (A) Evaluation of antibody production to injected sheep anti-GBM antibody by ELISA. Mouse anti–sheep IgG antibody was measured in sera of MMP9+/+ (hatched bars) and MMP9−/− (white bars) mice 24 h and 15 d after injection of the antibody. n = 11. Serum dilutions ranged from 1:25 to 1:10,000. The serum dilution shown on this graph is 1:200. MMP9−/− mice exhibited slightly lower absorbance values that were not statistically significant. (B–E) Estimation of antibody deposition by immunofluorescence 15 d after injection of the antibody. Deposition of injected (B and C) sheep anti-GBM antibody or (D and E) mouse anti–sheep IgG was compared in (B and D) MMP9+/+ and (C and E) MMP9−/− mice. A similar intensity of fluorescence was observed in MMP9−/− and control mice. Bar, 70 μm.
The proinflammatory cytokine IL-1β plays an important role in the development of glomerular lesions observed in anti-GBM nephritis 16 and can be degraded in vitro by metalloproteases 17. We next investigated whether defective IL-1β degradation could contribute to the severity of renal lesions observed in MMP9−/− mice. We used a rabbit polyclonal antibody directed against the entire protein which recognized a single band at the expected molecular size (17.5 kD) in Western blot analysis performed with recombinant mouse IL-1β (Fig. 5 A, middle). We verified that IL-1β could be cleaved in vitro by MMP9, leading to a degradation product of 14.5 kD (Fig. 5 A, middle). We further checked the specificity of these bands, which disappeared when the membrane was incubated with the appropriate control (polyclonal antibody preincubated with an excess of IL-1β) (Fig. 5 A, right). We did not observe any difference in the amounts of the intact (17.5 kD) and the degraded forms (14.5 kD) of IL-1β in isolated glomeruli from both MMP9−/− and MMP9+/+ mice (Fig. 5 A, left, and B).
Figure 5.
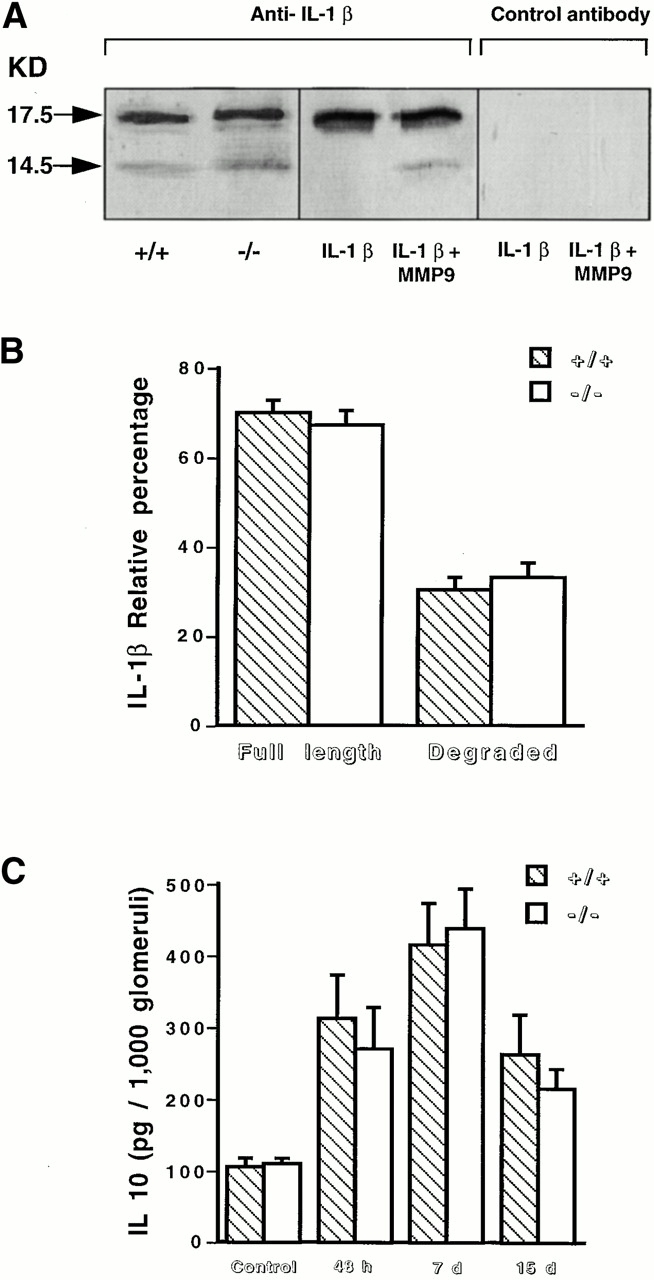
IL-1β degradation and IL-10 production. (A) The amount of IL-1β produced by glomeruli isolated from MMP9+/+ and MMP9−/− mice 15 d after injection of anti-GBM antibody was estimated in a semiquantitative manner by Western blot analysis. The polyclonal anti–mouse antibody yielded a single band with 100 ng recombinant mouse IL-1β and two bands corresponding to the intact (17.5 kD) and degraded (14.5 kD) forms of IL-1β after incubation with activated recombinant mouse MMP9 (IL-1β plus MMP9, middle). The specificity of these bands was further assessed by incubating the membrane with the polyclonal antibody preincubated with an excess of IL-1β (Control antibody, right). The intact and degraded forms of IL-1β were observed with the same intensity in glomerular extracts (100 μg of total proteins) from MMP9+/+ and MMP9−/− mice (left). (B) Quantitative analysis of the blots performed with glomeruli isolated from MMP9+/+ and MMP9−/− mice 15 d after injection of anti-GBM antibody. Note similar ratio of the two bands in both groups of mice. (C) IL-10 production by glomeruli isolated from MMP9+/+ (hatched bars) and MMP9−/− (white bars), noninjected mice (Control), or mice injected with anti-GBM antibody after 48 h, 7 d, and 15 d. IL-10 production was significantly increased in both groups of injected mice compared with their respective noninjected controls (P < 0.01), but it was not significantly different between MMP9−/− and MMP9+/+ glomeruli at the different time points after injection. Values are mean ± SEM.
We also measured IL-10, an antiinflammatory cytokine that plays a protective role in this model 18, because IL-10 production was shown to be reduced in a model of skin contact hypersensitivity in MMP9−/− mice 19. IL-10 production was increased in both groups of mice 48 h, 7 d, and 15 d after anti-GBM injection compared with their respective noninjected controls (Fig. 5 C). IL-10 normalized to total protein in glomerular lysate (data not shown) or to 1,000 glomeruli (Fig. 5 C) was similar in MMP9−/− and MMP9+/+ glomeruli 48 h, 7 d, and 15 d after anti-GBM injection.
MMP9 Is a Fibrinolytic Enzyme.
We next asked what the critical substrate for MMP9 is in kidney. Because fibrin accumulated in MMP9−/− mice, we hypothesized that MMP9 exerts a protective effect in the development of glomerular lesions by degrading fibrin. We performed three sets of experiments to address this hypothesis.
First, we demonstrated that MMP9 degrades fibrin by fibrin zymography. SDS-PAGE gel of activated recombinant mouse MMP9 loaded at different doses was applied to a fibrin gel (Fig. 6 A). The lytic zones corresponded to the migration of the active form of MMP9 with their intensity proportional to the quantity of MMP9. When recombinant human TIMP1, the natural MMP9 inhibitor, was incorporated in the fibrin gel, fibrinolysis was abolished. These results suggest that MMP9 is able to degrade fibrin in a dose-dependent manner.
Figure 6.
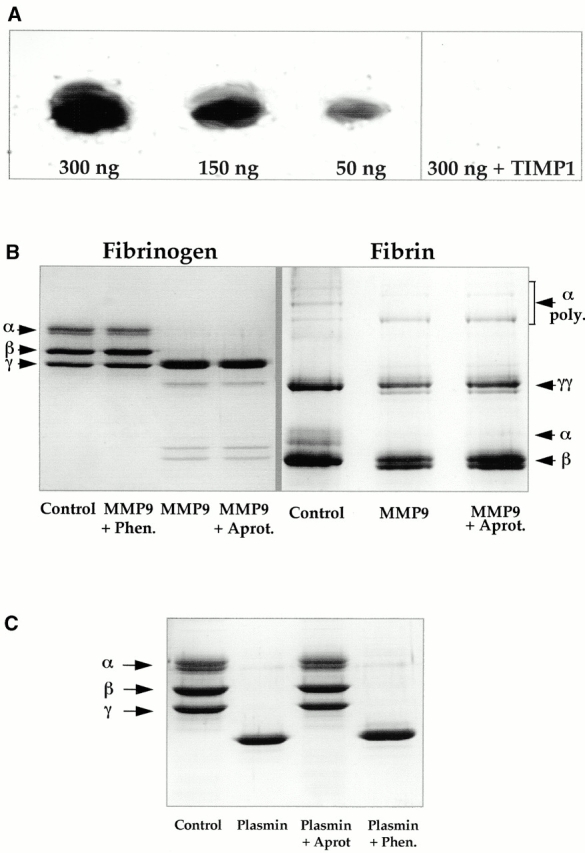
Fibrinolytic activity of MMP9. (A) Zymogram performed on fibrin gel. 300 ng, 150 ng, and 50 ng of activated recombinant mouse MMP9 were loaded on 8% SDS polyacrylamide gels that were applied to a fibrin gel. Note a lytic band at the expected molecular size for the active form of MMP9. Its intensity was proportional to the quantity of MMP9. Lytic activity was inhibited when 10 μg/ml of recombinant human TIMP1 was incorporated in the fibrin gel (300 ng plus TIMP1). (B) SDS-PAGE analysis of 10 μg fibrinogen and 3 μg fibrin after a 24- or 72-h incubation, respectively, with 300 ng of activated recombinant mouse MMP9 alone or in the presence of a metalloprotease inhibitor (1 mM 1,10-phenanthroline [Phen.]) or of a plasmin inhibitor (8 U aprotinin [Aprot.]). Note that MMP9 degraded Aα and Bβ chains of fibrinogen (compare MMP9 with Control), and that degradation was inhibited by 1,10-phenanthroline (MMP9 + Phen. versus MMP9) but not by aprotinin (MMP9 + Aprot. versus MMP9). The α chain and α polymers of fibrin chains were preferentially degraded by MMP9 (MMP9 versus Control). Degradation was not inhibited by aprotinin (MMP9 + Aprot. versus MMP9). The minor band forming a doublet with γγ and β chains, respectively, in the presence of MMP9 could result from partial degradation of these chains by MMP9 and/or from degradation of α chain and α polymers. (C) SDS-PAGE analysis of 10 μg fibrinogen after a 24-h incubation with 10 mU of plasmin alone or in the presence of a metalloprotease inhibitor (1 mM 1,10-phenanthroline [Phen.]) or of a plasmin inhibitor (8 U aprotinin [Aprot.]). Note that plasmin degraded the three chains of fibrinogen, and that degradation was inhibited by aprotinin (Plasmin + Aprot versus Plasmin) but not by 1,10-phenanthroline (Plasmin + Phen. versus Plasmin).
Second, we determined the sensitivity of fibrinogen and fibrin chains to MMP9 proteolysis. MMP9 degraded the Aα and Bβ chains of fibrinogen, and this proteolytic effect was inhibited by 1,10-phenanthroline, a zinc chelator (and thus a metalloprotease inhibitor), but not by aprotinin, a plasmin inhibitor (Fig. 6 B). Fibrinolytic products generated by plasmin were different since γγ chains were degraded. As expected, plasmin proteolytic effect was inhibited by aprotinin, but not by 1,10-phenanthroline (Fig. 6 C). The fibrin α chains and the α polymers were preferentially degraded by MMP9. Doublets observed for γγ and β chains were most likely degradation products of α polymers and α chains, but they might also result from partial degradation of γγ and β chains. Fibrin degradation did not occur when 1,10-phenanthroline was added along with MMP9 (data not shown). Although fibrin pellets were washed extensively, we could not rule out a possible contamination with plasminogen or plasminogen activators, tissue-type plasminogen activator (tPA) and urokinase-type plasminogen activator (uPA), since fibrin was prepared from plasma. To verify that MMP9 directly cleaved fibrin and had no effect upstream on plasmin activation, we added aprotinin, a plasmin inhibitor, to the incubation medium. The MMP9 degradation profile of fibrin was not altered by aprotinin (Fig. 6 B), which indicated that MMP9 degraded fibrin directly.
Finally, we showed that MMP9 could degrade fibrin in situ. Incubation with activated recombinant mouse MMP9 of serial kidney cryosections from MMP9−/− mice killed 15 d after anti-GBM injection strongly reduced glomerular fibrin deposition. This effect was abolished by 1,10-phenanthroline (Fig. 7 A). To further demonstrate that fibrin is a physiological substrate for MMP9 in vivo, we incubated serial kidney sections with control gelatinase substrate buffer (Fig. 7 B, 1) or glomerular extracts from injected MMP9+/+ mice treated either with plasmin inhibitor (Fig. 7 B, 2) or with plasmin and metalloproteinase inhibitor (Fig. 7 B, 3). Results show that glomerular fibrin deposits can be cleaved by glomerular extracts when plasmin is inhibited (Fig. 7 B, 2) and that this fibrinolytic activity is due to MMP9 since it is abolished in the presence of phenanthroline (Fig. 7 B, 3), or when MMP9+/+ glomerular extracts are replaced by MMP9−/− glomerular extracts (Fig. 7 B, 4). Fibrin deposits completely disappeared when plasmin activity was not blocked in sections incubated with untreated glomerular extracts from MMP9+/+ injected mice (data not shown). These results demonstrate that MMP9, like plasmin, contributes to fibrinolytic activity in this model.
Figure 7.

Fibrinolytic activity of MMP9 on tissue sections. (A) In situ degradation of glomerular fibrin deposits by MMP9. Serial kidney cryosections from MMP9−/− mice killed 15 d after anti-GBM injection were incubated with incubation medium (Control), 300 ng of activated recombinant mouse MMP9 alone (MMP9), or with 1,10-phenanthroline (1 mM 1,10-phenanthroline [MMP9 + Phenan.]) before fibrin detection by immunofluorescence. Note that MMP9 strongly reduced intensity of glomerular fibrin deposits, but phenanthroline prevented this effect. Bar, 400 μm. (B) In situ degradation of glomerular fibrin deposits by glomerular extracts. 1, gelatinase substrate buffer; 2, glomerular extracts from +/+ control mice (15 d after anti-GBM injection) treated with plasmin inhibitor (1,000 U/ml aprotinin); 3, glomerular extracts from +/+ control mice (15 d after anti-GBM injection) treated with plasmin inhibitor and metalloproteinase inhibitor (1 mM 1,10-phenanthroline); 4, glomerular extracts from MMP9−/− mice (15 d after anti-GBM injection) treated with plasmin inhibitor. Fibrin deposits on glomerular sections were detected by immunofluorescence. Note that glomerular extracts containing MMP9 activity strongly reduced the intensity of glomerular fibrin deposits in the absence of plasmin activity 2. This effect was abolished when MMP9 activity was inhibited 3 or absent 4. Bar, 400 μm.
Discussion
Our data show that MMP9 is an important regulator of renal function. We took advantage of the availability of MMP9−/− mice 8 to analyze their renal phenotype in an accelerated model of crescentic proliferative glomerulonephritis. MMP9−/− mice developed a more severe form of anti-GBM nephritis than their wild-type mates. We provide the first evidence that MMP9 protects mice from crescentic proliferative glomerulonephritis (a model where fibrin deposits play a major role) through its fibrinolytic activity.
We have previously shown that MMP9 is produced at the first stage of kidney embryogenesis in vivo and is required for branching morphogenesis in vitro 3. We postulated that MMP9 deficiency in mice would impede kidney growth. However, MMP9−/− mice are viable, suggesting the absence of major kidney defects. The histological appearance, immunohistochemical analysis of several basement membrane components, and renal function of these mice were normal up to 5 mo 15. We confirmed these results in MMP9−/− and control mice up to 6 mo of age (data not shown).
MMP9 expression is increased in various human and experimental models of nephropathies (reference 6; for a review, see reference 7). To establish its role, we investigated the susceptibility of MMP9−/− mice to crescentic proliferative glomerulonephritis in an accelerated model of anti-GBM disease. By analogy to the results observed in bullous pemphigoid 20, where the initial lesion affects the basement membrane as in anti-GBM nephritis, we hypothesized that MMP9−/− mice would be protected from the development of glomerular lesions. In the murine model of bullous pemphigoid, MMP9−/− mice showed deposition of injected antibodies to the membrane and neutrophil recruitment to the skin but epidermal blistering does not develop 20. This defect owes to a lack of inactivation of α1-proteinase inhibitor by MMP9, which does not allow neutrophil elastase to function 21. Unexpectedly, however, MMP9 deficiency was associated with functional and histological exacerbation of glomerular injury. MMP9−/− mice showed a significantly increased propensity to form crescents. Crescents result from accumulation, in the urinary space, of fibrin that is chemotactic for macrophages and favors proliferation of glomerular parietal epithelial cells 9 22. Indeed, we observed a decreased clearance or degradation of fibrin because of lack of MMP9 activity in MMP9−/− mice. The increased renal impairment, fibrin deposition, and crescent formation in those mice suggest that MMP9 plays a important role in protecting glomeruli against injury.
The extent of immune response to the exogenous antigen could not account for the severity of injury since circulating levels of mouse anti–sheep IgG antibodies and antibody deposition to the basement membrane were similar in MMP9−/− and MMP9+/+ mice. We observed a slight but not significant increase in the number of intra- and periglomerular macrophages. Results observed in the literature vary from one system to another. Macrophage infiltration and collagen accumulation in the heart-infarcted region of MMP9−/− mice were decreased in an experimental model of myocardial infarction 23, but were not modified in young and adult MMP9−/− mice that were resistant to the development of experimental autoimmune encephalomyelitis 24. Macrophages have a pivotal role in the transition between inflammation and repair through the balanced secretion of pro- and antiinflammatory cytokines. Although the number of glomerular and periglomerular macrophages was not significantly altered by MMP9 deficiency, it was still possible that macrophage phenotype was altered. We analyzed IL-1β and IL-10 for two reasons. First, IL-1β enhances glomerular fibrin deposition and crescent formation in this model 16 whereas IL-10 has a protective effect 18. Second, MMP9 degrades IL-1β in vitro 17 and increases IL-10 production in a model of skin contact hypersensitivity 19. We hypothesized that IL-1β degradation or IL-10 production could be altered in MMP9−/− mice with anti-GBM proliferative glomerulonephritis. However, we could not find significant differences in the production or degradation of these cytokines that could account for the increment of glomerular lesions.
The exacerbation of glomerular injury observed in MMP9−/− mice was reminiscent of the findings observed in tPA- or plasminogen-deficient mice 25 after induction of a similar crescentic proliferative glomerulonephritis. tPA-deficient mice and plasminogen-deficient mice, indeed, showed severe functional and histological exacerbation of glomerular injury, whereas neither uPA nor uPA receptor deficiency affected the expression of glomerulonephritis. We first verified, by fibrin zymography on serum and glomerular tissues, that tPA and uPA enzymatic activities were not downregulated during the course of glomerulonephritis in MMP9−/− mice (data not shown). We then hypothesized that MMP9 itself could be involved in fibrinolysis. Other members of the MMP family, MT1-MMP (MMP14), stromelysin-1 (MMP3), collagenase-1 (MMP1) MMP2 26, and MT4-MMP (MMP17) 11 are able to degrade fibrinogen and fibrin in vitro. Our data show that MMP9 not only can degrade fibrin in vitro, but also is responsible for its degradation in vivo during the course of glomerulonephritis. We found that MMP9 produced lysis bands in fibrin gel, degraded the Aα and Bβ chains of fibrinogen and the α chains and α polymers of fibrin, and partially solubilized in situ glomerular fibrin deposits in mice with anti-GBM proliferative glomerulonephritis. The action of MMP9 on fibrin was direct, and not plasmin mediated, since aprotinin, a plasmin inhibitor, does not prevent MMP9-induced fibrin degradation. MMP9 cleaves the same fibrin and fibrinogen chains as MT1-MMP, suggesting a similar fibrinolytic effect of these enzymes 26. In addition, we show for the first time that the fibrinolytic activity of MMP9 is critical in vivo. On the basis of the results obtained by Kitching et al. 25 and our own data, we anticipate that the severity of anti-GBM nephritis would be even more greater in double tPA-MMP9 knockout mice.
MMP9 was first described because of its ability to cleave extracellular matrix macromolecules including type IV and V collagens and denatured collagens (gelatin) in vitro (for a review, see reference 1). Subsequently, MMP9 has been shown to cleave other extracellular matrix substrates, such as BP 180, and nonmatrix components, like serpins, and to activate TGF-β in vitro (for reviews, see references 1, 21, 27). Thus, our data add to the increasing list of MMP9 substrates that are not conventional matrix components. They also show that fibrin is a critical substrate for MMP9 in vivo. Only one substrate, α1-proteinase inhibitor, had been previously identified as an MMP9 substrate in vivo 21.
In conclusion, our results show that MMP9 contributes to the breakdown of fibrin caps observed in glomeruli during the course of glomerulonephritis. Since fibrin is an important mediator of glomerular injury favoring the proliferation of epithelial cells (leading to crescent formation) and subsequently the invasion of fibroblasts, the fibrinolytic effect of MMP9 can partially account for its antifibrotic properties.
Acknowledgments
We are grateful to R. Monteiro for the anti–Mac-1 and F4/80 antibodies, B. Fouqueray for providing us with the anti-GBM antibody, and G. Murphy and P. Zaoui for their gift of recombinant murine MMP9 and recombinant human TIMP1, respectively. We are thankful to M. Galceran for IL-1β Western blot analysis, and M.A. Alyanakian and J. Bauchet for albumin and creatinine determination, respectively, and to J. Perez for help in glomeruli isolation. We thank B. Mougenot for critical review of the morphological data.
This work was supported by grants from the Association pour la Recherche sur le Cancer (5714), the Institut National de la Santé et de la Recherche Médicale, the Université Pierre et Marie Curie (Paris 6), the US Public Health Service, the National Institutes of Health (CA72006), and the Human Frontiers Science Program (RG 0051/1999M).
Footnotes
Abbreviations used in this paper: GBM, glomerular basement membrane; MMP, matrix metalloproteinase; PAS, periodic acid-Schiff; TIMP, tissue inhibitor of metalloproteinase; tPA, tissue-type plasminogen activator; uPA, urokinase-type plasminogen activator.
References
- Vu T.H., Werb Z. Gelatinase Bstructure, regulation and function. In: Parks W.C., Mercham R.P., editors. Matrix Metalloproteinases. Academic Press; San Diego: 1998. pp. 115–140. [Google Scholar]
- Reponen P., Sahlberg C., Munaut C., Thesleff I., Tryggvason K. High expression of the 92-kD type IV collagenase (gelatinase B) in the osteoclast lineage during mouse development. J. Cell Biol. 1994;124:1091–1102. doi: 10.1083/jcb.124.6.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelongt B., Trugnan G., Murphy G., Ronco P.M. Matrix metalloproteinases MMP2 and MMP9 are produced in early stages of kidney morphogenesis but only MMP9 is required for renal organogenesis in vitro. J. Cell Biol. 1997;137:1–11. doi: 10.1083/jcb.136.6.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett D.H., Sterzel R.B., Kashgarian M., Ryan J.L. Neutral proteinase activity produced in vitro by cells of the glomerular mesangium. Kidney Int. 1983;23:342–349. doi: 10.1038/ki.1983.25. [DOI] [PubMed] [Google Scholar]
- Piedagnel R., Murphy G., Ronco P.M., Lelongt B. Matrix metalloproteinase 2 (MMP2) and MMP9 are produced by kidney collecting duct principal cells but are differentially regulated by SV40 large-T, arginine vasopressin, and epidermal growth factor. J. Biol. Chem. 1999;274:1614–1620. doi: 10.1074/jbc.274.3.1614. [DOI] [PubMed] [Google Scholar]
- McMillan J.I., Riordan J.W., Couser W.G., Pollock A.S., Lovett D.H. Characterization of a glomerular epithelial cell metalloproteinase as matrix metalloproteinase-9 with enhanced expression in a model of membranous nephropathy. J. Clin. Invest. 1996;97:1094–1101. doi: 10.1172/JCI118502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman J.T., Lewis M. Matrix metalloproteinases (MMPs) in renal fibrosis. Kidney Int. 1996;49:S61–S63. [PubMed] [Google Scholar]
- Vu T.H., Shipley J.M., Bergers G., Berger J.E., Helms J.A., Hanahan D., Shapiro S.D., Senior R.M., Werb Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdsworth S.R., Thomson N.M., Glasgow E.F., Atkins R.C. The effect of defibrination on macrophage participation in rabbit nephrotoxic nephritisstudies using glomerular culture and electronmicroscopy. Clin. Exp. Immunol. 1979;31:38–43. [PMC free article] [PubMed] [Google Scholar]
- Fouqueray B., Suberville S., Isaka Y., Akagi Y., Gerard C., Velu T., Imai E., Baud L. Reduction of proteinuria in anti-glomerular basement membrane (GBM) nephritis by interleukine-10 gene transfer J. Am. Soc. Nephrol 7 1996. 1698(Abstr.). [Google Scholar]
- English W.R., Puente X.S., Freije J.M., Knauper V., Amour A., Merryweather A., Lopez-Otin C., Murphy G. Membrane type 4 matrix metalloproteinase (MMP17) has tumor necrosis factor-α convertase activity but does not activate pro-MMP2. J. Biol. Chem. 2000;275:14046–14055. doi: 10.1074/jbc.275.19.14046. [DOI] [PubMed] [Google Scholar]
- Makino H., Lelongt B., Kanwar Y.S. Nephritogenicity of proteoglycans. II. A model of immunecomplex nephritis. Kidney Int. 1988;34:195–208. doi: 10.1038/ki.1988.165. [DOI] [PubMed] [Google Scholar]
- Fouqueray B., Boutard V., Philippe C., Kornreich A., Marchant A., Perez J., Goldman M., Baud L. Mesangial cell-derived interleukin-10 modulates mesangial cell response to lipopolysaccharide. Am. J. Pathol. 1995;147:176–182. [PMC free article] [PubMed] [Google Scholar]
- Schwartz M.L., Pizzo S.V., Hill R.L., McKee P.A. The effect of fibrin-stabilizing factor on the subunit structure of human fibrin. J. Clin. Invest. 1971;50:1506–1513. doi: 10.1172/JCI106636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews K.L., Betsuyaku T., Rogers S., Shipley J.M., Senior R.M., Miner J.H. Gelatinase B (MMP-9) is not essential in the normal kidney and does not influence progression of renal disease in a mouse model of alport syndrome. Am. J. Pathol. 2000;157:303–311. doi: 10.1016/S0002-9440(10)64541-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolic-Paterson D.J., Main I.W., Tesch G.H., Lan H.Y., Atkins R.C. Interleukin-1 in renal fibrosis. Kidney Int. 1996;54:S88–S90. [PubMed] [Google Scholar]
- Ito A., Mukaiyama A., Itho Y., Nagase H., Thogersen I.B., Enghild J.J., Sasaguri Y., Mori Y. Degradation of interleukin-1β by matrix metalloproteinases. J. Biol. Chem. 1996;271:14657–14660. doi: 10.1074/jbc.271.25.14657. [DOI] [PubMed] [Google Scholar]
- Kitching A.R., Tipping P.G., Timoshanko J.R., Holdsworth S.R. Endogenous interleukin-10 regulates Th1 responses that induce crescentic glomerulonephritis. Kidney Int. 2000;57:518–525. doi: 10.1046/j.1523-1755.2000.00872.x. [DOI] [PubMed] [Google Scholar]
- Wang M., Qin X., Mudgett J.S., Ferguson T.A., Senior R.M., Welgus H.G. Matrix metalloproteinase deficiencies affect contact hypersensitivitystromelysin-1 deficiency prevents the response and gelatinase B deficiency prolongs the response. Proc. Natl. Acad. Sci. USA. 1999;96:6885–6889. doi: 10.1073/pnas.96.12.6885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Shipley J.M., Vu T.H., Zhou X., Diaz L.A., Werb Z., Senior R.M. Gelatinase B–deficient mice are resistant to experimental bullous pemphigoid. J. Exp. Med. 1998;188:475–482. doi: 10.1084/jem.188.3.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Zhou X., Shapiro S.D., Shipley J.M., Twining S.S., Diaz L.A., Senior R.M., Werb Z. The serpin α1-proteinase inhibitor is a critical substrate for gelatinase B/MMP-9 in vivo. Cell. 2000;102:647–655. doi: 10.1016/s0092-8674(00)00087-8. [DOI] [PubMed] [Google Scholar]
- Thomson N.M., Moran J., Simpson I.J., Peters D.K. Defibrination with ancrod in nephrotoxic nephritis in rabbits. Kidney Int. 1976;10:343–347. doi: 10.1038/ki.1976.120. [DOI] [PubMed] [Google Scholar]
- Ducharme A., Frantz S., Aikawa M., Rabkin E., Lindsey M., Rohde L.E., Schoen F.J., Kelly R.A., Werb Z., Libby P., Lee R.T. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J. Clin. Invest. 2000;106:55–62. doi: 10.1172/JCI8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois B., Masure S., Hurtenbach U., Paemen L., Heremans H., van den Oord J., Sciot R., Meinhardt T., Hammerling G., Opdenakker G., Arnold B. Resistance of young gelatinase B-deficient mice to experimental autoimmune encephalomyelitis and necrotizing tail lesions. J. Clin. Invest. 1999;104:1507–1515. doi: 10.1172/JCI6886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitching A.R., Holdsworth S.R., Ploplis V.A., Plow E.F., Collen D., Carmeliet P., Tipping P.G. Plasminogen and plasminogen activators protect against renal injury in crescentic glomerulonephritis. J. Exp. Med. 1997;185:963–968. doi: 10.1084/jem.185.5.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraoka N., Allen E., Apel I.J., Gyetko M.R., Weiss S.W. Matrix metalloproteinases regulate neovascularization by acting as pericellular fibrinolysins. Cell. 1998;95:365–377. doi: 10.1016/s0092-8674(00)81768-7. [DOI] [PubMed] [Google Scholar]
- Yu Q., Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-β and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]